
INTRODUCTION
The “two-signal theory” of lymphocyte activation explains the mechanism of T cell activation or anergy when a naive T cell makes contact with an antigen [1, 2]. Accordingly, efficient activation of antigen-specific lymphocytes needs specific antigen recognition by lymphocytes and an additional signal. Later, it was found that coinhibitory signals also exist. Receptors eliciting coinhibitory signals function as immune checkpoints and have an essential status in the maintenance of peripheral tolerance and inhibition of autoimmunity [3–6].
The best-studied pathway of T-cell costimulation involves the B7-CD28-CTLA-4 superfamily, of which PD-1 and its ligands belong, revealed that the immune system has developed several coinhibitory pathways for T cell activation and tolerance [7–11]. Costimulation has been of therapeutic interest in cancer therapy because the augmentation of costimulatory signals could promote T cell activation to enhance antitumor immune responses [12]. With the discovery of CTLA-4 as an effective immune checkpoint, blockade of CTLA-4 was found to promote antitumor immune reactions and gain notable clinical effectiveness as cancer therapy [13, 14]. The understanding of cancer immunotherapy was modified and strategies for removing other coinhibitory signals to activate the immune system were widely investigated. The tumor immunotherapy targeting PD-1/PD-L1 has achieved encouraging therapeutic outcomes, with response rates of 20% to 40% in various cancer types [15]. Thus far, five immune checkpoint inhibitors targeting PD-1/PD-L1 have been approved by the US Food and Drug Administration, such as PD-1-blocking monoclonal antibodies (mAb) pembrolizumab and nivolumab, and PD-L1-targeted mAb atezolizumab, avelumab, and durvalumab. Compared with anti-CTLA-4 mAb, immune-related toxicities induced by PD-1/PD-L1 blockade were much less frequent and the most frequently observed toxicity was fatigue [16, 17].
PD-1 PATHWAY
Expression of PD-1 and PD-1 ligands
PD-1 is expressed on activated CD4 and CD8 T cells, B cells, monocytes, natural killer (NK) cells, and dendritic cells (DCs) [18–21]. The expression of PD-1 on T cells can be induced by the common γ chain cytokines interleukin-2 (IL-2), IL-7, IL-15, and IL-21 [22]. PD-1 is encoded in the Pdcd1 gene. Nuclear factor of activated T-cells cytoplasmic 1 (NFATc1) is a significant transcription factor, which promotes the PD-1 expression [23]. Other established transcriptional activators such as forkhead box O 1 (Foxo1), Notch proteins, and interferon regulatory factor 9 (IRF9) can also promote PD-1 transcription, and T-box transcription factor TBX21 (T-bet) functions as a transcriptional repressor [24–27]. During chronic viral infection, PD-1 expression is enhanced and maintained on exhausted virus-specific T cells to prevent their proliferation and function [28, 29]. CpG oligodeoxynucleotides treatment induced PD-1 expression in human CD19+ B cells [30]. Environmental hyaluronan fragments from hepatoma cells produced PD-1high regulatory B cells via TLR4 activation, during which TLR4-mediated Bcl-6 upregulation was critical [31]. Interferon (IFN)-sensitive responsive element (ISRE) and STAT1/2 regulate PD-1 expression mediated by IFN-γ in macrophages [32].
PD-L1 (B7-H1 or CD274) and PD-L2 (B7-DC or CD273) are the ligands of PD-1, which are type I transmembrane glycoproteins. There is approximately 40% of the same acidic identity between PD-L1 and PD-L2 whereas the similarity between PD-Ls and B7s is 20% [33, 34]. PD-Ls have different patterns of expression. The expression of PD-L1 constitutively exists on T and B cells, DCs, macrophages, mesenchymal stem cells, and bone marrow-derived mast cells [19]. PD-L1 is also expressed on a large-scale in nonhematopoietic cells such as lung, vascular endothelial, fibroblastic reticular, liver nonparenchymal, and mesenchymal stem cells, and pancreatic islets, astrocytes, neurons, and keratinocytes [20]. In contrast with PD-L1, PD-L2 expression is restricted to activated DCs, macrophages, bone marrow-derived mast cells, and over 50% of peritoneal B1 cells [35]. PD-L1 expression can be induced by γ chain cytokines IL-2, IL-7, and IL-15 on T cells, and IL-21 promoted PD-L1 expression on CD19+ B cells. LPS or BCR activation also stimulate the expression of PD-Ls on B cells [36–38]. Treatment of interferon-gamma (IFN-γ) or IL-10 results in the expression of both ligands in monocytes, and IL-4 and granulocyte macrophage colony-stimulating factor (GM-CSF) induce PD-L2 expression on DCs [39]. In tumor cells, the PD-1 and PD-1 ligands ligation mediates inhibitory signals to cause a harmful effect on antitumor immunity, resulting in the escape from immunosurveillance [40–42].
The influence of epigenetic modification on PD-1 expression
Epigenetic modification, including DNA methylation, histone methylation/acetylation, and microRNA regulation, also controls the expression of PD-1. During acute infection, CD8+ T cell differentiation from naïve T cells was accompanied by transient DNA demethylation at Pdcd1 locus, which gained DNA methylation during further differentiation into functional memory T cells. In contrast, PD-1 promoter was dramatically demethylated in exhausted CD8+ T cells and imprinted during the effector phase of CD8 T cell exhaustion [43, 44]. In patients with chronic HIV, PD-1 promoter was demethylated in PD-1-high virus-specific T cells, and methylated in naïve, PD-1-low T cells from the same donors, while after anti-retroviral therapy, there is no remethylation of DNA at the PD-1 promoter, indicating that the poised epigenetic status for PD-1 remained after prolonged exposure to HIV virus [45]. Modifications to histone proteins also lead to changes in Pdcd1 transcription. Enhancers are marked by histone H3 lysine 4 monomethylation (H3K4me1) and H3K27 acetylation (ac) at the “active” status [46], and other activation marks such as H3K9ac and H3K27ac were enriched at the promoter when PD-1 expression was induced on CD8 T cells in vitro [47, 48]. MicroRNAs also take a part in PD-1 expression. In the melanoma-bearing mice, the expression of PD-1 was attenuated after transfection with miR-28 mimic [49]. In immunocompetent murine models, miR-138 treatment of GL261 gliomas reduced the expression of PD-1, CTLA-4, and FoxP3 in T cells, promoting tumor regression [50]. Moreover, in lymphocytes from patients with chronic hepatitis B virus, transfection of miR-4717 mimics significantly decreased PD-1 expression in concert with increased tumor necrosis factor-α (TNF-α) and IFN-γ production [51].
The PD-1 downstream signaling pathway
The mechanism of how PD-1 inhibits T-cell receptor signaling is a focus of investigation. Beginning with recruiting SHP-2 (SRC Homology 2-Domain-Containingprotein Tyrosine Phosphatase 2) proximate to the T-cell receptor, PD-1 ligation inhibits the activation of T-cell receptor proximal kinases, resulting in depression of the phosphorylation of ZAP-70 mediated by Lck and initiation of downstream events [52].
