
The FOX family and cancer
In recent years, a number of FOX family members have been linked to tumorigenesis, carcinogenesis, and the survival of malignant cell growth [1, 2]. Members of the FOXA, FOXC, FOXM, FOXO, and FOXP subclasses of FOX proteins, in particular, were found to have direct effects on the initiation, maintenance, progression, and drug resistance of cancers [2]. For example, the removal of FOXM1, which is known to play an integral role in G1-S and G2-M cell cycle progression and mitotic spindle integrity [3], results in the inability to commence mitosis in mice [4]. Furthermore, the overexpression of FOXM1 accelerates the proliferation and progression of prostate cancers in mouse models [5]. The widely studied FOXO proteins are key negative regulators of tumour suppression, as the simultaneous deletion of FOXO1, FOXO3, and FOXO4 alleles in somatic cells invokes thymic lymphomas and systemic haemangiomas in mouse models [6]. As such, many FOX family members are desirable new avenues for further research as possible therapeutic targets in cancer treatment. Of these, perhaps the most important is the FOXC1 forkhead transcription factor (TF), which has been shown recently to have profound and critical roles in several disparate cancer types following its initial identification as a key prognostic indicator of basal-like breast cancer (BLBC) [7–9]. Within this paper we will review FOXC1’s impact in cancer, focusing on FOXC1’s role in signaling pathways, gene regulation, and interactions with other proteins and how these factors affect the nature of this malignant disease.
The FOX family
The FOX gene family, otherwise known as the Forkhead box gene family, is a group of highly evolutionarily conserved genes [10] with a common DNA-binding domain of 110 amino acids known as the forkhead box or “winged helix” domain (FHD) (Figure 1) [3, 11]. The general structure of the FHD consists of three α-helices, three β-sheets, and two “wing” regions situated on either side of a third β-sheet – this produces the “butterfly-like” characteristic that inspired the moniker of the “winged helix domain” [10]. FOX transcription factors (TFs) have distinct roles in embryonic and adult development [12], and are connected to chromatin remodeling as well as nuclear delocalization [2, 11, 12]. The orthologue of this functionally diverse family was found nearly three decades ago in Drosphila melanogaster, in which a mutation in the homeotic gene forkhead (fkh) was found to inhibit gene expression and manifest aberrant head structures [13]. Since then, more than fifty different forkhead proteins have been discovered in humans, classified in subgroups ranging from FOXA to FOXS [2, 10, 11].
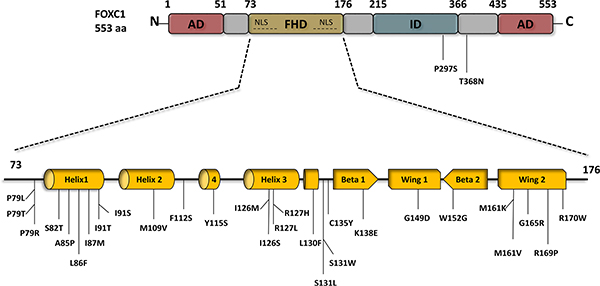
Figure 1: FOXC1 schematic structure and FOXC1 missense mutations. FOXC1 protein contains two activation domains (AD) that are located at the N-terminus 1-51 aa, and the C-terminus 435-553 aa, both of which play a main role in FOXC1 activation. Engineered FOXC1 proteins that lack either the N- or/and C- terminus have reduce activity and improper functions. FOXC1 protein localizes to the cell nucleus via two nuclear localization sequences (NLS), and binds to DNA via the forkhead domain (FHD) 73-176 aa. To date 28 point mutations have been identified in the FHD of FOXC1, most of which are linked to ocular defects and malformations. Deletion of the inhibitory domain (ID) 435-533 aa.significantly increases FOXC1 activity. In contrast to the two ADs that activate FOXC1, specific residues in the ID experience post-translational phosphorylation and as a result inhibit FOXC1function.
FOXC1: background, function, structure, and mutations
FOXC1, which is also known as Mf1, Fkh-1 [11] or FREAC3 [14], is a single exon gene located at 6p25 encoding a 533 aa protein that localizes to the nucleus, where it can bind to the DNA and regulate gene expression [15]. FOXC1 is an essential component of mesodermal [16], neural crest [17] and ocular development [18–20] and is often studied and discussed in relation to Axenfeld Rieger syndrome (ARS). ARS can be caused by FOXC1 mutations [3, 21] and involves the abnormal development of the anterior segment of the eye. Importantly, 50% of ARS patients go on to develop high ocular pressure [22]. FOXC1 is also associated with Dandy-Walker malformation, which is a condition in which patients suffer from an underdeveloped cerebellum and enlarged posterior fossa [21, 23]. While this gene is undoubtedly an integral developmental transcription factor – the deletion of both FOXC1 alleles in mice leads to not only issues in ocular development, but it also gives rise to hydrocephalic, cardiac, organogenesis, and skeletal anomalies, thus increasing the propensity for neonatal mortality [15, 16, 24]. More recently, FOXC1 has been found to play a role in carcinogenesis and tumorigenesis, most notably in BLBC [25]. FOXC1, however, is not only involved in BLBC – studies have shown that FOXC1 plays a role in the interleukin-8 inflammatory pathway associated with hepatocellular carcinoma [26, 27] while other studies reveal FOXC1’s involvement in endometrial cancer progression via miRNA 204 and miRNA 495 [28, 29].The relationships between FOXC1 and these cancers will be expanded upon later in this review.
Like others of the FOX family, the phosphoprotein FOXC1 [22] possesses the “winged-helix” structure in its DNA binding domain (Figure 1). The third α-helix of the “winged helix” crosses perpendicularly to the DNA helical axis, creating a sequence-specific contact with the major groove in the core base sequence GTAAATAAA-3’ [30–32] to which FOXC1 has a strong affinity, as determined through in vitro experiments [14]. There are additional protein-DNA contacts possible in the second wing region [32]. FOXC1 regulates transcription through its N- and C- terminal activation domains as well as a phosphorylated transcription inhibitory domain [15].
The transactivation of FOXC1 requires the N-terminal activation domain and a glutamine-rich/hydrophobic C-terminal activation domain, which are located at residues 1 – 51 and 435 – 553, respectively (Figure 1) [15, 33]. HeLa cells transfected with the full-length FOXC1 (1-553) cDNA were compared to empty vectors with a luciferase reporter and were found to have a ten-fold induction of luciferase activity compared to the latter [15]. In addition, when the 1-29 or 1-51 N-terminal amino acids were deleted, the luciferase activity decreased 50% to 55% respectively, leading to the proposal that these residues in the N-terminus are essential to the full activation of FOXC1 [15]. A FOXC1 protein expressed lacking both the N- and C-terminal regions yielded similar luciferase levels to the empty vectors [15]. Furthermore, the activity at these domains is mitigated by a phosphorylated inhibitory domain (ID). The phosphorylation of residues in the ID play a role in FOXC1 stability and activity [15]. Berry and his colleagues have shown that the phosphorylation of FOXC1 through the activation of the ERK1/2 mitogen-activated protein kinase (MAPK) pathway is critical in stabilizing FOXC1 in HeLa cells [22]. The trypsin digest pattern of FOXC1 is altered by its phosphorylation, further bolstering the proposal that FOXC1 is regulated through conformational change as altered conformation may affect the availability of protease-protein contact. It was suggested also that FOXC1 may be regulated by kinase and phosphatase activity [32].
FOXC1 point mutations have been reported and studied [34–41] . These mutations have been shown to reduce FOXC1 protein level, FOXC1 transactivation, and/or FOXC1’s DNA binding ability [36, 37, 41]. To date, 31 missense variants in ARS patients have been identified in FOXC1, 29 of which occur within the forkhead domain (Figure 1). Normally, FOXC1 is located in the nucleus where it binds to DNA to activate or inactivate other genes. Missense and nonsense mutations within the FOXC1 forkhead domain that alter FOXC1 translocation to the nucleus reduce its function. For example, Saleem and colleagues functionally characterized various mutations throughout the forkhead domain of FOXC1 (Figure 1). They found that FOXC1 with either the S82T, L86F, F112S, or I126M mutation displayed 80-100% nuclear localization compared to wild-type FOXC1, 61-80% for either P79L, P79T, or S131L, 41-60% for I91T, and 0-20% for either I91S or R127H [33, 37, 41]. These mutations had shown to reduce FOXC1 activity due to impaired FOXC1 translocation to the nucleus. Aside from nuclear translocation, mutations within the FHD of FOXC1 can impair binding activity of FOXC1 to its target genes. Specifically, the R127H and S131L mutations in α-helix3 reduced FOXC1 binding to DNA by 90 % compared to wild-type FOXC1 binding efficiency [33, 37, 42]. Moreover, some mutations in the FHD were reported to cause other molecular defects to FOXC1. In particular, the I87M, R127H, and H128R mutations reduce protein stability, alter binding specificity, and extend protein half-life, respectively [33, 37, 42]. Missense mutations that alter FOXC1 translocation to the nucleus, binding to DNA, and protein stability consequently reduce FOXC1 function. In addition, recently, gain of function mutations have also been found to be rare causes of dominant glaucoma [40]. Similarly, loss and gain of function mutations in FOXC2 have been shown to cause lymphedema-distichiasis [43]. Together these mutations consequences are likely to be responsible for the developmental anomalies in ARS (Figure 2) and lymphedema-distichiasis patients.
