
INTRODUCTION
Pancreatic cancer, one of the most lethal human cancers, is currently the fourth-most common cause of cancer-related death, and is estimated to be the second deadliest cancer by 2030 [1]. Recent increases in our understanding of cancer biology and the development of new treatment modalities, such as targeted therapy, immunotherapy and advanced radiologic techniques have helped to improve cancer survival. However, pancreatic cancer is still characterized by very poor prognosis with a 1 year overall survival rate of only about 10% and a 5-year overall survival rate of less than 5% [2]. Furthermore, no significant improvements in survival have been achieved during the last three decades (5-year survival, 4.4%), mainly owing to a lack of appropriate treatment modalities and proper methods for detecting pancreatic cancer in the early stage [3]. Less than 20% of patients are candidates for surgical resection; therefore, chemotherapy and radiation therapy remain the only treatment options [4].
To date, it has been estimated that, among cancer patients who have been cured, about 40% were treated with radiotherapy, with or without other modalities [5]. In the case of pancreatic cancer, patients with nonmetastatic pancreatic cancer benefit from the combination of radiation therapy and chemotherapy but radiation therapy in advanced-stage pancreatic cancer has remained in a matter of debate. In a Phase II study of induction chemotherapy followed by concurrent chemoradiotherapy in locally advanced pancreatic cancer patients, Kim et al. reported only modest survival gain [6]. This weak effect of radiation therapy in pancreatic cancer was mainly attributable to resistance to radiotherapy. The exact mechanism of radiation resistance has not yet been elucidated, but activity of the DNA damage-repair system is a problem that commonly underlies interruption of cancer radiotherapy. Accordingly, many researchers have investigated strategies for inhibiting DNA damage-response pathways. If radiation resistance could be overcome using radiosensitizers, it would enhance the efficacy of radiotherapy and increase survival, allowing initially unresectable primary tumors to be removed after radiotherapy in locally advanced pancreatic cancer. Therefore, new therapeutic options for overcoming radioresistance and enhancing radiotherapy are needed.
The phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) pathway plays a key role in cell growth, proliferation and survival and is known to be disrupted in many cancers, including pancreatic cancer. Radiation also activates the PI3K/AKT pathway, an effect that is associated with radiotherapy resistance. Inhibition of the PI3K/AKT pathway has been shown to increase radiosensitivity in various cancers including glioblastoma, non-small cell lung cancer, colorectal cancer and head and neck squamous cell carcinoma [7–10]. A number of studies have shown that PI3K inhibitors exert synergistic effects in combination with radiation, resulting in improved radiosensitization [11–13]. Additionally, PI3K/AKT targeting led to an inhibition of repair response of radiation-induced DNA-double strand breaks (DSBs) and subsequent enhancement of radiation sensitivity [14]. In particular, this enhanced radiosensitivity is at least partly attributable to catalytic inhibition of ATM and DNA-PKcs enzymes, the main mediators of the DNA damage-repair system [15]. Since inhibition of the PI3K/AKT pathway increases therapeutic efficacy and helps to overcome radioresistance, targeting the PI3K/AKT pathway is an effective strategy for improving radiotherapy in cancer treatment. We previously reported that HS-173, novel PI3K inhibitor, has shown therapeutic effect against pancreatic cancer cells and xenograft models [16–18]. Here, we determined whether HS-173 enhances radiation sensitivity and anti-cancer effect by inhibiting DNA damage-repair processes in human pancreatic cancer. We demonstrated that inhibition of the PI3K/AKT pathway by HS-173 blocked DNA damage-repair signaling through suppression of ATM and DNA-PKcs activation, leading to radiosensitization in vitro and in vivo.
RESULTS
The radiosensitizing effects of HS-173 on pancreatic cancer cells
To determine whether HS-173 might enhance radiosensitivity, we treated Miapaca-2 and PANC-1cells with HS-173, exposed them to different doses of radiation (2-6 Gy) for 24 h and then performed clonogenic assays. As shown in Figure 1, radiation caused a dose-dependent reduction in clonogenic survival in pancreatic cancer cells. Treatment with HS-173 for 24 h prior to irradiation resulted in a significant reduction in the clonogenic survival of pancreatic cancer cells at radiation doses of 2, 4, and 6 Gy, indicating that HS-173 induced synergic effects by enhancing the efficacy of radiation.
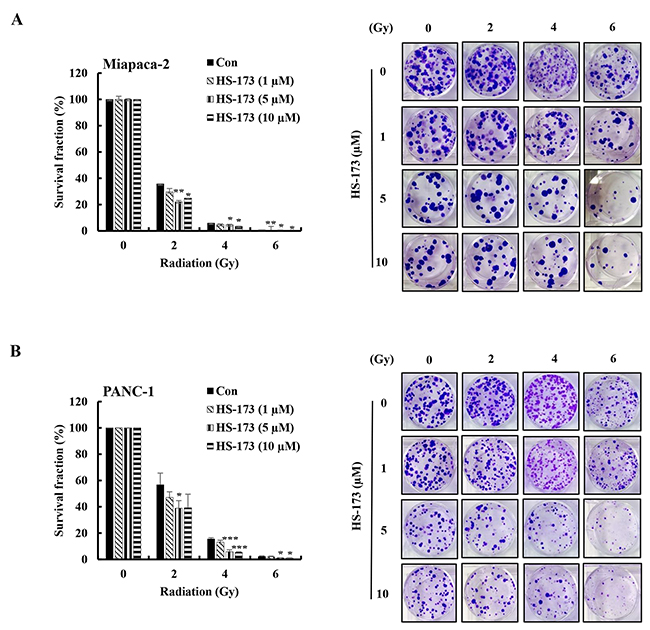
Figure 1: HS-173 significantly radiosensitizes pancreatic cancer cells. Miapaca-2 (A) and PANC-1 (B) pancreatic cancer cells were treated with different concentrations of HS-173 for 24 h and then irradiated with the indicated doses (0–6 Gy). After 2 h, media were changed and cells were processed for clonogenic survival assays at the end of experiments (10-14 days). Colonies were counted by eye, using a cut-off value of 50 viable cells per colony. Data are presented as means ± S.D. from triplicate experiments (*P < 0.05, **P < 0.01, and ***P < 0.001 vs. control).
