
INTRODUCTION
As a progressive neurodegenerative disease with a prevalence of 1-2% in the population aged above 65 years [1–3], Parkinson’s disease (PD) is characterized by the main features of tremor, bradykinesia and rigidity, which are present only when more than 70%-80% degeneration of dopaminergic (DA) neurons in the substantia nigra (SN) [4–6]. The profiles of gene expression, such as SNCA, Parkin, PINK1, LRRK2, UCHL1, TRIM24, MUL1, USP30, and DJ-1, have already been shown to be altered in the substantia nigra of PD patients, and may act as potential risk factors in PD diagnosis [2, 7–11]. Healy et al found that six mutations of LRRK2 were proven pathogenic to PD and the frequency of the common LRRK2 Gly2019 Ser mutation was 1% of sporadic PD patients and 4% of hereditary PD patients [2]. Maraganore’s research found that ubiquitin carboxy-terminal hydrolase L1 (UCHL1) was a susceptibility gene for PD and a potential target for disease-modifying therapies [12]. However, these genes expression profiles could not be analyzed in brain during life and were mainly obtained in PD subjects accompanied with classic motor symptoms [13]. Similarly, clinic diagnosis is hard to made before these signs and symptoms appear. Therefore, a wide agreement is that preclinical risk factors of PD are of benefit for earlier preventive therapy that might slow down or even prevent the progression of PD, making the identification of biomarkers for early diagnosis important and necessary. Currently, almost all the risk factors are obtained at some time points in which the PD symptoms are already present [14–16], and there is no developing risk factors obtained from completely normal situation to the onset or even advanced stage of PD in individual person to monitor the progress of PD. Animal models, especially the monkey model, are important tools in experimental medical science to better understand the pathogenesis and therapeutic strategies of PD [17]. Up to now, 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP), which is a neurotoxin specifically damage the DA neurons in SN and cause PD-like symptom [18, 19], has emerged unquestionably as a popular chemical reagent to induce PD model [20].
In the present study, MPTP was used to generate progressive PD monkey model with typical motor symptoms. Nine candidate genes including UCHL1, Parkin, PINK1, LRRK2, TRIM24, MUL1, USP30, DJ-1 and SNCA in whole blood samples were analyzed along with PD progress in an aim to identify specific biomarkers for monitor this progress, and diagnose PD at the early stage. The results showed that six genes’ expression (including Parkin, USP30, MUL1, PINK1, LRRK2 and UCHL1) in peripheral blood were significantly expressed before the present of PD symptoms and then changed along with the progression of PD pathogenesis, and the terminal results were consistent with those of in SN. Thus these results could provide an insight into the functions of these genes at different stages of PD, and may facilitate the development of biomarkers for early diagnosis and efficacy evaluation of new drugs on PD model and patients.
RESULTS
Behavioral assessment
The monkeys were divided into three groups, the sensitive group (total dose, 15.83±1.47mg/kg, MPTP, N=3), moderate sensitive group (total dose, 25.17±3.38 mg/kg, MPTP, N=3) and hyporesponsiveness group (total dose, 47.05±1.35 mg/kg, MPTP, N=2), according to their personal sensitivity and distinct clinical behavioral symptoms. The sensitive group were first revealed motor abnormalities, while the hyporesponsiveness group were the last. However, with the continued MPTP injection, all the monkeys showed mild PD symptoms at the 9th week and gradually reached a classic and stable parkinsonism stage at the 18th week, since then no significant variation between them were observed (Figure 1). In addition, all the monkeys reproduced classic motor phenotypes, including posture and resting tremor, rigidity, bradykinesia and gesture instability.
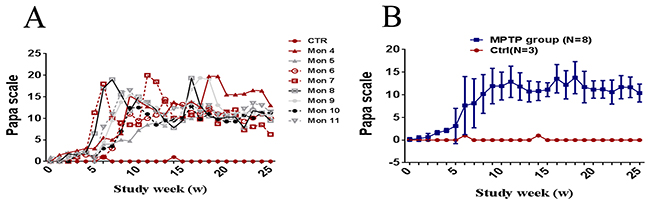
Figure 1: Evolution of motor score by Papa scale. (A) Longitudinal evolution of the clinical score (in weeks) through the protocol for each monkey. (B) Longitudinal evolution of the clinical score (in weeks) through the protocol for all cases. Data are expressed as the mean ± SD.
