
Introduction
Urothelial carcinoma of the bladder (UCC) is the fourth commonest male malignancy [1]. Despite its high prevalence and the cost of managing affected patients, there have been few advances in the treatment of this disease since the introduction of cisplatin based regimens [2, 3]. Clinical and molecular data suggest UCC is characterised by two distinct phenotypes with low and high-grade differentiation [4, 5]. Whilst the former has an excellent prognosis [6], high-grade tumors are aggressive cancers that progress to invasion, metastases and death [7]. Recent profiling reports have revealed invasive high-grade UCC can be clustered according to genes reflecting FGFR3 status (including papillary histology and FGFR3 related microRNAs), those seen in luminal breast cancer, and epithelial lineage and stem/progenitor cytokeratins (including a proportion of tumors with squamous histology) [8]. This clustering can reflect basal-like and luminal compartments in breast cancer, and may result in different sensitivities to cisplatin based chemotherapy [9], [10].
These data underlie the importance of FGFR3 in UCC biology. Whilst mutation of this oncogene occurs most commonly in low-grade non-invasive UCC [11], FGFR3 upregulation characterises the large chemosensitive sub-group of invasive cancers [10]. We were the first group to report FGFR3 upregulation in UCC through loss of microRNA-99a/100 expression (miRs-99a/100) [12]. We identified this appeared before FGFR3 mutation and may explain the high proportion of invasive UCC with upregulated wild type FGFR3 [8]. Importantly, the FGFR3 cluster of invasive UCC are also characterised by loss of expression of miRs-99a/100. MicroRNAs (miRs) are short non-coding RNA molecules that post-transcriptionally modulate protein expression [13]. Mature miRs are directed to mRNAs with a complementary seed sequence to nucleotides 1-8 of their 5’ structure. Modulation of protein expression appears proportional to the extent of complementation, the context, the frequency and proximity, and the evolutionary conservation of the seed sequence. As each miR may target many mRNAs, we hypothesized that miRs-99a/100 modulate the expression of other mRNAs important for UCC biology. Here we report a search for and validation of potential candidates.
Results
Identification of potential targets for miRs-99a/100
We extracted mRNA expression profiles from 7 microarray datasets [14-18] [19] [20] defining genes upregulated in n=958 low grade, superficial or non-muscle invasive UCC (n=12 Oncomine concepts, Supplementary table 1) when compared to either invasive (n=446) tumors or normal urothelial (n=135) samples. We selected the top 10% of upregulated genes (4,935 unique genes) and identified 20 that were predicted to be targets of miRs-99a/100 (from a total of 52 predicted targets). Upregulated genes appeared enriched for miR-99a/100 targets, when compared to the entire genome (20/4,935 vs 52/32,000, χ2=8.2, p<0.001). We ranked genes according to frequency of detection in the microarray comparisons (table 1). The commonest detected target was FGFR3 (found in 8/12 concepts), followed by the transcription factors FOXA1 and HOXA1 (in 6/12 and 3/12 concepts, respectively). We correlated expression profiles for miRs-99a/100 with these predicted targets using RNA profiles in the 72 samples from cohort 1 (table 2) and identified significant inverse correlations for FGFR3 (r=-0.48 to -0.52, p<0.01) and FOXA1 (r=-0.33 to -0.43, p<0.05). Non-significant negative correlations were seen for HOXA1.
Table 1: Predicted targets of microRNA-99a/100 found with increased expression in low grade or non-invasive in Urothelial carcinoma of the bladder. The frequency of detection (from 12 Oncomine concepts) is shown, together with the Pearson’s correlation coefficient to the expression of miRs-99a/100 in 72 urothelial samples
|
Freq. in Oncomine concepts |
Pearson's correlation of miR-mRNA expression |
|
Gene |
miR-99a |
miR-100 |
|
FGFR3 |
8 |
-0.48** |
-0.52** |
FOXA1 |
6 |
-0.43** |
-0.33* |
HOXA1 |
3 |
-0.22 |
-0.16 |
BMPR2 |
2 |
0.03 |
0.10 |
ICMT |
2 |
0.07 |
0.05 |
MTMR3 |
2 |
0.04 |
0.05 |
OGT |
2 |
0.04 |
0.05 |
ZBTB7A |
2 |
-0.11 |
-0.05 |
ADCY1 |
1 |
-0.09 |
0.12 |
C4orf16 |
1 |
0.32 |
0.14 |
EIF2C2 |
1 |
-0.18 |
-0.12 |
FRAP1 |
1 |
-0.01 |
0.04 |
HS3ST3B1 |
1 |
0.08 |
0.08 |
IGF1R |
1 |
-0.06 |
-0.07 |
NXF1 |
1 |
0.12 |
0.07 |
PI15 |
1 |
0.05 |
0.10 |
PPP1CB |
1 |
0.04 |
0.13 |
SMARCA5 |
1 |
0.07 |
0.02 |
SMARCD1 |
1 |
0.18 |
0.04 |
ZZEF1 |
1 |
0.20 |
0.03 |
* Significance p<0.05 |
|||
** Significance p<0.01 |
|||
Table 2: Description of the patient samples used to investigate miRs-99a/100 targeting of mRNAs in bladder cancer. The first cohort was used to investigate correlations between potential target genes and microRNA expression. The second cohort was used to explore the role of the predicted targets across the UCC spectrum
|
|
Cohort 1 |
|
Cohort 2 |
||
|
|
n |
% |
|
n |
% |
Tissue |
UCC |
52 |
72.2% |
207 |
94.1% |
|
Normal (UCC case) |
10 |
13.9% |
7 |
3.2% |
||
Normal (non-UCC) |
10 |
13.9% |
6 |
2.7% |
||
Gender |
Male |
40 |
76.9% |
161 |
73.2% |
|
Female |
12 |
23.1% |
59 |
26.8% |
||
Age |
Mean |
72.3 yrs |
71.9 yrs |
|||
Range |
46-90 yrs |
36-93 yrs |
||||
Phenotype |
Low Grade NMI |
22 |
42.3% |
60 |
27.3% |
|
High Grade NMI |
12 |
23.1% |
56 |
25.5% |
||
Invasive |
18 |
34.6% |
81 |
36.8% |
||
Not known |
8 |
3.6% |
||||
Stage |
pTa |
25 |
48.1% |
74 |
33.6% |
|
pTis |
2 |
3.8% |