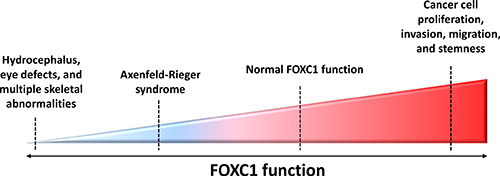
Figure 2: FOXC1 function and activity in human diseases. FOXC1 has been shown to play an integral role in development and adulthood, with both increased and decreased FOXC1 function linked to abnormal disease phenotypes. For example, due to profound defects in ocular development, hydrocephaly, cardiac organogenesis and skeletal anomalies, homozygous null Foxc1 mice do not survive past birth [16]. Mutations in FOXC1 are shown to hinder FOXC1-DNA binding activity, FOXC1 protein level and stability, as well as FOXC1 translocation to the nucleus – all of these defects resulting in Axenfeld-Rieger Syndrome (ARS). More recently, FOXC1 has been demonstrated to have a key role in cancer progression. Contrary to the reduced FOXC1 function observed in ARS, recent studies are linking escalated FOXC1 protein levels to the development of more aggressive phenotypes in cancers such as breast cancer, HCC, and endometrial cancer.
Interestingly, the increased function of FOXC1 has also been linked to malignancy. In contrast to how reduced FOXC1 underlies ARS, an increase in function and activity of FOXC1 is responsible for cancer cell proliferation, differentiation, survival and metastasis (Figure 2). The nature of FOXC1’s contribution to this malignancy is further discussed in this review.
FOXC1 and basal-like breast cancer (BLBC)
Currently, out of all the associations FOXC1 has with different forms of cancer, FOXC1’s relationship with breast cancer, specifically BLBC, is the most elucidated. BLBC is a form of triple negative breast cancer (TNBC), in which the receptors for estrogen (ER), progesterone (PR), and human epidermal growth factor receptor 2 (HER2), are all not overexpressed [44]. BLBC usually presents with high histologic grade, aggressive clinical features, poor prognosis, and a propensity to metastasize to the brain and lung [45]. Genes normally expressed in basal/myoepithelial cells are also expressed in the normal mammary gland in BLBC, and it must be noted that BLBC prognosis is usually determined with additional immunohistochemistry markers such as basal cytokeratins CK5/6, CK14, CK17, and epidermal growth receptor (EGFR) to increase accuracy [46]. BLBCs have been defined in different studies using differing sets of diagnostic markers. For example, Nielsen et al., 2004 defined BLBC on the basis of negative ER and HER2 expression but positive basal cytokeratin, EGFR, and/or c-kit expression [47], while other groups used the combination of negative ER and HER2 expression and positive CK5, P-cadherin, and p63 expression [48] or positive vimentin, EGFR, and CK5/6 expression [49]. Thus, there is no internationally accepted definition for basal-like cancers, and there is no genetic test available in clinical practice to identify these tumors. Although the gold standard for the diagnosis of BLBC is gene expression profiling, emerging data suggests that FOXC1 is a sensitive biomarker for triple negative breast cancers, and in particular, BLBC [50, 51].
FOXC1 is a key prognostic indictor for basal-like breast cancer
Recently, a central role in BLBC for FOXC1 has been clearly established [7, 8, 25, 50–52]. As indicated in (Figure 3), FOXC1 is associated with BLBC through critical signaling pathways [7, 8, 52] and is directly linked to tumor metastasis and invasion [25].
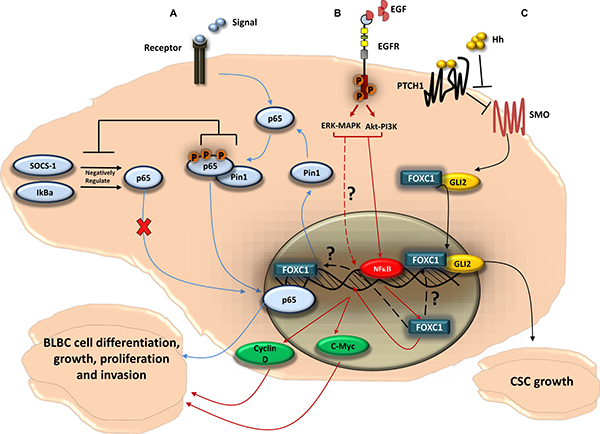
Figure 3: FOXC1-signaling pathways in BLBC. (A) FOXC1 regulates the function of the NF-κB pathway in BLBC cell; NF-κB pathway can be activated as a cellular response to stimuli. Once activated, the NF-κB subunit p65 get phosphorylated and translocated to the nucleus where it binds to DNA. The p65 activity is negatively regulated by the ubiquitin ligase cytokine signal inhibitor SOCS-1 [133] that sends p65 to the proteasome for degradation, and by IκBα that plays a role in the steady-state cytoplasmic localization of p65 dimers, thus preventing p65 nuclear localization and DNA binding [134]. The NF-κB pathway activity has been linked to tumorigenesis. In BLBC cell, FOXC1 regulates the expression of Pin1, a peptidyl-prolyl isomerase, that regulates the activity of p65 [133] and has been linked to tumor development [135]. Pin1 physically binds to p65 in the cytoplasm. This physical binding thus blocks p65 association with SOCS-1 and IκBα, as a result inhibits the p65 degradation. This then leads to p65 phosphorylation and p65 translocation to the nucleus. p65 binds to DNA and activates genes that enhances BLBC cell growth and proliferation. (B) EGFR, via MAPK-ERK and PI3K-Akt pathways, upregulates FOXC1 in BLBC; upon activation of EGFR by the ligand EGF, two of the classical pathways Mitogen-Activated Protein Kinase (MAPK) and Phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) can be activated. The PI3K and MAPK pathways thus upregulate FOXC1 protein and mRNA expression through the ERK and Akt proteins. It has been shown that Akt and ERK phosphorylate and activate NF-κB that leads to its translocation to the nucleus [130]. NF-κB then would bind to FOXC1 promoter region and increases FOXC1 transcription activity. FOXC1 then would enhance the expression of the transcription factor c-Myc and Cyclin D, in which both play a key role in BLBC cell growth, proliferation, and invasion. (C) FOXC1 activates Smoothened-independent Hedgehog Signaling; the ligand Hh binds to the receptor Patched 1 (PTCH1) which allow SMO to activate the transcription factor Glioma-Associated Oncogene Family Zinc Finger 2 (GLI2). FOXC1 can activate GLI2 independently from SMO, where the FOXC1 N-terminal domain (aa 1-68) binds directly to a certain internal region of GLI2 (aa 898-1168), increasing GLI2-DNA transcription activity. FOXC1 activation of the non-canonical Hh signaling can result in cancer stem cell growth and expansion, consequently produces the BLBC stem-like phenotype.
As a transcription factor of the functionally versatile FOX family, FOXC1 has a role in many gene regulatory pathways [7, 8, 11, 12, 23, 52]. Of these pathways, the most intriguing from the perspective of cancer biology are those involved in cell growth, proliferation, differentiation, invasion, and cancer stem cell growth (Figure 3). FOXC1 is consistently and exclusively over-expressed in BLBC when compared to other breast cancer molecular subtypes in multiple, independent, gene expression microarray datasets [50]. Ray and his colleagues determined a significant positive correlation between high FOXC1 activity and FOXC1 mRNA expression and BLBC [50]. Further expansion on these relationships yielded that brain metastasis-free survival was significantly tied to high FOXC1 mRNA levels. Moreover, the ectopic overexpression of FOXC1 invoked more aggressive breast cancer phenotypes, including epithelial-mesenchymal transition, increased cell proliferation, increased migration, and increased invasion [50]. This association of increased FOXC1 levels with BLBC and poor prognosis appears to be the result of the aggressive cell phenotypes that result from over-expression of FOXC1 [50, 51]. Knockdown of FOXC1 expression by siRNA in BLBC cell lines significantly decreased cell proliferation, migration, and invasion [50]. Furthermore, several studies have reported on the interaction between FOXC1 and signaling pathways. For example, FOXC1 can regulate the BLBC cells by activating the NF-ĸB signaling pathway (Figure 3) [52]. FOXC1 also mediates the function of EGFR [8], which has previously been suggested as a surrogate biomarker in BLBC [46]. While the activation of EGFR leads to the upregulation of FOXC1 expression through ERK- and AKT, FOXC1 is a necessary component in EGF-invoked cell proliferation, migration, and invasion (Figure 3) [8]. More recently, Han et al., 2015 found that FOXC1 interacts with Gli2 in different BLBC cell lines through direct binding, and that FOXC1 mediates the non-canonical Smoothened (SMO)-Independent Hedgehog (Hh) signaling that establishes the BLBC stem-like phenotype and anti-Hh sensitivity (Figure 3) [7]. These findings clearly suggest that FOXC1 is a specific biomarker for BLBC. Since FOXC1 has a critical role in the aggressive BLBC cellular phenotype, modulation of FOXC1 activity could lead towards effective BLBC treatment.