Pathological examination of SN
Anti-TH immunostaining showed that the number of DA neurons in SN was significantly decreased in PD monkeys (P<0.01, N=8) compared with control group (N=3) (Figure 2). The result indicate that the MPTP-induced monkey model is very successful.
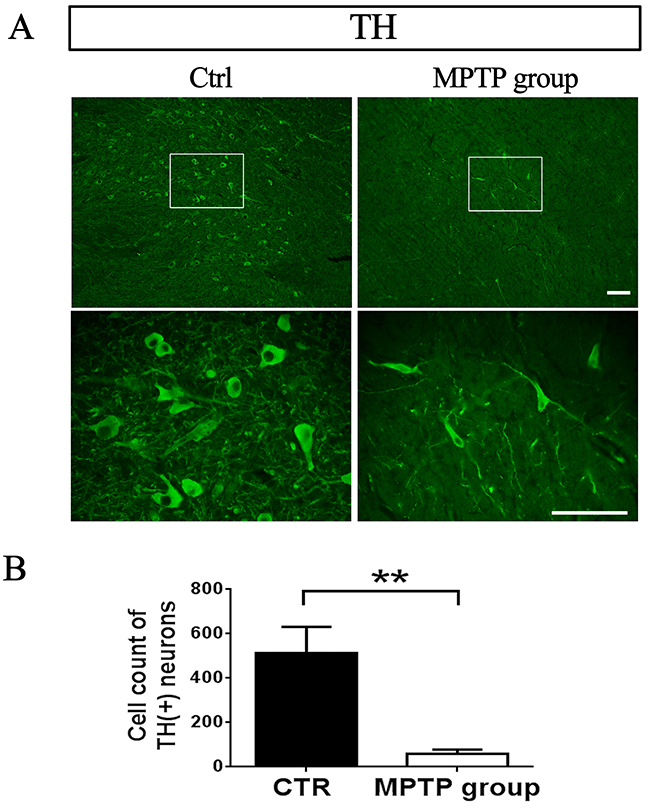
Figure 2: TH-positive neurons in SNpc decreased after MPTP injection. (A) Representative images from control monkeys injected with saline and PD monkeys injected with MPTP. Scale bar, 100μm. (B) DA neurons count in 4x.**P=0.000.
Changes of mRNA of interested genes in whole blood and SN
To identify the transcriptional profiles associated with PD process, we probed RNA extracted from whole blood of eight monkeys during the intoxication of MPTP. The expressions of UCHL1, Parkin, PINK1, LRRK2, TRIM24, MUL1, USP30, DJ-1 and SNCA mRNA were assessed by RT-qPCR in whole blood.
Compared with the pro-MPTP period (0W), the expression of Parkin mRNA significantly increased (P<0.01, N=8) at the 1st week of MPTP injection and remained at a high level (P<0.01, N=8) until the end (Figure 3A). The expression of USP30, MUL1, PINK1, and LRRK2 mRNA, significantly increased at the 3th, 3th, 5th, and 8th week and reached to the highest level at the 8th, 8th, 8th, and 10th week (P<0.01, N=8), respectively, and then remained high until the end (Figure 3B, 3C, 3D, 3E).
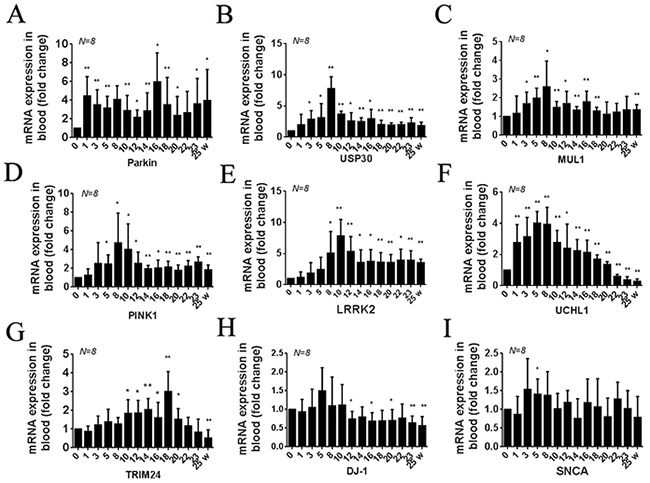
Figure 3: Summary of transcription (RNA) profiles in peripheral blood samples from MPTP-induced monkeys. Quantification shows the different times of Parkin (A) USP30 (B) MUL1 (C) PINK1 (D) LRRK2 (E) UCHL1 (F) TRIM24 (G) DJ-1 (H) and SNCA (I) mRNA expression in all monkeys. Data are expressed as mean ± SD. *P < 0.05, **P<0.01, compared with self-control (0W).
