
INTRODUCTION
Tetralogy of Fallot (TOF) is the most common cyanotic congenital heart disease (CHD) with an incidence rate of 1/3600 in live births and 10% in CHD [1]. TOF is characterised by cardiac outflow tract malformation caused by non-uniform separation between truncus arteriosus and bulbus arteriosus during embryo stage. TOF pathological features include ventricular septal defect, aortic overriding, right ventricular outflow tract stenosis or pulmonary artery stenosis and right ventricular hypertrophy. In neonatal period, children with TOF may manifest oxygen deficiency, pneumonia, intractable congestive heart failure and other complications with high early mortality. Despite TOF treatment in children, some complications will still occur because of poor prognosis. TOF brings heavy burden to the family and society. Therefore, to investigate TOF etiology, possible pathogenesis and risk factors for prenatal diagnosis and counselling and prognostic evaluation is of great significance.
Heart development is a complex and orderly process including cardiac tube formation, loop formation, intracardiac separation and vascular connection. Heart development is related to many genes, which express and interact in different spaces and at different times to form a precise regulatory mechanism. Abnormal expression occurring in any of these genes is likely to affect heart development, leading to heart malformation. For TOF, embryonic developmental mechanism is nearly definite. However, the molecular pathogenesis remains to be clarified. TOF is associated with gene mutation [2]. TOF-related genes include NKX2-5 [3], GATA4 [3, 4], and JAG-1 [5, 6]. Approximately 15% of TOF are from 22q11 microdeletion syndrome [7] caused by chromosome 22 long arm 1 zone 1 subzone deletion and manifest extracardiac abnormalities, such as abnormal face, thymic hypoplasia, cleft palate and hypocalcemia. In addition, TOF is also related to chromosomal aneuploidy, and approximately 3% of TOF is from 21-trisomy syndrome [8]. However, the pathogenesis of isolated TOF is still to be elucidated.
Whole exome sequencing (WES) is suitable to high-throughput sequencing for all genomic exon regions. Human exome accounting for approximately 1%–2% of genome contains important information of protein synthesis, which directly reflects gene function. In most diseases, related mutations are located in the exome region. WES may be used to find pathogenic gene and predisposing gene in complex diseases, monogenic diseases and cancer because it can investigate protein encoding information in several individuals; its data are also accurate [9]. The aim of this study was to find TOF-related pathogenic genes through WES technology performed among eight small families with TOF, providing a basis for studying TOF pathogenesis.
RESULTS
Clinical features
All the eight TOF children manifested cyanosis and rough systolic-ejection murmurs at the left sternal border between the second and fourth ribs. Echocardiography showed right ventricular enlargement, right ventricular anterior wall thickening, right ventricular outflow tract and pulmonary artery stenosis, ventricular septal defect and aortic overriding (Figure 1). The echocardiographic results of eight TOF children in the right ventricle, right ventricular anterior wall, right ventricular outflow tract, main pulmonary artery, left pulmonary artery, right pulmonary artery, ventricular septal defect and left ventricular ejection fraction (Table 1). The eight TOF children displayed no other malformations, and their parents showed no abnormalities.
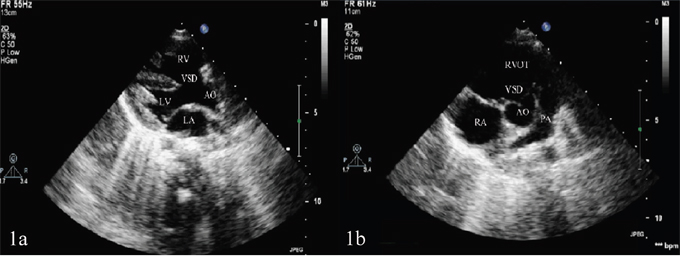
Figure 1: (A) Ventricular septal defect and aortic overriding shown in the left ventricular long-axis view of transthoracic echocardiography. (B) Pulmonary artery stenosis shown in the aorta short-axis view of transthoracic echocardiography.
LA: left atrium; LV: left ventricle; AO: aorta; VSD: ventricular septal defect; RA: right atrium; RV: right ventricle; PA: pulmonary artery; RVOT: right ventricular outflow tract.
