
Introduction
Although fluorine is an essential mineral element for all mammals, including humans, exposure to high fluorine levels has toxic effects on various organs and tissues [1]. Fluorine, which is added to drinking water, is rapidly absorbed by the gastrointestinal tract after ingestion [2]. Other common sources of fluorine include dental products, food, drugs, and industrial emissions [3]. Excessive fluoride intake can have many harmful effects, including damage to the teeth, bones, and other organs [4]. We have previously demonstrated that excessive fluorine intake can induce cytotoxicity, immunotoxicity, oxidative damage, and pathological injury in the thymus [5], spleen [6-8], bursa of Fabricius [9], cecal tonsil [10-14], liver [15, 16], kidney [17-19], peripheral blood [20-23], and intestine [24-28] in broiler chickens. Other studies have also demonstrated that fluoride induces cytotoxicity, apoptosis, and DNA damage in both humans and animals [1, 29-31].
Dysregulation of the cell cycle, which is crucial for the maintenance of homeostasis in multicellular organisms [32], may result in uncontrolled cell proliferation or excessive cell death; such changes can promote tumor formation and various other disease states [32-34]. DNA damage activates molecules that inhibit the cell cycle and promote cell death to prevent proliferation of genetically altered cells [32]. Excessive fluoride increases the G0/G1 cell cycle phase population in thymocytes and splenocytes [5, 8] and reduces numbers of T and B cells in young broiler chickens. However, fluoride increases the S phase and decreases the G2/M phase population in rat osteoblasts, but does not affect the size of the G0/G1 phase population [34]. We recently demonstrated that sodium fluoride (NaF) inhibits proliferation and induces apoptosis in mouse splenic lymphocytes both in vivo and in vitro [3, 8, 35, 36] and increases G0/G1 arrest in the broiler chicken kidney [17] and thymus [5]. Furthermore, the molecular mechanisms underlying the ability of NaF to inhibit proliferation in splenic lymphocytes include alterations in the expression of cytokine proteins and increases in cell cycle arrest [36]. The liver is a crucial metabolic organ that regulates the metabolism of trace elements, and excessive fluoride intake can also damage hepatic tissue. We have previously demonstrated that elevated fluorine levels result in oxidative damage, pathological injuries [16], and S phase arrest [37] in the livers of broiler chickens. Shashi et al. [2] reported that fluoride exposure activated hepatic enzymes during osteofluorosis. In addition, fluoride caused lesions and induced the synthesis of stress proteins in the livers and kidneys of mice [38-40]. Chen et al. [41] reported that excessive fluoride inhibited hepatocellular proliferation and differentiation, which might be related to increased S-phase arrest.
Although some studies have examined fluorine-induced cell cycle arrest, very few have examined this relationship in liver cells either in vivo or in vitro, and the mechanisms underlying fluoride-induced cell cycle arrest in the liver remain unclear. In this study, we examined hepatocellular cell cycle distribution and cell cycle regulatory protein levels in mice to determine how NaF suppresses hepatocellular proliferation. After the administration of different concentrations of NaF, hepatocellular cell cycle distribution and the expression of cell cycle control molecules, including phosphorylated ataxia-telangiectasia-mutated (p-ATM, Ser1981), phosphorylated ATM- and Rad3-related (p-ATR, Thr1989), p-p53(Ser15), p21, p-Chk1(Ser317), p-Cdc25A(Ser76), cyclin E/D/A/B, CDK1/2/4, and proliferating cell nuclear antigen (PCNA) were examined using flow cytometry (FCM), western blot (WB), and quantitative real-time polymerase chain reaction (qRT-PCR).
Results
Fluoride inhibited liver development
Liver development was evaluated based on liver growth index (GI) values. GI values were similar in the control and 12 mg/kg groups throughout the 42-day experiment and in the control and 24 mg/kg groups on day 21 of the experiment. However, GI was lower (P < 0.05 and P < 0.01) in the 48 mg/kg group on days 21 and 42 and in the 24 mg/kg group on day 42 than in the control group (Figure 1b).
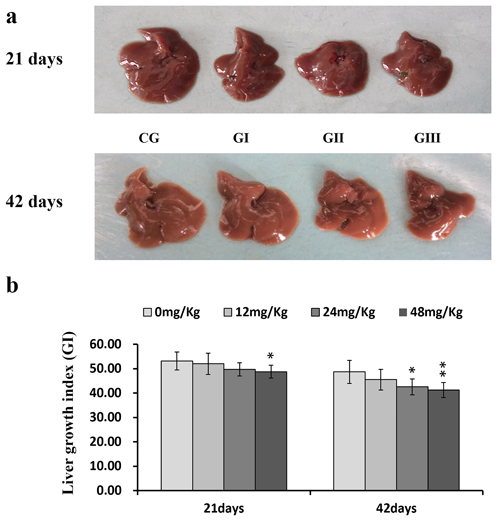
Figure 1: Changes of the liver (a) and changes of the growth index (GI, b) of liver at 21 and 42 days of experiment. Livers are smaller in the three NaF-treated groups than those in the control group. Hepatic growth index is decreased in the 24 and 48 mg/kg groups. CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. b changes of the growth index (GI) in liver, *P<0.05, compared with the control group **P<0.01, compared with the control group.
Fluoride induced pathological changes in the liver
Macroscopically, livers were smaller in the three NaF treatment groups than in the control group (Figure 1a). However, there were no obvious changes in hepatic color or texture.
As shown in Figures 2 and 3, fluoride administration increased the number of hepatocytes with granular and vacuolar degeneration in a dose- and time-dependent manner, and tiny particles as well as small or large vacuoles were visible in the cytoplasm of these hepatocytes. Necrotic hepatocytes were also observed in the 48 mg/kg group. There was no evidence of these lesions in the control group.
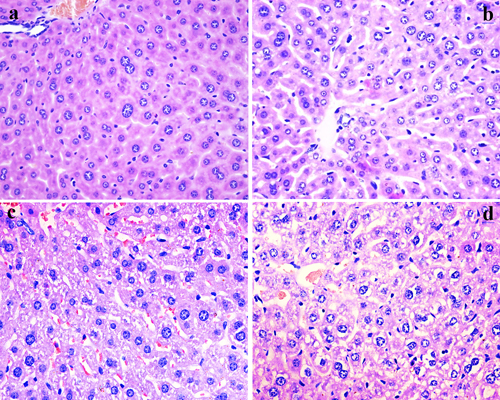
Figure 2: Histopathological changes in the liver at 21 days of experiment. (a) The control group (H&E ×400). (b) The 12 mg/kg group. Hepatocytes show slight granular and vacuolar degeneration (H&E ×400). (c) The 24 mg/kg group. Hepatocytes show granular and vacuolar degeneration (H&E ×400). (d) The 48 mg/kg group. Hepatocytes show obvious granular and vacuolar degeneration. Also, necrotic hepatocytes are observed (H&E ×400).
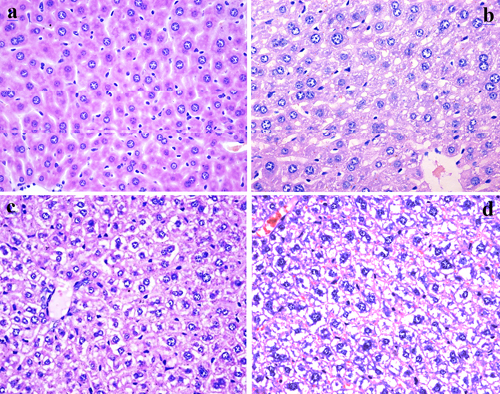
Figure 3: Histopathological changes in the liver at 42 days of experiment. (a) The control group (H&E ×400). (b) The 12 mg/kg group. Hepatocytes show granular and vacuolar degeneration (H&E ×400). (c) The 24 mg/kg group. Hepatocytes show obvious granular and vacuolar degeneration (H&E ×400). (d) The 48 mg/kg group. Hepatocytes show significant granular and vacuolar degeneration. Also, Necrotic hepatic cells are observed (H&E ×400).
NaF induced S-phase arrest in the liver
As shown in Figures 4-6, percentages of hepatocytes in S phase increased in a dose- and time-dependent manner, while percentages of hepatocytes in the G0/G1 and G2/M phases decreased.
Percentages of cells in the G0/G1 phase decreased (p < 0.05 or p < 0.01) in the 48 mg/kg group at both days 21 and 42 and in the 24 mg/kg group at day 21 of the experiment when compared to the control group. Percentages of cells in the G2/M phase were also lower (p < 0.05 or p < 0.01) in the 24 mg/kg and 48 mg/kg groups at day 42 of the experiment than in the control group. Finally, percentages of cells in S phase increased (p < 0.05 or p < 0.01) in the 24 mg/kg and 48 mg/kg groups at days 21 and 42 and in the 12 mg/kg group at day 42 of the experiment when compared to the control group.
