
INTRODUCTION
Ulcerative colitis (UC) and Crohn’s disease (CD) are collectively termed inflammatory bowel disease (IBD). In patients with IBD, long-term exposure to chronic inflammation is the primary risk factor for colorectal cancer development as analogous to liver cancer in chronic hepatitis and gastric cancer in chronic atrophic gastritis [1–3]. Malignancies in colorectum, anus, small intestine, or intestinal fistula which developed in IBD are described as colitis-associated cancer (CAC) or colitic cancer. In a meta-analysis, quantitative estimates of CAC risk in UC patients have been reported to be 2% after 10 years, 8% after 20 years, and 18% after 30 years of disease [4] and CAC risk was highly concordant with the location and extent of UC disease, with an incidence ratio of 1.7 for proctitis, 2.8 for left-sided colitis, and 14.8 for pan-colitis [5], which supports the strong association between chronic inflammation and cancer development. Similarly, CD increases the cumulative risk of CAC to 2.9% for 10 years, 5.6% for 20 years, and 8.3% for 30 years of disease [6]. Now intensive colonoscopic surveillance is clinically recommended to screen for CAC or its precursor dysplasia in patients suffering from longstanding IBD every 1–2 years [7], and cohort studies have demonstrated improved survival in patients with IBD undergoing colonoscopy [8].
The causal and clinical relationship between chronic inflammation in IBD and CAC is well established, but the molecular understanding of how chronic inflammation in IBD leads to CAC development is still incomplete. Sporadic colorectal cancer (CRC) is much better understood in terms of the molecular mechanism as explained by the adenoma-carcinoma sequence [9]. In this model, mutation of APC initiates adenoma, and subsequent mutations of KRAS and TP53 progressively lead to invasive carcinoma. However, this model for sporadic CRC has been considered not to be concordant with CAC development. Mutation of APC was reported to be much less frequent in CACs, whereas somatic mutation of TP53 was more prevalent and observed even in premalignant lesions [10]. A current model for CAC development is proposed to be the inflammation-dysplasia-carcinoma sequence although somatic alterations that advance the sequence are largely unclear [11].
The innovation of DNA sequencing technologies enabled cataloging somatic alterations in cancer comprehensively as already demonstrated by several cancer genome projects [12–15]. Two recent studies reported genomic analyses of 31 and 47 CAC samples, respectively [16, 17]. These studies found several recurrent mutations, including prevalent somatic mutations of TP53 and less frequent somatic mutations of APC and KRAS. However, the previous studies may have missed low-frequency driver mutations because of their limited sample size.
Here we performed targeted sequencing of total 90 CACs, following exome sequencing of 22 frozen fresh CACs. We found that RNF43 was somatically mutated in 11% of the CACs and the mutations preferentially occurred in patients with longer duration of IBD. RNA-Seq analysis showed that RNF43-mutated CACs had elevated expression of c-Myc and its target genes. Our study indicates that somatic mutation of RNF43 is the molecular link between chronic inflammation and cancer development in approximately 10% of CAC cases.
RESULTS
Somatic mutations of RNF43 were recurrently found in CAC
In order to search for driver genes of CAC development, we employed a two-step approach. The first step was exome sequencing of 22 CACs and enumeration of recurrently mutated genes. As the second step, we performed targeted sequencing of these genes and analyzed their somatic mutations in 90 CACs.
As the first step of our search, we collected 22 frozen samples of this rare type tumors with UC, and we performed exome sequencing of their fresh frozen tumor and normal colon tissues. Because the frozen tissues had low tumor purity, we sequenced the tumor exomes more deeply than the typical study. The average depth in the target region was 144-fold for tumor and 69-fold for normal tissues (Supplementary Table 3). Somatic single-nucleotide variants (SNVs) and short insertion/deletions (INDELs) were called as differences of tumor exomes from normal exomes by using VarScan2 [18]. One sample (RK378) was hypermutated, having 428 SNVs and 367 INDELs (Supplementary Figure 1A). This sample had a somatic missense mutation in a DNA mismatch repair gene MLH1 (p.Ser295Ile). The other 21 samples had 72.2 SNVs and 10.2 INDELs on average (Supplementary Table 3). Somatic SNVs and INDELs in the 22 CACs affected 2112 genes (median 78 genes per patient, Supplementary Table 4). Among them, 262 genes were recurrently mutated. The 30 most frequently mutated genes included known driver genes TP53, APC, KRAS and SMAD4 (Supplementary Figure 1B). This result suggests that these recurrently mutated genes are involved in CAC development and worth extensive investigation.
As the second step of our search, we performed targeted sequencing of these recurrently mutated genes and analyzed their somatic mutations in a cohort of increased size. We collected FFPE specimen from 90 CAC tissues, of which 58 are associated with UC and 32 are associated with CD (Table 1) and dissected tumor cells to increase tumor purity for these FFPE samples. Fifteen of the 58 UC patients were identical to the patients analyzed by the exome sequencing. We selected 43 genes that had recurrent somatic mutations in exome analyses of this study and a previous study [16] as the target of sequencing. The targeted genes are listed in Supplementary Table 5. The average sequence depth in the target region was 496-fold (Supplementary Table 6). Somatic SNVs and INDELs were called by comparing tumor reads with the reference human genome and filtering common variants in the dbSNP database. All variants were examined by capillary sequencing of normal DNA, and the germline variants were filtered out. As a result, 144 somatic SNVs and 23 somatic INDELS were detected (Supplementary Table 7). The number of somatic variants per sample range from 0 to 8 (mean 1.86), and at least one somatic variant was detected in 69 of 90 samples. The somatic variants were found in 33 of 43 target genes. The most commonly mutated gene was TP53, affecting 59 of 90 patients (66%, Figure 1). Other frequently mutated genes were APC, KRAS, SMAD4, RNF43, and LTBP4 (affecting 14, 10, 10, 10 and 5 patients, respectively). We examined the 33 genes that were somatically mutated in the 90 CACs for their association with tumor stages using the Wilcoxon rank sum test. However, no gene had statistically significant association with tumor stages after the Benjamini-Hochberg correction for multiple hypothesis testing.