Table 1: Results of echocardiogram in the 8 children with tetralogy of Fallot
ID |
Gender |
Age |
RV |
RVAW |
RVOT |
MPA |
LPA |
RPA |
VSD |
LVEF |
|---|---|---|---|---|---|---|---|---|---|---|
1 |
female |
3months |
8.0 |
5.0 |
5.4 |
5.6 |
4.3 |
4.8 |
9.5 |
73 |
2 |
male |
8months27days |
12.0 |
6.0 |
5.7 |
5.3 |
4.3 |
5.3 |
10.2 |
68 |
3 |
male |
1years11months |
13.9 |
6.8 |
7.2 |
10.5 |
6.6 |
6.7 |
13.5 |
72 |
4 |
female |
2months24days |
11.8 |
4.1 |
6.0 |
15.3 |
9.3 |
7.7 |
11.5 |
57 |
5 |
female |
6months |
11.1 |
6.1 |
9.0 |
11.8 |
7.6 |
7.6 |
12.9 |
67 |
6 |
male |
8months2days |
13.0 |
6.3 |
7.0 |
6.4 |
4.2 |
4.4 |
13.5 |
72 |
7 |
female |
11months26days |
13.4 |
5.9 |
5.8 |
12.4 |
6.7 |
7.1 |
13.1 |
69 |
8 |
male |
6months11days |
10.3 |
5.0 |
6.1 |
9.0 |
4.3 |
5.7 |
13.2 |
74 |
RV: right ventricle; RVAW: right ventricular anterior wall; RVOT: Right ventricular outflow tract; MPA: main pulmonary artery; LPA: left pulmonary artery; RPA: right pulmonary artery; VSD: ventricular septal defect; LVEF: left ventricular ejection fraction.
Mutation detection
A total of sixteen differential single nucleotide polymorphisms (SNPs) loci and eight differentially small InDels were discovered by sequencing all exons in the eight small families and alignment of SNPs and small InDels within families and between families (Table 2 and Table 3).
Table 2: SNPs identified by whole exome sequencing
Chr |
Position |
ID |
Ref |
Alt |
Mutation |
Symbol |
Amino |
SIFT |
PolyPhen |
|---|---|---|---|---|---|---|---|---|---|
chr12 |
56717682 |
rs745387506 |
T |
G |
missense |
NACA |
H/P |
- |
unknown(0) |
chr5 |
141123595 |
- |
T |
A |
missense |
PCDHB4 |
S/T |
tolerated (1) |
benign(0) |
chr12 |
7190555 |
- |
G |
C |
missense |
PEX5 |
A/P |
tolerated (0.22) |
benign(0.067) |
chr2 |
240042663 |
- |
C |
T |
missense |
PRR21 |
R/H |
tolerated (0.56) |
unknown(0) |
chr4 |
1237208 |
- |
A |
T |
stop_gained |
CTBP1 |
C/* |
- |
- |
chrX |
37010556 |
rs61999275 |
G |
C |
missense |
FAM47C |
A/P |
tolerated(1) |
benign(0) |
chr1 |
26282352 |
rs6667693 |
C |
A |
missense |
UBXN11 |
G/C |
tolerated (0.05) |
unknown(0) |
chr1 |
86368686 |
rs75376884 |
T |
C |
missense |
ODF2L |
M/V |
tolerated (0.14) |
benign(0.001) |
chr4 |
87614868 |
- |
G |
A |
missense |
DSPP |
D/N |
- |
unknown(0) |
chr4 |
87614877 |
rs150132251 |
A |
G |
missense |
DSPP |
N/D |
- |
unknown(0) |
chr4 |
88008301 |
rs117078377 |
G |
A |
missense |
PKD2 |
A/T |
tolerated(0.3) |
benign(0.395) |
chr11 |
71527593 |
rs199903176 |
A |
G |
missense |
KRTAP5-7 |
Y/C |
tolerated (0.22) |
unknown(0) |
chr12 |
51346625 |
rs117443541 |
T |
G |
missense |
CELA1 |
Y/S |
tolerated(0.13) |
benign(0.045) |
chr15 |
88856792 |
rs12899191 |
A |
G |
missense |
ACAN |
T/A |
tolerated(0.56) |
benign(0.084) |
chrX |
104250509 |
rs9697856 |
T |
G |
missense |
ESX1 |
T/P |
tolerated (0.54) |
unknown(0) |
chr22 |
11068057 |
- |
A |
T |
missense |
BAGE5 |
R/W |
tolerated (0.12) |
damaging(0.675) |
Chr: chromosome; Ref: reference sequence base; Alt: alternative base identified; SIFT: sorting intolerant from tolerant.