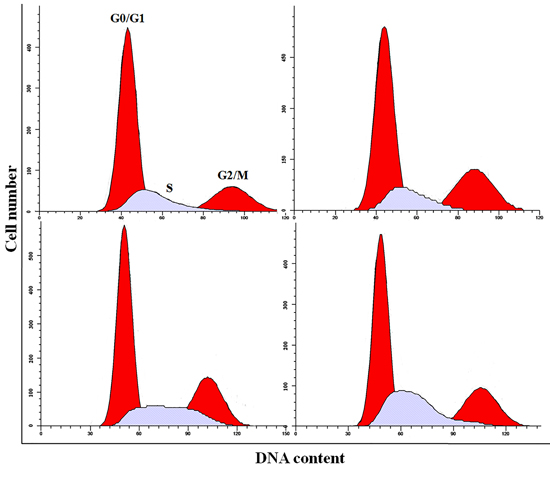
Figure 4: Cell cycle changes in the liver at 21 days of age by flow cytometry.
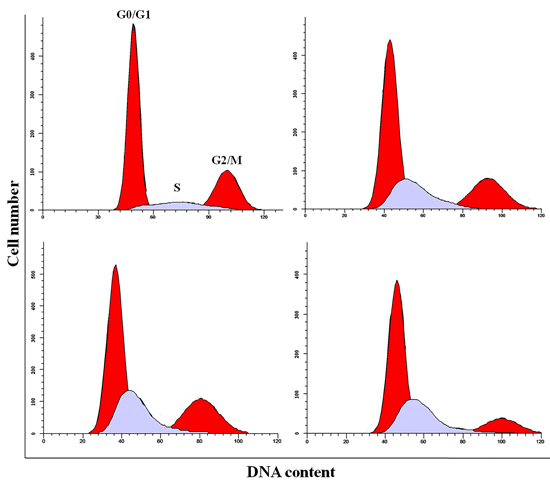
Figure 5: Cell cycle changes in the liver at 42 days of age by flow cytometry.
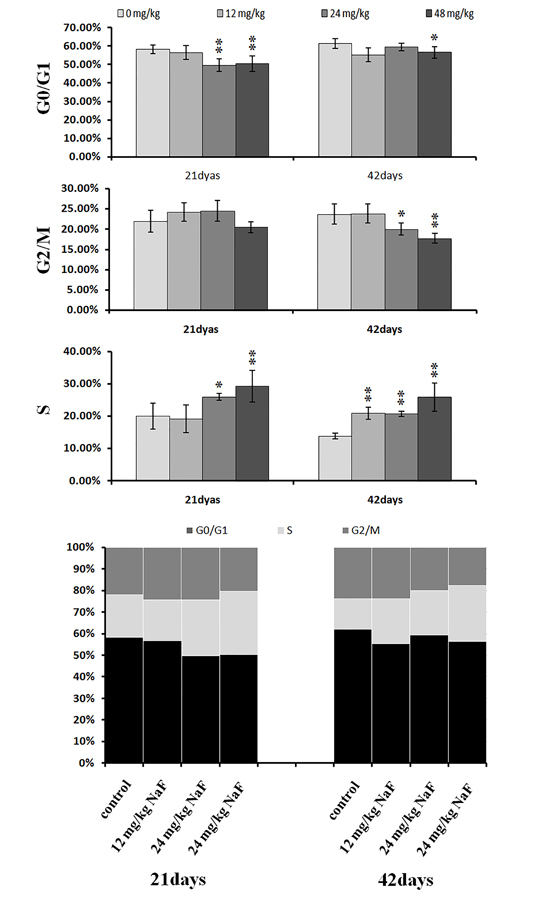
Figure 6: Changes of cell cycle phase distribution (%) in the liver. Data are presented with the mean ± standard deviation (n=8),*P<0.05, compared with the control group **P<0.01, compared with the control group.
Changes in expression of cyclins, CDKs, and PCNA in the liver
To understand the mechanisms underlying NaF-induced hepatocyte cell cycle arrest, we evaluated the expression of regulatory molecules associated with the G1/S phase, including cyclin E/D/A/B, CDK1/2/4, and PCNA, at the protein and mRNA levels. The results are shown in Figures 7-14.
Cyclin E and A protein levels were decreased (p < 0.05 or p < 0.01) in the 24 mg/kg and 48 mg/kg groups at day 21 and in all three NaF-treated groups at day 42 of the experiment when compared to the control group; cyclin E protein levels were also decreased (p < 0.05) in the 12 mg/kg group at day 21 of experiment in comparison to the control group (Figures 7c and 8c). Similarly, cyclin E and A mRNA expression was lower (p < 0.05 or p < 0.01) in the 24 mg/kg and 48 mg/kg groups at days 21 and 42 and in the 12 mg/kg group at day 42 of the experiment than in the control group (Figures 7a and 8a).
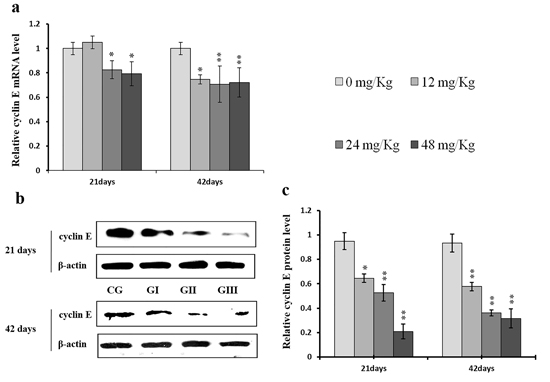
Figure 7: Changes of mRNA and protein expression levels of cyclin E in the liver at 21 and 42 days of experiment. (a) The relative mRNA expression levels of cyclin E. (b) The western blot assay of cyclin E. (c) The relative protein expression levels of cyclin E. CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. Data are presented with the means ± standard deviation (n=8), *p<0.05, compared with the control group; **p<0.01, compared with the control group.
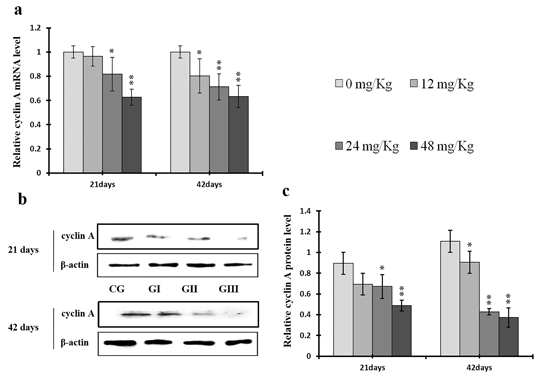
Figure 8: Changes of mRNA and protein expression levels of cyclin A in the liver at 21 and 42 days of experiment. (a) The western blot assay of cyclin A. (b) The relative mRNA expression levels of cyclin A. (c) The relative protein expression levels of cyclin A. CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. Data are presented with the means ± standard deviation (n=8), *p<0.05, compared with the control group; **p<0.01, compared with the control group.
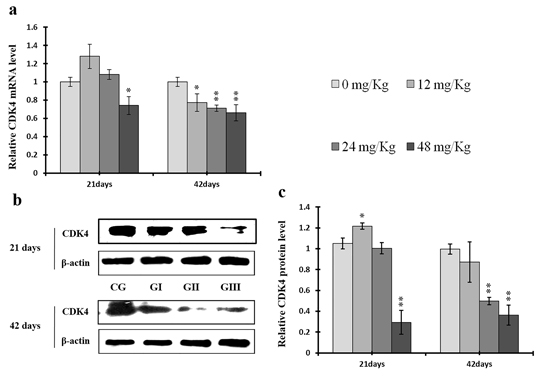
Figure 9: Changes of mRNA and protein expression levels of CDK4 in the liver at 21 and 42 days of experiment. (a) The relative mRNA expression levels of CDK4. (b) The western blot assay of CDK4. (c) The relative protein expression levels of CDK4. CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. Data are presented with the means ± standard deviation (n=8), *p<0.05, compared with the control group; **p<0.01, compared with the control group.