Table 1: Summary of clinical features
Underlying disease |
||
UC |
58 (64%) |
|
CD |
32 (36%) |
|
Sex |
||
Male |
56 (62%) |
|
Female |
34 (38%) |
|
Age at cancer diagnosis |
||
Median (range) |
49 (27–79) |
|
Age at IBD onset |
||
Median (range) |
28 (11–67) |
|
Duration of IBD |
||
Median (range) |
17.8 yrs (0.2–40.5) |
|
Stage |
||
0 |
4 (4%) |
|
I |
13 (14%) |
|
II |
36 (40%) |
|
III |
26 (29%) |
|
IV |
6 (7%) |
|
NA |
5 (6%) |
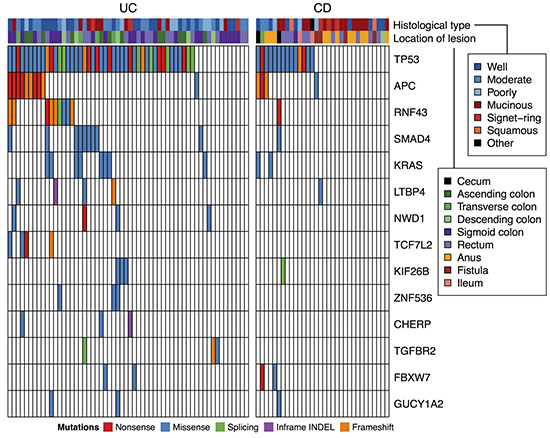
Figure 1: Recurrent somatic mutations in targeted sequencing of 90 CACs. Genes mutated in 3 or more cases were shown.
Interestingly, somatic mutations of RNF43 were frequently observed (10 of 90 patients; 11%). RNF43 negatively regulates the Wnt signaling pathway through its E3 ubiquitin ligase activity against frizzled receptors [19]. Frequent mutations of RNF43 has been reported in sporadic CRC, endometrial cancer, gastric adenocarcinoma, and intraductal papillary mucinous neoplasms of the pancreas [13, 20, 21]. LTBP4 (latent transforming growth factor beta binding protein 4) is an extracellular protein that regulates TGF-β bioavailability [22]. Mutations in LTBP4 are common in recurrent glioblastoma, and silencing of LTBP4 impairs the proliferation of glioma cell lines [23]. To examine the function of LTBP4 in colorectal cancer, we knocked down LTBP4 expression in a colon cancer cell line SW480 by siRNA and performed an invasion assay. Silencing of LTBP4 promoted invasion and migration of the cells (Supplementary Figure 2), suggesting that LTBP4 plays a role also in colorectal cancer.
Comparison of somatic mutations between CAC and sporadic CRC
We compared the frequency of somatic mutations between CAC and sporadic CRC (Figure 2A). APC had the largest difference, whose somatic mutation was found in 58% of sporadic CRCs but only in 16% of CACs. Other commonly mutated genes in sporadic CRCs, including KRAS, PIK3CA and BRAF, were less mutated in CACs. The only exception was TP53, which was more mutated in CAC (66%) than sporadic CRC (52%). Most of the somatic mutations of TP53 were missense mutations (38 of 60 mutations) and occurred in the DNA-binding domain, especially at the hotspot codons 175, 248, 273 and 282 (Figure 2B). A previous study reported that mutation distribution of TP53 was different between CAC and sporadic CRC [16]. We compared the types and distribution of TP53 mutations between CAC and sporadic CRC but were not able to find a significant difference between them.
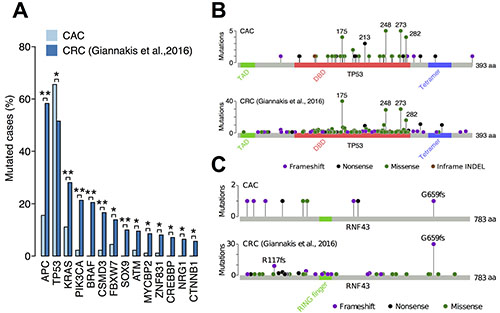
Figure 2: Comparison of somatic mutations between CAC and sporadic CRC. (A) The frequency of somatic mutations in 90 CACs and 619 sporadic CRCs. Only genes that were captured in both studies and had a significant difference between them were shown. **q-value < 0.01; *q-value < 0.05 using Fisher’s exact test and the Benjamini-Hochberg procedure. (B) Distribution of somatic TP53 mutations in CAC and sporadic CRC. TAD, transactivation domain; DBD, DNA-binding domain; Tetramer, tetramerization domain. (C) Distribution of somatic RNF43 mutations in CAC and sporadic CRC.