The PI3K/Akt signaling is an important target of the PD-1 downstream pathway [53]. First, PD-1 can inhibit the activation of PI3K by recruiting SHP-2 [54]. As a serine-threonine phosphatase, PTEN inhibits the activation of PI3K and suppresses the PI3K-Akt pathway signal transmission mediated by CK2. During T-cell activation, PTEN is phosphorylated by CK2 in the S380-T382-T383 cluster, resulting in PTEN stability and reducing PTEN phosphatase activity. PD-1 can target and inhibit CK2-mediated PTEN phosphorylation and promote its degradation while it induces PTEN phosphatase activity, thus repressing PI3K/Akt signaling [55–57].
The Ras/MEK/ERK pathway is another signaling pathway regulated by PD-1 [52, 53]. In T cells, the activation of RasGRP1 is critical for the activation of Ras and downstream MEK/ERK MAP kinase, and RasGRP1 is activated by calcium and diacylglycerol downstream of PLC-γ1 [58]. The activation of PLC-γ1 and Ras are inhibited by PD-1, resulting in diminished activation of the MEK/ERK pathway [53].
By similar mechanisms in B cells, PD-1 ligation with B-cell receptor (BCR) signaling results in SHP-2 recruitment to the ITSM tyrosine of PD-1, and inhibits BCR-mediated Ca2+ mobilization and tyrosine phosphorylation of effector molecules, including Ig β, Syk, phospholipase C-γ2 (PLC- 2), and ERK1/2 [59].
RESISTANCE TO PD-1/PD-L1 BLOCKADE
Immunotherapy has been viewed as one of the most promising methods for cancer treatment. Blocking of PD-1/PD-L1 interaction could overcome the counteraction and preserve the antitumor capacity of T cells to repress tumor cells [14, 15, 60]. Thus far, five PD-1/PD-L1 blocking mAbs have been approved by the U.S. FDA, and the superiority has been confirmed in over 15 cancer types [61]. Nevertheless, as compared to chemotherapy and molecular targeted therapy, a relatively higher rate of primary resistance with immune checkpoint inhibitors occurs and depresses the effectual clinical benefits. The efficacy of monotherapy for PD-1/PD-L1 blockade was rarely more than 40%, with a large proportion of partial responders [15, 62]. Moreover, after an initial response to PD-1/PD-L1 blockade, acquired resistance occurs in most patients, which leads to disease progression or relapse eventually.
The mechanisms of resistance are complex, dynamic, and interdependent. There are many tumor cell-intrinsic and -extrinsic factors relating to PD-1 blockade resistance, including PD-L1 expression, tumor neoantigen expression and presentation, associated cellular signaling pathways, tumor microenvironment (TME), related immune genes, and epigenetic modification (Table 1 and Figure 1).
Table 1: The mechanism of resistance to PD-1/PD-L1 blockade
Mechanism |
Description |
Reference |
|---|---|---|
PD-L1 expression |
PD-L1/2 expression could be associated with the higher local immune cytolytic activity and clinical response. |
|
Lack of effective antigen presentation |
The most straightforward reason why tumors would not respond to PD-1/PD-L1 blockade therapy is lack of recognition by T cells via the mechanisms such as absence of tumor antigens, loss of HLA expression, and Dysfunctional mutations in B2M. |
|
Cellular signaling pathways |
|
|
PI3K/AKT pathway |
Loss of PTEN-mediated PI3K/AKT activation significantly correlated with the decreased expression of IFN-γ, granzyme B, less CD8 T cell infiltration. |
[79] |
WNT/β-catenin pathway |
Stabilization of b-catenin resulting in constitutive WNT signaling pathway could induce T cell exclusion from cancers. |
[80] |
JAK/STAT/IFN-γ pathway |
Cancer cells could escape the effects of IFN-γ by downregulating or mutating molecules including IFNGR1/2, JAK2, and IRF1. |
|
MAPK pathway |
With the inducement of VEGF and IL-8, MAPK signaling has inhibitory effects on T cell recruitment and function. |
|
Tumor microenvironment |
|
|
Immunosuppressive cells |
|
|
Exhaustion T cells |
Disfunctional T cells and PD-1high phenotype exhaustion T cells cannot benefit from PD-1 blockade. |
[94] |
Tregs |
Suppress effector T cell (Teff) responses by secretion of IL-10, IL-35, and TGF-β. |
|
MDSCs |
Promote angiogenesis, tumor invasion and metastasis. |
|
TAMs |
Higher frequencies of TAMs are associated with poor prognosis. |
|
Immunosuppressive cytokines |
Often released by tumor or macrophages for local suppression of anti-tumor immune responses, including TGF-β, CCL5, CCL7, CXCL8, IDO, etc. |
|
Inhibitory receptors |
Apart from PD-1, over-expression of multiple inhibitory receptors including TIM3, CTLA4, LAG3 and BTLA is associated with inhibition of T-cell function and resistance to PD-1/PD-L1 blockade therapy. |
|
Immune related genes |
|
|
IPRES signatures |
Within TME, co-enrichment of a group of 26 transcriptomic signatures (named IPRES signatures) was also related to primary resistance to PD-1/PD-L1 blockade. |
[121] |
Epigenetic modification |
|
|
DNA methylation and histone acetylation |
Epigenetic modification may lead to changes in immune-related genes expression to impact antigen processing, presentation, immune evasion and T cell exhaustion, and DNA methyltransferase inhibitors and histone deacetylase inhibitors can reverseimmune suppression via several mechanisms. |
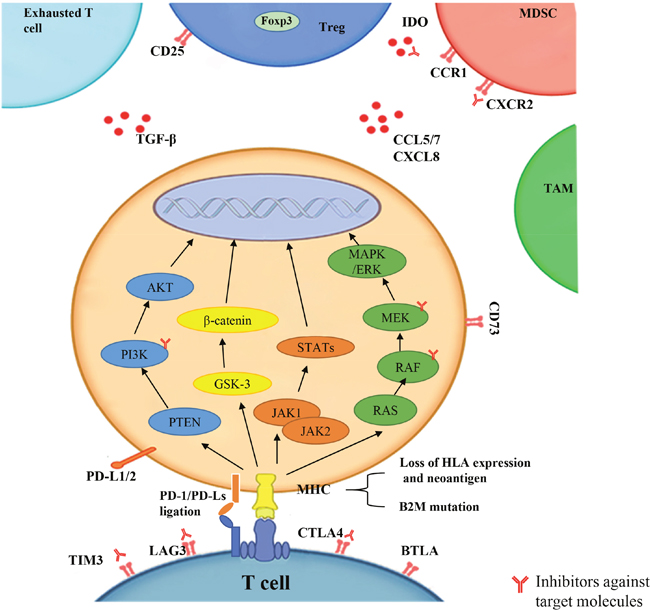
Figure 1: The factors that lead to resistance to PD-1 blockade include PD-L1 expression, tumor neoantigens expression and presentation, cellular signaling pathways (PI3K, WNT, IFN-γ, MAPK), tumor microenvironment (TME) (exhausted T cell, Treg, MDSC, TAM, other chemokines), and related immune genes (IPRES). The inhibitors against target molecules are indicated, which could enhance antitumor responses in in mouse models when combined with PD-1/PD-L1 blockade.