Induction of apoptotic cell death by the combination of HS-173 and radiation
Our previous studies showed that HS-173 induces apoptotic cell death in various cancer cell types [16–18]. Because combined treatment with HS-173 and radiation significantly reduced cell survival, we next investigated the apoptotic effects of this combination in pancreatic cancer cells using TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) assays. For these experiments, we pretreated Miapaca-2 and PANC-1 cells with HS-173 (1 and 10 μM) for 6 h before irradiation, and then exposed the cells to radiation (10 Gy). In accord with clonogenic assays, radiation induced apoptotic cell death, as evidence by an increase in TUNEL-positive cells (Figure 2). Notably, combined treatment with HS-173 and radiation resulted in more apoptotic cell death compared with radiation alone. These results were confirmed by an assessment of apoptosis-related proteins, which showed that combined treatment with HS-173 and radiation increased the levels of cleaved caspase-3 and cleaved PARP, but decreased the expression levels of the anti-apoptotic survivin compared with radiation alone, demonstrating that the HS-173-induced increase in radiosensitivity resulted in an increase in apoptosis in pancreatic cancer cells.
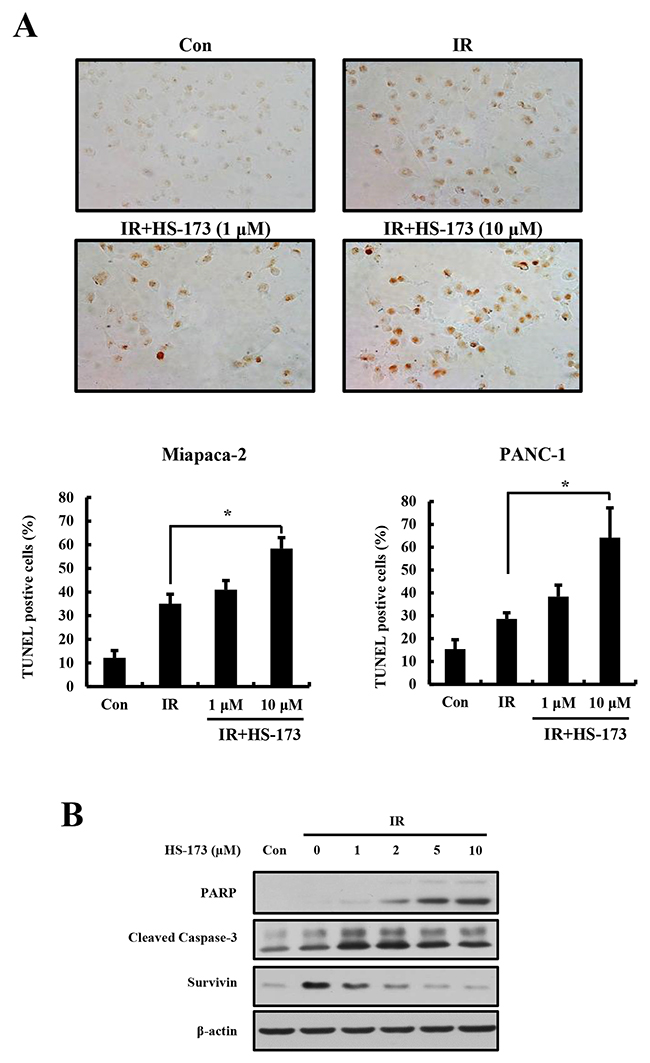
Figure 2: The combination of HS-173 and radiation synergistically induces apoptosis. (A) Miapaca-2 and PANC-1 cells were pretreated with HS-173 (0-10 μM) for 6 h and then irradiated (4 Gy). After 24 h, cells were fixed and analyzed for apoptotic cell death by TUNEL assay. (B) Miapaca-2 cells were treated with HS-173 for 6 h and then irradiated (10 Gy). After 24 h, cell lysates were prepared and analyzed by Western blotting for cleaved PARP, cleaved caspase-3, and survivin. Data are expressed as means ± S.D. from three experiments (*P < 0.05).
G2/M abrogation by the combination of HS-173 and radiation
To investigate the mechanisms by which the combination of HS-173 and radiation caused radiosensitization in pancreatic cancer cells, we analyzed the cell cycle distribution after combined treatment. On the basis of our previous study [16], we hypothesized that HS-173 would cause radiation-treated cells to accumulate in G2/M phase. As expected, treatment of Miapaca-2 cells with radiation for 24 h increased the G2/M phase cell population; when combined with HS-173, radiation induced a greater degree of G2/M arrest (91%) than radiation alone (75%) (Figure 3A). A further investigation showed that expression of p-Cdc2, which typically causes cell cycle arrest in the G2/M phase of cell cycle, was increased to a greater extent in these cells by combined treatment than by radiation alone (Figure 3B). Collectively, these results indicate that the combination of HS-173 with radiation exerted synergistic cell cycle effects, causing accumulation of cells in G2/M phase, with an attendant delay in cell division.
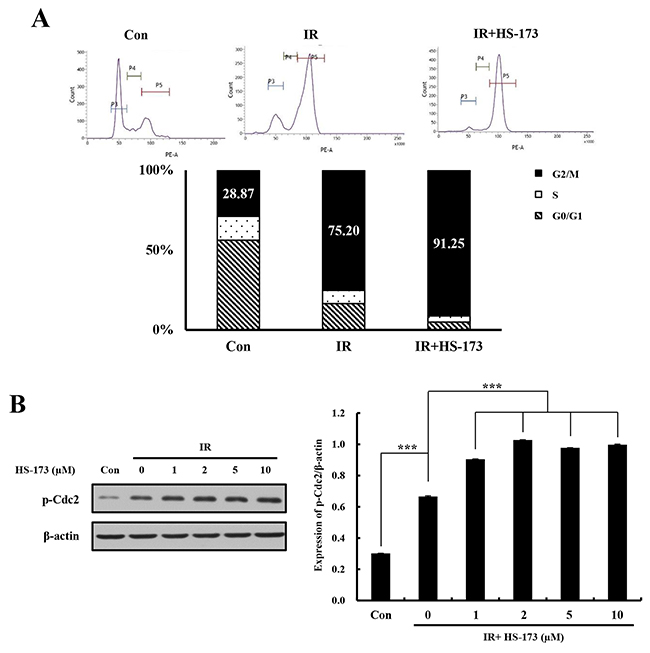
Figure 3: The combination of HS-173 and radiation increases G2/M arrest. (A) Miapaca-2 cells were treated with HS-173 (0-10 μM) for 6 h before irradiation (10 Gy). After 24 h, cells were stained with PI and analyzed by flow cytometry. Quantitative PI staining data is presented as a percentage of the cell cycle distribution. (B) Miapaca-2 cells were treated with HS-173 for 6 h before irradiation (10 Gy). After 24 h, p-Cdc2 levels were assayed by Western blotting. Data are expressed as means ± S.D. from three experiments (***P < 0.001).