In contrast, distribution of somatic RNF43 mutations was considerably different between CAC and sporadic CRC (Figure 2C). A large fraction of RNF43 mutations in sporadic CRCs was frameshift INDELs at codons 659 and 117 (41 of 90 mutations). In contrast, somatic mutations of RNF43 in CAC distributed differently, having only one mutation at the codon 659. This indicated that mutation mechanism of RNF43 in CAC is different from that in sporadic CRC. A previous study found that most RNF43 mutations in sporadic CRCs occur in cancers with high microsatellite instability (MSI) [20]. Therefore, somatic mutations of RNF43 in CAC may arose from a mechanism different from MSI.
Comparison of somatic mutations between UC and CD
Somatic mutations of our target genes were more frequent in UC-CAC than CD-CAC (Figure 1). TP53 was mutated in 14 of 32 CDs (44%) and 45 of 58 UCs (78%; p < 0.01 by Fisher’s exact test). Likewise, samples with no somatic mutation among the 43 target genes were significantly more frequent in CD (16 of 32; 50%) than in UC (5 of 58; 9%; p < 10−4 by Fisher’s exact test). Because the target genes were selected based on the exomes of UC-CAC, we were not able to conclude that the mutation burden was lower in CD-CAC compared to UC-CAC. However, these mutations could be used for stratification of CD-CACs.
Actually, we noticed that somatic mutations in our target genes was inversely associated with a specific subtype of CD-CAC. Asian patients with CD have a unique tendency to develop mucinous adenocarcinoma in anal canal and hemorrhoid fistula [24, 25]. Histology of such CD-CACs in this study was shown in Supplementary Figure 3A. We noticed that mucinous CD-CACs in the anus were less likely to have somatic mutations in our target genes (Figure 1). Among the 16 CD-CACs without somatic mutation in our target genes, 6 cases (38%) were mucinous carcinomas developed in the anus. In contrast, among the 16 CD-CACs with somatic mutations in our target genes, none was mucinous carcinomas in the anus (0%, p < 0.05 by Fisher’s exact test). To search for somatic driver mutations of the 6 mucinous CD-CACs in the anus, we performed their exome sequencing. We also exome-sequenced 4 other CD-CACs without somatic mutation in our target genes (2 signet ring carcinomas in the anus and 2 mucinous carcinomas in the rectum). To increase the tumor cell fraction, we dissected cancer cells from FFPE tissues before DNA extraction. A search for recurrently mutated genes showed that fourteen genes (ATRNL1, EPB41L3, GLI3, LOXL3, MROH2B, MUC3A, OTUD7B, RHOBTB1, RUNX2, SLC47A2, SLC6A3, TYMP, UNC13A, and ZDBF2) were somatically mutated in 2 of 10 samples. But well-known cancer genes rarely had somatic mutations except one TP53 mutation of low variant allele frequency and one RB1 mutation. We also analyzed somatic copy number alteration but no recurrent alteration was identified. Therefore, driver mutations of the mucinous CD-CACs in the anus remained largely unclear.
A prognostic analysis showed that patients with CD had significantly poor overall survival than patients with UC (Supplementary Figure 3B). A multivariate analysis using Cox proportional hazards model showed that overall survival in this cohort was largely determined by tumor stage (hazard ratio 2.5, 95% CI 1.7–3.8) and underlying disease (UC or CD; hazard ratio 5.9, 95% CI 2.2–16.0).
Somatic mutation of RNF43 was associated with chronic inflammation
To infer driver mutations that arose from chronic inflammation, we compared somatic mutations and clinico-pathological features of our CACs (Figure 3A). The duration of IBD is an index of chronic inflammation and one of the highest risk factor for CAC development [4]. We found that RNF43-mutated patients had longer duration of IBD than non-mutated patients (mean 328 months and 215 months, respectively; p < 0.01 by Wilcoxon rank sum test) (Figure 3B). The duration rather than age was relevant because somatic mutations of RNF43 did not associate with neither age at IBD onset nor age at cancer diagnosis (Supplementary Figure 4). This result raises the possibility that chronic inflammation is related with the somatic mutations of RNF43.
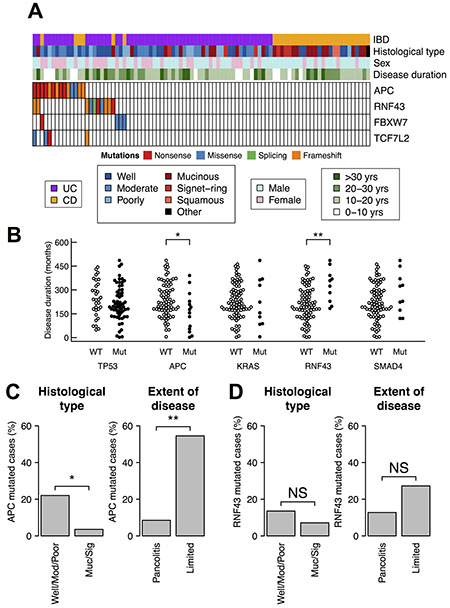
Figure 3: Somatic mutations of APC and RNF43 and clinico-pathological features. (A) Somatic mutations of APC, RNF43, and other two genes related to the Wnt pathway. (B) Somatic mutations and the duration of IBD. The statistical test was performed using Wilcoxon rank sum test. (C, D) Association of somatic mutations with histological type and the extent of disease. The extent of disease was available only in 58 UC cases. The statistical test was performed using Fisher’s exact test. (C) APC mutations. (D) RNF43 mutations. **p-value < 0.01; *p-value < 0.05.