PD-L1 expression
In Hodgkin’s lymphoma, frequent amplification of chromosome 9p24.1 that encodes PD-1 ligands PD-L1 and PD-L2 was observed, and the active JAK/STAT signaling further induced PD-L1/2 expression, which could be associated with the higher clinical response in Hodgkin lymphoma in response to PD-1/PD-L1 blockade [63]. In addition, genetic amplification of PD-L1/2 was positively correlated with high local immune cytolytic activity [64]. Other mechanisms to promote constitutive PD-L1 expression in cancer cells included PTEN deletions, PI3K and/or AKT mutations, EGFR mutations, MYC overexpression, CDK5 disruption, and an increase in PD-L1 transcripts stabilized by truncation of the 30 UTR of this gene [65–69]. It is currently undefined whether constitutive PD-L1 expression increases or reduces the sensitivity to PD-1/PD-L1 blockade therapy.
Lack of tumor antigens and effective antigen presentation
The most straightforward reason why tumors would not respond to PD-1/PD-L1 blockade therapy is lack of recognition by T cells because of absence of tumor antigens [70]. Human melanoma, renal cell carcinoma (RCC), and non small cell lung cancer (NSCLC) are highly immunogenic mutations, ranging from 5 to 10 per mega-base of DNA in the range of individual cell mutations [15, 71]. This finding is consistent with the clinical results that these tumors are most sensitive to PD-1/PD-L1 blockade therapy [72, 73]. Poorly immunogenic tumors that tend to have between 0.1-1 somatic mutation per mega-base of DNA largely show no response to PD-1/PD-L1 blockade, such as in the pancreas and prostate [71]. Meanwhile, mutational load is also associated with efficacy in specific tumor types. In two groups of patients with NSCLCs, a higher burden of nonsynonymous somatic mutations in tumors was detected, which was related to better clinical response and longer survival [72].
Decrease in neoantigens is also an important mechanism for acquired resistance to immunotherapy. In T cell-dependent immunoselection, Elimination of neoantigens has been considered as a mechanism of cancer immunoediting in mice [74]. A recent study revealed that the evolution of neoantigen loss could augment acquired resistance as an escape mechanism after PD-1/PD-L1 blockade treatment. In the NSCLC relapsed patients after initial response, a loss of 7 to 18 putative mutation-associated neoantigens in resistant clones was observed in the analysis of protein coding genes of matched pretreatment and resistant cancers [75].
Moreover, cancer cells may also develop mechanisms to avoid antigen processing and presenting to the cell surface, via silencing or altering the expression of antigen-presenting machinery, beta-2-microglobulin (B2M), or MHC molecules [76, 77]. B2M plays an essential role in supporting MHC class I molecules to present tumor specific peptides to T cells. Dysfunctional mutations in B2M have been viewed as an important mechanism of tumor resistance to T cell-mediated immune responses and lead to resistance to immunotherapy [78].
Cellular signaling pathways
The aberrant cellular signaling transduction is also a central factor contributing to the resistance to immunotherapy, such as PI3K/AKT pathway, WNT/β-catenin pathway, JAK/STAT/IFN-γ pathway, and mitogen-activated protein kinase (MAPK) pathway.
Oncogenic PI3K/AKT pathway has been proved to be associated with primary resistance to PD-1/PD-L1 checkpoint inhibition. PI3K/AKT signaling controls a variety of cellular processes including apoptosis, proliferation, motility, and metabolism, and contributes to tumor development and progression. Tumor suppressor PTEN, a lipid phosphatase, suppresses the activity of PI3K, and the loss of PTEN-mediated PI3K/AKT activation has been observed in many tumor types. In the Cancer Genome Atlas melanoma dataset, PTEN loss significantly correlated with the decreased expression of IFN-γ, granzyme B, less CD8 T cell infiltration, and further correlated with resistance to immune checkpoint therapy. In mice, the effectiveness of either PD-1/PD-L1 blockade or anti-CTLA4 mAb was enhanced by treatment with a selective PI3K inhibitor [79]. Whether the PI3K/AKT inhibitors would reverse the resistance to immune checkpoint blockade needs further clinical study.
In addition to the loss of PTEN, the potential of oncogenic signaling pathways to induce T cell exclusion from cancers has also been described through the stabilization of β-catenin resulting in constitutive WNT signaling pathway [80]. In a murine model, tumors with elevated β-catenin lacked a subset of CD103+ dendritic cells (DCs), due to decreased expression of CCL4, a chemokine that attracts CD103+ DCs. In addition, murine tumors lacking β-catenin responded effectively to immune checkpoint therapy whereas β-catenin-positive tumors did not. Non-T cell-inflamed human melanoma, which lacks T cells and CD103+ DCs in the tumor microenvironment, had significantly higher expression of tumor intrinsic β-catenin signaling genes. Therefore, cancer immune evasion and resistance to PD-1/PD-L1 blockade therapy could result from some crucial oncogenic signals, which might be new candidate targets for immune potentiation.
The IFN-γ pathway is emerging as a key player in primary, adaptive, and acquired resistance to checkpoint blockade therapy [81–83]. It has both favorable and detrimental effects on anti-tumor immune responses. Interferon-γ produced by tumor-specific T cells that have recognized their cognate antigen on cancer cells or APCs induces an effective anti-tumor immune response. However, continuous IFN-γ exposure can lead to immunoediting of cancer cells, resulting in immune escape [84, 85]. One mechanism by which cancer cells could escape the effects of IFN-γ is by decreasing the expression or mutations in molecules in IFN-γ signaling pathway, which goes through the IFN-γ receptors JAK1 and/or JAK2 and the signal transducer and activators of transcription (STATs) [86]. Analysis of tumors in patients who did not respond to therapy with the anti-CTLA-4 antibody ipilimumab revealed an enriched frequency of mutations in the IFN-γ pathway genes IFN-γ receptor 1 and 2 (IFNGR1 and IFNGR2), JAK2, and interferon regulatory factor 1 (IRF1) [81]. Any of these mutations would prevent signaling in response to IFN-γ and give an advantage to the tumor cells escaping from T cells, thereby resulting in primary resistance to anti-CTLA-4 therapy, and may also be a reason for the resistance to PD-1/PD-L1 blockade therapy. Mutations in this pathway would additionally result in lack of PD-L1 expression upon IFN-γ exposure, thereby resulting in cancer cells that would be genetically negative for inducible PD-L1 expression. In such a condition, blocking PD-L1 or PD-1 with therapeutic antibodies would not be useful, and these would be patients who are primarily resistant to PD-1/PD-L1 blockade therapy [87, 88].
The MAPK pathway plays an essential role in cell proliferation, and hyperactivation of MAPK signaling might also be related to the resistance to immunotherapy. With the inducing of VEGF and IL-8, MAPK signaling has inhibitory effects on T cell recruitment and function [89]. Inhibition of MAPK pathway promoted CD8+ T cell activation and infiltration, and induced the expression of tumor antigens as detected in human melanoma samples; in addition, acquired resistance to MAPK-targeted therapy was correlated with depletion of intratumor T cells, exhaustion of CD8 T cells, and loss of antigen presentation [90, 91]. Moreover, the combination of PD-1 blockade and short-term dual inhibition of BRAF and MEK could enhance tumor immune infiltration and improved tumor regression, suggesting that a nongenomic mechanism of MAPK inhibitor resistance might mediate cross-resistance to PD-1/PD-L1 blockade therapy [92].