Hepatocellular carcinomas and FOXC1
FOXC1 has recently also been shown to have key roles in other cancers as well. Hepatocellular carcinoma (HCC) is a subset of liver cancer [53], and is ranked as the fifth most common cancerous cause of death in men and the eighth most common cancerous cause of death in women [54]. HCC comprises 85% to 90% of all primary liver cancers [53]. The most common risk factor for HCC is cirrhosis, chronic deterioration of the liver with inflammation, cell degeneration, and fibrous thickening of tissue, as well as possibly jaundice, palmar erythema, and gynecomastia [55]. Other major risk factors include chronic consumption of alcoholic beverages, hepatitis B, hepatitis C, and non-alcoholic fatty liver disease [53, 55], while factors such as Wilson’s disease, hereditary hemochromatosis, alpha1-antitrypsin deficiency, primary biliary cirrhosis, and autoimmune hepatitis are less prevalent [56]. Methods of treatment include staging-guided treatment, surgical resection, liver transplantation, local ablation (especially radiofrequency ablation), trans-arterial chemoembolization and radio-embolization, and targeted molecular therapy [57].
In the past few years, FOXC1 has emerged as a transcription factor with a potentially crucial role in the metastasis of HCC [26, 27, 58]. Microvascular invasion (MIV) has been singled out as one of the most crucial clinicopathological risk factors to predict the carcinoma’s propensity for metastasis [59] and early recurrence despite curative liver resection and orthotopic liver transplantation [59, 60]. The epithelial-mesenchymal transition (EMT) is a process where polarized epithelial cells can acquire mesenchymal attributes such as fibroblastoid morphology, characteristic gene expression changes, increasing potential for motility, and increased invasion and metastasis in cancer [61], and is implicated in the MIV formation process [58]. Xu et al. discovered that the suppression of FOXC1 expression reverses the EMT process, as evidenced by the decreased expression of mesenchymal markers Vimentin and N-cadherin, the decreased translocation of β-catenin to the nucleus, and the increased expression of epithelial markers ZO-1 and Claudin-1 in response to FOXC1 knockdown [58]. Furthermore, the expression of FOXC1 was found to be elevated after the eighth day of a 14-day treatment of Huh7 (non-metastatic HCC cell line) cells with TGF-β1 and found that FOXC1 knockdown has no effect on TGF-β isomer expression [58], providing evidence that FOXC1 may operate downstream from TGF-β1. TGF-β1 is a multifunctional cytokine [62] that inhibits cell growth by arresting cells in any portion of the G0/G1 phase through various mechanisms, for example, through the suppression of retinoblastoma protein (Rb) phosphorylation by upregulating specific CDK inhibitors (i.e. P27/KIP1, P15/INK4B, and P21/CIP1) [62–64]. From the study of FOXC1’s interactions with other TFs involved in EMT regulation, FOXC1 is surmised to also operate downstream from other EMT regulators – such as snail, slug, or twist – and to help invoke the mesenchymal portion of the EMT process [58]. Increased N-cadherin mediation of heterotypic contacts between endothelial and melanoma cells as well as increased β-catenin translocation to the nucleus found in trans-endothelial migration [65] supports the argument that overexpressed FOXC1 favours MIV generation [58].
In a similar study conducted by Xia et al, upregulated levels of FOXC1 in HCC tissues were linked to poor prognosis in HCC patients [26]. FOXC1’s role in inducing the EMT process to increase cancer cell migration and invasion offers a possible explanation for how overexpressed FOXC1 mRNA was found to elevate HCC metastatic potential in vitro and encourage lung cell metastasis in vivo [26]. The inhibition of the cell adhesion mediator E-cadherin by FOXC1 transactivation of E-cadherin’s direct repressor, Snai1, as well as the overexpression FOXC1’s direct transcriptional target, NEDD9, have been positively correlated with increased cancer cell migration and invasion [26, 27] and may be involved in the FOXC1 regulation of EMT. As an inflammation-induced cancer, poor HCC prognosis may also be exacerbated via its inflammation signalling pathways. The pro-inflammatory CXC cytokine interleukin 8 (IL-8), secreted by tumour cells and tumour-associated macrophages (TAMs), are critical factors that bind to the receptors CXCR1 and CXCR2 to promote tumour angiogenesis and metastasis [66]. HIF1-α (hypoxia-inducible factor 1 alpha) binding sites are key factors in the IL-8 signaling pathway that are associated with the α-subunits [66–68] of the larger HIF1 αβ-heterodimeric DNA binding factor, which mediates hypoxia-inducible activity on the 3’ enhancer of erythropoietin [66, 69, 70]. HIF1-α is often overexpressed in cancer [67, 71, 72], and interactions between HIF1-α and the vascular endothelial growth factor (VEGF) were found to perform a role in angiogenesis [73–76]. VEGF is a known gene target for FOXC1, mainly operating in blood vessel maturation and lymph vessel sprouting [3, 77].
In HCC specifically, a mutation in the HIF1-α binding sites in the sequence between nt-960 and -635 in the FOXC1 promoter region of HCC cells leads to reduced FOXC1 promoter activity due to decreased IL8-mediated binding of HIF1-α to the FOXC1 promoter [27]. Out of four kinase inhibitors – the inhibitors of phosphatidylinositol-3-kinase (PI3K), extracellular signal-regulated kinase (ERK), c-Jun-N-terminal kinase (JNK), and p38 – only the PI3K inhibitor made significant changes to IL-8-induced FOXC1 expression by inhibiting HIF-α binding to the FOXC1 promoter region [27]. IL-8 is therefore likely to regulate FOXC1 through the PI3K/Akt/HIF-α signalling pathway in HCC [27, 58]. The transactivation of genes CXCR2 and CCL2 – which are correlated with tumour angiogenesis and metastasis as well as macrophage infiltration and breast metastasis promotion respectively [27, 78] – with upregulated FOXC1 also plays a role in inflammation-based HCC metastasis [27], indicating that there are many pathways through which FOXC1 influences HCC metastatic potential.
Therefore, overexpressed FOXC1 was found to not only aggravate the malignant development of HCC by favouring the EMT and MIV generation [26, 27, 57, 58], but also transactivate genes related to angiogenesis and metastasis, CXCR2 and CCL2, through the IL-8-regulated PI3K/Akt/HIF-α inflammatory signalling pathway. [27, 66, 78] Advances in the understanding of the underlying mechanisms involved in the relationships between high FOXC1 expression and increased HCC metastatic potential may yield effective targets for precise medical treatment for not only HCC, but other cancers as well.
Endometrial cancer and FOXC1
Endometrial cancer, a subset of uterine cancer, is not only the third most prevalent gynaecologic malignancy worldwide, but also the most common cancer pertaining to the female genital tract [79]. All tumours from the body of the uterus to the cervix, but not the adenocarcinomas of the endocervix spreading up to the body, are included in this definition [80]. There are two subsets of endometrial carcinomas: while Type I tumours are comprised mainly of endometrioid adenocarcinomas whose development is associated with estrogen hyperplasia and express steroid hormone receptors, Type II tumours, the mostly serous and clear-cell carcinomas, are usually negatively or weakly positive for steroid hormone receptors, poorly differentiated, and of a high grade [81]. The development of endometrial cancer is associated with increased coding errors and somatic mutations, thought to be caused by extensive endometrial cell proliferation from long-term exposure to estrogen [81, 82]. Other factors have also been associated with elevating the risk for endometrial cancer, such as early menstruation, late menopause, infertility, nulliparity, obesity, and estrogen replacement therapy without involvement of a progestin [82].