Table 3: Small InDels identified by whole exome sequencing
Chr |
Position |
ID |
Ref |
Alt |
Mutation |
Symbol |
Amino |
SIFT |
PolyPhen |
|---|---|---|---|---|---|---|---|---|---|
chr2 |
104856098 |
rs775150602 |
AGCC |
A |
inframe_deletion |
POU3F3 |
A/- |
- |
- |
chr12 |
111598972 |
rs769170503 |
C |
CTGT |
inframe_insertion |
ATXN2 |
Q/QQ |
- |
- |
chr4 |
13632182 |
. |
CT |
C |
splice_acceptor_variant&non_coding_transcript_variant |
- |
- |
||
chr9 |
136327352 |
. |
ACCCCCTC |
A |
TF_binding_site_variant&TFBS_ablation |
GPSM1 |
- |
- |
- |
chr17 |
75616380 |
rs371699907 |
CAGG |
C |
inframe_deletion |
MYO15B |
QE/Q |
- |
- |
chr19 |
9251064 |
rs201985790 |
CT |
C |
frameshift_variant |
OR7E24 |
F/X |
- |
- |
chr19 |
51694076 |
rs10689461 |
C |
CAG |
splice_acceptor_variant&non_coding_transcript_variant |
SPACA6P |
- |
- |
- |
chrX |
19345745 |
rs776856509 |
TC |
TCC, T |
frameshift_variant |
PDHA1 |
S/X |
- |
- |
Ten of the sixteen differential SNPs were reported, namely, Chr1 (rs6667693, UBXN11), Chr1 (rs75376884, ODF2L), Chr4 (rs150132251, DSPP), Chr4 (rs117078377, PKD2), Chr11 (rs199903176, KRTAP5-7), Chr12 (rs745387506, NACA), Chr12 (rs117443541, CELA1), Chr15 (rs12899191, ACAN), ChrX (rs61999275, FAM47C) and ChrX rs9697856, ESX1). Six of the 16 differential SNPs were not reported, namely, Chr 2 (PRR21), Chr 4 (CTBP1), Chr 4 (DSPP), Chr 5 (PCDHB4), Chr12 (PEX5) and Chr22 (BAGE5).
Six of the eight differentially small InDels were reported, namely, Chr2 (rs775150602, POU3F3), Chr12 (rs769170503, ATXN2), Chr17 (rs371699907, MYO15B), Chr19 (rs201985790, OR7E24), Chr19 (rs10689461, SPACA6P) and ChrX (rs776856509, PDHA1). Two of the eight differentially small InDels were not reported, namely, Chr4 (position: 13632182) and Chr9 (GPSM1).
SNPs and small InDel-related Online Mendelian Inheritance in Man (OMIM) genes and clinical phenotypes are shown in Table 4.
Table 4: SNPs- and small InDels-related OMIM genes and clinical phenotypes
Gene |
Chromosome |
OMIM |
Gene description |
Function |
|---|---|---|---|---|
NACA |
Chr12 |
601234 |
gene encodes a protein that associates with BTF3 to form NAC |
skeletal development |
PCDHB4 |
Chr5 |
606330 |
a member of the protocadherin beta gene cluster |
cell-cell neural connections |
PEX5 |
Chr12 |
600414 |
peroxisomal biogenesis factor 5 |
Peroxisome biogenesis disorder 2A, 2B; |
PRR21 |
Chr2 |
- |
- |
- |
CTBP1 |
Chr4 |
602618 |
gene encodes a protein that binds to the C-terminus of adenovirus E1A proteins |
transcriptional repressor and cellular proliferation |
FAM47C |
ChrX |
- |
- |
- |
UBXN11 |
Chr1 |
609151 |
a protein with a divergent C-terminal UBX domain |
affect the actin cytoskeleton and alter cell shape |
ODF2L |
Chr1 |
- |
- |
- |
DSPP |
Chr4 |
125485 |
a member of the small integrin-binding ligand N-linked glycoprotein family of proteins |
Deafness, autosomal dominant 39, with dentinogenesis; |
PKD2 |
Chr4 |
173910 |
a member of the polycystin protein family |
Polycystic kidney disease 2 |
KRTAP5-7 |
Chr11 |
- |
- |
- |
CELA1 |
Chr12 |
130120 |
Elastases form a subfamily of serine proteases that hydrolyze many proteins in addition to elastin |
evolutionarily silenced in pancreatic acinar cells |
ACAN |
Chr15 |
155760 |
a member of the aggrecan/versican proteoglycan family |
Osteochondritis dissecans, short stature, and early-onset osteoarthritis; |
ESX1 |
ChrX |
300154 |
gene encodes a dual-function 65 kDa protein that undergoes proteolytic cleavage to produce a 45 kDa N-terminal fragment with a paired-like homeodomain and a 20 kDa C-terminal fragment with a proline-rich domain |
placental development and spermatogenesis |
BAGE5 |
Chr22 |
- |
- |
- |
POU3F3 |
Chr2 |
602480 |
a member of the class III POU family of transcription factors |
expressed in the central nervous system |
ATXN2 |
Chr12 |
601517 |
gene belongs to a group of genes that is associated with microsatellite-expansion diseases, a class of neurological and neuromuscular disorders caused by expansion of short stretches of repetitive DNA |
Spinocerebellar ataxia 2; |
GPSM1 |
Chr9 |
609491 |
gene encodes a receptor-independent activator of G protein signaling, |
influence the basal activity of G-protein signaling systems |
MYO15B |
Chr17 |
- |
- |
- |
OR7E24 |
Chr19 |
- |
- |
- |
SPACA6P |
Chr19 |
- |
- |
- |
PDHA1 |
ChrX |
300502 |
a nuclear-encoded mitochondrial multienzyme complex that catalyzes the overall conversion of pyruvate to acetyl-CoA and CO(2), and provides the primary link between glycolysis and the tricarboxylic acid cycle |
Pyruvate dehydrogenase E1-alpha deficiency |
We discovered fourteen SNPs- and Small InDels-related OMIM genes, including: NACA, PCDHB4, PEX5, CTBP1, UBXN11, DSPP, PKD2, CELA1, ACAN, ESX1, POU3F3, ATXN2, GPSM1, PDHA1. PEX5 (OMIM:600414) is associated with Peroxisome biogenesis disorder 2A, 2B. NACA (OMIM:601234) is associated with skeletal development. ATXN2 (OMIM:601517) is associated with Spinocerebellar ataxia 2, susceptibility to Amyotrophic lateral sclerosis, susceptibility to Parkinson disease and late-onset. CELA1 (OMIM:130120) is associated with evolutionarily silenced in pancreatic acinar cells. PCDHB4 (OMIM:606330) cell-cell neural connections is associated with CTBP1(OMIM:602618) transcriptional repressor and cellular proliferation.