We also observed an inverse correlation between somatic mutations of APC and the duration of IBD. APC-mutated patients had shorter duration compared to non-mutated patients (mean 163 months and 239 months, respectively; p < 0.05 by Wilcoxon rank sum test) (Figure 3B), indicating that somatic mutations of APC were less related to chronic inflammation. Sporadic CRC can develop in patients of IBD, and discriminating such sporadic CRC from CAC is occasionally difficult. Our samples in this study may also contain sporadic CRCs in part. Because APC is the most frequently mutated gene in sporadic CRC, the inverse correlation suggests that many, if not all, of the APC-mutated CACs were actually sporadic CRCs.
This notion was further supported by two other clinico-pathological features. Firstly, mucinous adenocarcinoma and signet-ring cell carcinoma are known to be more frequent in CAC than in sporadic CRC [26, 27]. Secondly, the extent of IBD is reported to be a strong risk factor for CAC [5]. In our samples, APC was less frequently mutated in mucinous adenocarcinoma and signet-ring cell carcinoma (1 of 28; 4%) than in well, moderately, or poorly differentiated adenocarcinomas (13 of 59; 22%; p < 0.05 by Fisher’s exact test) (Figure 3C). Furthermore, APC was less frequently mutated in CACs originated from pancolitis (4 of 47; 9%) than in CAC with limited colitis (6 of 11; 55%; p < 0.01 by Fisher’s exact test).
Conversely, RNF43 did not have these two trends (Figure 3D). These results suggest that, although both RNF43 and APC are negative regulators of the Wnt signaling, only somatic mutation of RNF43 is associated with chronic inflammation of CACs.
Somatic mutation of RNF43 was associated with higher activity of c-Myc
To examine whether somatic mutations of RNF43 were associated with aberrant mRNA expression in CACs, we performed transcriptome profiling. Among the 22 fresh frozen UC-CACs, 17 tissues yielded high quality RNAs, and these RNAs were subjected to RNA-Seq analysis. A search for gene fusion using the RNA-Seq data identified a fusion between GOLIM4 and RSPO3 (R-Spondin 3) in one patient CC022 (Figure 4B–4D). RSPO3 activates the Wnt pathway by promoting membrane clearance of RNF43 and its functional homolog ZNRF3 [28]. Therefore, the RSPO3 fusion may attenuate RNF43 and activate the Wnt pathway as reported in sporadic CRC [29].
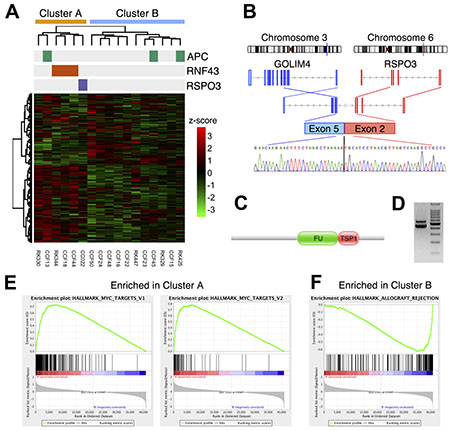
Figure 4: (A) Two-way clustering of transcriptome profiles in 17 UC-CAC samples with an annotation for somatic mutations. The clustering was performed by applying Ward’s method to log10(1+FPKM) values. (B–D) Gene fusion of GOLIM4 and RSPO3. (B) A schematic gene structure. (C) Domain structure of the fusion protein. FU: Furin-like repeat; TSP1: Thrombospondin type-1 (TSP1) repeat. (D) RT-PCR of the junction site. A chromatogram of capillary sequencing for the lower band is shown at the bottom of (B). (E, F) Gene set enrichment analysis. (E) Two gene sets regulated by MYC were more expressed in the cluster A than the cluster B. (F) A gene set involved in allograft rejection were more expressed in the cluster B than the cluster A.
Hierarchical clustering of the mRNA expression levels separated the 17 CACs into two clusters A and B, which consisted of 6 and 11 samples, respectively (Figure 4A). We found that the cluster A had significantly more somatic mutations in the Wnt pathway than the cluster B (5 of 6 in the cluster A; 2 of 11 in the cluster B; p < 0.05 by Fisher’s exact test). Three RNF43 mutations belonged to the cluster A in conjunction with one APC mutation and the RSPO3 fusion. The coherence of three RNF43 mutations (and one RSPO3 fusion) was intriguing because this suggested that these mutations affected common biological pathways in different patients.
To search for the biological pathways affected by the somatic mutations of RNF43, we analyzed differential gene expression between the clusters. The gene set enrichment analysis [30] showed that genes related to transplant rejection (including CD4, CD8A, GZMA and HLA class II) were highly expressed in the cluster B (q-value = 0.005; Figure 4F), suggesting intense infiltration of immune cells in the cluster B. Conversely, MYC and its target genes were expressed more in the cluster A than the cluster B (q-values < 0.05; Figure 4E). Considering the known regulatory relationship between RNF43, the Wnt/β-catenin signaling and c-Myc, the observed elevation of the c-Myc expression was consistently explained as a consequence of somatic mutations of RNF43, although contribution of many other mutations could not be ruled out. This result suggests that somatic mutations of RNF43 functioned as driver events of CAC through the activation of the Wnt/β-catenin signaling and its downstream c-Myc signaling.