As shown in Figure 9c, CDK4 protein levels were decreased (p < 0.05 or p < 0.01) in the 48 mg/kg group at days 21 and 42 and in the 24 mg/kg group at day 42 of the experiment, but were increased in the 12 mg/kg group at day 21 of the experiment, when compared to the control group. CDK2 protein levels were also lower (p < 0.05 or p < 0.01) in the 24 mg/kg and 48 mg/kg groups at days 21 and 42 and in the 12 mg/kg group at day 42 of the experiment than in the control group (Figure 10c). CDK4 mRNA expression was similarly decreased (p < 0.05 or p < 0.01) in the 48 mg/kg group at days 21 and 42 and in the 12 mg/kg and 24 mg/kg groups at day 42 of the experiment (Figure 9a). CDK2 mRNA expression was also lower (p < 0.05 or p < 0.01) in the 48 mg/kg group at days 21 and 42 and in the 24 mg/kg group at day 42 of the experiment than in the control group (Figure 10a). PCNA protein levels were decreased (p < 0.05 or p < 0.01) in the 24 mg/kg and 48 mg/kg groups at days 21 and 42 and in the 12 mg/kg group at day 21 of the experiment when compared to the control group. PCNA mRNA expression was also lower (p < 0.05 or p < 0.01) in the 48 mg/kg group at days 21 and 42 and in the 24 mg/kg group at day 42 of experiment than in the control group (Figure 11). In contrast, cyclin B/D and CDK1 protein and mRNA expression did not differ among any of the groups (Figures 12-14).
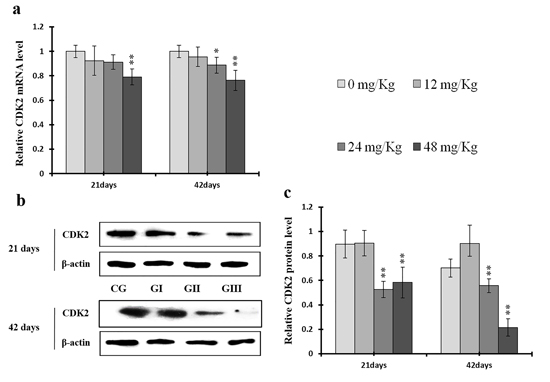
Figure 10: Changes of mRNA and protein expression levels of CDK2 in the liver at 21 and 42 days of experiment. (a) The relative mRNA expression levels of CDK2. (b) The western blot assay of CDK2. (c) The relative protein expression levels of CDK2. CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. Data are presented with the means ± standard deviation (n=8), *p<0.05, compared with the control group; **p<0.01, compared with the control group.
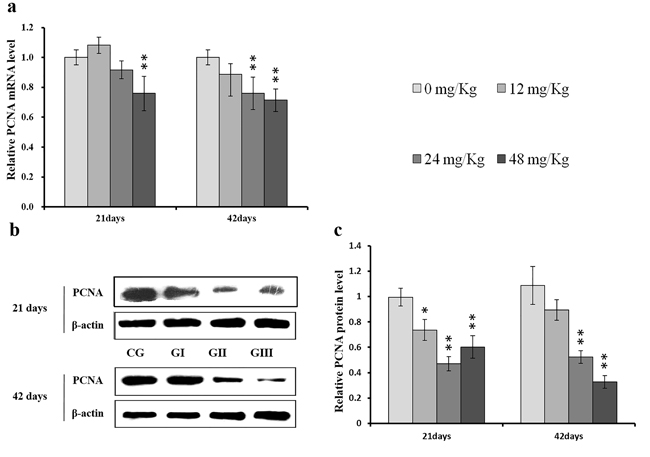
Figure 11: Changes of mRNA and protein expression levels of PCNA in the liver at 21 and 42 days of experiment. (a) The relative mRNA expression levels of PCNA. (b) The western blot assay of PCNA. (c) The relative protein expression levels of PCNA. CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. Data are presented with the means ± standard deviation (n=8), *p<0.05, compared with the control group; **p<0.01, compared with the control group.
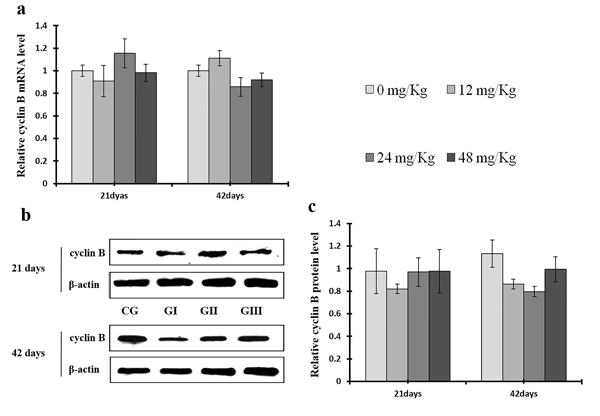
Figure 12: Changes of mRNA and protein expression levels of cyclin B in the liver at 21 and 42 days of experiment. (a) The relative mRNA expression levels of cyclin B. (b) The western blot assay of cyclin B. (c) The relative protein expression levels of cyclin B. CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. Data are presented with the means ± standard deviation (n=8), *p<0.05, compared with the control group; **p<0.01, compared with the control group.
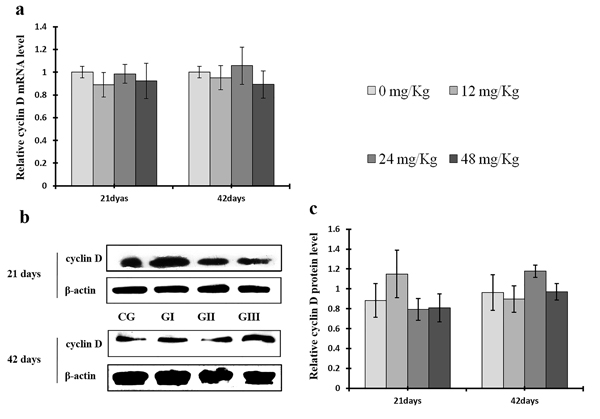
Figure 13: Changes of mRNA and protein expression levels of cyclin D in the liver at 21 and 42 days of experiment. (a) The relative mRNA expression levels of cyclin D. (b) The western blot assay of cyclin D. (c) The relative protein expression levels of cyclin D. CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. Data are presented with the means ± standard deviation (n=8), *p<0.05, compared with the control group; **p<0.01, compared with the control group.
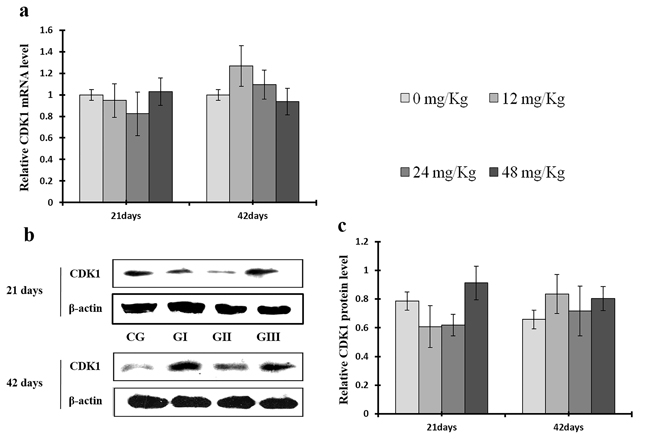
Figure 14: Changes of mRNA and protein expression levels of CDK1 in the liver at 21 and 42 days of experiment. (a) The relative mRNA expression levels of CDK1. (b) The western blot assay of CDK1. (c) The relative protein expression levels of CDK1. CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. Data are presented with the means ± standard deviation (n=8), *p<0.05, compared with the control group; **p<0.01, compared with the control group
Changes in protein and mRNA expression of cyclin/CDK regulators in the liver
Cdc25A promotes cell cycle progression by activating cyclin-dependent protein kinases, and its production is associated with entry of cells into the S phase [42-44]. Moreover, exposure to novel pactamycin analogs also triggers p53-dependent S-phase cell cycle arrest in human head and neck squamous cell carcinoma (HNSCC) [45]. To determine whether the Cdc25A and p53 pathway is involved in NaF-induced cell cycle arrest, we examined the expression of ATR-Chk1-Cdc25A and ATM-p53-p21 pathway members at both the protein and mRNA levels [43, 46, 47]. The results are shown in Figures 15-20.