Results verified by Sanger sequencing
We designed twenty-four pairs of primers to verify sixteen differential SNPs loci and eight differentially small InDels. The results of Sanger sequencing are consistent with that of WES (Figure 2).
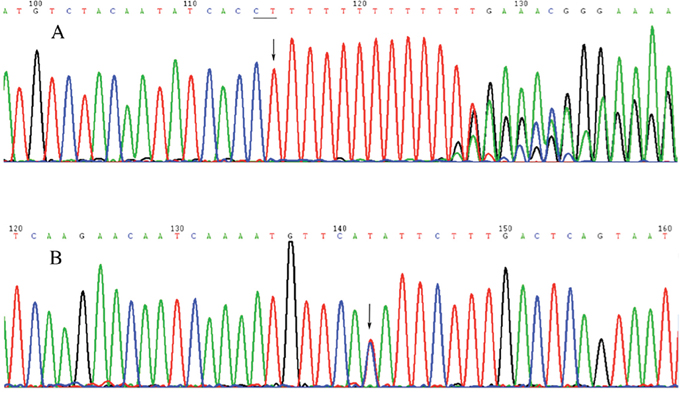
Figure 2: Diagram of the Sanger sequencing for two mutations. (A) chr4:13632182 (CT→C). (B) chr1:86368686 (T→C).
The verification of sixteen differential SNPs sites included NACA (rs745387506, T>G), FAM47C (rs61999275, G>C), UBXN11 (rs6667693, C>A), ODF2L (rs75376884, T>C), DSPP (rs150132251, A>G), PKD2 (rs117078377, G>A), KRTAP5-7 (rs199903176, A>G), CELA1 (rs117443541, T>G), ACAN (rs12899191, A>G), ESX1 (rs9697856, T>G), PCDHB4 (chr5, 141123595, T>G), PEX5 (chr12, 7190555, G>C), PRR21 (chr2, 240042663, C>T), CTBP1 (chr4, 1237208, A>T), DSPP (chr4, 87614868, G>A) and BAGE5 (chr22, 11068057, A>T) were investigated.
The verification of eight differentially small InDels sites included AGCC (rs775150602, AGCC>A), ATXN2 (rs769170503, C>CTGT), MYO15B (rs371699907, CAGG>C), OR7E24 (rs201985790, CT>C), SPACA6P (rs10689461, C>CAG), PDHA1 (rs776856509, TC>TCCT), GPSM1 (chr9, 136327352, ACCCCCTC>A), and (chr4, 13632182, CT>T) were investigated.
Gene ontology (GO)/pathway analysis
GO analysis indicated the percentages of 22 genes enriched in GO term (Figure 3). Growth was related to CHD, and it was associated with some genes including PEX5, NACA, ATXN2 and CELA1 (Table 5). PEX5, ATXN2 and CELA1 are responsible for multicellular organism growth (GO:0035264). PEX5, ATXN2, NACA and CELA1 are responsible for developmental growth (GO:0048589). PEX5, ATXN2 and NACA are responsible for regulation of developmental growth (GO:0048638) and growth regulation (GO:0040008). PEX5 and ATXN2 are responsible for regulation of multicellular organism growth (GO:0040014). PEX5 and NACA are responsible for positive regulation of developmental growth (GO:0048639) and positive growth regulation (GO:0045927). PEX5 is responsible for positive regulation of multicellular organism growth (GO:0040018). ATXN2 is responsible for negative regulation of multicellular organism growth (GO:0040015), epidermal growth-factor receptor binding (GO:0005154), negative regulation of developmental growth (GO:0048640), growth-factor receptor binding (GO:0070851) and negative growth regulation (GO:0045926).