Although the studies of FOXC1 in basal-like breast cancer and hepatocellular carcinoma are well underway, the investigation of FOXC1 in endometrial cancer has just begun. Studies in recent years have shed light onto the FOX transcription factor’s role in a variety of pathways that are involved in endometrial tumorigenesis, especially focusing on the oncogenic role of FOXC1 in pathways involving microRNAs (miRNA) [28, 29].
In 2007, Wong et al. reported FOXC1 as a newly found differentially regulated gene with a 5.21 fold upregulation in endometrioid endometrial cancer [83]. MicroRNAs – in particular, miRNA 204 (miR204) and miRNA 495 (miR495) – appear to play a part in bridging the observed relationship between FOXC1 and endometrial cancer [28, 29]. HEC1A and Ishikawa endometrial cancer cell lines treated with pre-miR-204 yielded minimized levels of FOXC1 protein and subsequently, reduced cell migration [28]. Through luciferase reporter assays, Chung et al. also demonstrated that the miR204 regulates FOXC1 expression by interacting with binding sites on the FOXC1 3’UTR (3’ untranslated region) [28]. Further studies by Chung et al. suggest that there may be a potential downstream pathway regulated by miR204 responsible for triggering endometrial cancer progression [28].
On the other hand, miR495 was initially demonstrated to be involved in a breast cancer stem cell pathway where it is activated by the transcription factors E12/E47 and down-regulates E-cadherin and REDD1 to promote oncogenesis and hypoxia [84]. Li et al. provide evidence that miR495 also plays a role in the inhibition of gastric cancer cell migration through direct interactions with a member of the PTP (protein tyrosine phosphatase) family, PRL-3 [85, 86]. In endometrial cancer, miR495 takes on the same inhibitory role as it does in gastric cancer; the miRNA suppresses cancer cell growth via cell apoptosis and was shown to inhibit migratory abilities in vitro with Matrigel-lacking transwell assays [29]. The miR495 binds to sites 667 and 1629 of the 3’ UTR region of FOXC1 and negatively regulates the endogenous expression of the FOX family member at the post-transcriptional level [29]. Further experiments in vivo asserted that miR495 suppressed carcinogenesis while downregulating FOXC1 [29]. Intriguingly, a rescue experiment involving the overexpression of FOXC1 abrogated miR495’s inhibition of cell growth and migration as well as promotion of apoptosis in AN3CA and KLE cells (endometrial cancer cells) [29]. These findings provide a strong argument for FOXC1’s role as a target of miR495 in the miR495-regulated malignancy phenotype found in endometrial cancer.
Thus far, FOXC1 appears primarily to be a potential oncogene in not only hepatocellular carcinoma, but in endometrial cancer as well. While MIV generation and IL-8-regulated PI3K/Akt/HIF-α inflammatory signalling pathway are the focus of FOXC1 regulation in HCC [26, 27, 57, 58], microRNAs take spotlight in FOXC1’s relationship with endometrial cancer. In particular, the upregulation of miRNA 204 and miRNA 495 was shown to invoke tumour suppression through decreased FOXC1 protein expression [28, 29]. Further research illuminating the pathways in which the miRNAs and FOXC1 interact in endometrial cancer will allow for an understanding of how to halt endometrial cancer progression and suppress endometrial cancer cell migration. With roles found in the development of other cancers such as breast cancer and gastric cancer, miR495 is also an viable avenue for deeper investigation – the existence of a common oncogenic pathway would be crucial to the development of a generalized but effective treatment plan that will be able to counteract a variety of cancers.
Lymphoma (hodgkin’s and non-hodgkin’s) and FOXC1
There are two main classifications of lymphoma: Hodgkin’s and Non-Hodgkin’s [87–90]. Hodgkin’s lymphoma (HL) can be further defined as nodular sclerosis, mixed cellularity, lymphocyte-rich, lymphocyte-depleted, and nodular lymphocyte predominant HL(NLPHL) – the first four together comprise what is known as ”classical HL” [89, 91, 92]. The organs implicated in HL include the peripheral lymph nodes, and sometimes the liver, the lungs, and bone marrow [89]. Conversely, Non-Hodgkin’s lymphomas (NHL) are a diverse group of lymphoproliferative disorders stemming from B-, T-, or natural killer (NK) lymphocytes [90]. In a pooled analysis of eight case-control studies of NHL, single nucleotide polymorphisms (SNPs) in tumour necrosis factor (TNF) and interleukin-10 (IL-10) genes –which were responsible for encoding key cytokines in inflammatory response and immune balance – were associated with a risk of NHL, particularly in diffuse large B-cell lymphoma [93, 94]. Patients with autoimmune diseases such as rheumatoid arthritis (RA), celiac disease, systemic lupus erythematosus (SLE) and Sjögren’s syndrome were also associated with a higher risk of NHL [90, 95].
The overexpression of FOXC1 has been consistently observed in the occurrence and development of Hodgkin’s lymphoma [96, 97]. For example, in the HL cell lines KM-H2 and U-HO1, Nagel and colleagues have identified elevated levels of FOXC1, suspected to be caused by chromosomal aberrations at 6p25 [97]. Further experiments support the possibility that FOXC1 directly regulates MSX1, a NKL homeobox gene downregulated during B-cell development [98] and overexpressed in cell lines derived from mantle cell lymphoma and acute myeloid leukemia [96]. A site found upstream of MSX1 at - 2661bp that is identical to the FOXC1-binding site found in the closely related MSX2 gene, predicts the direct binding of MSX1 by FOXC1 [96, 98].
ZHX2 is a B-cell specific factor that plays a role in differentiation and apoptosis [97, 99], where through expression analyses, it was found in hematopoietic cell lines and primary cells that ZHX2 acts as a tumour suppressor for HL and multiple myeloma [100]. Studies show that ZHX2 may have an influence over the NOTCH pathway [101], a pathway often active in HL and mediates apoptosis in a variety of B-cell related malignancies [102], as deduced from its activation of NOTCH-target genes HES4 and HOXA5 [100, 102, 103]. The FOXC1-provoked deregulation of MSX1 and shuttle-protein encoding gene, IPO7, is implicated in the downstream inhibition of ZHX2 – specifically, MSX1 and its co-repressor histone H1C inhibit ZHX2 expression while IPO7 encodes for a shuttle protein that transports histone H1 proteins into the nucleus and overexpression of these genes results in decreased ZHX2 levels [96, 97, 104, 105]. Therefore, aberrations in the chromosomal structure of FOXC1 produces abnormalities in MSX1 and IPO7 regulation, which in turn renders ZHX2 incapable of suppressing HL tumorigenesis.
While investigations into FOXC1’s connection to HL centered around MSX1, IPO7, and ZHX2 [96, 97, 104, 105], new studies of FOXC1 in NHL revolve around the activating protein (AP-1) Jun protein family. Jun proteins can exist as either homo- or heterodimers [106] that are usually activated in response to stress signals such as UV irradiation. Jun proteins also promote mitogen-induced cell cycle progression in growth factor pathways, or regulate apoptosis through the modulation of cancer suppressor p53 protein and cyclin D1 expression [107]. DLBCL (diffuse large B-cell lymphoma) is a common aggressive manifestation of non-Hodgkin’s lymphoma that has at least 3 molecular subtypes with distinct prognoses, each differing in the expression of hundreds of genes [108]. The knockdown of the genes encoding c-Jun and JunB in DLBCL cells results in an inability to produce the factors of IL-6 and IL-10, causing growth inhibition in neoplastic cells, especially in NHL [108–110]. Through gene expression profiling studies of cells with down-regulated c-Jun and JunB expression, the genes coding for matrix metalloproteinase 7, adhesion molecule CD44, vitronectin receptor (ITGAV), fractalkine receptor (CX3CRI), and most notably, FOXC1 – all known to encourage the metastasis and invasion of solid tumours [50, 111–118] – correspondingly displayed decreased expression [119]. Thus elevated Jun protein levels are linked to the increased migration and invasion of solid tumours in NHL through FOXC1 expression [119].
The activated B-cell-like subtype (ABC-DLBCL) is associated with the poorest prognosis, which is linked with the constitutive activation of the NF-κB pathway [119, 120]. The scaffold molecule CARD11, which is exclusively expressed in hematopoietic cells [121, 122] and plays a well-known role in antigen-induced NF-κB signaling activation [123–127], is correlated with the signaling induction of c-Jun and JunB in T cells. Elevated CARD11 activity drives the activation of c-Jun and JunB in DLBCL [118, 128] – the constant activation of CARD11 leads to decreased ubiquitination and degradation of c-Jun in human DLBCL cell lines, suggesting that CARD11 is responsible for the stabilization and accumulation of c-Jun [119]. Along with elevated JunB protein levels, the elevated c-Jun levels result in ectopic AP-1 activity that promotes lymphoma interaction with the microenvironment as well as lymphoma dissemination into extra-nodal sites such as the bone marrow in vivo [119], rendering more aggressive lymphoma conditions. Additional investigations examining the role of FOXC1 in the CARD11-Jun pathway are necessary to determine if FOXC1 plays a role in augmenting poor prognosis in DLBCL.