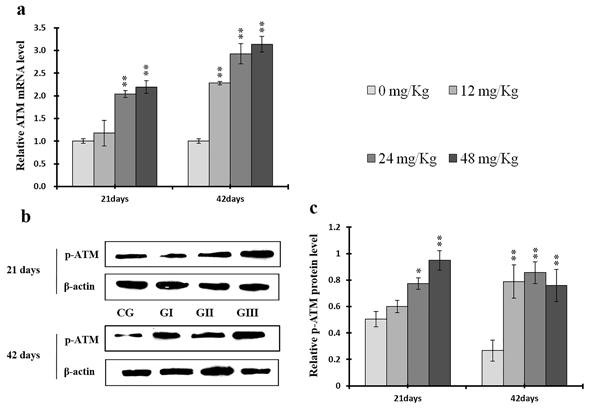
Figure 15: Changes of mRNA level of ATM and protein expression levels of p-ATM in the liver at 21 and 42 days of experiment. (a) The relative mRNA expression levels of ATM. (b) The western blot assay of p-ATM. (c) The relative protein expression levels of p-ATM. CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. Data are presented with the means ± standard deviation (n=8), *p<0.05, compared with the control group; **p<0.01, compared with the control group.
P-ATM protein and ATM mRNA levels were increased (p < 0.05 or p < 0.01) in the 24 mg/kg and 48 mg/kg groups at days 21 and 42 and in the 12 mg/kg group at day 42 of the experiment when compared to the control group (Figure 15). P-p53 protein and p53 mRNA levels were also increased (p < 0.05 or p < 0.01) in the 24 mg/kg and 48 mg/kg groups at days 21 and 42 and in the 12 mg/kg group at day 42 of the experiment in comparison to the control group (Figure 16). P21 protein levels were higher (p < 0.05 or p < 0.01) in all three NaF-treated groups than in the control group at days 21 and 42 of the experiment. Similarly, p21 mRNA expression was markedly increased (p < 0.05 or p < 0.01) in the 24 mg/kg and 48 mg/kg groups at days 21 and 42 of the experiment when compared to the control group (Figure 17).
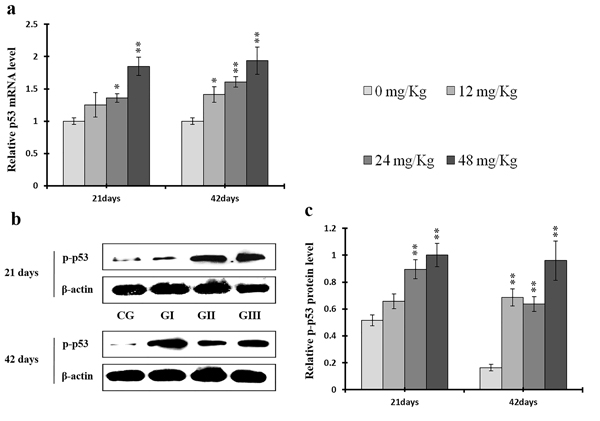
Figure 16: Changes of mRNA level of p53 and protein expression levels of p-p53 in the liver at 21 and 42 days of experiment. (a) The relative mRNA expression levels of p53. (b) The western blot assay of p-p53. (c) The relative protein expression levels of p-p53.CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. Data are presented with the means ± standard deviation (n=8), *p<0.05, compared with the control group; **p<0.01, compared with the control group.
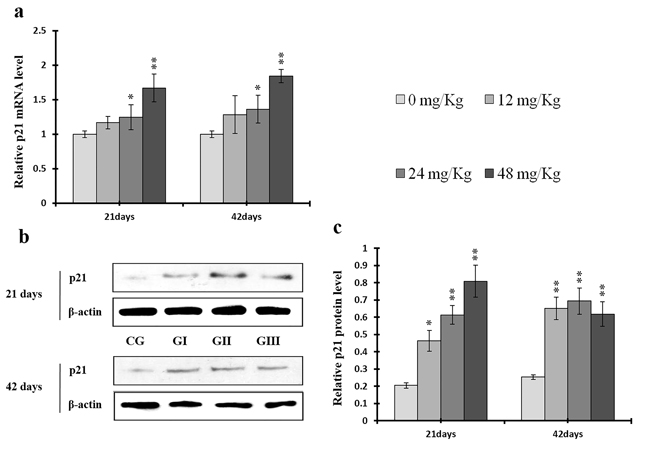
Figure 17: Changes of mRNA and protein expression levels of p21 in the liver at 21 and 42 days of experiment. (a) The relative mRNA expression levels of p21. (b) The western blot assay of p21. (c) The relative protein expression levels of p21. CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. Data are presented with the means ± standard deviation (n=8), *p<0.05, compared with the control group; **p<0.01, compared with the control group.
As shown in Figures 18c and 19c, p-ATR and p-Chk1 protein levels were increased (p < 0.05 or p < 0.01) in the 24 mg/kg and 48 mg/kg groups at days 21 and 42 and in the 12 mg/kg group at day 42 of the experiment when compared to the control group. P-Chk1 mRNA expression was also markedly higher (p < 0.05 or p < 0.01) in the 24 mg/kg and 48 mg/kg groups at days 21 and 42 and in the 12 mg/kg group at day 42 of the experiment than in the control group (Figure 19a). NaF treatment increased (p < 0.05 or p < 0.01) p-Cdc25A protein levels in the 48 mg/kg group at days 21 and 42 and in the 24 mg/kg group at day 42 of the experiment in comparison to the control group. However, Cdc25A mRNA expression was decreased (p < 0.05 or p < 0.01) in the 48 mg/kg group at days 21 and 42 of the experiment when compared to the control group (Figure 20).
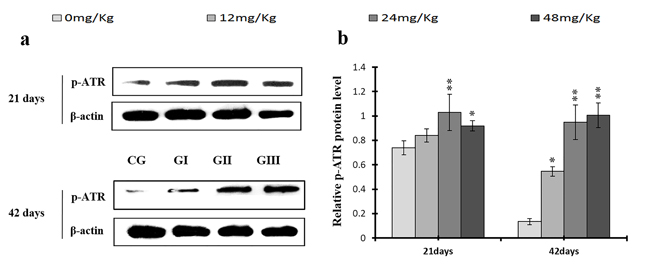
Figure 18: Changes of protein expression levels of p-ATR in the liver at 21 and 42 days of experiment. (a) The western blot assay of p-ATR. (b) The relative protein expression levels of p-ATR. CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. Data are presented with the means ± standard deviation (n=8), *p<0.05, compared with the control group; **p<0.01, compared with the control group.
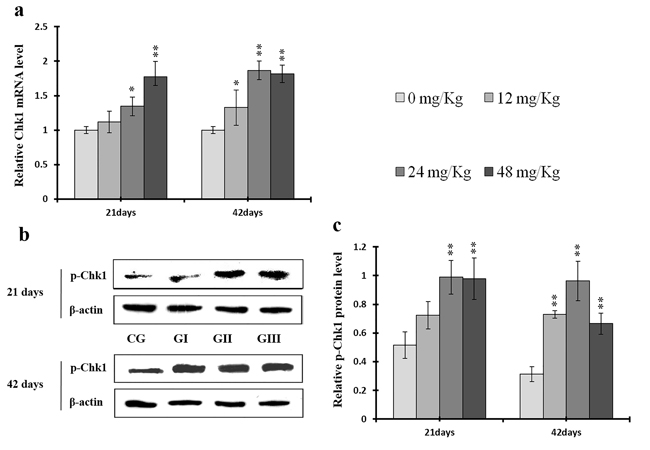
Figure 19: Changes of mRNA level of Chk1 and protein expression levels of p-Chk1 in the liver at 21 and 42 days of experiment. (a) The relative mRNA expression levels of Chk1. (b) The western blot assay of p-Chk1. c The relative protein expression levels of p-Chk1. CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. Data are presented with the means ± standard deviation (n=8), *p<0.05, compared with the control group; **p<0.01, compared with the control group.
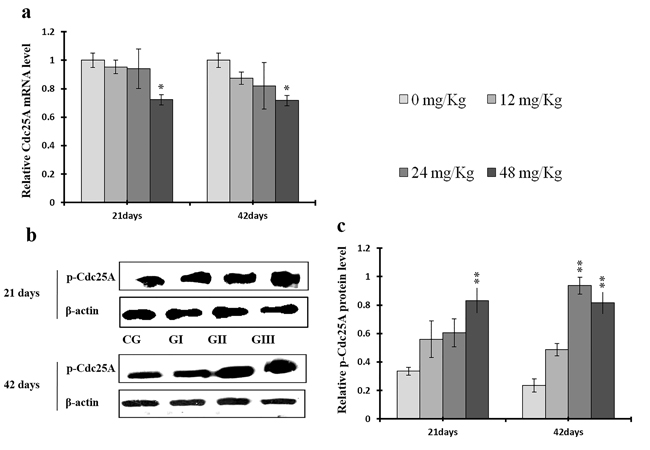
Figure 20: Changes of mRNA level of Cdc25A and protein expression levels of p-Cdc25A in the liver at 21 and 42 days of experiment. (a) The relative mRNA expression levels of Cdc25A. (b) The western blot assay of p-Cdc25A. (c) The relative protein expression levels of p-Cdc25A. CG: Control group; GI: 12mg/kg group; GII: 24mg/kg group; GIII: 48mg/kg group. Data are presented with the means ± standard deviation (n=8), *p<0.05, compared with the control group; **p<0.01, compared with the control group.