9 |
4.1% |
||
pT1 |
7 |
13.5% |
35 |
15.9% |
||
pT2-4 |
18 |
34.6% |
81 |
36.8% |
||
Not known |
8 |
3.6% |
||||
Recurrence |
Yes |
27 |
51.9% |
49 |
22.3% |
|
No |
25 |
48.1% |
149 |
67.7% |
||
Progression |
Yes |
18 |
34.6% |
61 |
27.7% |
|
No |
34 |
65.4% |
137 |
62.3% |
||
Follow up |
Mean |
35.6 months |
32.4 months |
|||
Range |
0-93 months |
0.6-111 months |
||||
Total |
|
72 |
100% |
|
220 |
100% |
Targeting of FOXA1 by miRs-99a/100
To explore targeting of FOXA1 and HOXA1 by miRs-99a/100, we examined protein expression in NHU cells following microRNA knockdown. For comparison we included FGFR3, phosphorylated-ERK 1/2 (as a marker of FGFR3 signaling pathway activity) and IGF1R (also potential target). Upregulation of FOXA1 (2.9 to 3.1 fold (±st. dev. 0.23 to 0.32)) and FGFR3 (1.5 to 2.1 fold (±0.15 to 0.23)) and ERK1/2 phosphorylation (1.3 to 2.5 fold (±0.1 to 0.4)) was seen following knockdown (figure 1a). In contrast, no change was seen for IGF1R and HOXA1. To investigate direct targeting of the FOXA1 3’ UTR we synthesized a luciferase reporter construct incorporating the miR-99a/100 seed sequence from FOXA1. We observed increases in luciferase fluorescence with anti-miRs to miR-99a (1.4 fold (±0.2)) and miR-100 (1.5 fold (±0.21), figure 1b).
Whilst direct targeting of the FOXA1 3’ UTR may occur through miRs-99a/100, we wondered whether a further indirect pathway may contribute to the 2-3 fold rise in protein expression seen. We investigated the potential for microRNA networks within a tumor [21]. We performed TLDA multiplex qrtPCR in triplicate on the NHU cells with anti-miRs to 99a/100 and a scrambled control. When normalized to this control, concordant reduced expression (<0.5 fold) was seen for 9 miRs (7 shared between miRs-99a/100) and increased expression (>2 fold) for 63 miRs (supplementary figure 1). We focused upon the 7 miRs with reduced expression for both miRs-99a/100, hypothesizing their loss could lead to upregulation of FOXA1 promoting factors. The expression of each miR was closely correlated (Pearson’s Coefficient r=0.43 to 0.74, p=8.0x10-14 to 0.08, table 3) with miRs-99a/100 in the 72 urothelial samples from cohort 1. Predicted mRNA targets of these 7 miRs (obtained from TargetScan) included 14 transcription factors with binding sites within the FOXA1 promoter, e.g. miR-485-5p predicted to target SOX5, JUN-D and FOXL1, and miR-486 proven to regulate FOXO1a [22]. As such, loss of expression of these miRs may lead to upregulation of these transcription factors and increased FOXA1.
Table 3: Correlation of microRNAs with loss of expression following miRs-99a/100 knock-down. (a). Expression is shown (as fold change) for each miR in NHU cells transfected with anti-miRs to miRs-99a/100, normalised to scrambled RNA control. For each, the correlation of expression is also shown in 72 urothelial samples (from cohort 1). In (b). we show predicted targeting by these microRNAs of transcription factors with binding sites within the FOXA1 promoter. For example, miR-485-5p is predicted to target 3 transcription factors. Targeting of FOXO1a by miR-486 has been reported [22]
a). Reciprocal loss of microRNA expression following miRs-99a/100 knock-down |
|||||||
|
miR-485-5p |
miR-500a |
miR-486 |
let-7e |
miR-657 |
miR-133b |
miR-139-5p |
Fold change (with anti miR-100) |
0.001 |
0.01 |
0.01 |
0.04 |
0.04 |
0.001 |
0.05 |
Fold change (with anti miR-99a) |
0.001 |
0.01 |
0.06 |
0.27 |
0.33 |
0.99 |
0.05 |
Correlation with miR-100 (Pearson, R) |
0.71 |
0.52 |
0.43 |
0.56 |
0.26 |
0.65 |
0.68 |
p value |
2.01E-09 |
6.20E-06 |
4.98E-05 |
5.14E-08 |
9.52E-02 |
4.03E-11 |
2.38E-11 |
Correlation with miR-99a (Pearson, R) |
0.68 |
0.57 |
0.46 |
0.6 |
0.27 |
0.65 |
0.74 |
p value |
1.61E-08 |
7.60E-07 |
1.23E-05 |
4.61E-09 |
8.87E-02 |
4.12E-11 |
8.00E-14 |
b). Transcription factors within the FOXA1 promoter |
|||||||
|
miR-485-5p |
miR-500a |
miR-486 |
let-7e |
miR-657 |
miR-133b |
miR-139-5p |
AHR |
|
1 |
|
|
|
|
|
AML1A |
1 |
||||||
BACH1 |
1 |
||||||
FOXL1 |
1 |
||||||
FOXO1a |
1 |
1 |
|||||
HOXA9 |
1 |
1 |
1 |
||||
JunD |
1 |
||||||
SOX5 |
1 |
1 |
|||||
Sp1 |
|
|
|
|
1 |
1 |
|
FOXA1 and FGFR3 expression in bladder cancer
Whilst recent data report upregulation of both FGFR3 and FOXA1 characterize the papillary (luminal breast cancer-like) molecular subtype of invasive UCC [8] [10], these reports did not include low-grade tumors. To examine expression throughout the UCC spectrum and to compare profiles with outcome, we measured mRNA using QrtPCR in 220 urothelial samples (cohort 2). For both mRNAs, highest expression was seen in non-invasive pTa and low-grade tumors (ANOVA p<0.02, Supplementary figure 2)). Upregulation of both FGFR3 and FOXA1 (when dichotomized around the mean) was seen in 69% of low-grade, in 47% of high-grade NMI and in 38% of muscle invasive cancers with high expression of either (figure 1c, χ2=18.0, p<0.001). Tumors with high expression had lower rates of progression to more advanced disease, when compared to those with low expression (figure 1d). This difference reached significance for FOXA1 (Log rank p=0.009, Bonferroni corrected) but not FGFR3 (p=0.08). Sub-group analysis revealed that this difference in progression free survival was only apparent in non-invasive cancers (supplementary figure 3) and in low grade tumors (data not shown, Log rank values for FOXA1; low grade p=0.002, high grade 3 p=0.24. Log rank values for FGFR3; low grade 1 p=0.003, high grade 3 p=0.29).