Although the pathways through which FOXC1 influences Hodgkin’s and non-Hodgkin’s lymphoma are different, there is one commonality that is observed: the overexpression of FOXC1 contributes to the further development of the lymphomas through either differentiation and apoptosis or migration and invasion [96, 97, 117]. In HL, FOXC1 is proposed to regulate the NKL homeobox gene involved in B-cell development, MSX1, which in conjunction with the shuttle-protein encoding gene IPO7, inhibits the B-cell specific factor involved in differentiation, apoptosis and the NOTCH signalling pathway, ZHX2 [96–98, 100, 104]. On the other hand, in NHL, the focus is placed on the relationship between FOXC1 and the CARD11-Jun pathway, where elevated Jun protein levels were linked to elevated FOXC1 levels, which in turn is linked to the increased occurrence of migration and invasion of solid tumours in NHL [118, 128]. However, the commonality observed is not only limited to HL and NHL. In all the cancers discussed thus far, the ectopic overexpression of FOXC1 is always linked to increased aggression in cancer disease phenotypes, indicating FOXC1’s potential as a major oncogene. The elucidation of existing FOXC1-related cancer pathways as well as the investigation into the role of FOXC1 in other cancers may yield not only a strong general prognostic biomarker for belligerent cancer phenotypes, but also precise genetic treatments for individual cases of malignancy.
CONCLUSIONS
FOXC1 is a master regulator of gene expression that plays a critical role in embryonic development, consistent with the fact that FOXC1 mutations are associated with developmental anomalies [15, 16, 129] (Figures 1 and 2). More recently, however, studies have linked FOXC1 activity to the aggressive phenotype in cancer cells. FOXC1 enhances cell invasion, proliferation, metastasis, EMT, and migration in BLBC [25]. however, the cross-talk between these pathways and the underlining mechanisms for their compensation still needs to be elucidated (Figure 3). Although the EGFR-MAPK-PI3K pathway upregulates the expression, activity, and protein level of FOXC1 [8] (Figure 3), the how and why of FOXC1 being exclusively expressed in BLBC rather than in other breast-cancer molecular subtypes has yet to be answered. Very recently, Chung and colleagues [130] have shown that NF-ĸB binds to the promoter region of FOXC1 once EGFR is activated by EGF. NF-ĸB binding to FOXC1 can increase FOXC1 transcription activity (Figure 3). It would be interesting to know if FOXC2 [131] is also involved in this cancer circuit. The factors that bind to and regulate FOXC1, for example in response to EGFR-MAPK-PI3K pathway activation, are still being discovered (Figure 3). Recently, FOXC1 has been shown to activate GLI2 in a SMO independent SHH pathway (Figure 3), which partly explains the aggressiveness of BLBC cell and adds a new role for FOXC1 in cancer cell stemness [7]. Moreover, EMT, which plays a key role in the generation and maintenance of cancer stem cells [132] was proposed to be activated by FOXC1 in breast cancer [25] which might explain part of this role of FOXC1 in cancer cell stemness.
In hepatocellular carcinoma, endometrial cancer, as well as both Hodgkin’s and Non-Hodgkin’s lymphoma, research support the role of FOXC1 as an oncogene, where upregulated FOXC1 expression is linked to increasingly aggressive disease phenotypes. FOXC1 has been implicated in numerous pathways that help determine the nature of different cancers, but the oncogenic mechanisms with which FOXC1 involved have yet to be completely elucidated. In HCC, increased FOXC1 expression was shown to encourage cell migration and invasion through its regulation of the EMT and MIV processes, hence elevating cell metastatic potential [58]. FOXC1 is also associated with the IL-8 signaling pathway [27] and the transactivation of genes responsible for tumour angiogenesis and metastasis, CXCR2 and CCL2 [27, 78]. For HCC, future studies on functionally characterizing factors that work with FOXC1 in EMT regulation will lead to a better understanding of how EMT and MIV contributes to HCC proliferation. Further investigation of how IL-8 regulates FOXC1 through the PI3K/Akt/HIF-α signaling pathway will also improve understanding of HCC pathology.
In endometrial cancer, the downregulation of FOXC1 by miRNA – specifically miRNA 204 and miRNA 495 – was revealed to inhibit cancer cell growth and migration while increasing the frequency of apoptosis [28, 29]. While currently, the mechanism by which miR204 interacts with FOXC1 is unclear [28], evidence suggests that miR495 interacts with FOXC1 through binding on the FOXC1 3’UTR [29]. Further exploration into the downstream regulation of FOXC1 by miR204 and miR495 as well as the pathways in which interactions are involved will lead to a greater understanding of how to mitigate more aggressive phenotypes of endometrial cancer with high metastatic potential. The regulatory role of microRNA 495 should be examined not only in depth in endometrial cancer, but laterally across other cancers as well; determining if the miRNA’s interaction with FOXC1 to mediate cell growth, migration and apoptosis is cancer-specific or common across a variety of cancers would be fruitful.
The overexpression of FOXC1 found in Hodgkin’s lymphoma was linked to abnormalities in the MSX1 and IPO7 regulation of ZHX2, a gene responsible for tumour suppression; elevated FOXC1 levels interfere with ZHX2 moderation of B-cell differentiation and apoptosis, leading to highly aberrant cell growth that may exacerbate HL lethality [96–98]. The FOXC1, MSX1, IPO7, and ZHX2 regulatory pathway of HL should thus be further explored; subsequent experiments that determine the molecular mechanisms through which FOXC1 dysregulates ZHX2 as well as other components that operate within the pathway would help develop specific methods to hinder increased HL aggression. On the other hand, research into FOXC1’s role in non-Hodgkin’s lymphoma centers around its interaction with Jun proteins in DLBCL, which play a role in lymphoma interaction with the microenvironment and dissemination into extra-nodal sites [119]. However, this research is still in the early stages, and although CARD11 and antigen-induced NF-κB signaling activation have been implicated in the regulation of Jun proteins in DLBCL [119], a clear picture of how each component is related to each other and what role FOXC1 plays has yet to be discerned.
In summary, recent investigations of FOXC1 are beginning to reveal a key protein at the juxtaposition of critical oncogenetic pathways for many cancers. Additional investigations of FOXC1 are likely to not only illuminate the regulation of key pathways in many different cancers, but may identify novel common entry points for treatments of these cancers.
ACKNOWLEDGMENTS
We would like to thank Mr. Tim Footz, Dr. Jamie Zagozewski, and Dr. Ping Wee for providing valuable input by critically going through this review.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Lehmann OJ, Sowden JC, Carlsson P, Jordan T, Bhattacharya SS. Fox’s in development and disease. Trends Genet. 2003; 19:339–44.
2. Lam EW, Brosens JJ, Gomes AR, Koo CY. Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer. 2013; 13:482–95.
3. Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 2007; 7:847–59.
4. Krupczak-Hollis K, Wang X, Kalinichenko VV, Gusarova GA, Wang IC, Dennewitz MB, Yoder HM, Kiyokawa H, Kaestner KH, Costa RH. The mouse Forkhead Box m1 transcription factor is essential for hepatoblast mitosis and development of intrahepatic bile ducts and vessels during liver morphogenesis. Dev Biol. 2004; 276:74–88.
5. Kalin TV, Wang IC, Ackerson TJ, Major ML, Detrisac CJ, Kalinichenko VV, Lyubimov A, Costa RH. Increased levels of the FoxM1 transcription factor accelerate development and progression of prostate carcinomas in both TRAMP and LADY transgenic mice. Cancer Res. 2006; 66:1712–20.
6. Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, Jiang S, Gilliland DG, Chin L, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007; 128:309–23.
7. Han B, Qu Y, Jin Y, Yu Y, Deng N, Wawrowsky K, Zhang X, Li N, Bose S, Wang Q, Sakkiah S, Abrol R, Jensen TW, et al. FOXC1 Activates Smoothened-Independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Reports. 2015; 13:1046–58.
8. Jin Y, Han B, Chen J, Wiedemeyer R, Orsulic S, Bose S, Zhang X, Karlan BY, Giuliano AE, Cui Y, Cui X. FOXC1 is a critical mediator of EGFR function in human basal-like breast cancer. Ann Surg Oncol. 2015; 19: 161–169.
9. Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, Miura N, Kutok JL, Hartwell K, Richardson AL, Weinberg RA. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci USA. 2007; 104:10069–74.
10. Hannenhalli S, Kaestner KH. The evolution of Fox genes and their role in development and disease. Nat Rev Genet. 2009; 10:233–40.