Discussion
In this study, we examined the molecular pathways underlying NaF-induced cell cycle arrest in the mouse liver. We found that, at does above 12 mg/kg, NaF induced cell cycle arrest in the S phase and decreased the percentages of cells in the G0/G1and G2/M phases (Figure 6).
Hayashi et al. [48] reported that NaF induced cell cycle-dependent cytotoxicity and clastogenicity in human diploid fibroblasts, and cells were especially sensitive to these effects during early and middle S phase. However, Aardema et al. [49] reported that NaF induced cell cycle was arrest in the G2/M phase and that cells in both the S and G2 phases were equally sensitive to the adverse effects of NaF [50]. Our results are consistent with those of Zhang et al. [51], who found that fluoride increased the proportion of cells in the S phase, decreased the proportions in both the G0/G1 and G2/M phases, and induced DNA damage in cultured primary cultured rat hippocampal neurons. Excessive fluoride levels can also increase the proportion of cells in the S phase, thereby affecting hepatocellular proliferation and differentiation [41].
Here, we examined the expression of S phase regulators to identify the molecular mechanisms underlying NaF-induced cell cycle arrest in the S phase in the liver. NaF increased p-ATM, p-p53, p21, p-ATR, p-Chk1, and p-Cdc25A protein levels, increased ATM, p53, p21, and Chk1 mRNA expression, and decreased Cdc25A mRNA expression (Figures 15-20). Li et al. [52] reported that fluoride induced p53 expression and cell cycle arrest in the S phase in human embryo hepatocytes in a dose-dependent manner; fluoride also increased or induced p53 expression in rat leukocytes [53] and human embryonic hepatocytes [54]. ATM plays a prominent role in checkpoint regulation at S phase transition and is associated with p53 phosphorylation after DNA damage-induced stresses [55-58]. Our results demonstrate conclusively that NaF up-regulates ATM and its downstream target p53, thus activating the ATM signal transduction pathways (Figures 15 and 16). These findings are similar to those of Guha et al., who demonstrated that novel pactamycin analogs can cause S-phase arrest in human head and neck squamous cell carcinoma (HNSCC) cells by increasing p53 activity, up-regulating expression of the cyclin kinase inhibitors p27 and p21, slightly reducing cyclin D1 expression, and moderately increasing cyclin E expression; no changes were observed in the expression of cyclin B, CDK2, or CDK4 [45]. Treatment with the novel anthraquinone derivative IMP1338 also increased p53-dependent cell cycle arrest in the S phase in human cancer cells [59]. P53 therefore may be a critical mediator of apoptosis, DNA repair, and cell cycle arrest in response to DNA damage and cellular stress [60]. In addition, p53 can up-regulate p21 expression, which also promotes cell cycle arrest in the S phase [61]. Here, NaF increased p21 protein levels and mRNA expression (Figure 17), which is consistent with studies demonstrating that various stimuli induce cell cycle arrest in the S phase through p53-dependent activation of p21 [45, 62-64]. P21 generally induces S-phase cell cycle arrest by inhibiting CDK2 activity [64-66] which, together with cyclin A, is necessary for the progression of cells through the S phase [67]; inactivation of cyclin A/CDK2 complexes prevents progression beyond the S phase checkpoint.
CDK2 is the major downstream target of Cdc25A, which activates the cyclin A/CDK2 complex [68]. Triptolide and simvastatin can induce S-phase arrest by inhibiting Cdc25A [69, 70]. In addition, Chk1, which is regulated by ATR, phosphorylates the Cdc25A protein at S76, which in turn up-regulates ubiquitin-mediated proteolysis of Cdc25A [47, 43]. Chk1 also regulates the S phase checkpoint by promoting proteolysis of Cdc25A, which in turn inhibits CDK2 activity, in response to some anticancer agents, such as infrared ray (IR) and ultraviolet (UV) light [71-73]. Our results demonstrate that NaF decreased cyclin A and CDK2 and increased ATR and Chk1 protein levels and mRNA expression (Figures 8, 10, 18, and 19). However, p-Cdc25A (S76) protein levels increased, while Cdc25A mRNA expression decreased, after NaF treatment (Figure 20). The increases in p-ATR, p-Chk1, and p-Cdc25A protein levels suggest that NaF activates the ATR signal transduction pathways. In addition, downregulation of Cdc25A induced S-phase arrest by inhibiting activation of the cyclin A/CDK2 complex. Cyclin D1, along with its kinase partners CDK4 and CDK6, is a major mitogen-induced regulator of cell cycle progression in the G1 phase. Cyclin E/CDK2 complexes are crucial for the transition from the G1 to the S phase [3]. As shown in Figures 7 and 9, NaF markedly reduced cyclin E and CDK4 protein levels and mRNA expression, thus inhibiting the transition from the G1 to the S phase. The decreases observed in cyclin E, cyclin A, Cdc25A, and CDK2 protein levels and mRNA expression are consistent with a previous report that triptolide induced S phase arrest in multiple myeloma cells by inhibiting cyclin E, cyclin A, Cdc25A, and CDK2 expression and up-regulating p21 and p27 expression [69].
The PCNA protein, a marker of proliferation that plays crucial roles in many cellular processes, is ubiquitously expressed in eukaryotic cells. For example, PCNA is involved in DNA replication, DNA repair, and chromatin assembly and maintenance [74-76]. We found that NaF decreased PCNA protein levels and mRNA expression (Figure 11), suggesting that NaF also inhibits hepatocellular proliferation by down-regulating PCNA expression. These decreases in PCNA protein levels and mRNA expression are consistent with a previous report that fluoride can decrease germ cell counts and damage the male reproductive system by inhibiting PCNA expression [77].
In conclusion, the results of this study demonstrate that NaF increases ATM, p53, p21, ATR, and Chk1 expression, decreases Cdc25A, cyclin E, cyclin A, CDK2, CDK4, and PCNA expression, activates the ATM-p53-p21 and ATR-Chk1-Cdc25A pathways, and ultimately leads to S-phase arrest in the mouse liver (Figure 21).
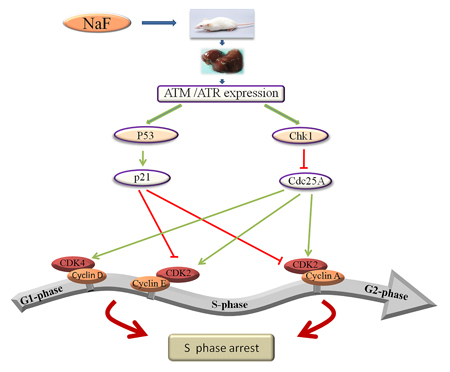
Figure 21: Schematic of NaF-induced S-phase cell cycle arrest in mouse liver. NaF can activate two signal transduction pathways: the ATM-p53-p21 and ATR-Chk1-Cdc25A pathways, which cause hepatocellular cell cycle arrest at S-phase.
Materials and Methods
Animals and treatments
240 healthy ICR mice were provided by the Experimental Animal Corporation of Dossy in Chengdu, China. Food and water were provided ad libitum. After 1 week of acclimatization, mice were divided randomly into four groups (N=60). The control group received intragastric doses of distilled water, while the experimental groups received intragastric doses of 12, 24, or 48 mg/kg NaF (Chengdu Kelong Chemical Co., Ltd., Chengdu, China). NaF was diluted with distilled water. The gavage doses for all four groups were 1 mL/100 g body weight once daily for 42 days. Liver samples were collected from mice on days 21 and 42 of the experiment.
The use of mice and all experimental procedures were approved by the Animal Care and Use Committee, Sichuan Agricultural University.
Determination of liver growth index
After body weights were recorded, eight mice per group were humanely killed on days 21 and 42 of the experiment. Gross observations and weights were recorded for each liver. Liver growth index (GI) was calculated according to the following formula:
Pathological observations
Liver samples were taken from eight mice in each group on days 21 and 42 of the experiment and gross observations were recorded. After gross examination, liver samples were fixed in 4% paraformaldehyde solution, dehydrated in ethanol, and embedded in paraffin. Serial slices at 5 μm thickness were prepared and stained with hematoxylin and eosin (H•E) for histopathological examination under a light microscope.