Compared with the pro-MPTP period (0W), the expression of UCHL1 mRNA significantly increased at the 1st week and reached to the highest level at the 8th week, since then gradually decreased and significantly lower than normal value at the 22th week (P<0.01, N=8) (Figure 3F). The expression of TRIM24 mRNA was slowly increased, significantly increased at the 10th week, and reached to the highest level at the 18th week, then gradually decreased and significantly lower than normal value at the 25th week (Figure 3G).
The expression of DJ-1 mRNA was not significantly changed before the week of ten, then significantly decreased and maintained low until the end (P<0.01, N=8) (Figure 3H). Meanwhile, the expression of SNCA mRNA was not significantly changed excepted at the 5th week (Figure 3I).
The six genes, including UCHL1, Parkin, MUL1, USP 30, PINK1 and LRRK2, changes were all earlier than clinical symptoms.
To investigate the validity of the gene measurements in blood, the genes mRNA expression in SN were also be detected by RT-qPCR. Compared with control group, the expression of UCHL1, TRIM24, DJ-1 and SNCA mRNA in SN was significantly decreased (P<0.01, N=8) (Figures 4F, 4G, 4H, 4I), and the expression of Parkin, USP30, MUL1, PINK1 and LRRK2 mRNA in SN was significantly increased (P<0.01, N=8) (Figure 4A, 4B, 4C, 4D, 4E). The terminal results of whole blood were highly consistent with those of in SN.
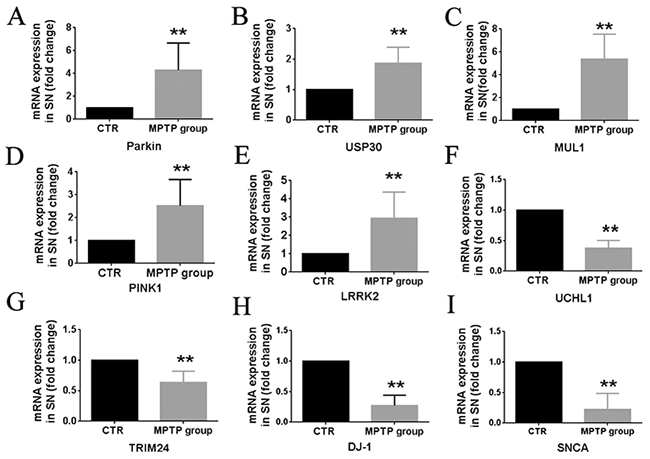
Figure 4: Summary of transcription (RNA) profiles in brain tissues from MPTP-induced monkeys and control monkeys. Quantification shows the Parkin (A) USP30 (B) MUL1 (C) PINK1 (D) LRRK2 (E) UCHL1 (F) TRIM24 (G) DJ-1 (H) and SNCA (I) mRNA expression in SN between MPTP group and control group. Data are expressed as mean ± SD. *P < 0.05, **P<0.01, compared with control monkeys.
DISCUSSION
PD is a progressive neurodegenerative disorder manifested by a broad spectrum of motor and non-motor features, some of which are present in other parkinsonian syndromes, such as progressive supranuclear palsy (PSP), multiple system atrophy (MSA), and corticobasal degeneration (CBD) [21]. Insufficient of perfect and definitive clinical diagnostic tests make the differentiating PD from related disorders a challenge. Therefore, further researches uncovering disease specific biomarkers allowing for its differentiation from other neurodegenerative disorders are required. Considerable damage occurs before onset of clinical symptoms in PD patients, the identification of biomarkers for the early stage is important. Not only will these biomarkers be useful for diagnosing the disease in affected persons, it will be useful for identifying family members or populations at risk, thus providing an opportunity to initiate neuroprotective therapy at an asymptomatic stage. Literatures have reported the transcription (RNA) profiles in peripheral blood samples might be of diagnostic value for estimating the risk of developing sporadic PD [16, 22]. Mitochondrial dysfunction, reactive oxygen species (ROS), proteasome dysfunction and neuroinflammation are implicated in PD [23–26], as the mutations or changes of genes related with these processes, such as PINK1, Parkin, DJ-1, SNCA, UCHL1, LRRK2, TRIM24, MUL1 and USP30, are found to be widely present in PD [27–29]. Thus, it’s reasonable to take these genes as potential markers for the PD diagnosis.