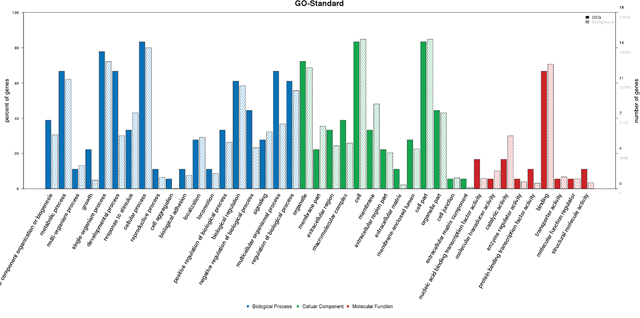
Figure 3: GO analysis of 22 genes.
Table 5: Genes involved by growth and possibly associated with CHD in GO term
Gene |
Term |
Database |
ID |
|---|---|---|---|
PEX5:5830|ATXN2:6311|CELA1:1990 |
multicellular organism growth |
Biological Process |
GO:0035264 |
PEX5:5830|ATXN2:6311|NACA:4666|CELA1:1990 |
developmental growth |
Biological Process |
GO:0048589 |
PEX5:5830|ATXN2:6311|NACA:4666 |
regulation of developmental growth |
Biological Process |
GO:0048638 |
PEX5:5830|ATXN2:6311 |
regulation of multicellular organism growth |
Biological Process |
GO:0040014 |
PEX5:5830|NACA:4666 |
positive regulation of developmental growth |
Biological Process |
GO:0048639 |
PEX5:5830|ATXN2:6311|NACA:4666|CELA1:1990 |
growth |
Biological Process |
GO:0040007 |
PEX5:5830|NACA:4666 |
positive regulation of growth |
Biological Process |
GO:0045927 |
PEX5:5830|ATXN2:6311|NACA:4666 |
regulation of growth |
Biological Process |
GO:0040008 |
PEX5:5830 |
positive regulation of multicellular organism growth |
Biological Process |
GO:0040018 |
ATXN2:6311 |
negative regulation of multicellular organism growth |
Biological Process |
GO:0040015 |
ATXN2:6311 |
epidermal growth factor receptor binding |
Molecular Function |
GO:0005154 |
ATXN2:6311 |
negative regulation of developmental growth |
Biological Process |
GO:0048640 |
ATXN2:6311 |
growth factor receptor binding |
Molecular Function |
GO:0070851 |
ATXN2:6311 |
negative regulation of growth |
Biological Process |
GO:0045926 |
Repression of Wnt target genes and Notch signalling were related to CHD, and was associated with PCDHB4 and CTBP1 genes (Table 6). Pathway analysis showed that twenty-two genes were enriched at the top fifty of pathway term (Figure 4). Genes involved in the Wnt (CTBP1 and PCDHB4) and Notch (CTBP1) signalling are important for pathway analysis in cardiac development. The mutation sites in the six genes were as follows: PEX5 (A60P), NACA (H1283P), ATXN2 (Q21QQ), CELA1 (Y5S), PCDHB4 (S533T) and CTBP1 (C35*). The transcript number of ATXN2 (Q21QQ) is ENST00000608853.5, and the transcript number of CTBP1 (C35*) is ENST00000629223.1.

Figure 4: Pathway analysis of 22 genes.
Table 6: Genes involved by pathway and possibly associated with CHD
Gene |
Term |
Database |
ID |
|---|---|---|---|
CTBP1:1487 |
repression of WNT target genes |
Reactome |
REACT_264567 |
CTBP1:1487 |
AXIN mutants destabilize the destruction complex, activating WNT signaling |
Reactome |
REACT_264496 |
CTBP1:1487 |
deletions in the AXIN genes in hepatocellular carcinoma result in elevated WNT signaling |
Reactome |
REACT_264286 |
CTBP1:1487 |
WNT signaling pathway |
KEGG PATHWAY |
hsa04310 |
CTBP1:1487 |
TCF dependent signaling in response to WNT |
Reactome |
REACT_264596 |
CTBP1:1487 |
RNF mutants show enhanced WNT signaling and proliferation |
Reactome |
REACT_264378 |
CTBP1:1487 |
Signaling by WNT |
Reactome |
REACT_11045 |
CTBP1:1487 |
Notch signaling pathway |
KEGG PATHWAY |
hsa04330 |
PCDHB4:56131|CTBP1:1487 |
WNT signaling pathway |
PANTHER |
P00057 |
Variant analysis
By alignment of protein sequences of PEX5, NACA, ATXN2, CELA1, PCDHB4 and CTBP1, S5333T of PCDHB4 was highly evolutionarily conserved in diverse species including Homo sapiens, Pan paniscus, Pongo abelii and gorilla (Figure 5a). In addition, Y5S of CELA1 was highly evolutionarily conserved in diverse species including H. sapiens, gorilla, Saimiri boliviensis, Papio anubis, Peromyscus maniculatus, Tapirus bairdii, Eptesicus fuscus and Sus scrofa (Figure 5b). The highly evolutionarily conserved protein sequences were not found in PEX5, NACA, ATXN2 and CTBP1 in other species.