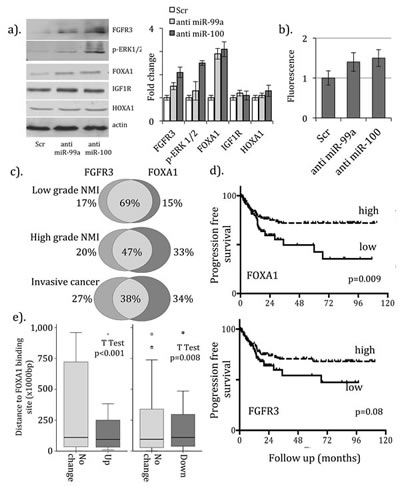
Figure 1: Regulation of FOXA1 expression by microRNAs-99a/100 in bladder cancer. (a). Western blotting for protein expression reveals upregulation of FGFR3, phosphorylation of ERK1/2 and FOXA1 following transfection of NHU cells with anti-miRs to miRs-99a/100 but not the scrambled RNA control. Image densitometry reveals 3-fold increases in expression for FOXA1 with anti-miRs-99a/100. (b). A reporter construct assay reveals 1.3/1.4 fold increases in luciferase expression in NHU cells treated with anti-miRs-99a/100, when normalized to the scrambled control. Expression profiling using QrtPCR in 220 benign and malignant urothelial samples revealed (c). overlap of upregulation for FOXA1 and FGFR3 in UCC, which is greatest in low-grade NMI cancers, and (d). lower rates of progression to more advanced disease for tumors with high FGFR3 and high FOXA1 when compared to those with low expression (the difference in greatest in low stage and low tumors (supplementary figure 3)). (e). Genes with upregulation following FOXA1 transfection in T24/EJ cells are located closer to known FOXA1 binding sites (in HepG2 cells) than those with no change in expression or those with reduced expression (down regulation).
Molecular events of FOXA1 upregulation in Bladder Cancer
Having identified high FOXA1 and high FGFR3 expression are correlated in a cohort of UCC and partially regulated through miRs-99a/100, we wondered about the molecular consequences of FOXA1 upregulation in UCC. For analysis, we transfected EJ/T24 cells (as they have low FOXA1 expression, have wild type FGFR3 and are not dependent upon FGF signaling) with FOXA1 or the empty plasmid (control)(supplementary figure 4) [23]. We analyzed whole genome mRNA expression and CpG methylation, as reports show that FOXA1 recruitment is associated with DNA demethylation and changes in chromatin conformation [24-26] [27]. Microarray data were filtered for probes concordant between duplicates and experimental replicates, before matching between platforms and normalizing to control (cells transfected with the empty plasmid). Our final dataset included 12,939 genes with 162,338 matching CpG probes across the promoter region (n=51,112), inside exon 1 and around transcription start sites (n=105,133), and downstream from the gene (n=6,093) (dataset is available at http://www.sheffield.ac.uk/oncology/units/urology/data). As expected, we observed increases in gene expression with the density of CpG hypomethylation and reductions in expression with hypermethylation (T test p<0.001, supplementary figure 5) in FOXA1 transfected cells [28]. These associations were closest for CpG probes within the gene promoter and around the transcription start site/exon 1 (annotated as inside the gene), when compared to probes downstream from the gene.
FOXA1 transfection produced upregulation of 5,455/12,939 (42%) transcripts including 1,650 (13%) with more than 1.2 fold increase (supplementary figure 5), when compared to controls. Differences in expression reached significance (FDR <5%) in 50 genes, including 14 up and 36 down-regulated mRNAs. These were involved in the negative regulation of signal transduction, chromatin alterations and nucleosome assembly, DNA methylation, DNA binding and mutagenesis repair and metabolic regulation (p<0.05, supplementary table 2). Identified members included those previously shown to be aberrantly silenced through DNA hypermethyaltion in UCC, e.g. IGFBP3 [29], and those found to charactize breast cancer sub-types (e.g. ERBB2) [10]. MEDIP-Chip identified increases in CpG hypomethylation for 32/111(29%) promoter, 32/96 (33%) inside and 10/13 (77%) downstream probes (chi sq. p=0.002) within 5 of the 14 upregulated genes. Contrastingly, reduced mRNA expression was seen in 7,484 (58%) genes following FOXA1 transfection, including 567 (4%) with less than 0.8 fold change. Increases in aberrant hypermethylation were seen at CpG loci in 1,708 (23%) and 134 (24%) of these down regulated genes, respectively. Motallebipour et al. mapped FOXA1 binding sites in the hepatocellular carcinoma cell line HepG2 [30]. We annotated our dataset with these loci and compared proximity for these 50 significantly altered genes. Genes with increased expression following FOXA1 transfection were significantly closer to FOXA1 binding sites (mean 217,515bp (95%CI=159,432-275,598)) than those without change (1,774,996bp (1,110,313-2,439,680), figure 1e, T Test p<0.001). Conversely, those with reduced expression following FOXA1 transfection were significantly more distant (2,048,425bp (1,015842-3,081,008) than those without change (802,277bp (425,626-1,178,928) p=0.008).