Increased expression of γ-H2AX and inhibition of ATM by HS-173 in irradiated pancreatic cancer cells
Radiation has been shown to induce PI3K/AKT activation in many cell types, and activation of the PI3K/AKT signaling pathway is correlated with radioresistance [19–20]. Accordingly, we assessed activation of AKT by comparing the levels of phosphorylated AKT (p-AKT) in Miapaca-2 and PANC-1 pancreatic cancer cells after treatment with 5 and 10 Gy radiation. Immunofluorescence analyses using antibodies specific for p-AKT confirmed that radiation increased expression of p-AKT in both Miapaca-2 and PANC-1 cells, implicating AKT in the cellular response to radiation in pancreatic cancer cells (Figure 4A). ATM is a principal regulator of the DNA damage response following irradiation [21]. In particular, ATM is a major determinant of AKT activation in response to radiation [22]. Thus, we investigated whether HS-173 was capable of inhibiting radiation-induced activation of ATM and AKT. Miapaca-2 and PANC-1 cells were pretreated with HS-173 for 6 h and then the cells were incubated for 30 min after radiation treatment (10 Gy). As shown in Figure 4B, HS-173 significantly attenuated radiation-induced increases in the levels of p-AKT and p-ATM, indicating that HS-173 inhibited critical components of the DNA damage response in human Miapaca-2 and PANC-1 pancreatic cancer cells. In addition, HS-173 increased the expression of γ-H2AX, which is frequently used as a marker for DNA double-strand breaks (DSBs) following irradiation (Figure 4C).
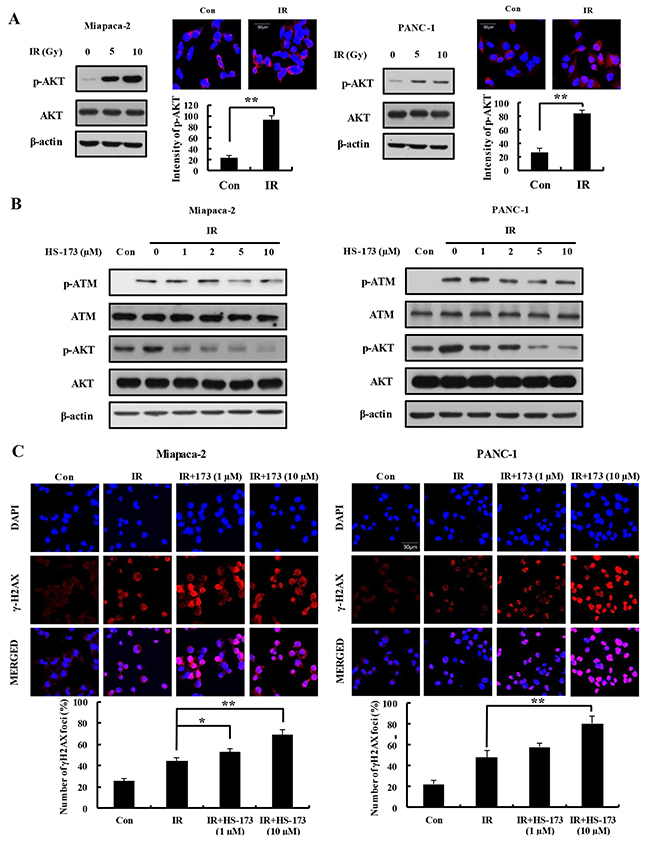
Figure 4: HS-173 increases expression of γ-H2AX through inhibition of ATM in irradiated pancreatic cancer cells. (A) Pancreatic cancer cells were irradiated with the indicated dose (5 and 10 Gy). In radiation condition, the high expression of p-AKT was observed in pancreatic cancer cells using Western blotting and immunoflorescence. (B) Miapaca-2 and PANC-1 cells were exposed to radiation (10 Gy) alone and/or together with different concentrations of HS-173 for 6 h. p-ATM and p-AKT levels were assessed by Western blotting. (C) Miapaca-2 and PANC-1 cells were pretreated with HS-173 for 6 h, irradiated (4 Gy) for 30 min, and then immunostained for γ-H2AX. γ-H2AX staining was quantified by analyzing the optical density of stained cells. Data are expressed as means ± S.D. from three experiments (*P < 0.05 and **P < 0.01).
Impairment of the DSB repair response by HS-173 through inhibition of DNA-PKcs in irradiated pancreatic cancer cells
Because PI3K/AKT signaling promotes DSB repair in various cancers primarily through processes involving DNA-PKcs [22–25], we assessed whether HS-173 radiosensitized human pancreatic cancer cells by inhibiting DNA repair. To this end, we treated Miapaca-2 and PANC-1 pancreatic cells with different concentrations of HS-173 for 6 h, harvested cells after 30 min exposure to radiation, and determined levels of DNA-PKcs, KAP1, and 53BP1 by Western blotting analysis. We found that HS-173 inhibited radiation-induced activation of DNA-PKcs in pancreatic cancer cells in dose-dependent manner, thereby blocking phosphorylation of the downstream substrates, KAP1, and 53BP1, a marker for DSBs (Figure 5A). Inhibition of KAP1 and 53BP1 phosphorylation was confirmed by immunostaining (Figure 5B). Collectively, these findings indicate that HS-173 inhibits DNA-PKcs and severely impairs the DSB repair response in irradiated pancreatic cancer cells.