Tumor microenvironment
Tumor cells closely interact with the stromal cells, immune cells, and extracellular in the immunosuppressive TME, protecting tumor cells from being detected and eradicated by immunosurveillance [93]. Within the TME, other than tumor cells, components that might be associated with primary or acquired resistance include exhausted T cells, Tregs, myeloid derived suppressor cells (MDSCs), M2 macrophages, and other inhibitory immune checkpoints and cytokines.
T cell exhaustion is manifested by dysfunction, sustained expression of inhibitory receptors, and different transcriptional status with functional effector or memory T cells. Exhausted CD8 T cells with intermediate expression of PD-1 can benefit from PD-1/PD-L1 blockade, whereas the severely exhausted CD8 T cells with PD-1high phenotype cannot benefit even impair the efficacy [94]. Thus, the ratio of partially exhausted PD-1intermediate CD8+ T cells to severely exhausted PD-1high CD8+ T cells might be a critical factor for the reversal of T cell exhaustion by PD-1/PD-L1 blockade.
Tregs are known to suppress effector T cell (Teff) responses by secretion of certain inhibitory cytokines, such as IL-10, IL-35, and transforming growth factor beta (TGF-β) [95–97]. In vivo studies have shown that depletion of Treg cells from the TME can strengthen the anti-tumor immune response [98–100]. Moreover, response to PD-1/PD-L1 blockade therapy was shown to be associated with increased ratio of Teff to Treg [101]. These data suggest that tumors without an increase of Teff and decrease of Treg following immunotherapy might be resistant to the treatment.
Myeloid-derived suppressor cells (MDSCs) have been considered as major regulators of immune responses in various pathological conditions. MDSCs could promote angiogenesis, tumor invasion, and metastasis [102]. Clinical findings have pointed out that the presence of MDSCs may be related to short survival in human cancers, such as breast and colorectal cancer [103]. Furthermore, the presence of MDSCs in TME contributed to decreased efficacy of immunotherapies, including immune checkpoint blockade [104]. In various tumor-bearing mice models, selective inhibition of MDSC by using PI3K inhibitors enhanced expression of proinflammatory cytokines and inhibited immunosuppressive factors, synergized with immune checkpoint inhibitors to promote tumor regression [105, 106]. These studies highlight inhibitors of PI3Ka as a potential therapeutic target for combination strategies with PD-1/PD-L1 blockade therapy.
Tumor-associated macrophages (TAMs) are another subset of cells that influence the responses to immunotherapy. TAMs include M1 macrophages, which are involved in promoting anti-tumor immunity, and M2 macrophages, which possess pro-tumorigenic properties [107]. Clinical studies have identified an association between higher frequencies of TAMs and poor prognosis in human cancers [108]. In a lung adenocarcinoma mice model, depletion of TAMs inhibited tumor growth as a result of decreased M2 TAM recruitment, possibly due to the inactivation of CCL2 and CCR2 signaling [109]. Reports suggest that macrophages can directly suppress T cell responses through PD-L1 in hepatocellular carcinoma [41]. To overcome the potential resistance of macrophages, blocking of CSF-1R, a receptor for macrophage colony-stimulating growth factor, in a pancreatic cancer-bearing mice model, decreased frequencies of TAMs, with subsequent increase in interferon production and tumor rejection. Importantly, CSF-1R blockade in combination with antibody against PD-1 or CTLA-4, in addition to gemcitabine, led to strengthened tumor regression [110].
Immunosuppressive cytokines are often released by tumor or macrophages for local suppression of anti-tumor immune responses. TGF-β plays an important role in immunosuppression by stimulating Tregs [111]. Increased TGF-β is associated with poor prognosis in multiple cancer types [112]. A preclinical study on radiation therapy combined with TGF-β inhibitor showed anti-tumor responses [113]. In addition, specific chemokines and chemokine receptors play a necessary role in trafficking of MDSCs and Tregs into tumors. For example, tumor secrete ligands CCL5, CCL7, and CXCL8, bind to their receptors CCR1 or CXCR2 expressed on subtypes of MDSCs, and attract MDSCs in the tumor microenvironment. Inhibitors of these chemokine receptors could abrogate immune evasion and improve anti-tumor T cell responses [114].
Indole 2,3-dioxygenase (IDO), which can be produced by tumors or immune cells, could improve the generation and activity of Tregs and MDSCs [115, 116]. IDO, as a rate-limiting enzyme, influences the catabolism of tryptophan, producing immunosuppressive metabolites to inhibit the proliferation of T cells and induce T cell anergy and apoptosis [117]. Holmgaard et al. found a marked delay in B16 melanoma tumor growth and increased overall survival in IDO knockout mice as compared with wild-type mice when treated with anti-CTLA-4/PD-1 antibody [118]. Based on the study, the combination of IDO inhibitors and PD-1/PD-L1 blockade may lead to more efficacious therapeutic anti-tumor immunity than applying the individual agent.
The immune response is dynamic, and the expression of immune molecules is fluctuated. Other than PD-1, overexpression of multiple inhibitory receptors such as T-cell immunoglobulin mucin 3 (TIM3), CTLA4, lymphocyte activation gene 3 (LAG3), and B and T lymphocyte attenuator (BTLA) is associated with inhibition of T-cell function and hamper to PD-1/PD-L1 blockade [119, 120]. Moreover, a recurrent tumor after PD-1/PD-L1 blockade treatment might be the result of increased expression of TIM-3 on T cells. Notably, preclinical studies showed that PD-1/PD-L1 blockade plus anti-TIM-3 led to improved responses in the tumor-bearing mice. Based on the fact that when one immune checkpoint is blocked, the other immune checkpoints may be induced, and hence, combination of PD-1/PD-L1 blockade with other immune checkpoint inhibitors may promisingly enhance antitumor responses.
IPRES signatures
IPRES signatures, a set of 26 transcriptomic signatures, were found co-enriched to improve resistance to PD-1/PD-L1 blockade in tumors form nonresponding melanoma patients and the upexpression of IPRES related to the regulation of mesenchymal transition, cell adhesion, extracellular matrix (ECM) remodeling, angiogenesis and wound-healing [121]. The confirmation of IPRES co-enrichment in other independent tumor such as pancreatic adenocarcinoma indicates a transcriptomic program among different type of cancers and it may be a new way to enhance the efficacy of PD-1/PD-L1 blockade by inhibit the IPRES relevant biological processes.
Epigenetic modification
Epigenetic modification in cancer cells may lead to changes in gene expression of immune-related genes, which can affect antigen processing, presentation, immune evasion, and T-cell exhaustion [122, 123]. Epigenetic modifying agents, DNA methyltransferase inhibitors and histone deacetylase inhibitors can reverse immune suppression via enhancing the expression of tumor-associated antigens, costimulatory molecules, components of antigen processing and presenting, other immune-related genes and chemokines [124, 125]. In addition, the low-dose DNA demethylating agent decitabine could directly promote T cell activation and cytolytic capacity with increased frequency of IFN-γ-expressing T cells [126]. Based on these findings, it is probably that epigenetic modification may ‘prime’ the host immune response for subsequent immunotherapy in combination therapy [124, 127, 128], and the efficacy of the combined strategy has been confirmed in both clinical studies and animal models [129–131]. Furthermore, immune priming by epigenetic therapy has been observed in combination with immune checkpoint inhibitors [132, 133]. Recently, it has been revealed that T cell exhaustion is associated with de novo DNA methylation, which can persist and be passed on to successive generations of T cells, whereas inhibition of DNA methylation by decitabine could reverse the exhausted state and promote T-cell rejuvenation [134]. These results establish a highly promising strategy for combination treatments by using epigenetic modifying agents and immune checkpoint blockade in cancer patients.