FOXA1 gene expression profiles in FGFR3 mutant and sporadic bladder tumors
Our data report correlated upregulation of FOXA1 and FGFR3 in UCC, partly through dual regulation by miRs-99a/100. Consequently, tumors with aberrant FGFR3 or FOXA1 expression may have symmetry of genotype. Lindgren et al. reported 380 genes upregulated and 468 down regulated in 46 UCC with mutant FGFR3 compared to 29 wild type controls [31]. We identified 222 and 325 of these, respectively, in our dataset. When compared, genes with upregulation in FGFR3 mutant cancers had significantly higher expression in the FOXA1 transfected cells (average 1.10±0.2 fold change, p<0.001, Figure 2a) than those without change. The difference was greater when genes with probes enriched for CpG hypomethylation were selected (relative fold change 1.12±0.2, p<0.001). In contrast, no difference in expression was seen for genes down regulated in FGFR3 mutant cancers in our FOXA1 transfected cells, even if those with hypomethylated or hypermethylated probes were selected (fold change 1.03±0.17, p=0.08). Comparison between the genes altered with FGFR3 mutation, with FOXA1 transfection and changes in DNA methylation revealed considerable overlap (figure 2c). Genes with increased expression in FGFR3 mutant cancers were significantly closer to FOXA1 binding sites (433,101bp (95%CI=331,878-534,324)) than those without change (837,209bp (814,843-859,575) T Test p<0.001). Those with reduced expression in FGFR3 mutants were also significantly closer (315,103bp (261,634-368,572)) than those without change (841,086bp (818,606-863,570), p<0.001).
For external validation, we examined the genes with symmetrical changes in FGFR3 mutant UCC and FOXA1 transfected cells (135 up and 170 down regulated) using a large microarray dataset of 256 sporadic UCC [32]. We identified 295 (97%) members and found significant aberrant expression, in the symmetrical manner seen in FOXA1 transfected cells and FGFR3 mutant UCC, for most gene members (89 (67%) up and 87 (54%) down-regulated genes, T Test p<0.05, suppl. figure 8a). There was significant correlation of expression between FOXA1 and FGFR3 (r=0.45 (95%CI 0.35-0.55), p<0.001), and once again the expression of each was highest in non-muscle invasive cancers (suppl. figure 8b, Χ2 p<0.01). Comparative analysis revealed 156/295 (54%) of these genes were significantly differentially expressed between low-grade NMI, high-grade NMI or invasive cancers (Figure 2c, t test p<0.05) in the manner expected from FGFR3 mutant UCC/FOXA1 transfected cells. The expression of many genes was significantly correlated with FGFR3, FOXA1 or both. In particular, 72 and 62 predicted up regulated genes were significantly positively correlated to increases in FOXA1 (r=0.12 to 0.82) and FGFR3 expression (r=0.12 to 0.82, Figure 2d). Conversely, 32 and 77 predicted down regulated genes were significantly negatively correlated with FOXA1 (r=-0.14 to -0.51, p<0.05) and FGFR3 (r=-0.12 to -0.66, p<0.05) expression, respectively.
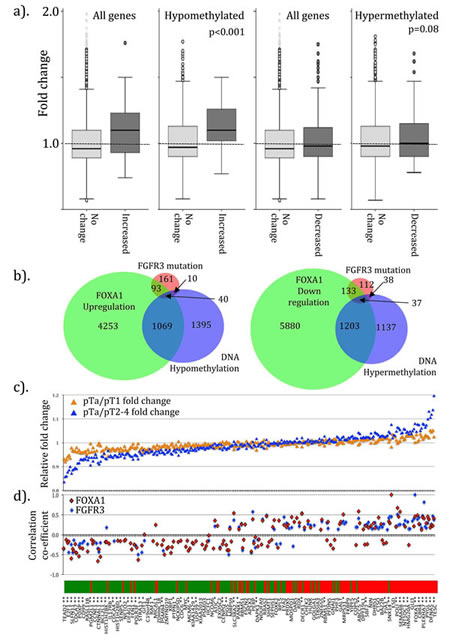
Figure 2: Symmetry of genotype between FOXA1 transfected cells, FGFR3 mutant tumors and in sporadic UCC. Gene expression changes following FOXA1 transfection in T24/EJ cells share many similarities with those found in FGFR3 mutant bladder UCC. In (a). we stratify changes in normalized gene expression following FOXA1 transfection within T24/EJ by those seen in FGFR3 mutant UCC. Genes upregulated in FGFR3 mutant tumors (labeled as Increased) have significantly higher expression in FOXA1 transfected cells than those with no change in FGFR3 mutants. The difference is largest for genes with associated DNA hypomethylation. No significant difference is seen for genes with decreased expression or DNA hypermethylation. (b). Area proportional Venn diagrams reveal the overlap for genes in FGFR3 mutant UCC and those upregulated following FOXA1 transfection or with increases in DNA hypomethylation following FOXA1 transfection (left). Overlap was also seen for genes downregulated in FGFR3 mutant UCC, and those down regulated or hypermethylated following FOXA1 transfection. We identified a 156 gene cohort with reciprocal, symmetrical expression in FOXA1 transfected cells and FGFR3 mutant UCC and analyzed this in a publically deposited microarray dataset [32]. In (c). we plot the fold change of these 156 genes as a ratio for low-grade NMI cancers relative to high-grade NMI cancer (orange triangle) and invasive cancers (blue). In (d). we plot the correlation coefficient vales (r) between expression of FGFR3 and FOXA1 in the 256 sporadic UCC [32]. The 156 genes in (c) and (d) are ordered according to expected fold changes (bar: green is loss of expression, red is increased expression) as seen in FOXA1 transfected cells. Significance of difference in fold change (low grade NMI versus more aggressive cancers) is shown as *p<0.05 and **p<0.01.