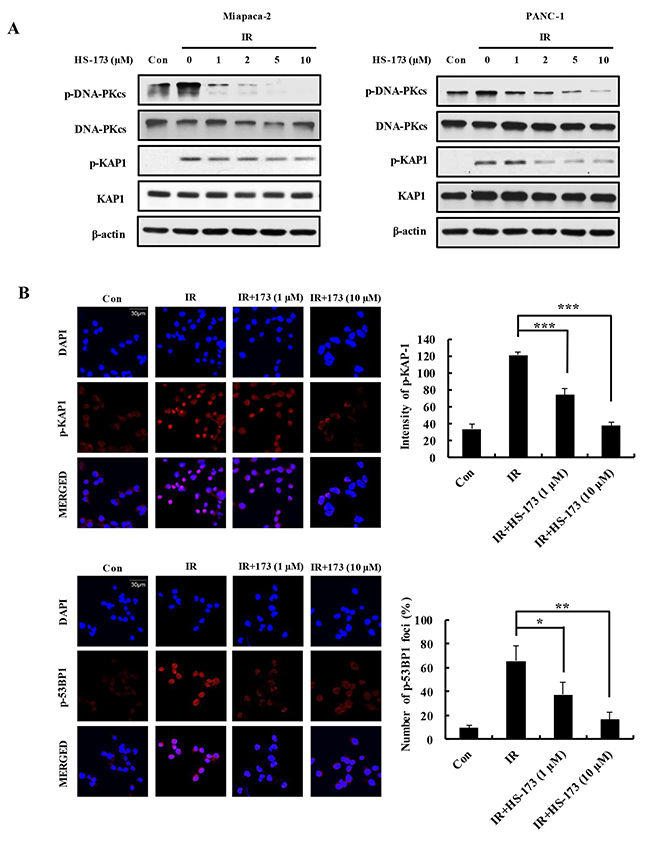
Figure 5: HS-173 impairs DSB repair responses by inhibiting DNA-PKcs activation in irradiated pancreatic cancer cells. (A) Miapaca-2 and PANC-1 cells were pretreated for 6 h with or without different concentrations of HS-173, and then exposed to radiation (10 Gy) for 30 min. p-DNA-PKcs, p-KAP1, and p-53BP1 levels were determined by Western blotting. (B) Miapaca-2 and PANC-1 cells were pretreated for 6 h with or without different concentrations of HS-173, then exposed to radiation (4 Gy) for 30 min. Cells were then stained for p-KAP1 and p-53BP1 (red) by immunofluorescence, and immunostaining was quantified densitometrically. Data are expressed as means ± S.D. from three experiments (*P < 0.05, **P < 0.01 and ***P < 0.001).
Delayed tumor growth by radiosensitization and inhibition of DNA repair responses through HS-173 in irradiated pancreatic cancer xenograft models
To determine if inhibition of DSB repair by HS-173 results in radiosensitization and tumor regression, we generated subcutaneous tumors using Miapaca-2 pancreatic cancer cells, and allowed the tumors to grow a to volume of 100 mm3 before initiating treatment. To compare the ability of either a single dose or a fractionated dose schedule of local ionizing radiation to control the growth of tumors, we exposed mice, with or without HS-173 (10 mg/kg) pretreatment, to two radiation therapy schedules: IR1, six exposures to 2 Gy; and IR2, a single exposure to 8 Gy (Figure 6A). Using the IR1 paradigm (fractionated dose), tumor growth was modestly delayed in mice treated with radiation alone compared with the control group; HS-173 alone also inhibited tumor growth. In contrast, combined treatment with ionizing radiation and HS-173 dramatically enhanced radiosensitization, as evidenced by a significantly greater inhibition of tumor growth. Mice treated with both HS-173 and radiation also showed no obvious changes in body weight or ALT (alanine transaminase) levels, indicating that this combination treatment is relatively nontoxic (Figure 6B). Combination treatment also caused enhanced inhibition of tumor growth in the IR2 paradigm (single dose) (Figure 6C). To determine if the tumor regression caused by combination treatment was associated with a DSB repair response, we assessed the expression of the DSB repair pathway-related proteins, ATM, DNA-PKcs and KAP1. As shown in Figure 6D, the combination of HS-173 and radiation significantly inhibited the phosphorylation of ATM and DNA-PKcs. Consequently, the combination treatment showed a significant reduction in phosphorylation of KAP1 and 53BP1, a marker for DSB repair (Figure 6E), implying that inhibition of ATM and DNA-PKcs is sufficient to inhibit downstream signaling through KAP-1 in pancreatic tumors.
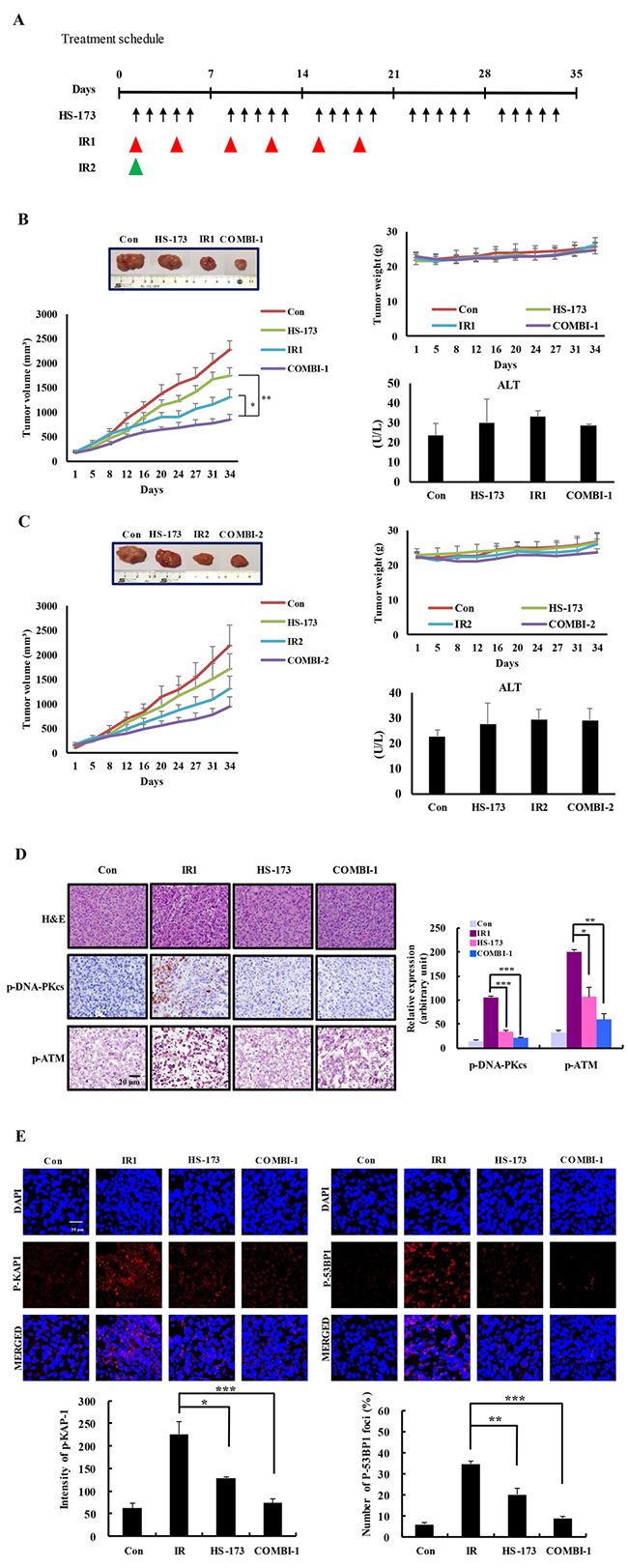
Figure 6: HS-173 inhibits tumor growth and DNA repair responses, thereby inducing radiosensitization in irradiated xenograft models. (A) Experiment schedule. Nude mice were intraperitoneally administered HS-173 (10 mg/kg, 5 times per week) and then irradiated (IR1: 2 Gy, twice a week for 3 weeks; IR2: 8 Gy, once). (B and C) Tumor size and body weight were measured every 2 days. After 34 days, tumors were excised and blood was collected for evaluation of ALT levels. A set of tumors excised at the end of the treatment period illustrates the marked reduction in tumor size with combination treatment. (D and E) Isolated pancreatic tumors were sectioned (8-μm) and stained for p-ATM, p-53BP1 and p-KAP1 by immunofluorescence and immunohistochemistry. Data are presented as means ± S.D. (*P < 0.05, **P < 0.01, and ***P < 0.001).