Animal models are important aid to study pathogenesis and therapeutic strategies in human diseases. So far, several chemical materials, including reserpine, methamphetamine, 6-OHDA, MPTP, paraquat-maneb, rotenone, 3-nitrotyrosine and transgenic α-synuclein have been used to generate Parkinson’s disease in mice, monkey and some other animals [17]. Among these models, MPTP induced monkey model is probably the most relevant model available that can generate clinical symptoms remarkably similar to sporadic PD in humans. MPTP cross the blood-brain barrier after administration and is metabolized into its active metabolite 1-methyl-4-phenylpyridinium (MPP+) in astrocytes. Then MPP+ is selectively absorbed into DA neurons via dopamine transporter (DAT), and is thus selectively inhibit mitochondrial complex I [30]. In this study, we developed a progressive and stabilized PD monkey model by intramuscularly injection of MPTP for 22 weeks, with no obvious damage on blood biochemistry (detected every one or three weeks, see Supplementary Figure 1), and kidney and liver architecture (detected at the end by hematoxylin- eosin staining, see Supplementary Figure 2), but with a gradual destruction of DA neurons in substantia nigra and a corresponding progressive appearance of PD symptom. And the selected biomarkers were detected in the blood of monkeys at different stages during the PD progress.
Both PINK1 (PTEN-induced putative kinase) and Parkin (E3-ubiquitin ligase) are located in mitochondria, share a common pathway in PD pathogenesis: The clinical symptoms of Parkin-linked parkinsonism could be influenced by PINK1 mutation [31]; Overexpression of Parkin rescues the muscle damage in PINK1-deficient drosophila [32, 33]. MUL1, which function to guard mitochondrial homeostasis and promote fragmentation of mitochondria, acts in parallel with PINK1 and Parkin, suppresses PINK1 or Parkin mutant phenotypes in Drosophila [34]. MUL1, PINK1, and Parkin were obviously increased in stage I (0W-8W), while all monkeys only showed mild PD symptoms. Thus, this results suggested damages to mitochondrial related degradation at this stage. UCHL1 protein is a deubiquitinating enzyme to cleave the carboxy-terminal peptide bond of polyubiquitine chains, and its I93M mutant mice exhibits decreased enzymatic activity and significant loss of dopaminergic cells [35, 36]. USP30, also one mitochondria-localized deubiquitinase enzyme, suppression of which maintaines mitochondrial health, improves climbing capability, and preventes dopamine depletion in PINK1 or Parkin mutant Drosophilia [33]. So, suppression of this gene may be a promising strategy for PD treatment [37]. LRRK2 (PARK8), which is highly expressed throughout the brain in the dopaminoceptive regions instead of dopaminergic neurons, is suggested to be involved in maintaining functions of autophagy and lysosome [38], regulating neurite growth and cytoskeleton dynamics [39], modifying protein translation and vesicle trafficking [35, 40]. So, the three proteasome related genes (UCHL1, USP30, LRRK2) highly expressed in the present study indicate activation of ubiquitin related degradation since stage I, increased UCHL1 and LRRK2 may act as protect role, increased USP30 may act as a damage role, however.
At stage II (8W-16W), all monkeys showed moderate PD symptoms and gradually reached a state of stable parkinsonism. In the state, mitochondria and proteasome related genes stayed increase, while antioxidant gene (DJ-1) was gradually decreased, suggesting an overwhelm of antioxidant defense by increased oxidative stress. At stage III (17W-22W), all monkeys showed classic and stable PD symptoms, meanwhile, the antioxidative gene DJ-1 in blood remained significantly decreased compared with self-control (0W). At the post-MPTP stage (23W-25W), the clinical symptoms of PD were stabled, at which stage the degeneration of DA neurons and neuritis reached to 80-90%, and the expression of interested genes was similar with stage III. It is noteworthy that the mRNA level of SNCA was not significantly elevated (excepted at the 5th week) during the administration of MPTP, which could be explained by that SNCA mainly expresses in brain and the changes of its level in blood was not enough to be sensitively detected by RT-qPCR [41]. However, the SNCA mRNA significantly decreased in SN at last, rather than accumulation, which can be explained by that 90% of DA neurons were already degenerated at last. TRIM24, one of nuclear localization protein involved in the transcription control of some nuclear receptors, has been found reduced in the peripheral blood and skin explants from parkinsonians [42], directly regulates genes in lipid metabolic, inflammation and damage pathways [43], acts negatively as a p53 (repressed by parkin in PD) regulator and leads to p53-induced apoptosis when depletion of TRIM24 in animal model [44–46]. So the changes of TRIM24 expression in the present study can indicate aberrantly regulation of lipid metabolic, inflammation and apoptosis since stage II. Furthermore, the accurate functions of the above genes remain poorly understood and some of the gene products may share a common pathway that protect or damage nigral neurons during the progress of PD. However, in this study, the longitudinal expression profiles can help us know more about the genes associated with sporadic PD and the relationship between each gene.