Flow cytometry cell cycle assay
Eight mice per group were humanely killed on days 21 and 42 of the experiment; liver samples were immediately removed and cut into pieces to form a cell suspension that was filtered through a 300-mesh nylon screen. The cells were washed twice with cold PBS (phosphate buffer solution, pH 7.2-7.4) and then suspended in PBS at a concentration of 1×106 cells/mL using the normal counting method for blood cells. 100 μL portions of the cell suspension were transferred into new 5 mL culture tubes. The cells were then incubated for 30 min at room temperature in the dark with 0.25% Triton X-100 and 5 μL propidium iodide (PI) (BD, Cat. No.51–66211E, USA). Cells were resuspended in 0.5 mL PBS and run on a BD FACS Calibur flow cytometer. The results were analyzed using Mod Fit LT for Mac V3.0.
Cell cycle regulatory protein expression by western blot
On days 21 and 42 of the experiment, liver samples were collected from eight mice per group and the expression of cell cycle regulatory proteins was examined by western blot. The liver was homogenized and proteins were extracted with RIPA lysis buffer and kept in Laemmli loading buffer. Protein samples were resolved on SDS-PAGE gels (5%–15%) and transferred to nitrocellulose filter membranes. Membranes were blocked with 5% fat-free milk for 1h and incubated with primary antibodies overnight at 4°C. The primary antibodies were cyclin D/E/B/A, CDK1/2/4, p-ATM, p-p53, p21, p-ATR, p-Chk1, and p-Cdc25A (Table 1). The membranes were then washed with PBS-Tween, incubated with biotin-conjugated secondary antibodies (Santa Cruz, USA) for 1h, and washed again with PBS-Tween. Blots were visualized by ECL™ (Bio-Rad, Hercules, CA, USA) and X-ray film. Statistical analysis of protein expression was performed using ImageJ2x software.
Table 1: Antibodies used in western blot
Name |
Company |
Cat# |
dilution |
cyclin E |
Abcam, China |
ab52189 |
1/1000 |
Cyclin A |
Abcam, China |
ab181591 |
1/1000 |
Cyclin D |
Abcam, China |
ab134175 |
1/1000 |
Cyclin B |
Abcam, China |
ab181593 |
1/1000 |
CDK1 |
Abcam, China |
ab32384 |
1/1000 |
CDK2 |
Abcam, China |
ab76146 |
1/1000 |
CDK4 |
Santa Cruz, China |
sc260 |
1/50 |
p-ATR |
Gene Tex, China |
GTX128145 |
1/500 |
p-Chk1 |
Cell Signaling, China |
12302T |
1/100 |
p-Cdc25A |
Santa Cruz, China |
sc101655 |
1/50 |
p-ATM |
Biolegend, China |
651201 |
1/400 |
p-p53 |
Cell Signaling, China |
9284T |
1/1000 |
p21 PCNA |
Boster, China Abcam, China |
BA0271 Ab92552 |
1/100 1/1000 |
Cell cycle regulatory molecule mRNA expression by quantitative real-time polymerase chain reaction (qRT-PCR)
On days 21 and 42 of the experiment, liver samples were collected from eight mice per group and stored in liquid nitrogen. They were then homogenized in liquid nitrogen using a mortar and pestle. Total RNA was extracted from frozen liver powders using RNAiso plus (9108/9109, Takara, China) following the manufacturer’s instructions. cDNA was then synthesized using a PrimScript™ RT reagent Kit (RR047A, Takara, China) according to the manufacturer’s instructions. The cDNA product was used as a template for qRT-PCR analysis. Specific oligonucleotide primers were designed and synthesized by Sangon Biotech (Shanghai, China; Table 2) according to the Mus musculus sequences. All qRT-PCR was performed on a Model C1000 Thermal Cycler (Bio Rad, USA) using the SYBR® Premix Ex Taq™Ⅱ system (RR820A, Takara, China) according to the standard protocols.
Mice β-actin expression was detected as an internal reference housekeeping gene. Gene expression from control group samples on days 21 and 42 of the experiment was used to calibrate gene expression in samples from the experimental groups. The 2-ΔΔCT method was used to analyze data from the qRT-PCR experiments [78].
Table 2: Sequence of primers used in qRT-PCR
Gene symbol |
Accession number |
Primer |
Primer sequence(5’-3’) |
Product size |
cyclin E |
NM_007633 |
Forward |
GTTACAGATGGCGCTTGCTC |
104 |
Reverse |
AGCCAGGACACAATGGTCAG |
|||
Cyclin A |
NM_009828 |
Forward |
CTTGTAGGCACGGCTGCTAT |
451 |
Reverse |
CATGTTGTGGCGCTTTGAGG |
|||
Cyclin D |
NM_007631 |
Forward |
AAGTGTGACCCGGACTGC |
174 |
Reverse |
GATGTCCACATCTCGCACG |
|||
Cyclin B |
NM_172301 |
Forward |
AGCGAAGAGCTACAGGCAAG |
141 |
Reverse |
CTCAGGCTCAGCAAGTTCCA |
|||
CDK1 |
NM_007659 |
Forward |
TCGGCTCGTTACTCCACTC |
154 |
Reverse |
GCCACACTTCGTTGTTAGG |
|||
CDK2 |
NM_016756 |
Forward |
TTGGAGTCCCTGTCCGAACT |
142 |
Reverse |
CGGGTCACCATTTCAGCAAAG |
|||
CDK4 |
NM_009870 |
Forward |
CAATGTTGTACGGCTGATGG |
120 |
Reverse |
GGAGGTGCTTTGTCCAGGTA |
|||
Chk1 |
NM_007691.5 |
Forward |
GCAAACTTTGGGAGAAGGTGC |
103 |
Reverse |
TATGGCCCGCTTCATGTCTAC |
|||
Cdc25A |
NM_007658 |
Forward |
AGAACCCTATTGTGCCTACTG |
124 |
Reverse |
TACTCATTGCCGAGCCTATC |
|||
ATM |
NM_007499.2 |
Forward |
CCTTCCCACTCCAGAAACAG |
123 |
Reverse |
CTCCGCATAACTTCCATCGT |
|||
p53 |
NM_011640.3 |
Forward |
AGAGACCGCCGTACAGAAGA |
227 |
Reverse |
GCATGGGCATCCTTTAACTC |
|||
p21 |
U24173.1 |
Forward |
CAAAGTGTGCCGTTGTCTCTT |
111 |
Reverse |
TCAAAGTTCCACCGTTCTCG |
|||
PCNA |
NM_011045 |
Forward |
ATCCCAGAACAGGAGTACAGC |
92 |
Reverse |
ACAGCATCTCCAATGTGGCT |
|||
β-actin |
NM_007393 |
Forward |
GCTGTGCTATGTTGCTCTAG |
117 |
Reverse |
CGCTCGTTGCCAATAGTG |
Statistical analysis
One-way analysis of variance (ANOVA) procedure in SPSS 18.0 software was carried out to analyze all the data. All the results were expressed as mean ± standard deviation. p < 0.05 was significant, and p < 0.01 was markedly significant.
Acknowledgments
This research was supported by the program for Changjiang scholars and the university innovative research team (IRT 0848), and the Shuangzhi project of Sichuan Agricultural University (03570327; 03571198).
Conflicts of Interest
The authors declare that there are no conflicts of interest.
References
1. Song G, Wang RL, Chen ZY, Zhang B, Wang HL, Liu ML, Gao JP, Yan XY. Toxic effects of sodium fluoride on cell proliferation and apoptosis of Leydig cells from young mice. J Physiol Biochem. 2014; 70:761–68.
2. Shashi A, Bhardwaj M. Study on blood biochemical diagnostic indices for hepatic function biomarkers in endemic skeletal fluorosis. Biol Trace Elem Res. 2011; 143:803–14.
3. Kuang P, Deng H, Cui H, Chen L, Fang J, Zuo Z, Deng J, Wang X, Zhao L. Sodium fluoride (NaF) causes toxic effects on splenic development in mice. Oncotarget. 2017; 8:4703–17. https://doi.org/10.18632/oncotarget.13971.
4. Perumal E, Paul V, Govindarajan V, Panneerselvam L. A brief review on experimental fluorosis. Toxicol Lett. 2013; 223:236–51.
5. Chen T, Cui H, Cui Y, Bai C, Gong T, Peng X. Cell-cycle blockage associated with increased apoptotic cells in the thymus of chickens fed on diets high in fluorine. Hum Exp Toxicol. 2011; 30:685–92.