Figure 5: (A) Alignment of multiple PCDHB4 protein sequences in different species reveals that the S533T amino acid is located in the highly conserved amino acid region in different species. (B) Alignment of multiple CELA1 protein sequences in different species reveals that the Y33T amino acid is located in the highly conserved amino acid region in different species.
DISCUSSION
TOF as the most common cyanotic CHD is characterised by right ventricular outflow tract obstruction, pulmonary stenosis, ventricular septal defect and aortic overriding caused by non-uniform separation between the truncus arteriosus and bulbus arteriosus at embryo stage. Currently, surgical repair is usually performed in the first month after birth to relieve the stenosis of the right outflow tract and close ventricular septal defect, enabling an exclusive ejection of oxygenated blood via the left ventricle. Cumulative survival and event-free survival were 72% and 25%, respectively, 40 years after follow-up [12].
TOF etiology still needs to be completely clarified because of TOF complexity attributed by both genetic and nongenetic effectors. Next-generation sequencing technology, such as WES with rapid, high throughput and cost-effective features, can meet the requirements for medical studies. WES has been used to obtain information about genetic alterations and potential predispositions possibly associated with TOF occurrence.
Eight small families were tested through WES, and differential SNP loci and InDels were found. From the comparison within each family and among the families, sixteen differential SNPs loci and eight differential Small InDels were screened out. Corresponding genes exist in the database, wherein ten of the sixteen differential SNPs have been reported and six of the sixteen differential SNPs have not been reported. Among the eight Small InDels, seven small Indels have corresponding genes in the database and one Small InDels does not have. Six of the eight differential Small InDels have been reported, whereas two of them have not. Among them, no gene in the database corresponds to the mutation of chr4, position: 13632182. It might be located among the genes or among noncoding regions, and the function requires further study. Go/Pathway analysis of the sixteen differential SNPs loci and eight differential Small InDels were conducted and PEX5, NACA, ATXN2, CELA1, PCDHB4 and CTBP1 were finally screened out and were associated with TOF.
Similar to some genes found to be related to TOF by detection of chromosome karyotype, gene copy number variation and de novo mutation (Table 7), Greenway et al. [13] found four genes (PRKAB2, CHD1L, BCL9 and GJA5) with the highest expression in the right ventricular outflow tract, which is malformed in TOF. Bittel et al. [14] included both idiopathic TOF patients and three patients, which harbour the syndromic 22q11.2 deletion. The expression of the genes A2BP1, VEGF, NPPA and NPPB located in the 22q11.2 region was half-reduced among syndromic patients as expected, whereas none of these genes was differentially expressed in any of the idiopathic TOF subjects. Notch pathway was also suppressed in patients with 22q11.2 deletions. Sharma et al. [15, 16] found that TOF expression of the majority of the genes associated with Wnt and Notch signalling pathways was significantly reduced. Their results also confirmed an upregulated expression of VEGF and proteins of the extracellular matrix reported in previous studies. Feiner et al. [17] found a novel locus at 1q32.2 in two unrelated TOF who displayed loss of copy number variations overlapping the PLXNA2 gene. Knockout mice deficient for PLXNA2 display CHD including TOF [18]. Amarillo et al. [19] found a 3.76 Mb de novo contiguous gain of 9q34.2-q34.3 by chromosome microarray analysis and then also confirmed it by karyotype analysis and fluorescence in situ hybridization. This duplicated interval disrupted retinoid X receptor alpha (OMIM #180245) at intron 1. To our knowledge, PEX5, PCDHB4, CTBP1, NACA, ATXN2 and CELA1 are novel SNPs associated with TOF, which have not been reported.