Pattern of base substitutions in CAC
To gain insight into the mechanism of somatic mutation in CAC, we analyzed the patterns of base substitutions in the exome data of the fresh frozen tissues. Among the six possible patterns of base substitutions, the most frequent was C-to-T transitions, constituting 716 of 1517 SNVs (47%) in the 21 non-hypermutated CACs. We then analyzed the trinucleotide context of base substitutions. Among the 716 C-to-T transitions, 450 occurred in the CpG dinucleotide (63%, Figure 5A). It is commonly considered that C-to-T transitions in the CpG context is the result of spontaneous deamination of 5-methylcytosine. To corroborate this possibility, we compared somatic mutation rate at CpG dinucleotides with their methylation levels in the ENCODE sigmoid colon data. Methylation levels of CpG sites correlated positively with their C-to-T mutation rate in CAC (p < 0.001 by the Cochran–Armitage trend test; Figure 5B), further supporting a large contribution by 5-methylcytosine deamination to mutations in CAC. However, these observations were not specific to CAC. Three independent data sets of sporadic CRC also had similar trends (11 exomes sequenced in this study, 500 exomes published by Giannakis et al. [31], and 209 exomes by the TCGA project [12]) (Figure 5A, 5B).
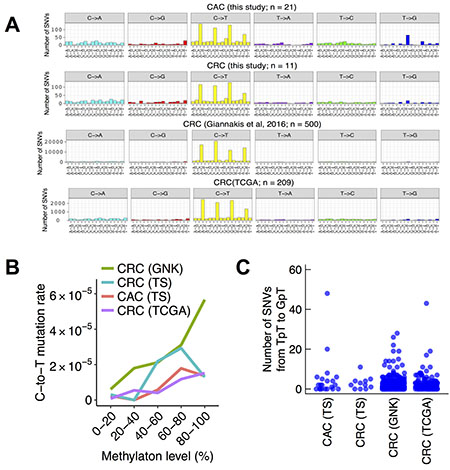
Figure 5: Trinucleotide context of base substitutions in CACs and sporadic CRCs. (A) Number of trinucleotide substitution patterns in non-hypermutated colorectal cancers. Six possible substitutions from pyrimidine bases were further subdivided into 96 patterns based on the neighboring nucleotides. The four panels represent 21 CACs, 11 sporadic CRCs sequenced in this study, 500 sporadic CRCs in a previous study, and 209 sporadic CRCs in TCGA. (B) Methylation levels and somatic C-to-T mutation rate at CpG dinucleotides. Methylation levels were obtained from the ENCODE sigmoid colon data. (C) The number of SNVs from TpT to GpT in CAC and sporadic CRC. TS, this study; GNK, Giannakis et al., 2016.
A previous study reported that T-to-G transversions in the CpTpT context, which is a common mutation pattern in esophageal and gastric adenocarcinomas [13, 14], showed an excess in CAC but not in sporadic CRC [16]. Consistently with the previous observation, we found an excess of somatic SNVs from TpT to GpT in CAC but not in sporadic CRC (Figure 5A). Most of the 110 somatic SNVs from TpT to GpT were contributed by 2 of 21 CAC samples (48 and 20 SNVs by CC022 and RK447, respectively; Figure 5C). In contrast, sporadic CRC rarely had 20 or more somatic SNVs from TpT to GpT (this study, 0 of 11 samples; Giannakis et al, 3 of 500 samples; TCGA, 1 of 209 samples).
DISCUSSION
In this study, we searched for recurrently mutated genes in 90 CACs employing a two-step approach. We first compiled a list of genes using the exome sequencing of 22 fresh frozen CACs and then performed targeted sequencing of 90 CAC archive specimens. We observed recurrent somatic mutations of TP53, APC, KRAS and SMAD4, consistently with previous reports [16, 17]. We also confirmed the previous findings that TP53 was commonly mutated whereas the other three genes were less frequently mutated. This highlights the difference of CAC from sporadic CRC. In addition to these core genes, we also found that RNF43 and LTBP4 were somatically mutated in 11% and 6% of cases, respectively. A previous study of CAC also reported somatic mutations of RNF43 in 6% of cases (3 of 47) [17].
Clinico-pathological analysis revealed that APC-mutated CACs were more likely to be sporadic CRCs. CAC tends to originate from pancolitis and more likely to show mucinous or signet-ring cell phenotypes compared to sporadic CRC [26, 27, 32]. However, somatic mutations of APC negatively associated with these features (Figure 3C). Moreover, APC-mutated CACs had significantly shorter duration of IBD than APC-wildtypes (Figure 3B). These data indicate that many of the APC-mutated CACs were sporadic CRC arose in the IBD background, although all the evidence is relative not absolute and there is always chance that these are indeed CACs.
In contrast to APC-mutated CACs, RNF43-mutated CACs did not resemble sporadic CRC. Somatic mutations of RNF43 did not show bias toward sporadic CRC (Figure 3D). The distribution of mutated sites was also different between CAC and sporadic CRC (Figure 2C). More importantly, somatic mutations of RNF43 correlated with the duration of IBD, indicating a relationship between chronic inflammation and the mutations (Figure 3B). RNA-Seq analysis showed that RNF43-mutated CACs belonged to a distinct subtype, in which target genes of c-Myc were upregulated. Considering these results, we propose that RNF43 is the driver gene of CAC development in about 10% of cases.