Discussion
Here we report FOXA1 as a regulatory target of miRs-99a/100 in UCC. This follows our previous findings identifying FGFR3 as a target of miRs-99a/100 [12] and supports data revealing these microRNAs discriminate of UCC genotype [8]. Our work provides a direct mechanistic link between miRs-99a/100, FGFR3 and FOXA1. The central role of miRs-99a/100 in this association suggests epigenetic events either precede the development of tumor genotype or act as a link between these pathways in tumors when either event occurs in isolation. For example, FGFR3 mutation may lead to increased mRNA expression, annealing and sequestration of miRs-99a/100 leading to FOXA1 upregulation [33]. There was considerable overlap between the genotypes of tumors with FOXA1 upregulation and FGFR3 mutation. This symmetry appears mediated through miRs-99a/100 and FOXA1, given the proximity of affected genes to FOXA1 binding sites and to changes in DNA hypomethylation, suggesting this axis plays a key role in determining genetic events in low-grade and papillary-type invasive high-grade UCC. Genotype often impacts upon phenotype, and we observed tumors with high expression of both FOXA1 and FGFR3 were most commonly low grade non-invasive and had lower rates of progression, when compared to those with low expression. Outcome differences were only seen in non-invasive cancers, suggesting consequential changes subsequent to these genes become more important with tumor evolution.
Our data provide an explanation of previous CpG methyl profiling reports. Wolff et al. identified regional hypomethylation across 16% of CpG probes in low grade UCC, when profiling a large UCC cohort [34]. FOXA1 is a transcription factor recruited to enhancers that acts to influence chromatin interactions through DNA demethylation and H3K4 methylation [24, 27]. The role of FOXA1 in malignancy has been best studied in endocrine-dependent cancers, where it is known to act as a transcriptional co-factor and to modulate hormone receptor activity through chromatin and DNA modifications [35].
Our findings also reflect those in a previous report of FOXA1 in UCC [36], although we make a different interpretation. Specifically, we propose microRNA-mediated upregulation of FOXA1 in papillary type UCC, rather than loss of FOXA1 expression in aggressive, squamous tumors. Our findings mirror those seen in other malignancies, e.g. FOXA1 upregulation confering a good prognosis on cancers with high expression [37, 38] and is a representative marker of luminal-type cancers [39], and are compatible with DeGraff et al. given recent evidence of a dual role for up and down regulation of FOXA1 in cancer [40, 41].
In summary, we identify microRNA mediated upregulation of both FGFR3 and FOXA1 in UCC. We propose this is a determinant of the papillary genotype of these cancers and that FOXA1 is a key mediator of this evolution.
Materials and methods
In Silico identification of miR-99a/100 targets in UCC
To identify potential targets of miRs-99a/100 in low grade UCC we created a microarray meta-database from publically deposited UCC mRNA expression datasets (from www.oncomine.org). We selected datasets in which we could identify genes upregulated in low grade, superficial or non-muscle invasive UCC, when compared to controls and/or high grade or invasive tumors. We searched for predicted targets of miRs-99a/100 (obtained from TargetScan (Version 4.2, www.targetscan.org) and PicTar (http://pictar.mdc-berlin.de)) across this meta-database and ranked according to the frequency of detection.
Patients and tumours
To evaluate potential mRNA targets we studied 292 freshly frozen urothelial samples (Table 1) from two cohorts. The first was used to compare the expression of miRs-99a/100 and putative mRNA targets. The second examined these putative targets in a larger unrelated population. UCC were classified using the 2004 WHO/ISUP criteria and treated according to standard care [5, 42]. Histologically normal urothelial samples were obtained from patients with UCC (distant to any tumor) and disease-free controls (at prostatectomy). We analyzed UCC cell lines representing the disease spectrum (RT4, RT112 and EJ/T24, respectively, purchased from ATCC) and normal non-immortalized human urothelial (NHU) cells [43].
RNA extraction, cDNA synthesis and rtPCR
We extracted total RNA using the mirVanaTM kit (Ambion, TX) from 10 x 10uM microdissected frozen tumor sections (>90% pure cell populations). We measured the expression of potential mRNA target using quantitative rtPCR (Taqman commercial assays purchased from Applied Biosystems, UK). cDNA was made using 100ug whole RNA, random primers, RT buffer, dNTP (100mM), RNase inhibitor and MultiScribe Reverse Transcriptase (cDNA Reverse Transcription kit, Applied Biosystems, Warrington, UK). Realtime quantified PCR with 2 µL cDNA, gene specific primers with FAM-TAMRA labeled probes, water and 2x Taqman Universal PCR MasterMix (Applied Biosystems, Warrington, UK) was performed on the ABI 7900HT system according to manufacturers guidelines. Relative mRNA quantification was determined with respect to the mean of GAPDH and β-Actin. We also used quantitative rtPCR to determine microRNA concentrations, with reagents specific to the mature 23bp sequence, in isolation and using the commercial Taqman low density microarray (n=365 miRs, as detailed in [12]). Reverse transcription using stem loop primers was performed with 50ng small RNA, MultiScribe Reverse Transcriptase (Applied Biosystems, Warrington, UK), RNase inhibitor, 100nm dNTPs and nuclease free water. Quantification with MGB labeled probes specific to the reverse transcribed product was performed according to the manufacturer’s guidelines (Applied Biosystems, Warrington, UK). Relative miR quantification was determined with respect to the average of two snoRNAs (RNU44 and RNU48) or the mean of the entire TLDA.
MicroRNA manipulation and Luciferase reporter construct
To examine functional implications of miR expression, we manipulated expression using specific anti-miRs and a scrambled RNA sequence controls (Ambion, TX). All experiments were performed in triplicate using non-immortalized NHU cells at 70% confluence, as detailed [44]. Briefly, each well of a six-well tissue culture plate was transfected with 100 pmol anti-miR using 5 µl siPORT (Life Technologies) in 200 µl Optimem (Life Technologies). Knockdown of relevant miR was confirmed by miR-specific taqman PCR (Applied Biosystems) after 48 hours.