Hyperaggressive disease with anti-PD-1/PD-L1 therapy
Besides primary and acquired resistance to immunotherapy, it was recently reported that PD-1/PD-L1 blockade might develop “hyperprogressive disease” in some patients, which means accelerated tumor growth after anti-PD-1/PD-L1 inhibitors [135]. A trial of 131 patients with PD-1 blockade therapies reported that 12 patients (9%) were considered as hyperprogressive disease; moreover, the hyperprogressive status seemed to be more common in elder patients over 65 year old [136]. Kurzrock et al reported that 6 patients out of 155 had extra copies of MDM2 or MDM4 genes and experienced time-to-treatment failure (TTF) of less than 2 months after immune checkpoint blockade therapy. Among these 6 patients, 4 patients developed hyperprogressive disease. In addition, patients with mutations in EGFR were also likely to experience short TTF and cause hyperprogressive disease [137]. Thus, exploring the mechanisms of resistance and hyperprogressive status to PD-1/PD-L1 blockade is particularly crucial. Actually, there is still not enough evidence to confirm that the accelerated tumor growth is pinned on immunotherapy, the hyperprogressive status could only occur with PD-1/PD-L1 blockade therapy in certain patients.
PROSPECTIVE
PD-1 has now been proved to be an important checkpoint inhibitory receptor that impacts the T-cell stimulation, differentiation, and anti-tumor immune function. Both primary and acquired resistance to anti-PD-1/PD-L1 antibodies inspired us to investigate novel strategies to augment the efficiency of PD-1/PD-L1 blockade. First, radiotherapy, chemotherapy, epigenetic therapy, and other immune stimulatory agents combined with PD-1/PD-L1 blockade could enhance the sensitivity of immunotherapy in tumors with low immunogenicity. Second, modulating the immunosuppressive tumor microenvironment and breaking the inhibitory status, such as depletion of Tregs, interference with the suppressive cytokines, and silencing of co-inhibitory receptors could also help increase the therapeutic responses of PD-1/PD-L1 blockade. Third, further understanding and exploring the mechanisms of both the upstream regulators of PD-1 and its downstream biological events and targets will be necessary for the design of combination therapies, to illuminate the resistance mechanisms, and identify the reliable predictive biomarkers of PD-1/PD-L1 blockade. Last, with the progress made in human genome sequencing and bioinformation analysis, detection and understanding of the patients’ genetic and epigenetic information of tumors and immunocytes will help establish individualized immunotherapy, to obtain clinical benefits for more patients.
CONFLICTS OF INTEREST
No potential conflicts of interest are disclosed.
FUNDING
This study is supported by the grants from the National Key R&D Program of China (2016YFC1303504 and 2016YFC1303501 to WDH), National Natural Science Foundation of China (31671338, 81472838 and 81650025) and Science and Technology Planning Project of Beijing City (Z151100003915076).
REFERENCES
1. Bretscher P, Cohn M. A theory of self-nonself discrimination. Science. 1970; 169: 1042-9.
2. Bretscher PA. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci U S A. 1999; 96: 185-90.
3. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995; 3: 541-7.
4. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995; 270: 985-8.
5. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999; 11: 141-51.
6. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001; 291: 319-22. https://doi.org/10.1126/science.291.5502.319.
7. Karandikar NJ, Vanderlugt CL, Bluestone JA, Miller SD. Targeting the B7/CD28:CTLA-4 costimulatory system in CNS autoimmune disease. J Neuroimmunol. 1998; 89: 10-8.
8. Oosterwegel MA, Greenwald RJ, Mandelbrot DA, Lorsbach RB, Sharpe AH. CTLA-4 and T cell activation. Curr Opin Immunol. 1999; 11: 294-300.
9. Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol. 2001; 19: 225-52. https://doi.org/10.1146/annurev.immunol.19.1.225.
10. Sansom DM. CD28, CTLA-4 and their ligands: who does what and to whom? Immunology. 2000; 101: 169-77.
11. Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol. 2001; 19: 565-94. https://doi.org/10.1146/annurev.immunol.19.1.565.
12. Chen L, Linsley PS, Hellstrom KE. Costimulation of T cells for tumor immunity. Immunol Today. 1993; 14: 483-6. https://doi.org/10.1016/0167-5699(93)90262-j.
13. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996; 271: 1734-6.
14. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363: 711-23. https://doi.org/10.1056/NEJMoa1003466.
15. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366: 2443-54. https://doi.org/10.1056/NEJMoa1200690.
16. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015; 372: 2521-32. https://doi.org/10.1056/NEJMoa1503093.
17. Valsecchi ME. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015; 373: 1270. https://doi.org/10.1056/NEJMc1509660#SA1.
18. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, Honjo T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996; 8: 765-72.
19. Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, Shin T, Tsuchiya H, Pardoll DM, Okumura K, Azuma M, Yagita H. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol. 2002; 169: 5538-45.
20. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008; 26: 677-704. https://doi.org/10.1146/annurev.immunol.26.021607.090331.
21. Finger LR, Pu J, Wasserman R, Vibhakar R, Louie E, Hardy RR, Burrows PD, Billips LG. The human PD-1 gene: complete cDNA, genomic organization, and developmentally regulated expression in B cell progenitors. Gene. 1997; 197: 177-87.
22. Kinter AL, Godbout EJ, McNally JP, Sereti I, Roby GA, O’Shea MA, Fauci AS. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol. 2008; 181: 6738-46.
23. Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 regulates PD-1 expression upon T cell activation. J Immunol. 2008; 181: 4832-9.
24. Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, Honjo T. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. 2011; 186: 2772-9. https://doi.org/10.4049/jimmunol.1003208.
25. Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, Cui G, Li MO, Kaech SM. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity. 2014; 41: 802-14. https://doi.org/10.1016/j.immuni.2014.10.013.
26. Mathieu M, Cotta-Grand N, Daudelin JF, Thebault P, Labrecque N. Notch signaling regulates PD-1 expression during CD8(+) T-cell activation. Immunol Cell Biol. 2013; 91: 82-8. https://doi.org/10.1038/icb.2012.53.
27. Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, Intlekofer AM, Boss JM, Reiner SL, Weinmann AS, Wherry EJ. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol. 2011; 12: 663-71. https://doi.org/10.1038/ni.2046.
28. Watanabe T, Bertoletti A, Tanoto TA. PD-1/PD-L1 pathway and T-cell exhaustion in chronic hepatitis virus infection. J Viral Hepat. 2010; 17: 453-8. https://doi.org/10.1111/j.1365-2893.2010.01313.x.
29. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006; 443: 350-4. https://doi.org/10.1038/nature05115.
30. Buermann A, Romermann D, Baars W, Hundrieser J, Klempnauer J, Schwinzer R. Inhibition of B-cell activation and antibody production by triggering inhibitory signals via the PD-1/PD-ligand pathway. Xenotransplantation. 2016; 23: 347-56. https://doi.org/10.1111/xen.12261.