Table 7: Summary of genes with a potential relevance to TOF
References |
Subjects |
Platform |
Genes |
Conclusions |
|---|---|---|---|---|
Steven C et al |
114 TOF Trios |
Affymetrix Human Genome Wide SNP Array 6.0 |
PRKAB2, CHDIL, BCL9, GJA5 |
potential TOF candidate gene |
Douglas C Bittel et al |
19 TOF |
Applied Microarrays Inc. CodeLink |
A2BP1, VEGF, NPPA, NPPB, |
potential relevance for TOF |
Amy Rodemoyer et al |
16 TOF |
Affymetrix Human Exon 1.0 ST v2 |
NEFH, SST, TF, KRT6A, KRT7, |
dysregulated network in TOF |
Goodship et al |
362 TOF |
SEQUENOM MALDI-TOF |
PTPN11 |
risk allele for TOF |
Candice K et al |
433 TOF |
Affymetrix Genome-Wide Human |
GJA5, PLXNA2 |
potential TOF candidate gene |
Rachel Soemedi et al |
283 TOF Trios |
Illumina 660W SNP Array 6.0 |
GJA5, HAND2, EDIL3, CNOT6 |
potential TOF candidate gene |
Heather J.Cordell et al |
798 TOF |
Illumina 600W QUAD array |
PTPN11, GPC5, NRP1 |
risk allele for TOF |
Marcel Grunert et al |
22 TOF |
NimbleGen sequence capture 365K array |
deleterious SNPs in BARX1, BCCIP, DAG1, EDN1, FANCL, FANCM, FMR1, FOXK1, HCN2, MYOM2, PEX6, ROCK1, TCEB3, TP53BP2, TTN, WBSCR16 |
imbalance of functional networks in TOF |
Tan ZP et al |
1 TOF |
Illumina Hiseq2000 |
SCN5A |
a possible cause of TOF |
LaHaye S et al |
2 TOF |
Illumina Hiseq2000 |
MYBPC3, SOS1 |
uncertain significance |
Our study aimed to identify other monogenic causes of TOF. GO analysis indicated that PEX5 was related to multicellular organism growth (GO:0035264), developmental growth (GO:0048589), regulation of developmental growth (GO:0048638), regulation of multicellular organism growth (GO:0040014), positive regulation of developmental growth (GO:0048639), positive regulation of growth (GO:0045927), growth (GO:0040007) and positive regulation of multicellular organism growth. In the 26 cases with TOF, Grunert M et al. [20] found deleterious SNPs in 16 genes, including BARX1, BCCIP, DAG1, EDN1, FANCL, FANCM, FMR1, FOXK1, HCN2, MYOM2, PEX6, ROCK1, TCEB3, TP53BP2, TTN and WBSCR16 which resulted in the imbalance of function networks in TOF. Huffnagel IC et al.[21] found that two of the 21 genetically proven Rhizomelic chondrodysplasia punctata patients suffered from TOF and showed PEX7 mutations. Plasmalogens were not detected in cardiac tissue of PEX7 knock-out mice, which is a model for RCDP type 1. The above-mentioned PEX5, PEX6 and PEX7 are all PEX family gene named Peroxins (PEXs) which are proteins essential for the assembly of functional peroxisomes. Therefore, PEX5 might participate in TOF growth and development process. PCDHB4 is located on Chr5q31.3. This gene is a member of the protocadherin beta gene cluster and is one of the three related and tandemly linked gene clusters at chromosome 5. Their specific functions are unknown but they most likely play a critical role in the establishment and function of specific cell–cell neural connections. Alazami AM et al. [22] performed WES on 143 multiplex consanguineous families, in which known disease genes had been excluded by autozygosity mapping. Patients, whose clinical phenotype is featured by global developmental delay and brain atrophy, have mutation in PCDHB4 (NM_021908:c.489T>G:p.Y163X) based on the candidate gene analysis, and the function of which participated in the growth and development process. Base on our research, Go analysis indicated that PCDHB4 (P00057) was related to WNT signaling pathway. Thus, PCDHB4 (P00057) may be a potential TOF candidate gene. NACA is located on Chr12q13.3. The protein encoded by NACA is associated with basic transcription factor 3, which forms the nascent polypeptide-associated complex. In this study, GO analysis indicated that NACA was related to positive regulation of developmental growth (GO:0048639) and positive regulation of growth (GO:0045927). Thus, NACA may be a potential TOF candidate gene. CELA1 is located on Chr12q13.13. This gene forms a subfamily of serine proteases that hydrolyze many proteins in addition to elastin. Humans possess six elastase genes, which encode the structurally similar elastases 1, 2, 2A, 2B, 3A and 3B. Unlike other elastases, pancreatic elastase 1 is not expressed in the pancreas. GO analysis indicated that CELA1 was related to multicellular organism growth (GO:0035264), developmental growth (GO:0048589) and growth (GO:0040007). Thus, CELA1 may have a potential relevance to TOF. ATXN2 is located on Chr12q24.12 with a transcript number of ENST00000608853.5. This gene belongs to a group of genes that are associated with microsatellite-expansion diseases, a class of neurological and neuromuscular disorders caused by expansion of short stretches of repetitive deoxyribonucleic acid (DNA). Genome-wide association studies indicate that loss-of-function mutations in this gene may be associated with susceptibility to type I diabetes, obesity and hypertension. Alternative splicing leads to multiple transcript variants. GO analysis indicated that ATXN2 was related to negative regulation of multicellular organism growth (GO:0040015), epidermal growth-factor receptor binding (GO:005154), negative regulation of developmental growth (GO:0048640), growth-factor receptor binding (GO:0070851) and negative regulation of growth (GO:0045926). Thus, ATXN2 may have a potential relevance to TOF. CTBP1 is located on Chr4p16.12 with a transcript number of ENST00000629223.1. This gene encodes a protein that binds to the C-terminus of adenovirus E1A proteins. This phosphoprotein is a transcriptional repressor and may play a role in cellular proliferation. This protein and the product of a second closely related gene, CTBP1, can dimerise. Both proteins can also interact with a polycomb group protein complex, which participates in regulation of gene expression during development. Alternative splicing of transcripts from this gene results in multiple transcript variants. GO analysis indicated that CTBP1 was related to Wnt signalling pathway and Notch signalling pathway. Thus, CTBP1 may be a potential TOF candidate gene.