6. Chen T, Cui H, Cui Y, Bai C, Gong T. Decreased antioxidase activities and oxidative stress in the spleen of chickens fed on high-fluorine diets. Hum Exp Toxicol. 2011; 30:1282–86.
7. Chen T, Cui Y, Bai C, Gong T, Peng X, Cui H. Increased apoptotic lymphocyte population in the spleen of young chickens fed diets high in fluorine. Fluoride. 2009; 42:94–100.
8. Chen T, Cui Y, Gong T, Bai C, Peng X, Cui H. Inhibition of splenocyte proliferation and spleen growth in young chickens fed high fluoride diets. Fluoride. 2009; 42:203–09.
9. Chen T, Gong T, Bai C, Peng X, Cui H. Effect of dietary high fluorine on the morphologic structure, cell cycle and apoptosis of bursa of fabricius in broilers. Chinese Journal of Animal & Veterinary Sciences. 2009; 40:1235–43.
10. Liu J, Cui H, Peng X, Fang J, Zuo Z, Wang H, Wu B, Deng Y, Wang K. Changes induced by high dietary fluorine in the cecal tonsil cytokine content of broilers. Fluoride. 2012; 45:102–06.
11. Liu J, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang H, Wu B, Deng Y, Wang K. Decreased IgA+ B cells population and IgA, IgG, IgM contents of the cecal tonsil induced by dietary high fluorine in broilers. Int J Environ Res Public Health. 2013; 10:1775–85.
12. Liu J, Cui H, Peng X, Fang J, Zuo Z, Wang H, Wu B, Deng Y, Wang K. Dietary high fluorine induces apoptosis and alters Bcl-2, Bax, and caspase-3 protein expression in the cecal tonsil lymphocytes of broilers. Biol Trace Elem Res. 2013; 152:25–30.
13. Liu J, Cui H, Peng X, Fang J, Zuo Z, Wang H, Wu B, Deng Y, Wang K. Decreased percentages of T-cell subsets and IL-2 contents in the cecal tonsil of broilers fed diets high in fluorine. Fluoride. 2012; 45:53–57.
14. Liu J, Cui H, Peng X, Fang J, Zuo Z, Wang H, Wu B, Deng Y, Wanga K. High dietary fluorine induction of oxidative damage in the cecal tonsil of broilers. Fluoride. 2012; 45:47–52.
15. Cui H, Zhu K, Peng X, Deng J, Zhi Y. Effect of dietary high copper on the cell cycle and apoptosis of liver in chickens. Acta Veterinaria Et Zootechnica Sinica. 2006; 37:908–13.
16. Gong T, Bai C, Chen T, Peng X, Cui H. Effect of high fluorine on the antioxidant function and ultrastructure of liver in chickens. Chinese Journal of Animal & Veterinary Sciences. 2009; 40:1389–94.
17. Bai C, Chen T, Cui Y, Gong T, Peng X, Cui HM. Effect of high fluorine on the cell cycle and apoptosis of renal cells in chickens. Biol Trace Elem Res. 2010; 138:173–80.
18. Bai C, Chen T, Gong T, Peng X, Cui H. Pathological effect of high fluorine on kidney and the related biochemical parameters of serum in the chicken. Chin J Vet Sci. 2010; 30:505–09.
19. Bai C, Peng X, Gong T, Chen T, Cui H. Effect of high fluorine on the antioxygen function of kidney in chickens. Chin J Vet Sci. 2010; 30:825–28.
20. Chen T, Cui Y, Bai C, Gong T, Peng X, Cui H. Decreased percentages of the peripheral blood T-cell subsets and the serum IL-2 contents in chickens fed on diets excess in fluorine. Biol Trace Elem Res. 2009; 132:122–28.
21. Deng Y, Cui H, Peng X, Fang J, Zuo Z, Deng J, Luo Q. Effects of high dietary fluoride on serum biochemistry and oxidative stress parameters in broiler chickens. Health. 2014; 06:1840–48.
22. Deng Y, Cui H, Peng X, Fang J, Zuo Z, Deng J, Luo Q. Effects of high dietary fluorine on erythrocytes and erythrocyte immune adherence function in broiler chickens. Biol Trace Elem Res. 2013; 155:247–52.
23. Deng Y, Cui H, Peng X, Fang J, Zuo Z, Deng J, Luo Q. High dietary fluorine alteration of serum cytokine and immunoglobulin in broilers. Fluoride. 2013; 46:118–27.
24. Luo Q, Cui H, Peng X, Fang J, Zuo Z, Deng J, Liu J, Deng Y. Dietary high fluorine alters intestinal microbiota in broiler chickens. Biol Trace Elem Res. 2016; 173:483–91.
25. Luo Q, Cui H, Peng X, Fang J, Zuo Z, Deng J, Liu J, Deng Y. Intestinal IgA⁺ cell numbers as well as IgA, IgG, and IgM contents correlate with mucosal humoral immunity of broilers during supplementation with high fluorine in the diets. Biol Trace Elem Res. 2013; 154:62–72.
26. Luo Q, Cui H, Peng X, Fang J, Zuo Z, Liu J, Wu B, Wang H, Deng Y, Huang J. Intestinal oxidative stress in broilers caused by high dietary fluorine. Fluoride. 2012; 45:349–56.
27. Luo Q, Cui H, Peng X, Fang J, Zuo Z, Deng J, Liu J, Deng Y. Suppressive effects of dietary high fluorine on the intestinal development in broilers. Biol Trace Elem Res. 2013; 156:153–65.
28. Luo Q, Cui H, Peng X, Fang J, Zuo Z, Liu J, Wu B, Deng Y. The association between cytokines and intestinal mucosal immunity among broilers fed on diets supplemented with fluorine. Biol Trace Elem Res. 2013; 152:212–18.
29. Wang AG, Xia T, Chu QL, Zhang M, Liu F, Chen XM, Yang KD. Effects of fluoride on lipid peroxidation, DNA damage and apoptosis in human embryo hepatocytes. Biomed Environ Sci. 2004; 17:217–22.
30. Nguyen Ngoc TD, Son YO, Lim SS, Shi X, Kim JG, Heo JS, Choe Y, Jeon YM, Lee JC. Sodium fluoride induces apoptosis in mouse embryonic stem cells through ROS-dependent and caspase- and JNK-mediated pathways. Toxicol Appl Pharmacol. 2012; 259:329–37.
31. Matsui H, Morimoto M, Horimoto K, Nishimura Y. Some characteristics of fluoride-induced cell death in rat thymocytes: cytotoxicity of sodium fluoride. Toxicol In Vitro. 2007; 21:1113–20.
32. Guo H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B, Chen K, Deng J. Dietary NiCl₂ causes G₂/M cell cycle arrest in the broiler’s kidney. Oncotarget. 2015; 6:35964–77. https://doi.org/10.18632/oncotarget.5934.
33. Méchali M, Lutzmann M. The cell cycle: now live and in color. Cell. 2008; 132:341–43.
34. Zhang Y, Sun G, Jin Y, Wang Y. [Effects of fluoride on cell cycle and apoptosis in cultured osteoblasts of rats]. [Article in Chinese]. Wei Sheng Yan Jiu. 2003; 32:432–33..
35. Deng H, Kuang P, Cui H, Chen L, Fang J, Zuo Z, Deng J, Wang X, Zhao L. Sodium fluoride induces apoptosis in cultured splenic lymphocytes from mice. Oncotarget. 2016; 7:67880–900. https://doi.org/10.18632/oncotarget.12081.
36. Kuang P, Deng H, Cui H, Chen L, Guo H, Fang J, Zuo Z, Deng J, Wang X, Zhao L. Suppressive effects of sodium fluoride on cultured splenic lymphocyte proliferation in mice. Oncotarget. 2016; 7:61905–15. https://doi.org/10.18632/oncotarget.11308.
37. Gong T, Chen T, Bai C, Peng X, Cui H. Effect of high fluorine on the cell cycle and apoptosis of liver cells in chickens. Chinese Journal of Animal and Veterinary Sciences. 2006; 37:1675–80.
38. Chattopadhyay A, Podder S, Agarwal S, Bhattacharya S. Fluoride-induced histopathology and synthesis of stress protein in liver and kidney of mice. Arch Toxicol. 2011; 85:327–35.
39. Nabavi SF, Daglia M, Sureda A, Nabavi SM. CHAPTER 16:Fluoride-Induced Oxidative Stress in the Liver. Fluoride. 2015; 56:271–91.
40. Shah BA, Nisa G, Farzana F, Bhat GM. A study of fluoride induced toxic effects on the microanatomy of liver in albino rats. Int J Curr Res. 2016; 8:33686–90.