In conclusion, MPTP as a specific DA neuronal insult can induce classic clinical symptom of PD accompanied with progressive change of PD-related genes in blood, which reflect pathology progress at different stages. So, the genes UCHL1, Parkin, USP30, MUL1 and PINK1 mRNA may act as early biomarkers, and LRRK2, TRIM24, and DJ-1 mRNA in blood may act as late biomarkers, to mirror the disease process, as well as the treatment effects of sporadic PD.
MATERIALS AND METHODS
Animals
Eleven rhesus monkeys (Macaca mulatta lasiotis) between 3-4.5 years (young adults) and weighting 3.5-5 kg at the beginning of the experiment were obtained from the Sichuan Primed Biological Technology Co., Ltd / National (Sichuan) Experimental Rhesus Monkey Resources Base (Certificate No SCXK(Chuan): 2013-105). Animals were housed in individual primate cages (one animal per cage) and their care and treatment were in strict accordance with the “the National Institutes of Health Guide for the Care and Use of Laboratory Animal” of the United States, and all the experimental protocols had been reviewed and approved by the Animal Welfare and Use Committee.
MPTP-induced parkinsonism
At the beginning of the research, eight monkeys from MPTP group were respectively daily administrated with a small dose (0.2 mg/(kg·d)) of 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP-HCl, Product Number: M0896, Sigma, St. Louis, MO), and then accepted the reagent by every 2-4 days to stable the clinical phenotype after appeared typical motor symptoms. Another three monkeys from control group were accepted the injection of saline instead, and the other conditions were the same with MPTP group.
Behavioral assessment
Before and after two weeks of the experimental protocol, assessment was carried out all day by video tape. The severity of behavioural phenotype of PD was evaluated by using the Papa scale [47, 48]. Briefly, the Papa scale including: posture (0 – 2); gait (0 – 2); tremor (0 – 2); general mobility (0 – 4), hand movements (0 – 2), climbing (0 – 4), holding food (0 – 1), eating (0 – 1), social interaction (0 – 2). Total score can vary from 0 to 20.
Blood collection
Blood collection was carried out by vein around 8:30-9:00 AM before breakfast every two or three weeks. Then more fresh fruits and vegetables than normally were given to supplemental nutrition.
Total RNA extraction and complimentary DNA synthesis
Total RNA was isolated from fresh peripheral blood samples by using RNAiso Blood Extractor reagent (Takara Biotechnology Co., Ltd. Dalian, China) according to the manufacturer’s instructions. RNA integrity was conducted by 1.2% formaldehyde agarose gel electrophoresis, and the 28S/18S ratio was >1.5. In addition, the concentration of total RNA was measured by spectrophotometer, and the A260/A280 ratio was typically >1.9. Total RNA samples were digested with DNase-I to remove contaminated genomic DNA, and subsequently the standard reverse transcription was performed using the PrimeScriptTM RT reagent Kit (Takara Biotechnology Co., Ltd. Dalian, China) and each RT reaction contained 0.5 μg of total RNA according to the manufacturer’s instructions, then stored at -70°C for further use.
Quantitative real-time RT-qPCR analysis
Quantitative real-time RT-qPCR was conducted for 10 genes (Table 1): α-synuclein (SNCA); PTEN-induced putative kinase 1 (PINK1); Parkin; DJ-1; leucine-rich repeat kinase 2 (LRRK2); MUL1; tripartite motif containing 24 (TRIM24); ubiquitin C-terminal hydrolase (UCHL-1); ubiquitin-proteasome system 30 KDa protein (USP30); β-actin (used as house-keeping gene).