DISCUSSION
Despite decades of research, pancreatic cancer remains among the most lethal tumors for which no effective treatment exists [26]. Because of the atypical clinical symptoms of pancreatic cancer, most patients are diagnosed too late to have the opportunity of surgery to remove the cancer [27]. Radiotherapy and/or chemotherapy are the common methods used to manage patients with local advanced pancreatic cancer and prolong survival. Radiotherapy is the treatment of choice for pancreatic cancer, but the effectiveness of treatment is limited by radiation resistance. Given that pancreatic cancer show high rate of resistance to radiotherapy, it needs novel therapeutic approaches to enhance radiotherapy for pancreatic cancer treatment. The PI3K/AKT pathway is uniformly activated in human pancreatic ductal adenocarcinoma and mouse models of K-Ras-driven pancreatic cancer [28]. Additionally, this pathway is highly associated with radiotherapy resistance [29]. Given the identification of the PI3K/AKT axis as a potential radiation-modulation target in cancer, we performed a preclinical investigation of the radiosensitizing effects of the PI3K inhibitor HS-173. We found that inhibition of PI3K/AKT by HS-173 treatment significantly radiosensitized pancreatic cancer to the subsequent effects of radiation, as demonstrated by the increase in apoptosis and altered cell cycle resulting from inhibition of the DNA repair response.
The PI3K pathway is commonly activated in various human cancers including pancreatic cancer [30]. In particular, K-Ras mutation has been found in more than 90% of pancreatic cancer patients [31], which activate Ras-dependent downstream effectors pathways including PI3K signaling. Many studies have reported that inhibition of the PI3K pathway causes cell cycle arrest, apoptosis, and decreased cell growth [32–34]. Therefore, we tested the hypothesis that inhibition of the PI3K/AKT pathway by HS-173 would sensitize pancreatic cancer cells to radiation. Indeed, we showed that the combination of HS-173 and radiation synergistically increased therapeutic efficacy by blocking cell proliferation compared with either treatment alone (Figure 1). HS-173 also enhanced radiation-induced apoptosis, as evidenced by increased levels of cleaved PARP and caspase-3 as well TUNEL-positive cells. Some studies have shown that the combination of a PI3K inhibitor and radiation or chemotherapy can improve the efficacy of both therapies [11, 15, 35]. Georgia et al. reported that the PI3K inhibitor BEZ-235 exerted synergistic effects with chemoradiation in the treatment of non-small lung cancer harboring K-Ras mutations [15].
We further found that the combination of HS-173 and radiation significantly increased the proportion of cells in G2/M phase and reduced the proportion of cells in S phase. The synergic effects of HS-173 and radiation not only inhibited the proliferation of pancreatic tumor cells, they also caused arrest of living cells in the G2/M phase of the cell cycle. These effects were confirmed by the demonstration that combined treatment increased the expression of p-Cdc2 in Miapaca-2 cells, in associated with delayed cell division. The G2/M phase plays a very important role in the synthesis and repair of DNA. Cells arrested in this phase resist proliferation and differentiation, but are also unable to repair the DNA damage induced by chemotherapy and radiation [35, 36]. The synergistic effect of HS-173 and radiation on the induction of cell cycle arrest in G2/M phase could enhance apoptosis in pancreatic cancer cells. Thus, HS-173 may exert its biological effects by promoting apoptosis and blocking the cell cycle in the G2/M phase, preventing cells from entering the next cycle and thereby inhibiting cell proliferation.
Induction of G2/M arrest by radiation was initially considered a passive consequence of damaged DNA, but extensive subsequent investigations have led to speculation that DNA repair plays an active role in this process [37, 38]. Indeed, a long G2 delay in a DNA repair-proficient background has been linked to radioresistance [39]. In G2-irradiated cells, about 20% of IR-induced DSBs are repaired with slow kinetics representing those DSBs that undergo resection and repair [40]. Accordingly, even if radiation induces greater DNA damage, radioresistance is inevitable if DNA repair occurs more rapidly than DNA damage accumulation. Accordingly, several studies have validated the effects of radiosensitizers in inhibiting DNA damage repair signaling pathways—including those involving ATM and DNA-PKcs—both in vitro and in vivo. Attenuation of the PI3K/AKT pathway increases therapeutic efficacy in radioresistant cancers [41, 42]. This has been demonstrated using several different PI3K inhibitors that can inhibit the DNA damage repair process. Indeed, it has been shown that the PI3K/AKT inhibitor BEZ-235 inhibits DNA damage response proteins, including ATM and DNA-PKcs, in glioma [15], and the PI3K inhibitor LY294002 enhances the efficacy of radiotherapy in cervical cancer [43]. As noted above, a major component of this radiosensitization is secondary to off-target effects through DNA-PKcs and ATM [15, 44]. In this study, we found that HS-173 inhibited radiation-induced activation of both ATM and DNA-PKcs, the two major kinases that respond to DSBs. HS-173 also inhibited expression of P-KAP1, downstream target of these kinases, in irradiated pancreatic cancer cells. Cross-inhibition of ATM and DNA-PKcs by PI3K inhibitors is not surprising because these kinases have homologous catalytic domains [45, 46]. In accord with our results, a previous study reported that the PI3K inhibitor BEZ-235 radiosensitized non-small cell lung carcinoma cells expressing oncogenic K-Ras, an effect that was correlated with higher levels of radiation-induced DNA breaks [47, 15]. ZSTK474, another PI3K inhibitor, has also been shown to inhibit DNA-PKcs activity [48]. We further observed that HS-173 inhibited tumor growth by enhancing the efficacy of radiation through inhibition of elements of the DNA damage repair response pathway, including ATM and DNA-PKcs. Gupta et al., examining radiosensitization by the PI3K inhibitor LY294002 in bladder cancer xenograft models, reported similar results [49].