To investigate direct targeting of the FOXA1 3’UTR we synthesized a Luciferase reporter construct (methods detailed [44]). We cloned 800 bases around the miR-99a/100 seed sequence in the FOXA1 3’ UTR (chr14: 37,129,203 - 37,129,210) into EJ cells and ligated into pMIR REPORT (Invitrogen, UK). Dual luciferase assays were conducted in a 6 well plate format at 70% confluence. Forty-eight hours post transfection, firefly and renilla luciferase were quantified sequentially using the Dual Luciferase Assay kit (Promega, UK) and luminescence was measured using the manufacturers recommended luminometer (Promega Glomax). Firefly luciferase expression was quantified and normalized to Renilla luciferase expression.
FOXA1 transfection
EJ cells were transfected with a pcDNA3 vector containing FOXA1 or an empty vector control. 24 µg DNA / 90 µl Lipofectamine LTX (Life Technologies) was used to transfect each 90mm dish at 70% confluence. Experiments were performed in triplicate. A small portion (20%) of transfected cells was used to confirm FOXA1 expression by western blot.
Genome wide profiling of DNA methylation
Methylated DNA immunoprecipitation and tiling CpG island microarrays (Human CpG Island Microarray, Agilent, CA) (MeDIP-CHIP) were used to determine genome wide methyl-cytosine profiles, as detailed elsewhere [28]. Genomic DNA was sonicated and incubated with antibodies raised to either 5mC (anti-5-methylcytidine, Eurogentec, Hampshire, UK) or murine IgG (negative control). The antibody-antigen complex was captured with magnetic beads conjugated to anti-mouse-IgG (Santa Cruz Biotechnology), washed, unbound, non-specific DNA removed, before methylated DNA elution. Immunoprecipitated (Cyanine 5-dUTP) and reference DNA (Cyanine 3-dUTP) were labeled (Genomic DNA Enzymatic Labeling Kit, Agilent), cleaned (Amicon filters, Millipore) and quantified. Competitive hybridization onto the CpG microarray was performed (ChIP-on-Chip Hybridization Kit, Agilent) in a rotating SureHyb chamber at 67oC for 40 hours. Washed slides were scanned (High-Resolution C Scanner, Agilent) and fluorescence obtained using Feature Extraction software. The microarray contains 244,000 probes that tile through 27,800 CpG features at an average of 100bp separation. We identified concordant probes within the highest (>80%) and lowest quintiles (<20% of Cy-5) fluorescence and defined these as enriched for hyper or hypo-methylated, respectively. Finally, as each experiment was performed in triplicate, we excluded probes without concordance in 2 or 3 replicates.
Whole genome mRNA expression
Whole genome mRNA expression was determined by microarray (HG-U133 Plus 2.0, Affymetrix, Cal.) [28]. This platform contains 54,000 probesets, including 33,000 to known coding genes. RNA was prepared using the Affymetrix protocol (enzymes from Invitrogen) and annealed to an oligo-d(T) primer with a T7 polymerase binding site. cDNA was generated using superscript II and E. coli DNA ligase and polymerase I. The reaction was completed with T4 DNA polymerase and EDTA. Amplified cDNA was cleaned, biotin-labeled, fragmented and hybridized to the microarray for 16 hours at 45°C in a rotating oven at 60rpm. After washing and staining, the arrays were scanned (GC3000 scanner) and data processed using Gene Chip Operating System software. mRNA expression was determined using Microarray Analysis Suite 5 (Affymetrix) and defined as expressed (perfect match probeset intensity greater than mismatch intensity) or absent (mismatch probeset intensity greater or equal to perfect match intensity). Expression data were exported into Expression console (Affymetrix), RMA log2 converted and analyzed within Significance Analysis of Microarrays [45]. This microarray data is deposited on line at GEO datasets (GEO accession number GSE56037).
Protein expression: western blotting
Cells were lysed in RIPA buffer (20m M Tris·HCl, 135 mM NaCl, 10% glycerol, 1% Igepal, 0.1% SDS, 0.5% deoxycholic acid, 2 mM EDTA) containing protease and phosphatase inhibitors (Complete EDTA-free protease inhibitor cocktail and PhosSTOP phosphatase inhibitor cocktail; Roche, Mannheim, Germany), and protein content was quantified using the DC-protein assay reagent (Bio-Rad, Hercules, CA). Protein lysates (50µg) were loaded onto 8% gels, fractionated, and electroblotted onto nitrocellulose membranes. After blocking with 5% non-fat milk powder and 0.1% Tween, the membranes were incubated overnight with the relevant primary antibody (listed in supplementary table 3) overnight at 4°C, washed, and incubated at room temperature for 1h with an appropriate HRP-conjugated secondary antibody (1:1000; Cell Signaling Technologies Inc.). . The immune complexes were visualized by enhanced chemiluminescence (GE Healthcare, Buckinghamshire, UK) and quantified using Image J for band densitometry. The final values were plotted relative to the negative control and normalized to the corresponding Beta-Actin value.
Statistical analysis
Relative mRNA and miR concentrations were calculated using the median of their respective reference molecules (∆Ct = Ct miR – Ct median control) and expression fold changes computed using 2-∆∆Ct calculations [46]. MiR-mRNA expression was correlated using Pearson’s coefficient. RNA expression was compared with clinicopathological data using the χ2, T test or Mann Whitney U test where appropriate. Disease progression was defined when a non-muscle invasive tumor became invasive or a muscle invasive tumor developed metastases. Progression-specific survival probability following tumor resection was analyzed using the Kaplan-Meier method and compared with the Log rank test, for which significance was adjusted using a Bonferroni correction. Patients without progression were censored at last reviewed or when they died of other causes. All analyses were two tailed and carried out using SPSS (version 14, SPSS Inc).
Funding
JWFC was supported by a GSK Clinician Scientist fellowship and grants from Yorkshire Cancer Research (Grant number S305PA and S310), Astellas Educational Foundation and the European Union (European Community’s Seventh Framework Programme. Grant Numbers: FP7/2007-2013, HEALTH-F2-2007-201438). HEB was supported by an RCUK research fellowship. SM was supported by fellowship funding from The Urological Foundation.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget
Disclosure of potential conflicts of interest
The authors declare no competing conflicts of interest
References
1. Chavan S, Bray F, Lortet-Teulent J, Goodman MM and Jemal A. International Variations in Bladder Cancer Incidence and Mortality. Eur Urol. 2014; 66(1):59-73.