11. Zhu H. Forkhead box transcription factors in embryonic heart development and congenital heart disease. Life Sci. 2016; 144:194–201.
12. Carlsson P, Mahlapuu M. Forkhead transcription factors: key players in development and metabolism. Dev Biol. 2002; 250:1–23.
13. Weigel D, Jürgens G, Küttner F, Seifert E, Jäckle H. The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell. 1989; 57:645–58.
14. Pierrou S, Hellqvist M, Samuelsson L, Enerbäck S, Carlsson P. Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J. 1994; 13:5002–12.
15. Berry FB, Saleem RA, Walter MA. FOXC1 transcriptional regulation is mediated by N- and C-terminal activation domains and contains a phosphorylated transcriptional inhibitory domain. J Biol Chem. 2002; 277:10292–97.
16. Kume T, Deng KY, Winfrey V, Gould DB, Walter MA, Hogan BL. The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell. 1998; 93:985–96.
17. Hong HK, Lass JH, Chakravarti A. Pleiotropic skeletal and ocular phenotypes of the mouse mutation congenital hydrocephalus (ch/Mf1) arise from a winged helix/forkhead transcriptionfactor gene. Hum Mol Genet. 1999; 8:625–37.
18. Mears AJ, Jordan T, Mirzayans F, Dubois S, Kume T, Parlee M, Ritch R, Koop B, Kuo WL, Collins C, Marshall J, Gould DB, Pearce W, et al. Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomaly. Am J Hum Genet. 1998; 63:1316–28.
19. Nishimura DY, Searby CC, Alward WL, Walton D, Craig JE, Mackey DA, Kawase K, Kanis AB, Patil SR, Stone EM, Sheffield VC. A spectrum of FOXC1 mutations suggests gene dosage as a mechanism for developmental defects of the anterior chamber of the eye. Am J Hum Genet. 2001; 68:364–72.
20. Doucette LP, Rasnitsyn A, Seifi M, Walter MA. The interactions of genes, age, and environment in glaucoma pathogenesis. Surv Ophthalmol. 2015; 60:310–26.
21. Mortemousque B, Amati-Bonneau P, Couture F, Graffan R, Dubois S, Colin J, Bonneau D, Morissette J, Lacombe D, Raymond V. Axenfeld-Rieger anomaly: a novel mutation in the forkhead box C1 (FOXC1) gene in a 4-generation family. Arch Ophthalmol. 2004; 122:1527–33.
22. Berry FB, Mirzayans F, Walter MA. Regulation of FOXC1 stability and transcriptional activity by an epidermal growth factor-activated mitogen-activated protein kinase signaling cascade. J Biol Chem. 2006; 281:10098–104.
23. Somerville TD, Somervaille TC. Tissue-inappropriate derepression of FOXC1 is frequent and functional in human acute myeloid leukemia. Mol Cell Oncol. 2016; 3:e1131355.
24. Kume T, Deng K, Hogan BL. Murine forkhead/winged helix genes Foxc1 (Mf1) and Foxc2 (Mfh1) are required for the early organogenesis of the kidney and urinary tract. Development. 2000; 127:1387–95.
25. Han B, Bhowmick N, Qu Y, Chung S, Giuliano AE, Cui X. FOXC1: an emerging marker and therapeutic target for cancer. Oncogene. 2017; 36:3957–63.
26. Xia L, Huang W, Tian D, Zhu H, Qi X, Chen Z, Zhang Y, Hu H, Fan D, Nie Y, Wu K. Overexpression of forkhead box C1 promotes tumor metastasis and indicates poor prognosis in hepatocellular carcinoma. Hepatology. 2013; 57:610–24.
27. Huang W, Chen Z, Zhang L, Tian D, Wang D, Fan D, Wu K, Xia L. Interleukin-8 Induces Expression of FOXC1 to Promote Transactivation of CXCR1 and CCL2 in Hepatocellular Carcinoma Cell Lines and Formation of Metastases in Mice. Gastroenterology. 2015; 149:1053–67.e14.
28. Chung TK, Lau TS, Cheung TH, Yim SF, Lo KW, Siu NS, Chan LK, Yu MY, Kwong J, Doran G, Barroilhet LM, Ng AS, Wong RR, et al. Dysregulation of microRNA-204 mediates migration and invasion of endometrial cancer by regulating FOXC1. Int J Cancer. 2012; 130:1036–45.
29. Xu YY, Tian J, Hao Q, Yin LR. MicroRNA-495 downregulates FOXC1 expression to suppress cell growth and migration in endometrial cancer. Tumour Biol. 2016; 37:239–51.
30. Pierrou S, Enerbäck S, Carlsson P. Selection of high-affinity binding sites for sequence-specific, DNA binding proteins from random sequence oligonucleotides. Anal Biochem. 1995; 229:99–105.
31. Clark KL, Halay ED, Lai E, Burley SK. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature. 1993; 364:412–20.
32. Lines MA, Kozlowski K, Walter MA. Molecular genetics of Axenfeld-Rieger malformations. Hum Mol Genet. 2002; 11:1177–84.
33. Saleem RA, Banerjee-Basu S, Berry FB, Baxevanis AD, Walter MA. Structural and functional analyses of disease-causing missense mutations in the forkhead domain of FOXC1. Hum Mol Genet. 2003; 12:2993–3005.
34. Ito YA, Footz TK, Murphy TC, Courtens W, Walter MA. Analyses of a novel L130F missense mutation in FOXC1. Arch Ophthalmol. 2007; 125:128–35.
35. Ito YA, Footz TK, Berry FB, Mirzayans F, Yu M, Khan AO, Walter MA. Severe molecular defects of a novel FOXC1 W152G mutation result in aniridia. Invest Ophthalmol Vis Sci. 2009; 50:3573–79.
36. Murphy TC, Saleem RA, Footz T, Ritch R, McGillivray B, Walter MA. The wing 2 region of the FOXC1 forkhead domain is necessary for normal DNA-binding and transactivation functions. Invest Ophthalmol Vis Sci. 2004; 45:2531–38.
37. Saleem RA, Banerjee-Basu S, Berry FB, Baxevanis AD, Walter MA. Analyses of the effects that disease-causing missense mutations have on the structure and function of the winged-helix protein FOXC1. Am J Hum Genet. 2001; 68:627–41.
38. Saleem RA, Banerjee-Basu S, Murphy TC, Baxevanis A, Walter MA. Essential structural and functional determinants within the forkhead domain of FOXC1. Nucleic Acids Res. 2004; 32:4182–93.
39. Fetterman CD, Mirzayans F, Walter MA. Characterization of a novel FOXC1 mutation, P297S, identified in two individuals with anterior segment dysgenesis. Clin Genet. 2009; 76:296–99.
40. Medina-Trillo C, Sánchez-Sánchez F, Aroca-Aguilar JD, Ferre-Fernández JJ, Morales L, Méndez-Hernández CD, Blanco-Kelly F, Ayuso C, García-Feijoo J, Escribano J. Hypo- and hypermorphic FOXC1 mutations in dominant glaucoma: transactivation and phenotypic variability. PLoS One. 2015; 10:e0119272.
41. Saleem RA, Murphy TC, Liebmann JM, Walter MA. Identification and analysis of a novel mutation in the FOXC1 forkhead domain. Invest Ophthalmol Vis Sci. 2003; 44:4608–12.
42. Seifi M, Footz T, Taylor SA, Walter MA. Comparison of Bioinformatics Prediction, Molecular Modeling, and Functional Analyses of FOXC1 Mutations in Patients with Axenfeld-Rieger Syndrome. Hum Mutat. 2017; 38:169–79.
43. Tavian D, Missaglia S, Maltese PE, Michelini S, Fiorentino A, Ricci M, Serrani R, Walter MA, Bertelli M. FOXC2 disease-mutations identified in lymphedema-distichiasis patients cause both loss and gain of protein function. Oncotarget. 2016; 7:54228–39. https://doi.org/10.18632/oncotarget.9797.
44. Badve S, Dabbs DJ, Schnitt SJ, Baehner FL, Decker T, Eusebi V, Fox SB, Ichihara S, Jacquemier J, Lakhani SR, Palacios J, Rakha EA, Richardson AL, et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Mod Pathol. 2011; 24:157–67.
45. Smid M, Wang Y, Zhang Y, Sieuwerts AM, Yu J, Klijn JG, Foekens JA, Martens JW. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008; 68:3108–14.
46. Jensen TW, Ray T, Wang J, Li X, Naritoku WY, Han B, Bellafiore F, Bagaria SP, Qu A, Cui X, Taylor CR, Ray PS. Diagnosis of Basal-Like Breast Cancer Using a FOXC1-Based Assay. J Natl Cancer Inst. 2015; 107:1–9.
47. Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler L, Akslen LA, Ragaz J, Gown AM, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004; 10:5367–74.
48. Elsheikh SE, Green AR, Rakha EA, Samaka RM, Ammar AA, Powe D, Reis-Filho JS, Ellis IO. Caveolin 1 and Caveolin 2 are associated with breast cancer basal-like and triple-negative immunophenotype. Br J Cancer. 2008; 99:327–34.
49. Livasy CA, Karaca G, Nanda R, Tretiakova MS, Olopade OI, Moore DT, Perou CM. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod Pathol. 2006; 19:264–71.
50. Ray PS, Wang J, Qu Y, Sim MS, Shamonki J, Bagaria SP, Ye X, Liu B, Elashoff D, Hoon DS, Walter MA, Martens JW, Richardson AL, et al. FOXC1 is a potential prognostic biomarker with functional significance in basal-like breast cancer. Cancer Res. 2010; 70:3870–76.
51. Ray PS, Bagaria SP, Wang J, Shamonki JM, Ye X, Sim MS, Steen S, Qu Y, Cui X, Giuliano AE. Basal-like breast cancer defined by FOXC1 expression offers superior prognostic value: a retrospective immunohistochemical study. Ann Surg Oncol. 2011; 18:3839–47.
52. Wang J, Ray PS, Sim MS, Zhou XZ, Lu KP, Lee AV, Lin X, Bagaria SP, Giuliano AE, Cui X. FOXC1 regulates the functions of human basal-like breast cancer cells by activating NF-κB signaling. Oncogene. 2012; 31:4798–802.
53. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007; 132:2557–76.
54. American Cancer Society. Cancer Facts & Figures 2016. Cancer Facts Fig. 2016; 2016:1–9.
55. El-Serag HB, Marrero JA, Rudolph L, Reddy KR. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology. 2008; 134:1752–63.
56. Heidelbaugh JJ, Bruderly M. Cirrhosis and chronic liver failure: part I. Diagnosis and evaluation. Am Fam Physician. 2006; 74:756–62.
57. El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011; 365:1118–27.
58. Xu ZY, Ding SM, Zhou L, Xie HY, Chen KJ, Zhang W, Xing CY, Guo HJ, Zheng SS. FOXC1 contributes to microvascular invasion in primary hepatocellular carcinoma via regulating epithelial-mesenchymal transition. Int J Biol Sci. 2012; 8:1130–41.
59. Hu J, Wang Z, Fan J, Dai Z, He YF, Qiu SJ, Huang XW, Sun J, Xiao YS, Song K, Shi YH, Sun QM, Yang XR, et al. Genetic variations in plasma circulating DNA of HBV-related hepatocellular carcinoma patients predict recurrence after liver transplantation. PLoS One. 2011; 6:e26003.
60. Yao FY, Ferrell L, Bass NM, Bacchetti P, Ascher NL, Roberts JP. Liver transplantation for hepatocellular carcinoma: comparison of the proposed UCSF criteria with the Milan criteria and the Pittsburgh modified TNM criteria. Liver Transpl. 2002; 8:765–74.
61. Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, Hollier BG, Ram PT, Lander ES, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci USA. 2010; 107:15449–54.
62. Hill CS. Signalling to the nucleus by members of the transforming growth factor-beta (TGF-beta) superfamily. Cell Signal. 1996; 8:533–44.
63. Datto MB, Yu Y, Wang XF. Functional analysis of the transforming growth factor beta responsive elements in the WAF1/Cip1/p21 promoter. J Biol Chem. 1995; 270:28623–28.
64. Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994; 371:257–61.
65. Qi J, Chen N, Wang J, Siu CH. Transendothelial migration of melanoma cells involves N-cadherin-mediated adhesion and activation of the β-catenin signaling pathway. Mol Biol Cell. 2005; 16:4386–97.
66. Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008; 14:6735–41.
67. Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003; 9:677–84.
68. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003; 3:721–32.
69. Wiesener MS, Turley H, Allen WE, Willam C, Eckardt KU, Talks KL, Wood SM, Gatter KC, Harris AL, Pugh CW, Ratcliffe PJ, Maxwell PH. Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1alpha. Blood. 1998; 92:2260–68.
70. Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996; 16:4604–13.
71. Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992; 12:5447–54.
72. Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. The expression and distribution of the hypoxia-inducible factors HIF-1α and HIF-2α in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000; 157:411–21.
73. Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999; 59:5830–35.
74. Jośko J, Gwóźdź B, Jedrzejowska-Szypułka H, Hendryk S. Vascular endothelial growth factor (VEGF) and its effect on angiogenesis. Med Sci Monit. 2000; 6:1047–52.
75. Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999; 13:9–22.
76. Conway EM, Collen D, Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001; 49:507–21.
77. Seo S, Fujita H, Nakano A, Kang M, Duarte A, Kume T. The forkhead transcription factors, Foxc1 and Foxc2, are required for arterial specification and lymphatic sprouting during vascular development. Dev Biol. 2006; 294:458–70.
78. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011; 475:222–25.
79. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013; 63:11–30.
80. Rose PG. Endometrial carcinoma. N Engl J Med. 1996; 335:640–49.
81. Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol. 1983; 15:10–17.
82. Dizon DS. Treatment options for advanced endometrial carcinoma. Gynecol Oncol. 2010; 117:373–81.
83. Wong YF, Cheung TH, Lo KW, Yim SF, Siu NS, Chan SC, Ho TW, Wong KW, Yu MY, Wang VW, Li C, Gardner GJ, Bonome T, et al. Identification of molecular markers and signaling pathway in endometrial cancer in Hong Kong Chinese women by genome-wide gene expression profiling. Oncogene. 2007; 26:1971–82.
84. Hwang-Verslues WW, Chang PH, Wei PC, Yang CY, Huang CK, Kuo WH, Shew JY, Chang KJ, Lee EY, Lee WH. miR-495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of E-cadherin and REDD1. Oncogene. 2011; 30:2463–74.
85. Bessette DC, Qiu D, Pallen CJ. PRL PTPs: mediators and markers of cancer progression. Cancer Metastasis Rev. 2008; 27:231–52.
86. Li Z, Cao Y, Jie Z, Liu Y, Li Y, Li J, Zhu G, Liu Z, Tu Y, Peng G, Lee DW, Park SS. miR-495 and miR-551a inhibit the migration and invasion of human gastric cancer cells by directly interacting with PRL-3. Cancer Lett. 2012; 323:41–47.
87. Boffetta PI. Epidemiology of adult non-Hodgkin lymphoma. Ann Oncol. 2011; 22:27–31.
88. Canellos GP, Rosenberg SA, Friedberg JW, Lister TA, Devita VT. Treatment of Hodgkin lymphoma: a 50-year perspective. J Clin Oncol. 2014; 32:163–68.
89. Küppers R, Engert A, Hansmann ML. Hodgkin lymphoma. J Clin Invest. 2012; 122:3439–47.
90. Zelenetz AD, Abramson JS, Advani RH, Andreadis CB, Bartlett N, Bellam N, Byrd JC, Czuczman MS, Fayad LE, Glenn MJ, Gockerman JP, Gordon LI, Harris NL, et al. Non-Hodgkin’s lymphomas. J Natl Compr Canc Netw. 2011; 9:484–560.
91. Eichenauer DA, Engert A, Andre M, Federico M, Illidge T, Hutchings M, Ladetto M. André M, Federico M, Illidge T, Hutchings M, Ladetto M. Hodgkin’s lymphoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014; 25:70–75.
92. Lukes RJ, Butler JJ. The pathology and nomenclature of Hodgkin’s disease. Cancer Res. 1966; 26:1063–83.
93. Swerdlow SH, Camop E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. World Health Organization Calssification of Tumours of Haematopoietic and Lymphoid Tissue. 2008; 2.
94. Skibola CF, Bracci PM, Nieters A, Brooks-Wilson A, de Sanjosé S, Hughes AM, Cerhan JR, Skibola DR, Purdue M, Kane E, Lan Q, Foretova L, Schenk M, et al. Tumor necrosis factor (TNF) and lymphotoxin-α (LTA) polymorphisms and risk of non-Hodgkin lymphoma in the InterLymph Consortium. Am J Epidemiol. 2010; 171:267–76.
95. Rothman N, Skibola CF, Wang SS, Morgan G, Lan Q, Smith MT, Spinelli JJ, Willett E, De Sanjose S, Cocco P, Berndt SI, Brennan P, Brooks-Wilson A, et al. Genetic variation in TNF and IL10 and risk of non-Hodgkin lymphoma: a report from the InterLymph Consortium. Lancet Oncol. 2006; 7:27–38.