41. Chen B, Zhong Y, Zhong W, Zou Z, Chen Z, Wei X, Deng R, Lin Q, Dong C. Time-effect and dose-effect of fluoride on cell cycle arrest in rat liver and kidney cells. J Environ Health. 2013; 30:317–20.
42. Blomberg I, Hoffmann I. Ectopic expression of Cdc25A accelerates the G(1)/S transition and leads to premature activation of cyclin E- and cyclin A-dependent kinases. Mol Cell Biol. 1999; 19:6183–94.
43. Kang T, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung MC, Piwnica-Worms H. GSK-3 β targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3 β inactivation correlates with Cdc25A overproduction in human cancers. Cancer Cell. 2008; 13:36–47.
44. Molinari M, Mercurio C, Dominguez J, Goubin F, Draetta GF. Human Cdc25 A inactivation in response to S phase inhibition and its role in preventing premature mitosis. EMBO Rep. 2000; 1:71–79.
45. Guha G, Lu W, Li S, Liang X, Kulesz-Martin MF, Mahmud T, Indra AK, Ganguli-Indra G. Novel pactamycin analogs induce p53 dependent cell-cycle arrest at S-phase in human head and neck squamous cell carcinoma (HNSCC) cells. PLoS One. 2015; 10:e0125322.
46. Nam EA, Zhao R, Glick GG, Bansbach CE, Friedman DB, Cortez D. Thr-1989 phosphorylation is a marker of active ataxia telangiectasia-mutated and Rad3-related (ATR) kinase. J Biol Chem. 2011; 286:28707–14.
47. Myers JS, Cortez D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J Biol Chem. 2006; 281:9346–50.
48. Hayashi N, Tsutsui T. Cell cycle dependence of cytotoxicity and clastogenicity induced by treatment of synchronized human diploid fibroblasts with sodium fluoride. Mutat Res. 1993; 290:293–302.
49. Aardema MJ, Gibson DP, LeBoeuf RA. Sodium fluoride-induced chromosome aberrations in different stages of the cell cycle: a proposed mechanism. Mutat Res. 1989; 223:191–203.
50. Aardema MJ, Tsutsui T. Sodium fluoride-induced chromosome aberrations in different cell cycle stages. Mutat Res. 1995; 331:171–72.
51. Zhang M, Wang A, Xia T, He P. Effects of fluoride on DNA damage, S-phase cell-cycle arrest and the expression of NF-kappaB in primary cultured rat hippocampal neurons. Toxicol Lett. 2008; 179:1–5.
52. Li X, Chu Q, Xia T. Effects of Fluoride on Cell Cycle, Apoptosis and p53 Expression in Human Embryo Hepatocytes. Acta Universitatis Medictnae Tangji. 2004; 33:550–52.
53. Gutiérrez-Salinas J, Morales-González JA, Madrigal-Santillán E, Esquivel-Soto J, Esquivel-Chirino C, González-Rubio MG, Suástegui-Domínguez S, Valadez-Vega C. Exposure to sodium fluoride produces signs of apoptosis in rat leukocytes. Int J Mol Sci. 2010; 11:3610–22.
54. Wang AG, Chu QL, He WH, Xia T, Liu JL, Zhang M, Nussler AK, Chen XM, Yang KD. Effects on protein and mRNA expression levels of p53 induced by fluoride in human embryonic hepatocytes. Toxicol Lett. 2005; 158:158–63.
55. Lim DS, Kim ST, Xu B, Maser RS, Lin J, Petrini JH, Kastan MB. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000; 404:613–17.
56. Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY, Qin J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002; 16:571–82.
57. Saito S, Goodarzi AA, Higashimoto Y, Noda Y, Lees-Miller SP, Appella E, Anderson CW. ATM mediates phosphorylation at multiple p53 sites, including Ser(46), in response to ionizing radiation. J Biol Chem. 2002; 277:12491–94.
58. Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000; 1:179–86.
59. Choi HK, Ryu H, Son AR, Seo B, Hwang SG, Song JY, Ahn J. The novel anthraquinone derivative IMP1338 induces death of human cancer cells by p53-independent S and G2/M cell cycle arrest. Biomed Pharmacother. 2016; 79:308–14.
60. Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996; 274:1664–72.
61. Radhakrishnan SK, Feliciano CS, Najmabadi F, Haegebarth A, Kandel ES, Tyner AL, Gartel AL. Constitutive expression of E2F-1 leads to p21-dependent cell cycle arrest in S phase of the cell cycle. Oncogene. 2004; 23:4173–76.
62. Shenberger JS, Dixon PS. Increased p21 and p53 levels are associated with S phase growth arrest in hyperoxia-treated human bronchial smooth muscle cells. Pediatr Res. 1998; 43:335.
63. Shenberger JS, Dixon PS. Oxygen induces S-phase growth arrest and increases p53 and p21(WAF1/CIP1) expression in human bronchial smooth-muscle cells. Am J Respir Cell Mol Biol. 1999; 21:395–402.
64. Huang Z, Wang L, Chen L, Zhang Y, Shi P. Induction of cell cycle arrest via the p21, p27-cyclin E,A/Cdk2 pathway in SMMC-7721 hepatoma cells by clioquinol. Acta Pharm. 2015; 65:463–71.
65. He G, Siddik ZH, Huang Z, Wang R, Koomen J, Kobayashi R, Khokhar AR, Kuang J. Induction of p21 by p53 following DNA damage inhibits both Cdk4 and Cdk2 activities. Oncogene. 2005; 24:2929–43.
66. Bilodeau JF, Faure R, Piedboeuf B, Mirault ME. Hyperoxia induces S-phase cell-cycle arrest and p21(Cip1/Waf1)-independent Cdk2 inhibition in human carcinoma T47D-H3 cells. Exp Cell Res. 2000; 256:347–57.
67. Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J Clin Oncol. 2005; 23:9408–21.
68. Sampath D, Shi Z, Plunkett W. Inhibition of cyclin-dependent kinase 2 by the Chk1-Cdc25A pathway during the S-phase checkpoint activated by fludarabine: dysregulation by 7-hydroxystaurosporine. Mol Pharmacol. 2002; 62:680–88.
69. Hung FM, Chen YL, Huang AC, Hsiao YP, Yang JS, Chung MT, Chueh FS, Lu HF, Chung JG. Triptolide induces S phase arrest via the inhibition of cyclin E and CDC25A and triggers apoptosis via caspase- and mitochondrial-dependent signaling pathways in A375.S2 human melanoma cells. Oncol Rep. 2013; 29:1053–60.
70. Tu YS, Kang XL, Zhou JG, Lv XF, Tang YB, Guan YY. Involvement of Chk1-Cdc25A-cyclin A/CDK2 pathway in simvastatin induced S-phase cell cycle arrest and apoptosis in multiple myeloma cells. Eur J Pharmacol. 2011; 670:356–64.
71. Liu YJ, Wang XG, Tang YB, Chen JH, Lv XF, Zhou JG, Guan YY. Simvastatin ameliorates rat cerebrovascular remodeling during hypertension via inhibition of volume-regulated chloride channel. Hypertension. 2010; 56:445–52.
72. Sørensen CS, Syljuåsen RG, Falck J, Schroeder T, Rönnstrand L, Khanna KK, Zhou BB, Bartek J, Lukas J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell. 2003; 3:247–58.
73. Xiao Z, Chen Z, Gunasekera AH, Sowin TJ, Rosenberg SH, Fesik S, Zhang H. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem. 2003; 278:21767–73.
74 Bravo R, Frank R, Blundell PA, Macdonald-Bravo H. Cyclin/PCNA is the auxiliary protein of DNA polymerase-delta. Nature. 1987; 326:515–17.
75. Dillehay K, Dong Z, Tan Z, Wortman M. Identification of pcna targeting compounds for cancer therapy and pcna function regulation. Google Patents. 2012: WO2012033938 A2
76. Punchihewa C, Inoue A, Hishiki A, Fujikawa Y, Connelly M, Evison B, Shao Y, Heath R, Kuraoka I, Rodrigues P, Hashimoto H, Kawanishi M, Sato M, et al. Identification of small molecule proliferating cell nuclear antigen (PCNA) inhibitor that disrupts interactions with PIP-box proteins and inhibits DNA replication. J Biol Chem. 2012; 287:14289–300.
77 . Qin J, Zhu J, Jiang C, Cheng X, Ba Y, Fan Q, Zhuang D, Cui L. Effect of fluoride on expression of proliferating cell nuclear antigen in germ cells of rats’ testes. Zhonghua Difangbingxue Zazhi. 2005; 24:520–22.
78. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–08.