31. Xiao X, Lao XM, Chen MM, Liu RX, Wei Y, Ouyang FZ, Chen DP, Zhao XY, Zhao Q, Li XF, Liu CL, Zheng L, Kuang DM. PD-1hi identifies a novel regulatory B-cell population in human hepatoma that promotes disease progression. Cancer Discov. 2016; 6: 546-59. https://doi.org/10.1158/2159-8290.cd-15-1408.
32. Cho HY, Lee SW, Seo SK, Choi IW, Choi I, Lee SW. Interferon-sensitive response element (ISRE) is mainly responsible for IFN-alpha-induced upregulation of programmed death-1 (PD-1) in macrophages. Biochim Biophys Acta. 2008; 1779: 811-9. https://doi.org/10.1016/j.bbagrm.2008.08.003.
33. Lazar-Molnar E, Yan Q, Cao E, Ramagopal U, Nathenson SG, Almo SC. Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc Natl Acad Sci U S A. 2008; 105: 10483-8. https://doi.org/10.1073/pnas.0804453105.
34. Lin DY, Tanaka Y, Iwasaki M, Gittis AG, Su HP, Mikami B, Okazaki T, Honjo T, Minato N, Garboczi DN. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc Natl Acad Sci U S A. 2008; 105: 3011-6. https://doi.org/10.1073/pnas.0712278105.
35. Zhong X, Tumang JR, Gao W, Bai C, Rothstein TL. PD-L2 expression extends beyond dendritic cells/macrophages to B1 cells enriched for V(H)11/V(H)12 and phosphatidylcholine binding. Eur J Immunol. 2007; 37: 2405-10. https://doi.org/10.1002/eji.200737461.
36. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000; 192: 1027-34.
37. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001; 2: 261-8. https://doi.org/10.1038/85330.
38. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999; 5: 1365-9. https://doi.org/10.1038/70932.
39. Selenko-Gebauer N, Majdic O, Szekeres A, Hofler G, Guthann E, Korthauer U, Zlabinger G, Steinberger P, Pickl WF, Stockinger H, Knapp W, Stockl J. B7-H1 (programmed death-1 ligand) on dendritic cells is involved in the induction and maintenance of T cell anergy. J Immunol. 2003; 170: 3637-44.
40. Currie AJ, Prosser A, McDonnell A, Cleaver AL, Robinson BW, Freeman GJ, van der Most RG. Dual control of antitumor CD8 T cells through the programmed death-1/programmed death-ligand 1 pathway and immunosuppressive CD4 T cells: regulation and counterregulation. J Immunol. 2009; 183: 7898-908. https://doi.org/10.4049/jimmunol.0901060.
41. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009; 206: 1327-37. https://doi.org/10.1084/jem.20082173.
42. Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. 2009; 114: 1545-52. https://doi.org/10.1182/blood-2009-03-206672.
43. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, Wei Z, Lu P, Austin JW, Riley JL, Boss JM, Ahmed R. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity. 2011; 35: 400-12. https://doi.org/10.1016/j.immuni.2011.06.015.
44. Ahn E, Youngblood B, Lee J, Lee J, Sarkar S, Ahmed R. Demethylation of the PD-1 promoter is imprinted during the effector phase of CD8 T cell exhaustion. J Virol. 2016; 90: 8934-46. https://doi.org/10.1128/jvi.00798-16.
45. Youngblood B, Noto A, Porichis F, Akondy RS, Ndhlovu ZM, Austin JW, Bordi R, Procopio FA, Miura T, Allen TM, Sidney J, Sette A, Walker BD, et al. Cutting edge: prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J Immunol. 2013; 191: 540-4. https://doi.org/10.4049/jimmunol.1203161.
46. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010; 107: 21931-6. https://doi.org/10.1073/pnas.1016071107.
47. McPherson RC, Konkel JE, Prendergast CT, Thomson JP, Ottaviano R, Leech MD, Kay O, Zandee SE, Sweenie CH, Wraith DC, Meehan RR, Drake AJ, Anderton SM. Epigenetic modification of the PD-1 (Pdcd1) promoter in effector CD4(+) T cells tolerized by peptide immunotherapy. Elife. 2014; 3: e03416. https://doi.org/10.7554/eLife.03416.
48. Lu P, Youngblood BA, Austin JW, Mohammed AU, Butler R, Ahmed R, Boss JM. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J Exp Med. 2014; 211: 515-27. https://doi.org/10.1084/jem.20130208.
49. Li Q, Johnston N, Zheng X, Wang H, Zhang X, Gao D, Min W. miR-28 modulates exhaustive differentiation of T cells through silencing programmed cell death-1 and regulating cytokine secretion. Oncotarget. 2016; 7: 53735-50. https://doi.org/10.18632/oncotarget.10731.
50. Wei J, Nduom EK, Kong LY, Hashimoto Y, Xu S, Gabrusiewicz K, Ling X, Huang N, Qiao W, Zhou S, Ivan C, Fuller GN, Gilbert MR, et al. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro Oncol. 2016; 18: 639-48. https://doi.org/10.1093/neuonc/nov292.
51. Zhang G, Li N, Li Z, Zhu Q, Li F, Yang C, Han Q, Lv Y, Zhou Z, Liu Z. microRNA-4717 differentially interacts with its polymorphic target in the PD1 3’ untranslated region: a mechanism for regulating PD-1 expression and function in HBV-associated liver diseases. Oncotarget. 2015; 6: 18933-44. https://doi.org/10.18632/oncotarget.3662.
52. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, Qiu Y, Jussif JM, Carter LL, Wood CR, Chaudhary D. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004; 574: 37-41. https://doi.org/10.1016/j.febslet.2004.07.083.
53. Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. 2012; 5: ra46. https://doi.org/10.1126/scisignal.2002796.
54. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005; 25: 9543-53. https://doi.org/10.1128/mcb.25.21.9543-9553.2005.
55. Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000; 20: 5010-8.
56. Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001; 276: 993-8. https://doi.org/10.1074/jbc.M009134200.
57. Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol. 2013; 33: 3091-8. https://doi.org/10.1128/mcb.00319-13.
58. Bivona TG, Perez De Castro I, Ahearn IM, Grana TM, Chiu VK, Lockyer PJ, Cullen PJ, Pellicer A, Cox AD, Philips MR. Phospholipase Cgamma activates Ras on the Golgi apparatus by means of RasGRP1. Nature. 2003; 424: 694-8. https://doi.org/10.1038/nature01806.
59. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A. 2001; 98: 13866-71. https://doi.org/10.1073/pnas.231486598.
60. Chen DS, Irving BA, Hodi FS. Molecular pathways: next-generation immunotherapy--inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res. 2012; 18: 6580-7. https://doi.org/10.1158/1078-0432.ccr-12-1362.
61. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015; 348: 56-61. https://doi.org/10.1126/science.aaa8172.
62. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013; 369: 134-44. https://doi.org/10.1056/NEJMoa1305133.
63. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, Rodig SJ, Chapuy B, Ligon AH, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015; 372: 311-9. https://doi.org/10.1056/NEJMoa1411087.
64. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015; 160: 48-61. https://doi.org/10.1016/j.cell.2014.12.033.
65. Lastwika KJ, Wilson W 3rd, Li QK, Norris J, Xu H, Ghazarian SR, Kitagawa H, Kawabata S, Taube JM, Yao S, Liu LN, Gills JJ, Dennis PA. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016; 76: 227-38. https://doi.org/10.1158/0008-5472.can-14-3362.
66. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, Wilkerson MD, Fecci PE, Butaney M, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013; 3: 1355-63. https://doi.org/10.1158/2159-8290.cd-13-0310.
67. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gutgemann I, Eilers M, Felsher DW. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016; 352: 227-31. https://doi.org/10.1126/science.aac9935.
68. Dorand RD, Nthale J, Myers JT, Barkauskas DS, Avril S, Chirieleison SM, Pareek TK, Abbott DW, Stearns DS, Letterio JJ, Huang AY, Petrosiute A. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science. 2016; 353: 399-403. https://doi.org/10.1126/science.aae0477.
69. Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, Maeda T, Nagata Y, Kitanaka A, Mizuno S, Tanaka H, Chiba K, Ito S, et al. Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature. 2016; 534: 402-6. https://doi.org/10.1038/nature18294.
70. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, Mulder GE, Toebes M, Vesely MD, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014; 515: 577-81. https://doi.org/10.1038/nature13988.
71. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015; 348: 69-74. https://doi.org/10.1126/science.aaa4971.
72. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015; 348: 124-8. https://doi.org/10.1126/science.aaa1348.
73. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJ, Behjati S, Hilkmann H, El Atmioui D, Nieuwland M, Stratton MR, Kerkhoven RM, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013; 31: e439-42. https://doi.org/10.1200/jco.2012.47.7521.
74. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, Hundal J, Wendl MC, Demeter R, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012; 482: 400-4. https://doi.org/10.1038/nature10755.
75. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, Hruban C, Guthrie VB, Rodgers K, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 2017; 7: 264-76. https://doi.org/10.1158/2159-8290.CD-16-0828.
76. Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol. 2000; 74: 181-273.
77. Sucker A, Zhao F, Real B, Heeke C, Bielefeld N, Mabetaen S, Horn S, Moll I, Maltaner R, Horn PA, Schilling B, Sabbatino F, Lennerz V, et al. Genetic evolution of T-cell resistance in the course of melanoma progression. Clin Cancer Res. 2014; 20: 6593-604. https://doi.org/10.1158/1078-0432.ccr-14-0567.
78. Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, Rosenberg SA. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J Natl Cancer Inst. 1996; 88: 100-8.
79. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, Williams LJ, Deng W, Chen G, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016; 6: 202-16. https://doi.org/10.1158/2159-8290.cd-15-0283.
80. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015; 523: 231-5. https://doi.org/10.1038/nature14404.
81. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, Chen PL, Hwu P, Allison JP, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016; 167: 397-404.e9. https://doi.org/10.1016/j.cell.2016.08.069.
82. Ribas A. Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov. 2015; 5: 915-9. https://doi.org/10.1158/2159-8290.cd-15-0563.
83. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, Saco J, Homet Moreno B, Mezzadra R, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016; 375: 819-29. https://doi.org/10.1056/NEJMoa1604958.
84. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001; 410: 1107-11. https://doi.org/10.1038/35074122.
85. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DS, Pauken KE, Huang AC, Gangadhar TC, Amaravadi RK, Schuchter LM, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 2016; 167: 1540-54.e12. https://doi.org/10.1016/j.cell.2016.11.022.
86. Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994; 264: 1415-21.
87. Shin DS, Ribas A. The evolution of checkpoint blockade as a cancer therapy: what’s here, what’s next? Curr Opin Immunol. 2015; 33: 23-35. https://doi.org/10.1016/j.coi.2015.01.006.
88. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, Palaskas N, Rodriguez GA, Parisi G, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017; 7: 188-201. https://doi.org/10.1158/2159-8290.cd-16-1223.
89. Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, Chen JQ, Li HS, Watowich SS, Yang Y, Tompers Frederick D, Cooper ZA, Mbofung RM, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013; 19: 393-403. https://doi.org/10.1158/1078-0432.CCR-12-1626.
90. Donia M, Fagone P, Nicoletti F, Andersen RS, Hogdall E, Straten PT, Andersen MH, Svane IM. BRAF inhibition improves tumor recognition by the immune system: potential implications for combinatorial therapies against melanoma involving adoptive T-cell transfer. Oncoimmunology. 2012; 1: 1476-83. https://doi.org/10.4161/onci.21940.
91. Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB, Sosman JA, Ribas A, Lo RS. Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell. 2015; 162: 1271-85. https://doi.org/10.1016/j.cell.2015.07.061.
92. Deken MA, Gadiot J, Jordanova ES, Lacroix R, van Gool M, Kroon P, Pineda C, Geukes Foppen MH, Scolyer R, Song JY, Verbrugge I, Hoeller C, Dummer R, et al. Targeting the MAPK and PI3K pathways in combination with PD1 blockade in melanoma. Oncoimmunology. 2016; 5: e1238557. https://doi.org/10.1080/2162402x.2016.1238557.
93. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012; 21: 309-22. https://doi.org/10.1016/j.ccr.2012.02.022.
94. Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A. 2008; 105: 15016-21. https://doi.org/10.1073/pnas.0801497105.
95. Oida T, Zhang X, Goto M, Hachimura S, Totsuka M, Kaminogawa S, Weiner HL. CD4+CD25- T cells that express latency-associated peptide on the surface suppress CD4+CD45RBhigh-induced colitis by a TGF-beta-dependent mechanism. J Immunol. 2003; 170: 2516-22.
96. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008; 133: 775-87. https://doi.org/10.1016/j.cell.2008.05.009.
97. Sundstedt A, O’Neill EJ, Nicolson KS, Wraith DC. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J Immunol. 2003; 170: 1240-8.
98. Chaudhary B, Elkord E. Regulatory T cells in the tumor microenvironment and cancer progression: role and therapeutic targeting. Vaccines (Basel). 2016; 4: E28. https://doi.org/10.3390/vaccines4030028.
99. Linehan DC, Goedegebuure PS. CD25+ CD4+ regulatory T-cells in cancer. Immunol Res. 2005; 32: 155-68. https://doi.org/10.1385/ir:32:1-3:155.
100. Viehl CT, Moore TT, Liyanage UK, Frey DM, Ehlers JP, Eberlein TJ, Goedegebuure PS, Linehan DC. Depletion of CD4+CD25+ regulatory T cells promotes a tumor-specific immune response in pancreas cancer-bearing mice. Ann Surg Oncol. 2006; 13: 1252-8. https://doi.org/10.1245/s10434-006-9015-y.
101. Dyck L, Wilk MM, Raverdeau M, Misiak A, Boon L, Mills KH. Anti-PD-1 inhibits Foxp3+ Treg cell conversion and unleashes intratumoural effector T cells thereby enhancing the efficacy of a cancer vaccine in a mouse model. Cancer Immunol Immunother. 2016; 65: 1491-8. https://doi.org/10.1007/s00262-016-1906-6.
102. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC, Moses HL. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008; 13: 23-35. https://doi.org/10.1016/j.ccr.2007.12.004.
103. Solito S, Falisi E, Diaz-Montero CM, Doni A, Pinton L, Rosato A, Francescato S, Basso G, Zanovello P, Onicescu G, Garrett-Mayer E, Montero AJ, Bronte V, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood. 2011; 118: 2254-65. https://doi.org/10.1182/blood-2010-12-325753.
104. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, Michielin O, Romano E, Speiser DE. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014; 63: 247-57. https://doi.org/10.1007/s00262-013-1508-5.
105. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, Budhu S, Ghosh A, Pink M, Tchaicha J, Douglas M, Tibbitts T, Sharma S, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature. 2016; 539: 443-7. https://doi.org/10.1038/nature20554.
106. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, Schmid MC, Pink M, Winkler DG, et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature. 2016; 539: 437-42. https://doi.org/10.1038/nature19834.
107. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). 2014; 6: 1670-90. https://doi.org/10.3390/cancers6031670.
108. Hu W, Li X, Zhang C, Yang Y, Jiang J, Wu C. Tumor-associated macrophages in cancers. Clin Transl Oncol. 2016; 18: 251-8. https://doi.org/10.1007/s12094-015-1373-0.
109. Fritz JM, Tennis MA, Orlicky DJ, Lin H, Ju C, Redente EF, Choo KS, Staab TA, Bouchard RJ, Merrick DT, Malkinson AM, Dwyer-Nield LD. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front Immunol. 2014; 5: 587. https://doi.org/10.3389/fimmu.2014.00587.
110. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014; 74: 5057-69. https://doi.org/10.1158/0008-5472.can-13-3723.
111. Lebrun JJ. The dual role of TGFbeta in human cancer: from tumor suppression to cancer metastasis. ISRN Mol Biol. 2012; 2012: 381428. https://doi.org/10.5402/2012/381428.
112. Lin RL, Zhao LJ. Mechanistic basis and clinical relevance of the role of transforming growth factor-beta in cancer. Cancer Biol Med. 2015; 12: 385-93. https://doi.org/10.7497/j.issn.2095-3941.2015.0015.
113. Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, Barcellos-Hoff MH, Demaria S. TGFbeta is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. 2015; 75: 2232-42. https://doi.org/10.1158/0008-5472.can-14-3511.
114. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, Kaplan RN, Mackall CL. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. 2014; 6: 237ra67. https://doi.org/10.1126/scitranslmed.3007974.
115. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013; 34: 137-43. https://doi.org/10.1016/j.it.2012.10.001.
116. Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, Muller AJ. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. 2014; 63: 721-35. https://doi.org/10.1007/s00262-014-1549-4.
117. Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012; 72: 5435-40. https://doi.org/10.1158/0008-5472.can-12-0569.
118. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013; 210: 1389-402. https://doi.org/10.1084/jem.20130066.
119. Thommen DS, Schreiner J, Muller P, Herzig P, Roller A, Belousov A, Umana P, Pisa P, Klein C, Bacac M, Fischer OS, Moersig W, Savic Prince S, et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol Res. 2015; 3: 1344-55. https://doi.org/10.1158/2326-6066.cir-15-0097.
120. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, Jones RE, Kulkarni MM, Kuraguchi M, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016; 7: 10501. https://doi.org/10.1038/ncomms10501.
121. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E, Lomeli S, Kong X, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016; 165: 35-44. https://doi.org/10.1016/j.cell.2016.02.065.
122. Karpf AR, Jones DA. Reactivating the expression of methylation silenced genes in human cancer. Oncogene. 2002; 21: 5496-503. https://doi.org/10.1038/sj.onc.1205602.
123. Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011; 3: 166-79.
124. Heninger E, Krueger TE, Lang JM. Augmenting antitumor immune responses with epigenetic modifying agents. Front Immunol. 2015; 6: 29. https://doi.org/10.3389/fimmu.2015.00029.
125. Wang LX, Mei ZY, Zhou JH, Yao YS, Li YH, Xu YH, Li JX, Gao XN, Zhou MH, Jiang MM, Gao L, Ding Y, Lu XC, et al. Low dose decitabine treatment induces CD80 expression in cancer cells and stimulates tumor specific cytotoxic T lymphocyte responses. PLoS One. 2013; 8: e62924. https://doi.org/10.1371/journal.pone.0062924.
126. Li X, Zhang Y, Chen M, Mei Q, Liu Y, Feng KC, Jia H, Dong L, Shi L, Liu L, Nie J, Han W. Increased IFN-gamma+ T cells are responsible for the clinical responses of low-dose DNA demethylating agent decitabine anti-tumor therapy. Clin Cancer Res. 2017. https://doi.org/10.1158/1078-0432.CCR-17-1201.
127. Sigalotti L, Fratta E, Coral S, Maio M. Epigenetic drugs as immunomodulators for combination therapies in solid tumors. Pharmacol Ther. 2014; 142: 339-50. https://doi.org/10.1016/j.pharmthera.2013.12.015.
128. Terracina KP, Graham LJ, Payne KK, Manjili MH, Baek A, Damle SR, Bear HD. DNA methyltransferase inhibition increases efficacy of adoptive cellular immunotherapy of murine breast cancer. Cancer Immunol Immunother. 2016; 65: 1061-73. https://doi.org/10.1007/s00262-016-1868-8.
129. Bao L, Dunham K, Lucas K. MAGE-A1, MAGE-A3, and NY-ESO-1 can be upregulated on neuroblastoma cells to facilitate cytotoxic T lymphocyte-mediated tumor cell killing. Cancer Immunol Immunother. 2011; 60: 1299-307. https://doi.org/10.1007/s00262-011-1037-z.
130. Xu P, Hu G, Luo C, Liang Z. DNA methyltransferase inhibitors: an updated patent review (2012-2015). Expert Opin Ther Pat. 2016; 26: 1017-30. https://doi.org/10.1080/13543776.2016.1209488.
131. Lucarini V, Buccione C, Ziccheddu G, Peschiaroli F, Sestili P, Puglisi R, Mattia G, Zanetti C, Parolini I, Bracci L, Macchia I, Rossi A, D’Urso MT, et al. Combining type I interferons and 5-aza-2’-deoxycitidine to improve anti-tumor response against melanoma. J Invest Dermatol. 2017; 137: 159-69. https://doi.org/10.1016/j.jid.2016.08.024.
132. Jazirehi AR, Nazarian R, Torres-Collado AX, Economou JS. Aberrant apoptotic machinery confers melanoma dual resistance to BRAF(V600E) inhibitor and immune effector cells: immunosensitization by a histone deacetylase inhibitor. Am J Clin Exp Immunol. 2014; 3: 43-56.
133. Yao Y, Zhou J, Wang L, Gao X, Ning Q, Jiang M, Wang J, Wang L, Yu L. Increased PRAME-specific CTL killing of acute myeloid leukemia cells by either a novel histone deacetylase inhibitor chidamide alone or combined treatment with decitabine. PLoS One. 2013; 8: e70522. https://doi.org/10.1371/journal.pone.0070522.
134. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, Carter R, Awad W, Neale G, Thomas PG, Youngblood B. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell. 2017; 170: 142-57.e19. https://doi.org/10.1016/j.cell.2017.06.007.
135. Berger KN, Pu JJ. PD-1 pathway and its clinical application: a 20year journey after discovery of the complete human PD-1 gene. Gene. 2018; 638: 20-5. https://doi.org/10.1016/j.gene.2017.09.050.
136. Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, Chaput N, Eggermont A, Marabelle A, Soria JC, Ferte C. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin Cancer Res. 2017; 23: 1920-8. https://doi.org/10.1158/1078-0432.ccr-16-1741.
137. Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res. 2017; 23: 4242-50. https://doi.org/10.1158/1078-0432.ccr-16-3133.