96. Nagel S, Ehrentraut S, Meyer C, Kaufmann M, Drexler HG, MacLeod RA. Oncogenic deregulation of NKL homeobox gene MSX1 in mantle cell lymphoma. Leuk Lymphoma. 2014; 55:1893–903.
97. Nagel S, Meyer C, Kaufmann M, Drexler HG, MacLeod RA. Deregulated FOX genes in Hodgkin lymphoma. Genes Chromosomes Cancer. 2014; 53:917–33.
98. Nagel S, Schneider B, Meyer C, Kaufmann M, Drexler HG, Macleod RA. Transcriptional deregulation of homeobox gene ZHX2 in Hodgkin lymphoma. Leuk Res. 2012; 36:646–55.
99. Mirzayans F, Lavy R, Penner-Chea J, Berry FB. Initiation of early osteoblast differentiation events through the direct transcriptional regulation of Msx2 by FOXC1. PLoS One. 2012; 7:e49095.
100. Armellini A, Sarasquete ME, García-Sanz R, Chillón MC, Balanzategui A, Alcoceba M, Fuertes M, López R, Hernández JM, Fernández-Calvo J, Sierra M, Megido M, Orfão A, et al. Low expression of ZHX2, but not RCBTB2 or RAN, is associated with poor outcome in multiple myeloma. Br J Haematol. 2008; 141:212–15.
101. Nagel S, Schneider B, Rosenwald A, Meyer C, Kaufmann M, Drexler HG, MacLeod RA. t(4;8)(q27;q24) in Hodgkin lymphoma cells targets phosphodiesterase PDE5A and homeobox gene ZHX2. Genes Chromosomes Cancer. 2011; 50:996–1009.
102. Zweidler-McKay PA, He Y, Xu L, Rodriguez CG, Karnell FG, Carpenter AC, Aster JC, Allman D, Pear WS. Notch signaling is a potent inducer of growth arrest and apoptosis in a wide range of B-cell malignancies. Blood. 2005; 106:3898–906.
103. Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003; 194:237–55.
104. Weerkamp F, Luis TC, Naber BA, Koster EE, Jeannotte L, van Dongen JJ, Staal FJ. Identification of Notch target genes in uncommitted T-cell progenitors: no direct induction of a T-cell specific gene program. Leukemia. 2006; 20:1967–77.
105. Nagel S, Ehrentraut S, Meyer C, Kaufmann M, Drexler HG, MacLeod RA. Aberrantly expressed OTX homeobox genes deregulate B-cell differentiation in Hodgkin lymphoma. PLoS One. 2015; 10:e0138416.
106. Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004; 117:5965–73.
107. Mathas S, Hinz M, Anagnostopoulos I, Krappmann D, Lietz A, Jundt F, Bommert K, Mechta-Grigoriou F, Stein H, Dörken B, Scheidereit C. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-κ B. EMBO J. 2002; 21:4104–13.
108. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000; 403: 503–511.
109. Voorzanger N, Touitou R, Garcia E, Delecluse HJ, Rousset F, Joab I, Favrot MC, Blay JY. Interleukin (IL)-10 and IL-6 are produced in vivo by non-Hodgkin’s lymphoma cells and act as cooperative growth factors. Cancer Res. 1996; 56:5499–505.
110. Rousset F, Garcia E, Defrance T, Péronne C, Vezzio N, Hsu DH, Kastelein R, Moore KW, Banchereau J. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc Natl Acad Sci USA. 1992; 89:1890–93.
111. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010; 140:883–99.
112. Viana LS, Affonso RJ Jr, Silva SR, Denadai MV, Matos D, Salinas de Souza C, Waisberg J. Relationship between the expression of the extracellular matrix genes SPARC, SPP1, FN1, ITGA5 and ITGAV and clinicopathological parameters of tumor progression and colorectal cancer dissemination. Oncology. 2013; 84:81–91.
113. Drillenburg P, Pals ST. Cell adhesion receptors in lymphoma dissemination. Blood. 2000; 95:1900–10.
114. Jamieson WL, Shimizu S, D’Ambrosio JA, Meucci O, Fatatis A. CX3CR1 is expressed by prostate epithelial cells and androgens regulate the levels of CX3CL1/fractalkine in the bone marrow: potential role in prostate cancer bone tropism. Cancer Res. 2008; 68:1715–22.
115. Nevo I, Sagi-Assif O, Meshel T, Ben-Baruch A, Jöhrer K, Greil R, Trejo LE, Kharenko O, Feinmesser M, Yron I, Witz IP. The involvement of the fractalkine receptor in the transmigration of neuroblastoma cells through bone-marrow endothelial cells. Cancer Lett. 2009; 273:127–39.
116. Shiomi T, Okada Y. MT1-MMP and MMP-7 in invasion and metastasis of human cancers. Cancer Metastasis Rev. 2003; 22:145–52.
117. Sizemore ST, Keri RA. The forkhead box transcription factor FOXC1 promotes breast cancer invasion by inducing matrix metalloprotease 7 (MMP7) expression. J Biol Chem. 2012; 287:24631–40.
118. Yao X, Qi L, Chen X, Du J, Zhang Z, Liu S. Expression of CX3CR1 associates with cellular migration, metastasis, and prognosis in human clear cell renal cell carcinoma. Urol Oncol. 2014; 32:162–70.
119. Blonska M, Zhu Y, Chuang HH, You MJ, Kunkalla K, Vega F, Lin X. Jun-regulated genes promote interaction of diffuse large B-cell lymphoma with the microenvironment. Blood. 2015; 125:981–91.
120. Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001; 194:1861–74.
121. Lam LT, Davis RE, Pierce J, Hepperle M, Xu Y, Hottelet M, Nong Y, Wen D, Adams J, Dang L, Staudt LM. Small molecule inhibitors of IkappaB kinase are selectively toxic for subgroups of diffuse large B-cell lymphoma defined by gene expression profiling. Clin Cancer Res. 2005; 11:28–40.
122. Blonska M, Lin X. NF-κB signaling pathways regulated by CARMA family of scaffold proteins. Cell Res. 2011; 21:55–70.
123. Bertin J, Wang L, Guo Y, Jacobson MD, Poyet JL, Srinivasula SM, Merriam S, DiStefano PS, Alnemri ES. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J Biol Chem. 2001; 276:11877–82.
124. Wang D, You Y, Case SM, McAllister-Lucas LM, Wang L, DiStefano PS, Nuñez G, Bertin J, Lin X. A requirement for CARMA1 in TCR-induced NF-kappa B activation. Nat Immunol. 2002; 3:830–35.
125. Egawa T, Albrecht B, Favier B, Sunshine MJ, Mirchandani K, O’Brien W, Thome M, Littman DR. Requirement for CARMA1 in antigen receptor-induced NF-κ B activation and lymphocyte proliferation. Curr Biol. 2003; 13:1252–58.
126. Hara H, Wada T, Bakal C, Kozieradzki I, Suzuki S, Suzuki N, Nghiem M, Griffiths EK, Krawczyk C, Bauer B, D’Acquisto F, Ghosh S, Yeh WC, et al. The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity. 2003; 18:763–75.
127. Newton K, Dixit VM. Mice lacking the CARD of CARMA1 exhibit defective B lymphocyte development and impaired proliferation of their B and T lymphocytes. Curr Biol. 2003; 13:1247–51.
128. Gaide O, Favier B, Legler DF, Bonnet D, Brissoni B, Valitutti S, Bron C, Tschopp J, Thome M. CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-kappa B activation. Nat Immunol. 2002; 3:836–43.
129. Tamimi Y, Lines M, Coca-Prados M, Walter MA. Identification of target genes regulated by FOXC1 using nickel agarose-based chromatin enrichment. Invest Ophthalmol Vis Sci. 2004; 45:3904–13.
130. Chung S, Jin Y, Han B, Qu Y, Gao B, Giuliano AE, Cui X. Identification of EGF-NF-κB-FOXC1 signaling axis in basal-like breast cancer. Cell Commun Signal. 2017; 15:22.
131. Hollier BG, Tinnirello AA, Werden SJ, Evans KW, Taube JH, Sarkar TR, Sphyris N, Shariati M, Kumar SV, Battula VL, Herschkowitz JI, Guerra R, Chang JT, et al. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res. 2013; 73:1981–92.
132. Pradella D, Naro C, Sette C, Ghigna C. EMT and stemness: flexible processes tuned by alternative splicing in development and cancer progression. Mol Cancer. 2017; 16:8.
133. Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, Rottapel R, Yamaoka S, Lu KP. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. 2003; 12:1413–26.
134. Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012; 26:203–34.
135. Lu KP. Prolyl isomerase Pin1 as a molecular target for cancer diagnostics and therapeutics. Cancer Cell. 2003; 4:175–80.