In this study, we demonstrated that HS-173 is a potent inhibitor of PI3K/AKT signaling that responds to radiation-induced DNA breaks and repair by inhibiting ATM and DNA-PKcs, resulting in profound radiosensitization in pancreatic cancer cells. Moreover, the radiosensitizing effects of HS-173 appear to be associated with G2/M cell cycle arrest and induction of apoptosis in irradiated cells. Therefore, we suggest that HS-173 could be a potential radiosensitizer option for pancreatic cancer in combination with radiotherapy.
MATERIALS AND METHODS
Cells and materials
The human pancreatic cancer cell lines, PANC-1 and Miapaca-2, were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). PANC-1 and Miapaca-2 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cell culture media, FBS, penicillin-streptomycin, and other supplementary reagents were purchased from GIBCO (Uxbridge, UK). All cell lines were maintained at 37°C in a CO2 incubator with a controlled humidified atmosphere composed of 95% air and 5% CO2.
Preparation of HS-173
The imidazopyridine derivative, ethyl 6-(5-(phenylsulfonamido)pyridin-3-yl)imidazo[1,2-a]pyridine-3-carboxylate (HS-173), is a new PI3Kα inhibitor that was synthesized as described in our previous study [16]. For all in vitro studies, HS-173 was dissolved in dimethylsulfoxide (DMSO) at a concentration of 10 mM before use.
Irradiation
Cells were irradiated with γ-rays using a 137Cs irradiation source (Model 68; J.L. Shepherd and Associates, Glenwood, CA, USA) at a dose rate of 3-4 Gy/min.
Clonogenic assay
Pancreatic cancer cells were plated at appropriate densities in collagen-coated 6-well plates and incubated overnight. Cells were treated with different concentrations of HS-173 for 24 h, and then irradiated with the indicated doses. After a 2 h exposure to radiation, the medium was changed and cells were cultured for 14 days in a 5% CO2 incubator at 37°C. Colonies were fixed with 4% paraformaldehyde (PFA) and stained with 0.5% crystal violet. Colonies containing more than 50 cells were counted, and averages of triplicate dishes were obtained for each sample. The results were normalized with respect to the plating efficiencies of the corresponding non-irradiated cells, and the surviving fractions were calculated.
Western blotting
After the cells were treated with different concentrations of HS-173 and irradiated for various times, they were collected and washed with cold phosphate-buffered saline (PBS). The cells were then lysed with RIPA buffer containing protease and phosphatase inhibitor cocktails (GenDEPOT, Barker, TX, USA). Proteins in whole-cell lysates were resolved by sodium dodecyl sulfate protease and phosphatase inhibitor (SDS-PAGE), and transferred onto nitrocellulose membranes. The blots were first incubated with the appropriate primary antibodies, and then with horseradish peroxidase (HRP)-conjugated secondary antibodies. Antibody binding was detected using enhanced chemiluminescence reagents (Bio-Rad, Hercules, CA, USA). Primary monoclonal antibodies against the following proteins were used: cleaved PARP, cleaved caspase-3, survivin, p-AKT, AKT (Cell Signaling Technology, MA, USA) p-Cdc2, p-ATM, ATM, p-DNA-PKcs, DNA-PKcs, p-KAP1, p-53BP1 (Abcam, Cambridge, UK) KAP1, 53BP1(Santa Cruz, Dallas, TX, USA) and β-actin (Sigma-Aldrich, OH, USA) Secondary antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA).
TUNEL assay
Miapaca-2 and PANC-1 cells were pretreated with HS-173 (0-10 μM) for 6 h and then irradiated (4 Gy) for 24 h. After treatment, cells were fixed in ice-cold 4% PFA, washed with PBS, and then analyzed for apoptotic cells using a TUNEL assay kit (Chemicon, Temecula, CA, USA), following the manufacturer’s instructions.
Immunofluorescence
After treating with HS-173 for 6 h, Miapaca-2 and PANC-1 cells were irradiated with 4 Gy for 30 min, washed twice with PBS, and fixed by incubating in an acetone:methanol solution (1:2) for 10 min at –20°C. Fixed cells were incubated in blocking buffer for 1 h at room temperature, and then incubated overnight with primary antibody (p-AKT, γ-H2AX, p-KAP1, p-53BP1) at 4°C. After washing several times with PBS, cells were incubated with rhodamine isothiocyanate (RITC)-conjugated rabbit secondary antibody (1:100; Dianova, Hamburg, Germany) for 1 h at room temperature, and then counterstained with 4,6-diamidino-2-phenylindole (DAPI) to visualize nuclei. Slides were then washed twice with PBS, covered with the anti-fading agent, 1,4-diazobicyclo (2,2,2)-octane (DABCO, Sigma-Aldrich), and viewed with a confocal laser-scanning microscope (Olympus) at 488 and 568 nm.