The risk of CAC increases over the duration of IBD, but the molecular mechanism behind this phenomenon has been unclear. A likely explanation is that chronic inflammation somehow causes somatic alterations of genome, and some of the alterations subsequently drive CAC development. Therefore, which gene is altered by chronic inflammation and drives CAC is an important question for understanding CAC. An attractive candidate would be the gene that is frequently mutated only after the long duration of IBD, and RNF43 appeared to have this association (Figure 3B). Therefore, we propose a hypothesis that chronic inflammation of IBD somatically mutates RNF43 and the mutation drives CAC development, thereby increasing the CAC risk over disease duration. However, we have to admit that our hypothesis has two downsides. First, this hypothesis accounts for only 10% of CAC cases. Other mechanisms may operate on the remaining cases. Secondly, the association between mutations and disease duration alone is not conclusive evidence that somatic mutation of RNF43 is sufficient to drive CAC development. Further studies of RNF43 mutations, especially in premalignant mucosa, will be required to clarify the relationship between chronic inflammation, somatic mutation of RNF43, and CAC development.
LTBP4 was the another recurrently mutated gene in this study. To the best of our knowledge, somatic mutations of LTBP4 has not been linked to colon cancer. In particular, we confirmed that silencing LTBP4 promotes invasion of a colon cancer cell line. In conjunction with LTBP4, genes in the TGFβ pathway (SMAD4, SMAD2, TGFBR2, ACVR1B and LTBP4) were somatically mutated in 19 of 90 patients (21%), suggesting that this pathway also contributes significantly to CAC development.
This study could not detect any recurrent driver mutation in a half of CD-CACs. In Japan, common type of CD-CACs are mucinous carcinoma in anal canal and hemorrhoid fistula [24, 25] and unknown genomic alterations could affect this distinct CD-CAC phenotype specific in Asia. We found that 14 genes were recurrently mutated in these CD-CACs, but unknown other genetic or epigenetic events could drive the carcinogenesis in this type of CACs in Asian CD disease. Overall, genetic events of CAC appear to have heterogeneity depending on the background disease, histology and ethnicity.
The patterns of base substitutions provide an insight into mutation mechanisms in cancer. A previous study analyzed FFPE samples of CAC and showed that base substitutions in CAC were dominated by C-to-T transitions at the CpG dinucleotides, indicating the effect of spontaneous deamination of 5-methylcytosine [16]. The authors also reported a slight excess of SNVs from TpT to GpT, resembling esophageal and gastric adenocarcinomas. We analyzed the patterns of base substitutions in the exome sequences of 21 fresh frozen non-hypermutated CACs, which had no bias of FFPE, and obtained essentially the same result to the previous study. However, the substitution patterns in CAC were largely similar to that of sporadic CRC. Although the excess of SNVs from TpT to GpT was specific to CAC, the excess was contributed by only a part of samples (2 of 21). We concluded that the mutation patterns of most CAC samples were identical to that of sporadic CRC.
This study provides a detailed profile of somatic mutations in 90 CACs. The profile revealed that RNF43 was mutated and functioned as a cancer driver in about 10% of cases. The profile also confirmed that somatic mutations of APC could be informative for discriminating CAC from sporadic CRC in patients with IBD. These findings may have diagnostic and therapeutic utility for the treatment of CAC.
MATERIALS AND METHODS
Clinical samples
Fresh-frozen cancer tissues and normal tissues from UC patients (n = 22) and sporadic colorectal cancers without IBD background (n = 11) in Hyogo College of Medicine Hospital underwent exome and RNA-Seq. The Institutional Review Boards at RIKEN and Hyogo College of Medicine approved this work. All subjects agreed with informed consent to participate in the study following ICGC guidelines. Their clinico-pathological features are in Supplementary Table 1. DNA and RNA were extracted from fresh-frozen tumor specimens and adjacent normal tissues. FFPE (formalin-fixed paraffin-embedded) tissues (n = 90) of CACs in Hyogo College of Medicine Hospital were sliced, and after reviewing their HE-stained slides, tumor regions and normal regions were dissected and DNAs/RNAs were extracted by Qiagen Allprep kit.
Whole exome and targeted sequencing
Exome capture was carried out using SureSelect XT Exome V5 Custom kit (Agilent Technologies). The exome-captured libraries were sequenced on HiSeq2000/2500 with paired reads of 100–125 bp. For DNAs from FFPE samples, we constructed sequencing libraries by using KAPA HyperPrep Plus kit. We selected 43 genes and performed target capturing by SureSelect XT Target Enrichment System. Targeted sequencing was performed on HiSeq2000/2500 with paired reads of 125 bp.