Table 1: Sequences of primer pairs used for RT-qPCR
Genes |
Accession number (Gene Bank) |
Primer pairs sequence (5’-3’) |
Location (bp) |
Size (bp) |
|---|---|---|---|---|
SNCA |
XM_001095402.2 |
AGCAAGTGACAAATGTTGGAGGA |
152-174 |
128bp |
ATTCTTGCCCAACTGGTCCTT |
259-279 |
|||
PINK1 |
JU325788.1 |
TCCTCGTTATGAAGAACTATCCCTG |
855-879 |
163bp |
AGGATGTTGTCGGATTTCAGGT |
996-1017 |
|||
Parkin |
XM_001100436.2 |
TGTGGGTTTGCCTTCTGCC |
1078-1096 |
116bp |
GCGGCTCTTTCATCAACTCTGT |
1172-1193 |
|||
DJ-1 |
JV046088.1 |
GAGCAGAGGAAATGGAGACGG |
38-58 |
139bp |
TCAAGACTGGCATCAGGACAAAT |
154-176 |
|||
LRRK2 |
XM_002798570.1 |
ATGCACTCACGAGCTTTCCAC |
286-306 |
137bp |
AGAGACATCAAGGTTAGCAACACAA |
398-422 |
|||
MUL1 |
NM_001261655.1 |
CTTGCTGTGGACTTGGTGGAG |
1410-1430 |
102bp |
TGCGGCAGAACCTCTTTGG |
1493-1511 |
|||
TRIM24 |
NM_001260835.1 |
ATACCACGACAAGCAATAAAGCC |
1636-1658 |
139bp |
TGGGACTGGAAGGAGTAGAGGAT |
1752-1774 |
|||
UCHL1 |
JV044711.1 |
GCTGCTGTTTCCACTCACGG |
150-169 |
146bp |
CTGCGTGTATAAGTCCGATTGTG |
273-295 |
|||
USP30 |
NM_001257829.1 |
GTTCGATTTGATACCTTTGATAGCC |
727-751 |
120bp |
AACATCCCGCACTGATTCTGA |
826-846 |
|||
β-actin |
NM_001033084.1 |
GTGACGTGGACATCCGTAAAGAC |
854-876 |
162bp |
CAGAGTACTTGCGCTCAGGAGG |
994-1015 |
All the RT-qPCR procedures were performed by the Bio-rad CFX96 Sequence Detection System in 25 μl reaction containing 12.5 μl of SYBR®Premix Ex Taq II, 2μl of cDNA, 1 μl of each primer, and 8.5 μl of dH2O. Polymerase chain reactions were carried out using the following temperature profile: at 95°C for 3 minutes, 95°C for 10 s, 53-59°C for 30 s, and 72°C for 10 s, 40 cycles, and 72°C for 10min. The relative expression analysis was quantified by the comparative Ct method using the Bio-rad CFX96 system program. Finally, the delta Ct was converted into fold changes. All PCR reactions were run in three duplicates.
Histopathologic evaluation
At the end of experiment, all monkeys were anaesthetized and their brain were immediately dissected. Left cerebral hemisphere of every monkey was cut into coronal slices (400 μm) fixed in 4% paraformaldehyde for at least 24h, right cerebral hemisphere was cut out the SN stored in liquid nitrogen until use. After continuous coronal slices (15 μm), anti-TH (1:1000, ENZO, USA) immunofluorescence staining was used to observed the degeneration of DA neurons in SN. Then the photographs were captured with fluorescence microscopy (OLYMPUS, BX43, Japan), and the number of DA neurons was counted by Image-Pro Plus 5.1 (USA) in 4×.
Statistical analysis
Data analysis was performed using Graphpad Prism 5.0, independent sample test was analyzed using SPSS 20.0 software (IBM Corp, USA), and results were expressed as mean ± standard deviation (x ± SD). *P<0.05 and **P<0.01 were considered to be significantly differences compared with self-control (0W) or compared with control monkeys injected with saline.
ACKNOWLEDGMENTS
This work was supported by the National Key Technology Support Program (2014BAI03B01) and National Key Scientific Instrument and Equipment Development Project of China (2013YQ49085906).
CONFLICTS OF INTEREST
All authors have no actual or potential conflicts of interest.
REFERENCES
1. Scherzer CR, Eklund AC, Morse LJ, Liao Z, Locascio JJ, Fefer D, Schwarzschild MA, Schlossmacher MG, Hauser MA, Vance JM. Molecular markers of early Parkinson’s disease based on gene expression in blood. Proceedings of the National Academy of Sciences. 2007; 104: 955-960.
2. Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, Ferreira JJ, Tolosa E, Kay DM, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurology. 2008; 7: 583-590.