Flow cytometry
Miapaca-2 cells were treated with HS-173 (0-10 μM) for 6 h before irradiation (10 Gy), then fixed by incubating at –25°C overnight in cold 70% ethanol. After washing with PBS, the cells were stained with 50 μg/mL propidium iodide (PI) and 100 μg/mL RNase A for 30 min at room temperature in the dark, and then analyzed by flow cytometry to determine the percentage of cells in specific phases of the cell cycle using a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA). Flow cytometry data were analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Immunohistochemistry
Tumor tissue was excised, cut into 8-μm-thick sections, deparaffinized, and subjected to antigen retrieval by microwaving in citrate buffer (pH 6.0) for 5 min. After quenching peroxidase with 0.3% hydrogen peroxide (H2O2) in PBS for 10 min, sections were washed in water and pre-blocked with normal goat or horse serum for 1 h. Tissue sections were then incubated overnight at 4°C with anti-p-ATM antibody (Abcam, Cambridge, UK), diluted 1:50. After washing with PBS, sections were incubated with biotinylated secondary antibodies (1:100) for 1 h, and then streptavidin-HRP was applied. Finally, immunoreactive proteins were developed by incubating sections with diaminobenzidine tetrahydrochloride substrate (DAB) for 10 min, followed by counterstaining sections with hematoxylin. At least three random fields of each section were examined at a magnification of ×400.
Tumor xenograft studies
A mouse Miapaca-2 cells tumor xenograft model was established by injecting the right flank of 6-week old athymic BALB/c nude mice (Central Lab. Animal Inc. Seoul, Korea) with 5 × 106 Miapaca-2 cells (in PBS). After tumors reached a volume of 50-100 mm3, mice were intraperitoneally administered HS-173 (10 mg/kg, 5 times/week) and then irradiated using fractionated dosing (IR1: 2 Gy, twice a week for 3 weeks) or a single dose (IR2: 8 Gy, once). Body weight and tumor dimensions were recorded twice per week. Tumor size was calculated according to the formula, 0.5 × long axis × (short axis)2. Animal care and experimental procedures were conducted in accordance with the Guide for Animal Experiments, published by the Korean Academy of Medical Sciences, and the protocols used were approved by the Institutional Animal Care and Use Committee of Inha University Hospital (Approval No. 151119-388-1).
Statistical analysis
Data are expressed as means ± S.D., and were analyzed by analysis of variance (ANOVA) or unpaired Student’s t-test, as appropriate. Duncan post hoc tests were applied to assess significant differences between groups. A p-value ≤ 0.05 was considered statistically significant. Comparisons of results were performed using Student’s t-test.
CONFLICTS OF INTEREST
The authors do not have any conflicts of interest to disclose.
GRANT SUPPORT
This research was supported by the National Research Foundation of Korea (NRF) grant (2015R1A2A1A10054108, 2014M3C1A3051476, 2014009392M, 2014R1A1A1004621), Republic of Korea.
REFERENCES
1. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014; 74:2913–2921.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2016; 66:7–30.
3. Sun H, Ma H, Hong G, Sun H, Wang J. Survival improvement in patients with pancreatic cancer by decade: a period analysis of the SEER database, 1981-2010. Sci Rep. 2014; 4:6747.
4. Wason MS, Colon J, Das S, Seal S, Turkson J, Zhao J, Baker CH. Sensitization of pancreatic cancer cells to radiation by cerium oxide nanoparticle-induced ROS production. Nanomedicine (Lond). 2013; 9:558–569.
5. Sharma RA, Plummer R, Stock JK, Greenhalgh TA, Ataman O, Kelly S, Clay R, Adams RA, Baird RD, Billingham L, Brown SR, Buckland S, Bulbeck H, et al, and NCRI CTRad Academia-Pharma Joint Working Group. Clinical development of new drug-radiotherapy combinations. Nat Rev Clin Oncol. 2016; 13:627–642.
6. Kim JS, Lim JH, Kim JH, Im SA, Chie EK, Hwang JH, Kim TY, Bang YJ, Ha SW, Yoon YB. Phase II clinical trial of induction chemotherapy with fixed dose rate gemcitabine and cisplatin followed by concurrent chemoradiotherapy with capecitabine for locally advanced pancreatic cancer. Cancer Chemother Pharmacol. 2012; 70:381–389.
7. McDowell KA, Riggins GJ, Gallia GL. Targeting the AKT pathway in glioblastoma. Curr Pharm Des. 2011; 17:2411–2420.
8. Fumarola C, Bonelli MA, Petronini PG, Alfieri RR. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem Pharmacol. 2014; 90:197–207.
9. Pandurangan AK. Potential targets for prevention of colorectal cancer: a focus on PI3K/Akt/mTOR and Wnt pathways. Asian Pac J Cancer Prev. 2013;14:2201–2205.
10. Simpson DR, Mell LK, Cohen EE. Targeting the PI3K/AKT/mTOR pathway in squamous cell carcinoma of the head and neck. Oral Oncol. 2015; 51:291–298.
11. Kim KW, Myers CJ, Jung DK, Lu B. NVP-BEZ-235 enhances radiosensitization via blockade of the PI3K/mTOR pathway in cisplatin-resistant non-small cell lung carcinoma. Genes Cancer. 2014; 5:293–302. https://doi.org/10.18632/genesandcancer.27.
12. Fokas E, Im JH, Hill S, Yameen S, Stratford M, Beech J, Hackl W, Maira SM, Bernhard EJ, McKenna WG, Muschel RJ. Dual inhibition of the PI3K/mTOR pathway increases tumor radiosensitivity by normalizing tumor vasculature. Cancer Res. 2012; 72:239–248.
13. Potiron VA, Abderrahmani R, Giang E, Chiavassa S, Di Tomaso E, Maira SM, Paris F, Supiot S. Radiosensitization of prostate cancer cells by the dual PI3K/mTOR inhibitor BEZ235 under normoxic and hypoxic conditions. Radiother Oncol. 2013; 106:138–146.
14. Toulany M, Kehlbach R, Florczak U, Sak A, Wang S, Chen J, Lobrich M, Rodemann HP. Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Mol Cancer Ther. 2008; 7:1772–1781.
15. Mukherjee B, Tomimatsu N, Amancherla K, Camacho CV, Pichamoorthy N, Burma S. The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM- and DNA-PKCs-mediated DNA damage responses. Neoplasia. 2012; 14:34–43.
16. Yun SM, Jung KH, Lee H, Son MK, Seo JH, Yan HH, Park BH, Hong S, Hong SS. Synergistic anticancer activity of HS-173, a novel PI3K inhibitor in combination with sorafenib against pancreatic cancer cells. Cancer Lett. 2013; 331:250–261.
17. Lee H, Jung KH, Jeong Y, Hong S, Hong SS. HS-173, a novel phosphatidylinositol 3-kinase (PI3K) inhibitor, has anti-tumor activity through promoting apoptosis and inhibiting angiogenesis. Cancer Lett. 2013; 328:152–159.