2. Svatek RS, Hollenbeck BK, Holmang S, Lee R, Kim S, Stenzl A and Lotan Y. The economics of bladder cancer: Costs and considerations of caring for this disease. Eur Urol 2014; In Press: doi: 10.1016/j.eururo.2014.01.006.
3. Meeks JJ, Bellmunt J, Bochner BH, Clarke NW, Daneshmand S, Galsky MD, Hahn NM, Lerner SP, Mason M, Powles T, Sternberg CN and Sonpavde G. A systematic review of neoadjuvant and adjuvant chemotherapy for muscle-invasive bladder cancer. Eur Urol. 2012; 62(3):523-533.
4. Knowles MA. Molecular subtypes of bladder cancer: Jekyll and Hyde or chalk and cheese? Carcinogenesis. 2006; 27(3):361-373.
5. Catto JW, Azzouzi AR, Rehman I, Feeley KM, Cross SS, Amira N, Fromont G, Sibony M, Cussenot O, Meuth M and Hamdy FC. Promoter hypermethylation is associated with tumor location, stage, and subsequent progression in transitional cell carcinoma. J Clin Oncol. 2005; 23(13):2903-2910.
6. Linton KD, Rosario DJ, Thomas F, Rubin N, Goepel J, Abbod MF and Catto JW. Disease specific mortality in patients with low risk bladder cancer and the impact of cystoscopic surveillance. J Urol. 2013; 189(3):828-883.
7. Rink M, Ehdaie B, Cha EK, Green DA, Karakiewicz PI, Babjuk M, Margulis V, Raman JD, Svatek RS, Fajkovic H, Lee RK, Novara G, Hansen J, Daneshmand S, Lotan Y, Kassouf W, et al. Stage-specific impact of tumor location on oncologic outcomes in patients with upper and lower tract urothelial carcinoma following radical surgery. Eur Urol.. 2012; 62(4):677-684.
8. The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014; 507: 315-322
9. Damrauer JS, Hoadley KA, Chism DD, Fan C, Tiganelli CJ, Wobker SE, Yeh JJ, Milowsky MI, Iyer G, Parker JS and Kim WY. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci U S A. 2014; 111(8):3110-3115.
10. Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, Roth B, Cheng T, Tran M, Lee IL, Melquist J, Bondaruk J, Majewski T, Zhang S, Pretzsch S, Baggerly K, et al. Identification of distinct Basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014; 25(2):152-165.
11. van Oers JM, Zwarthoff EC, Rehman I, Azzouzi AR, Cussenot O, Meuth M, Hamdy FC and Catto JW. FGFR3 mutations indicate better survival in invasive upper urinary tract and bladder tumours. Eur Urol. 2009; 55(3):650-657.
12. Catto JW, Miah S, Owen HC, Bryant H, Dudziec E, Larre S, Milo M, Rehman I, Rosario DJ, DiMartino E, Knowles MA, Meuth M, Harris AL and Hamdy FC. Distinct microRNA alterations characterize high and low grade bladder cancer. Cancer Res. 2009; 69(21):8472-8481.
13. Catto JW, Alcaraz A, Bjartell AS, De Vere White R, Evans CP, Fussel S, Hamdy FC, Kallioniemi O, Mengual L, Schlomm T and Visakorpi T. MicroRNA in Prostate, Bladder, and Kidney Cancer: A Systematic Review. Eur Urol. 2011; 59(5):671-681.
14. Stransky N, Vallot C, Reyal F, Bernard-Pierrot I, de Medina SG, Segraves R, de Rycke Y, Elvin P, Cassidy A, Spraggon C, Graham A, Southgate J, Asselain B, Allory Y, Abbou CC, Albertson DG, et al. Regional copy number-independent deregulation of transcription in cancer. Nat Genet. 2006; 38(12):1386-1396.
15. Blaveri E, Simko JP, Korkola JE, Brewer JL, Baehner F, Mehta K, Devries S, Koppie T, Pejavar S, Carroll P and Waldman FM. Bladder cancer outcome and subtype classification by gene expression. Clin Cancer Res. 2005; 11(11):4044-4055.
16. Modlich O, Prisack HB, Pitschke G, Ramp U, Ackermann R, Bojar H, Vogeli TA and Grimm MO. Identifying superficial, muscle-invasive, and metastasizing transitional cell carcinoma of the bladder: use of cDNA array analysis of gene expression profiles. Clin Cancer Res. 2004; 10(10):3410-3421.
17. Dyrskjot L, Thykjaer T, Kruhoffer M, Jensen JL, Marcussen N, Hamilton-Dutoit S, Wolf H and Orntoft TF. Identifying distinct classes of bladder carcinoma using microarrays. Nat Genet. 2003; 33(1):90-96.
18. Dyrskjot L, Zieger K, Kruhoffer M, Thykjaer T, Jensen JL, Primdahl H, Aziz N, Marcussen N, Moller K and Orntoft TF. A molecular signature in superficial bladder carcinoma predicts clinical outcome. Clin Cancer Res. 2005; 11(11):4029-4036.
19. Sanchez-Carbayo M, Socci ND, Lozano J, Saint F and Cordon-Cardo C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol. 2006; 24(5):778-789.
20. Dyrskjot L, Zieger K, Real FX, Malats N, Carrato A, Hurst C, Kotwal S, Knowles M, Malmstrom PU, de la Torre M, Wester K, Allory Y, Vordos D, Caillault A, Radvanyi F, Hein AM, et al. Gene expression signatures predict outcome in non-muscle-invasive bladder carcinoma: a multicenter validation study. Clin Cancer Res. 2007; 13(12):3545-3551.
21. Forman JJ, Legesse-Miller A and Coller HA. A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc Natl Acad Sci U S A. 2008; 105(39):14879-14884.