3. Lim KL, Zhang CW. Molecular events underlying Parkinson’s disease - an interwoven tapestry. Front Neurol. 2013; 4: 33.
4. Agid Y. Parkinson’s disease: pathophysiology. The Lancet. 1991; 337: 1321-1324.
5. Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM. Compensations after lesions of central dopaminergic neurons: some clinical and basic implications. Trends in neurosciences. 1990; 13: 290-296.
6. Riederer P. Time course of nigrostriatal degeneration in Parkinson’s disease. Journal of neural transmission. 1976; 38: 277-301.
7. Kumaran R, Vandrovcova J, Luk C, Sharma S, Renton A, Wood NW, Hardy JA, Lees AJ, Bandopadhyay R. Differential DJ-1 gene expression in Parkinson’s disease. Neurobiology of disease. 2009; 36: 393-400.
8. Lu L, Neff F, Alvarez-Fischer D, Henze C, Xie Y, Oertel WH, Schlegel J, Hartmann A. Gene expression profiling of Lewy body-bearing neurons in Parkinson’s disease. Experimental neurology. 2005; 195: 27-39.
9. Shehadeh L, Mitsi G, Adi N, Bishopric N, Papapetropoulos S. Expression of Lewy body protein septin 4 in postmortem brain of Parkinson’s disease and control subjects. Movement Disorders. 2009; 24: 204-210.
10. Aguiar PM, Severino P. Biomarkers in Parkinson Disease: global gene expression analysis in peripheral blood from patients with and without mutations in PARK2 and PARK8; Biomarcadores na doença de Parkinson: avaliação da expressão gênica no sangue periférico de pacientes com e sem mutações nos genes PARK2 e PARK8. Einstein (Säo Paulo). 2010; 8.
11. Mutez E, Leprêtre F, Le Rhun E, Larvor L, Duflot A, Mouroux V, Kerckaert JP, Figeac M, Dujardin K, Destée A. SNCA locus duplication carriers: from genetics to Parkinson disease phenotypes. Human mutation. 2011; 32: E2079-E2090.
12. Maraganore DM, Lesnick TG, Elbaz A, Chartier-Harlin MC, Gasser T, Kruger R, Hattori N, Mellick GD, Quattrone A, Satoh J, Toda T, Wang J, Ioannidis JP, et al. UCHL1 is a Parkinson’s disease susceptibility gene. Ann Neurol. 2004; 55: 512-521.
13. Bhidayasiri R. Dystonia: genetics and treatment update. Neurologist. 2006; 12: 74-85.
14. Foulds PG, Diggle P, Mitchell JD, Parker A, Hasegawa M, Masuda-Suzukake M, Mann DM, Allsop D. A longitudinal study on alpha-synuclein in blood plasma as a biomarker for Parkinson’s disease. Sci Rep. 2013; 3: 2540.
15. Molochnikov L, Rabey JM, Dobronevsky E, Bonucelli U, Ceravolo R, Frosini D, Grunblatt E, Riederer P, Jacob C, Aharon-Peretz J. A molecular signature in blood identifies early Parkinson’s disease. Mol Neurodegener. 2012; 7.
16. Grünblatt E, Zehetmayer S, Jacob CP, Müller T, Jost WH, Riederer P. Pilot study: peripheral biomarkers for diagnosing sporadic Parkinson’s disease. Journal of neural transmission. 2010; 117: 1387-1393.
17. Betarbet R, Sherer TB, Greenamyre JT. Animal models of Parkinson’s disease. Bioessays. 2002; 24: 308-318.
18. Langston JW, Forno LS, Rebert CS, Irwin I. Selective nigral toxicity after systemic administration of 1-methyl-4-phenyl-1, 2, 5, 6-tetrahydropyrine (MPTP) in the squirrel monkey. Brain research. 1984; 292: 390-394.
19. Burns RS, Chiueh CC, Markey SP, Ebert MH, Jacobowitz DM, Kopin IJ. A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine. Proceedings of the National Academy of Sciences. 1983; 80: 4546-4550.
20. Beal MF. Experimental models of Parkinson’s disease. Nature Reviews Neuroscience. 2001; 2: 325-334.
21. Seppi K, Schocke M, Esterhammer R, Kremser C, Brenneis C, Mueller J, Boesch S, Jaschke W, Poewe W, Wenning G. Diffusion-weighted imaging discriminates progressive supranuclear palsy from PD, but not from the parkinson variant of multiple system atrophy. Neurology. 2003; 60: 922-927.