18. Lee H, Kim SJ, Jung KH, Son MK, Yan HH, Hong S, Hong SS. A novel imidazopyridine PI3K inhibitor with anticancer activity in non-small cell lung cancer cells. Oncol Rep. 2013; 30:863–869.
19. Tomioka A, Tanaka M, De Velasco MA, Anai S, Takada S, Kushibiki T, Tabata Y, Rosser CJ, Uemura H, Hirao Y. Delivery of PTEN via a novel gene microcapsule sensitizes prostate cancer cells to irradiation. Mol Cancer Ther. 2008; 7:1864–1870.
20. Tanno S, Yanagawa N, Habiro A, Koizumi K, Nakano Y, Osanai M, Mizukami Y, Okumura T, Testa JR, Kohgo Y. Serine/threonine kinase AKT is frequently activated in human bile duct cancer and is associated with increased radioresistance. Cancer Res. 2004; 64:3486–3490.
21. Bernstein C, Bernstein H, Payne CM, Garewal H. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat Res. 2002; 511:145–178.
22. Golding SE, Morgan RN, Adams BR, Hawkins AJ, Povirk LF, Valerie K. Pro-survival AKT and ERK signaling from EGFR and mutant EGFRVIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol Ther. 2009; 8:730–738.
23. Jiang Z, Pore N, Cerniglia GJ, Mick R, Georgescu MM, Bernhard EJ, Hahn SM, Gupta AK, Maity A. Phosphatase and tensin homologue deficiency in glioblastoma confers resistance to radiation and temozolomide that is reversed by the protease inhibitor nelfinavir. Cancer Res. 2007; 67:4467–4473.
24. Kao GD, Jiang Z, Fernandes AM, Gupta AK, Maity A. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J Biol Chem. 2007; 282:21206–21212.
25. Mukherjee B, McEllin B, Camacho CV, Tomimatsu N, Sirasanagandala S, Nannepaga S, Hatanpaa KJ, Mickey B, Madden C, Maher E, Boothman DA, Furnari F, Cavenee WK, et al. EGFRVIII and DNA double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res. 2009; 69:4252–4259.
26. Carpelan-Holmström M, Nordling S, Pukkala E, Sankila R, Lüttges J, Klöppel G, Haglund C. Does anyone survive pancreatic ductal adenocarcinoma? A nationwide study re-evaluating the data of the Finnish Cancer Registry. Gut. 2005; 54:385–387.
27. Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, Hruban RH. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013; 63:318–348.
28. Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS, McBryan T, Doyle B, McKay C, Oien KA, Enders GH, Zhang R, Sansom OJ, Adams PD. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell. 2011; 42:36–49.
29. Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, Kearsley JH, Li Y. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014; 5:e1437.
30. Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005; 24:7455–7464.
31. Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS, McBryan T, Doyle B, McKay C, Oien KA, Enders GH, Zhang R, Sansom OJ, Adams PD. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell. 2011; 42:36–49.
32. Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003; 22:8983–8998.
33. Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Rheb promotes cell growth as a component of the insulin/tor signalling network. Nat Cell Biol. 2003; 5:566–571.
34. Martínez-Gac L, Alvarez B, García Z, Marqués M, Arrizabalaga M, Carrera AC. Phosphoinositide 3-kinase and forkhead, a switch for cell division. Biochem Soc Trans. 2004; 32:360–361.
35. Wardman P. Chemical radiosensitizers for use in radiotherapy. Clin Oncol (R Coll Radiol). 2007; 19:397–417.
36. Xu JM, Azzariti A, Colucci G, Paradiso A. The effect of gefitinib (Iressa, ZD1839) in combination with oxaliplatin is schedule-dependent in colon cancer cell lines. Cancer Chemother Pharmacol. 2003; 52:442–448.
37. Walters RA, Gurley LR, Tobey RA. Effects of caffeine on radiation-induced phenomena associated with cell-cycle traverse of mammalian cells. Biophys J. 1974; 14:99–118.
38. Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989; 246:629–634.
39. Smeets MF, Mooren EH, Abdel-Wahab AH, Bartelink H, Begg AC. Differential repair of radiation-induced dna damage in cells of human squamous cell carcinoma and the effect of caffeine and cysteamine on induction and repair of dna double-strand breaks. Radiat Res. 1994; 140:153–160.
40. Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Löbrich M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009; 28:3413–3427.
41. Dong P, Konno Y, Watari H, Hosaka M, Noguchi M, Sakuragi N. The impact of microrna-mediated PI3K/AKT signaling on epithelial-mesenchymal transition and cancer stemness in endometrial cancer. J Transl Med. 2014; 12:231.
42. Chang L, Graham PH, Ni J, Hao J, Bucci J, Cozzi PJ, Li Y. Targeting PI3K/Akt/mTOR signaling pathway in the treatment of prostate cancer radioresistance. Crit Rev Oncol Hematol. 2015; 96:507–517.
43. Liu Y, Cui B, Qiao Y, Zhang Y, Tian Y, Jiang J, Ma D, Kong B. Phosphoinositide-3-kinase inhibition enhances radiosensitization of cervical cancer in vivo. Int J Gynecol Cancer. 2011; 21:100–105.
44. Gil del Alcazar CR, Hardebeck MC, Mukherjee B, Tomimatsu N, Gao X, Yan J, Xie XJ, Bachoo R, Li L, Habib AA, Burma S. Inhibition of DNA double-strand break repair by the dual PI3K/mTOR inhibitor NVP-BEZ235 as a strategy for radiosensitization of glioblastoma. Clin Cancer Res. 2014; 20:1235–1248.
45. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009; 8:627–644.
46. Abraham RT. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amst). 2004; 3:883–887.
47. Konstantinidou G, Bey EA, Rabellino A, Schuster K, Maira MS, Gazdar AF, Amici A, Boothman DA, Scaglioni PP. Dual phosphoinositide 3-kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res. 2009; 69:7644–7652.
48. Kong D, Yaguchi S, Yamori T. Effect of ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor, on DNA-dependent protein kinase. Biol Pharm Bull. 2009; 32:297–300.
49. Gupta AK, Cerniglia GJ, Mick R, Ahmed MS, Bakanauskas VJ, Muschel RJ, McKenna WG. Radiation sensitization of human cancer cells in vivo by inhibiting the activity of PI3K using LY294002. Int J Radiat Oncol Biol Phys. 2003; 56:846–853.