22. Small EM, O’Rourke JR, Moresi V, Sutherland LB, McAnally J, Gerard RD, Richardson JA and Olson EN. Regulation of PI3-kinase/Akt signaling by muscle-enriched microRNA-486. Proc Natl Acad Sci U S A. 2010; 107(9):4218-4223.
23. Williams SV, Hurst CD and Knowles MA. Oncogenic FGFR3 gene fusions in bladder cancer. Hum Mol Genet. 2013; 22(4):795-803.
24. Serandour AA, Avner S, Percevault F, Demay F, Bizot M, Lucchetti-Miganeh C, Barloy-Hubler F, Brown M, Lupien M, Metivier R, Salbert G and Eeckhoute J. Epigenetic switch involved in activation of pioneer factor FOXA1-dependent enhancers. Genome research. 2011; 21(4):555-565.
25. Ung M, Ma X, Johnson KC, Christensen BC and Cheng C. Effect of estrogen receptor alpha binding on functional DNA methylation in breast cancer. Epigenetics : official journal of the DNA Methylation Society. 2014; 9(4).
26. Kerschner JL, Gosalia N, Leir SH and Harris A. Chromatin remodeling mediated by the FOXA1/A2 transcription factors activates expression in intestinal epithelial cells. Epigenetics : official journal of the DNA Methylation Society. 2014; 9(4).
27. Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS and Brown M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008; 132(6):958-970.
28. Dudziec E, Gogol-Doring A, Cookson V, Chen W and Catto J. Integrated epigenome profiling of repressive histone modifications, DNA methylation and gene expression in normal and malignant urothelial cells. PloS one. 2012; 7(3):e32750.
29. Yates DR, Rehman I, Abbod MF, Meuth M, Cross SS, Linkens DA, Hamdy FC and Catto JW. Promoter hypermethylation identifies progression risk in bladder cancer. Clin Cancer Res. 2007; 13(7):2046-2053.
30. Motallebipour M, Ameur A, Reddy Bysani MS, Patra K, Wallerman O, Mangion J, Barker MA, McKernan KJ, Komorowski J and Wadelius C. Differential binding and co-binding pattern of FOXA1 and FOXA3 and their relation to H3K4me3 in HepG2 cells revealed by ChIP-seq. Genome biology. 2009; 10(11):R129.
31. Lindgren D, Liedberg F, Andersson A, Chebil G, Gudjonsson S, Borg A, Mansson W, Fioretos T and Hoglund M. Molecular characterization of early-stage bladder carcinomas by expression profiles, FGFR3 mutation status, and loss of 9q. Oncogene. 2006; 25(18):2685-2696.
32. Kim WJ, Kim EJ, Kim SK, Kim YJ, Ha YS, Jeong P, Kim MJ, Yun SJ, Lee KM, Moon SK, Lee SC, Cha EJ and Bae SC. Predictive value of progression-related gene classifier in primary non-muscle invasive bladder cancer. Molecular cancer. 2010; 9:3.
33. Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ and Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010; 465(7301):1033-1038.
34. Wolff EM, Chihara Y, Pan F, Weisenberger DJ, Siegmund KD, Laird PW, Jones PA and Liang G. Unique DNA methylation patterns distinguish superficial and invasive bladder cancers and establish an epigenetic field defect in premalignant tissue. Cancer Res 2010; 70: 8169-8178.
35. Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D and Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nature genetics. 2011; 43(1):27-33.
36. DeGraff DJ, Clark PE, Cates JM, Yamashita H, Robinson VL, Yu X, Smolkin ME, Chang SS, Cookson MS, Herrick MK, Shariat SF, Steinberg GD, Frierson HF, Wu XR, Theodorescu D and Matusik RJ. Loss of the urothelial differentiation marker FOXA1 is associated with high grade, late stage bladder cancer and increased tumor proliferation. PloS one. 2012; 7(5):e36669.
37. Hu Q, Luo Z, Xu T, Zhang JY, Zhu Y, Chen WX, Zhong SL, Zhao JH and Tang JH. FOXA1: a Promising Prognostic Marker in Breast Cancer. Asian Pacific journal of cancer prevention : APJCP. 2014; 15(1):11-16.
38. Robinson JL, Hickey TE, Warren AY, Vowler SL, Carroll T, Lamb AD, Papoutsoglou N, Neal DE, Tilley WD and Carroll JS. Elevated levels of FOXA1 facilitate androgen receptor chromatin binding resulting in a CRPC-like phenotype. Oncogene. 2013 Dec 2. doi: 10.1038/onc.2013.508
39. Powe DG, Dhondalay GK, Lemetre C, Allen T, Habashy HO, Ellis IO, Rees R and Ball GR. DACH1: its role as a classifier of long term good prognosis in luminal breast cancer. PloS one. 2014; 9(1):e84428.
40. Jin HJ, Zhao JC, Ogden I, Bergan RC and Yu J. Androgen receptor-independent function of FoxA1 in prostate cancer metastasis. Cancer research. 2013; 73(12):3725-3736.
41. Sahu B, Laakso M, Pihlajamaa P, Ovaska K, Sinielnikov I, Hautaniemi S and Janne OA. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer research. 2013; 73(5):1570-1580.
42. Catto JW and Rosario DJ. The road to cystectomy: who, when and why? . Eur Urol: EAU Update series. 2005; 3(4):171-179.
43. Southgate J, Hutton KA, Thomas DF and Trejdosiewicz LK. Normal human urothelial cells in vitro: proliferation and induction of stratification. Lab Invest. 1994; 71(4):583-594.
44. Drayton RM, Dudziec E, Peter S, Bertz S, Hartmann A, Bryant HE and Catto JW. Reduced expression of microRNA-27a modulates cisplatin resistance in bladder cancer by targeting the cystine/glutamate exchanger SLC7A11. Clin Can Res. 2014; 20(7):1990-2000.
45. Tusher VG, Tibshirani R and Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001; 98(9):5116-5121.
46. Schmittgen TD, Lee EJ, Jiang J, Sarkar A, Yang L, Elton TS and Chen C. Real-time PCR quantification of precursor and mature microRNA. Methods (San Diego, Calif. 2008; 44(1):31-38.