Variant calling
Reads were adaptor-trimmed using Cutadapt [33] and mapped to GRCh37 using BWA [34]. PCR duplicates were removed using Picard (https://broadinstitute.github.io/picard/). Low-quality reads were filtered based on mapping quality, number of mismatches and INDELs. Improper reads were filtered based on discordance in chromosome, direction and distance of paired-end reads. For the exome data of freshly frozen samples, somatic single-nucleotide variants (SNVs) and short insertions/deletions (INDELs) were called using VarScan2 [18] with a minimum read depth of 20, a minimum variant allele frequency of 5%, minimum supporting reads of 4, and a p-value threshold of 0.05. For the targeted sequencing data of FFPE samples, somatic variants were called as follows. The numbers of matches, mismatches and INDELs at each chromosomal position were counted while skipping bases of quality <20 and alignments of mapping quality <10 using SAMtools (version 1.3.1). Common variants in dbSNP version 138 were discarded. SNVs were called when mismatches had read depth >50, variant allele frequency >5%, base quality bias >0.01, mapping quality bias >0.01, and read position bias >0.01. INDELs were required to have read depth >50, variant allele frequency >10% and support reads >20. Variants were discarded if identical alleles were detected in more than 8 samples. Copy number analysis was performed using VarScan2 and DNAcopy.
RNA-Seq
RNA-Seq was carried out for 17 frozen CAC samples for which high-quality RNA was available. Total RNA was extracted by Qiagen Allprep kit from the frozen CACs and quantity were evaluated by Bioanalyzer (Agilent). The high-quality RNA was subject to polyA+ selection and chemical fragmentation, and 100–200 base RNA fraction was used to construct cDNA libraries according to Illumina’s protocol. RNA-Seq was performed on HiSeq2500 using the standard paired-end 125 bp protocol. RNA-Seq reads were adaptor-trimmed using Cutadapt [33] and mapped to GRCh37 using STAR [35]. Reads mapped onto exons were counted using featureCounts [36] and converted into FPKM-UQ. For biclustering of samples and genes, the FPKM-UQ values of the genes expressed in all of the 17 samples were log-transformed with pseudocount 1.0, scaled for each gene, and clustered using the Euclidean distance and Ward’s method implemented in R. Gene fusion was detected using fusionfusion (https://github.com/Genomon-Project/fusionfusion).
Knockdown experiments and invasion assays
A colon cancer cell line SW480 was used for knockdown experiments of a new target gene (LTBT4). The cell lines were obtained from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan). Cells were seeded on 24-well plates a day before transfection (1 × 105 cells/well for growth assay of LTBP4), and transfected with two pre-designed siRNAs or negative control SIC, respectively, using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. Sequence information of siRNAs are listed in Supplementary Table 2. The knockdown efficiency was assessed by quantitative RT-PCR using primers: 5′-GTCTC CAACGAGAGCCAGAG-3′ and 5′-GGCAGCAGCAC TCTGTGTAG-3′. For the cell proliferation assay, cells were seeded in 24-well plate format a day before transfection. The cell numbers in triplicate wells were assessed by water-soluble tetrazolium salt (WST) assay (Dojindo, Kumamoto Japan) 96 hours after siRNA transfection. Invasion assay was performed in 24-well modified Boyden chambers pre-coated with Matrigel (BD Transduction, Franklin Lakes, NJ, USA). Cells were transfected with siRNAs in 6-well plate format using Lipofectamine RNAiMAX according to the manufacturer’s instructions. A day after siRNA transfection, appropriate numbers of each cells were transferred into the upper chamber. Following 24 hours of incubation, the migrated or invasive cells on the lower surface of filters were fixed and stained with the Diff-Quik stain (Sysmex, Kobe, Japan), and stained cells were counted directly with three different field of microscopy. The numbers of viable cells in each condition were assessed by WST assay at the same time point and the numbers of cells on invasion or migration assay were normalized with the ratio of viability score in SIC transfectants. Differences between subgroups were tested by Student’s t-test and considered significant at the p-value < 0.05 level.
Author contributions
Study concept and design: NM, NT, HI and HN. Acquisition of clinical samples: NM, IM, TM, MT, SH, and HI. Acquisition of data: M. Fujita, KM, AO, K. Nakano, AOS, HT, SM, and HN. Analysis and interpretation of data: M. Fujita, NM, KM, AF, M. Furuta, YS, RM, K. Nakai, SM and HN. Pathological analysis: IM and SH. Drafting of the manuscript: M. Fujita and HN. Critical revision of the manuscript for important intellectual content: all co-authors. Obtained funding: HN. Study supervision: NM, HI and HN.
ACKNOWLEDGMENTS AND FUNDING
This work was partly supported by JSPS KAKENHI Grant Number JP15K15301 awarded to HN. The super-computing resource ‘SHIROKANE’ was provided by the Human Genome Center, The University of Tokyo (http://sc.hgc.jp/shirokane.html).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest associated with this manuscript.
REFERENCES
1. Axelrad JE, Lichtiger S, Yajnik V. Inflammatory bowel disease and cancer: The role of inflammation, immunosuppression, and cancer treatment. World J Gastroenterol. 2016; 22:4794–801.
2. Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007; 117:60–9.
3. Berasain C, Castillo J, Perugorria MJ, Latasa MU, Prieto J, Avila MA. Inflammation and liver cancer: new molecular links. Ann N Y Acad Sci. 2009; 1155:206–21.
4. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001; 48:526–35.
5. Ekbom A, Helmick C, Zack M, Adami HO. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990; 323:1228–33.
6. Canavan C, Abrams KR, Mayberry J. Meta-analysis: Colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment Pharmacol Ther. 2006; 23:1097–104.
7. Rutter MD, Saunders BP, Wilkinson KH, Rumbles S, Schofield G, Kamm MA, Williams CB, Price AB, Talbot IC, Forbes A. Cancer surveillance in longstanding ulcerative colitis: endoscopic appearances help predict cancer risk. Gut. 2004; 53:1813–6.