22. Grünblatt E. Parkinson’s disease: Molecular risk factors. Parkinsonism & Related Disorders. 2012; 18: S45-S48.
23. Rocha EM, De Miranda B, Sanders LH. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis. 2017.
24. Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci. 2006; 7: 207-219.
25. Sun F, Kanthasamy A, Anantharam V, Kanthasamy AG. Environmental neurotoxic chemicals-induced ubiquitin proteasome system dysfunction in the pathogenesis and progression of Parkinson’s disease. Pharmacol Ther. 2007; 114: 327-344.
26. Manoharan S, Guillemin GJ, Abiramasundari RS, Essa MM, Akbar M, Akbar MD. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxid Med Cell Longev. 2016; 2016: 8590578.
27. Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004; 44: 595-600.
28. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004; 304: 1158-1160.
29. Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003; 299: 256-259.
30. Heikkila RE, Hess A, Duvoisin RC. Dopaminergic neurotoxicity of 1-methyl-4-phenyl-1, 2, 5, 6-tetrahydropyridine in mice. Science. 1984; 224: 1451-1453.
31. Funayama M, Li Y, Tsoi TH, Lam CW, Ohi T, Yazawa S, Uyama E, Djaldetti R, Melamed E, Yoshino H, Imamichi Y, Takashima H, Nishioka K, et al. Familial Parkinsonism with digenic parkin and PINK1 mutations. Mov Disord. 2008; 23: 1461-1465.
32. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006; 441: 1162-1166.
33. Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006; 441: 1157-1161.
34. Yun J, Puri R, Yang H, Lizzio MA, Wu C, Sheng ZH, Guo M. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. eLife. 2014; 3: e01958.
35. Gehrke S, Imai Y, Sokol N, Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010; 466: 637-641.
36. Yasuda T, Nihira T, Ren YR, Cao XQ, Wada K, Setsuie R, Kabuta T, Wada K, Hattori N, Mizuno Y, Mochizuki H. Effects of UCH-L1 on alpha-synuclein over-expression mouse model of Parkinson’s disease. J Neurochem. 2009; 108: 932-944.
37. Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014; 510: 370-375.
38. Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ 3rd, Shen J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A. 2010; 107: 9879-9884.
39. MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006; 52: 587-593.
40. MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013; 77: 425-439.
41. Hatano T, Kubo S, Sato S, Hattori N. Pathogenesis of familial Parkinson’s disease: new insights based on monogenic forms of Parkinson’s disease. J Neurochem. 2009; 111: 1075-1093.
42. Nenasheva VV, Novosadova EV, Makarova IV, Lebedeva OS, Grefenshtein MA, Arsenyeva EL, Antonov SA, Grivennikov IA, Tarantul VZ. The Transcriptional Changes of trim Genes Associated with Parkinson’s Disease on a Model of Human Induced Pluripotent Stem Cells. Mol Neurobiol. 2016.
43. Jiang S, Minter LC, Stratton SA, Yang P, Abbas HA, Akdemir ZC, Pant V, Post S, Gagea M, Lee RG, Lozano G, Barton MC. TRIM24 suppresses development of spontaneous hepatic lipid accumulation and hepatocellular carcinoma in mice. J Hepatol. 2015; 62: 371-379.
44. da Costa CA, Sunyach C, Giaime E, West A, Corti O, Brice A, Safe S, Abou-Sleiman PM, Wood NW, Takahashi H, Goldberg MS, Shen J, Checler F. Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson’s disease. Nat Cell Biol. 2009; 11: 1370-1375.
45. Allton K, Jain AK, Herz HM, Tsai WW, Jung SY, Qin J, Bergmann A, Johnson RL, Barton MC. Trim24 targets endogenous p53 for degradation. Proc Natl Acad Sci U S A. 2009; 106: 11612-11616.
46. Jain AK, Allton K, Duncan AD, Barton MC. TRIM24 is a p53-induced E3-ubiquitin ligase that undergoes ATM-mediated phosphorylation and autodegradation during DNA damage. Mol Cell Biol. 2014; 34: 2695-2709.
47. Papa SM, Chase TN. Levodopa–induced dyskinesias improved by a glutamate antagonist in parkinsonia monkeys. Annals of neurology. 1996; 39: 574-578.
48. Imbert C, Bezard E, Guitraud S, Boraud T, Gross CE. Comparison of eight clinical rating scales used for the assessment of MPTP-induced parkinsonism in the Macaque monkey. Journal of neuroscience methods. 2000; 96: 71-76.