8. Ananthakrishnan AN, Cagan A, Cai T, Gainer VS, Shaw SY, Churchill S, Karlson EW, Murphy SN, Kohane I, Liao KP. Colonoscopy is associated with a reduced risk for colon cancer and mortality in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2015; 13:322–9.
9. Leslie A, Carey FA, Pratt NR, Steele RJC. The colorectal adenoma-carcinoma sequence. Br J Surg. 2002; 89:845–60.
10. Brentnall TA, Crispin DA, Rabinovitch PS, Haggitt RC, Rubin CE, Stevens AC, Burmer GC. Mutations in the p53 gene: an early marker of neoplastic progression in ulcerative colitis. Gastroenterology. 1994; 107:369–78.
11. Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol. 2004; 287:G7–17.
12. Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, Kovar CL, Lewis LR, Morgan MB, Newsham IF, Reid JG, Santibanez J, Shinbrot E, et al; Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–7.
13. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014; 513:202–9.
14. Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C, Bandla S, Imamura Y, Schumacher SE, Shefler E, McKenna A, Carter SL, Cibulskis K, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013; 45:478–86.
15. Fujimoto A, Furuta M, Totoki Y, Tsunoda T, Kato M, Shiraishi Y, Tanaka H, Taniguchi H, Kawakami Y, Ueno M, Gotoh K, Ariizumi S, Wardell CP, et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016; 48:500–9.
16. Robles AI, Traverso G, Zhang M, Roberts NJ, Khan MA, Joseph C, Lauwers GY, Selaru FM, Popoli M, Pittman ME, Ke X, Hruban RH, Meltzer SJ, et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease-Associated Colorectal Cancers. Gastroenterology. Elsevier, Inc; 2016; 150:931–43.
17. Yaeger R, Shah MA, Miller VA, Kelsen JR, Wang K, Heins ZJ, Ross JS, He Y, Sanford E, Yantiss RK, Balasubramanian S, Stephens PJ, Schultz N, et al. Genomic Alterations Observed in Colitis-Associated Cancers Are Distinct From Those Found in Sporadic Colorectal Cancers and Vary by Type of Inflammatory Bowel Disease. Gastroenterology. Elsevier, Inc; 2016; 151:278–87.
18. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012; 22:568–76.
19. Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, van Es JH, Mohammed S, Heck AJ, Maurice MM, Clevers H. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012; 488:665–9.
20. Giannakis M, Hodis E, Jasmine Mu X, Yamauchi M, Rosenbluh J, Cibulskis K, Saksena G, Lawrence MS, Qian ZR, Nishihara R, Van Allen EM, Hahn WC, Gabriel SB, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet. Nature Publishing Group; 2014; 46:1264–6.
21. Wu J, Jiao Y, Dal Molin M, Maitra A, de Wilde RF, Wood LD, Eshleman JR, Goggins MG, Wolfgang CL, Canto MI, Schulick RD, Edil BH, Choti MA, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci U S A. 2011; 108:21188–93.
22. Todorovic V, Rifkin DB. LTBPs, more than just an escort service. J Cell Biochem. 2012; 113:410–8.
23. Bass AJ, Thorsson V, Shmulevich I, Reynolds SM, Miller M, Bernard B, Hinoue T, Laird PW, Curtis C, Shen H, Weisenberger DJ, Schultz N, Shen R, et al; Cancer Genome Atlas Research Network. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016; 48:768–76.
24. Sasaki H, Ikeuchi H, Bando T, Hirose K, Hirata A, Chohno T, Horio Y, Tomita N, Hirota S, Ide Y, Tsuchida Y, Uchino M. Clinicopathological characteristics of cancer associated with Crohn’s disease. Surg Today. 2017; 47:35–41.
25. Higashi D, Katsuno H, Kimura H, Takahashi K, Ikeuchi H, Kono T, Nezu R, Hatakeyama K, Kameyama H, Sasaki I, Fukushima K, Watanabe K, Kusunoki M, et al. Current state of and problems related to cancer of the intestinal tract associated with Crohn’s disease in Japan. Anticancer Res. 2016; 36:3761–6.
26. Delaunoit T, Limburg PJ, Goldberg RM, Lymp JF, Loftus EV. Colorectal cancer prognosis among patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2006; 4:335–42.
27. Choi PM, Zelig MP. Similarity of colorectal cancer in Crohn’s disease and ulcerative colitis: implications for carcinogenesis and prevention. Gut. 1994; 35:950–4.
28. Hao HX, Jiang X, Cong F. Control of Wnt receptor turnover by R-spondin-ZNRF3/RNF43 signaling module and its dysregulation in cancer. Cancers (Basel). 2016; 8:1–12.
29. Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS, Guillory J, Ha C, Dijkgraaf GJP, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012; 488:660–4.
30. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102:15545–50.
31. Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, Sukawa Y, Stewart C, Rosenberg M, et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016; 15:857–65.
32. Rubin DT, Huo D, Kinnucan JA, Sedrak MS, McCullom NE, Bunnag AP, Raun-Royer EP, Cohen RD, Hanauer SB, Hart J, Turner JR. Inflammation is an independent risk factor for colonic neoplasia in patients with ulcerative colitis: A case-control study. Clin Gastroenterol Hepatol. 2013; 11:1601–8.
33. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011; 17:10–2.
34. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–60.
35. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21.
36. Liao Y, Smyth GK, Shi W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923–30.