This study has some limitations, and the sample size is small. In the future, the number of samples will be further increased and an in-depth research will be conducted. The identified mutation sites have not been verified based on large TOF sample (n>50). Thus, whether the identified mutation sites may occur in a large group of TOF people has not been confirmed. Moreover, further study is necessary to verify relevant function genes and accordingly illustrate the pathogenesis of TOF.
In summary, TOF pathogenesis is complex because TOF is associated with more than one genetic variant. Eight small families were observed through WES and found that TOF-related genes were PEX5, NACA, ATXN2, CELA1, PCDHB4 and CTBP1, whose potential function might participate in TOF growth and development process. Although none of the three variants were predicted to be highly influential, they are in genes belonging to networks involved in relevant developmental pathways. Further study on the potential developmental mechanisms will be conducted based on these results.
MATERIALS AND METHODS
Subjects
A total of eight small families whose Chinese children received surgical TOF treatment at the Children’s Heart Center of Henan Provincial People’s Hospital between January 2016 and June 2016 were enrolled in this study. The eight children ageing 3–11 months included four male babies and four female babies. All manifested isolated TOF. Their parents were normal. All the eight children were diagnosed with TOF by echocardiogram, clinical symptoms and signs and intraoperative findings.
Methods
DNA extraction
In each member of the eight small families including 24 individuals, 600 μl peripheral blood was collected to extract genomic DNA using genomic DNA Extraction Kit (Qiage, Germany). The remaining blood samples were stored at −80°C.
WES and mutation screening
WES and subsequent variant annotation were performed on genomic DNA derived from the eight TOF children and their parents. Paired-end libraries were prepared according to the manufacturer’s protocols (Agilent). Exome libraries for the eight families were constructed using Agilent SureSelectXT Target Enrichment System according to Illumina Paired End Sequencing Protocol (Agilent Technologies, CA, USA). Capturing of whole exon was carried out according to the protocol of Agilent’s Sure Select Human All 1 UTRs 71 MB v4 kit. The flow cells were sequenced as paired-end 150 base pair reads on an Illumina HiSeq ×10 platform to a minimum depth of 50× targeted region coverage using TruSeq SBS sequencing kit version 3 and HiSeq data collection version 2.0.12.0 software (Illumina, Inc., San Diego, CA, USA).
The raw sequence reads we obtained were aligned to the human genome reference sequence (hg19) using Burrows–Wheeler Aligner (BWA) with standard parameters [10]. The BWA-aligned reads were statistically calculated using PICARD software to exclude polymerase chain reaction duplicates. Regional realignment and quality score recalibration were carried out using Genome Analysis Toolkit [11] with recommended parameters, which included local realignment of the sequences around InDels, base quality score recalibration, variant calling and variant quality score recalibration.
Variant detection in the eight TOF children by Sanger sequencing
BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and ABI 3130 Genetic Analyzer were used to detect variants among the eight TOF children.
Ethics statement
Approval was obtained from local ethics committees of Henan Provincial People’s Hospital, Zhengzhou University People’s Hospital. Informed consents were provided to all the participants of this study.
Abbreviations
TOF: tetralogy of Fallot; CHD: congenital heart disease; WES: whole-exome sequencing; SNPs: single nucleotide polymorphisms; CNVs: copy number variations; DNA: deoxyribonucleic acid; BWA: Burrows–Wheeler Aligner; GATK: Genome Analysis Toolkit; BTF3: basic transcription factor 3; NAC: nascent polypeptide-associated complex ; SRP: signal recognition particle.
Author contributions
Lin Liu designed the experiments, recruited patients, collected samples, analysed the data, performed statistics and wrote the manuscript. Hong-dan Wang designed the experiments, recruited the patients, collected samples, analysed the data, performed statistics and wrote the manuscript. Cun-ying Cui recruited the patients, collected the samples, analysed the data and wrote the manuscript. Yun-yun Qin collected samples, analysed the data and wrote the manuscript. Tai-bing Fan collected the samples. Bang-tian Peng collected the samples. Lian-zhong Zhang performed the statistics. Cheng-zeng Wang designed the experiments, recruited patients and critically reviewed the manuscript.
ACKNOWLEDGMENTS
The authors are grateful to the patients and families who participated in this research. Team members who supported the subject recruitment and sequencing are as follows: Yan-ze Li (CapitalBio Technology), Qian Zhang, Hao-ju Dong, Shu-bo Song, Yan-wei Zhang, Jia-yong Zheng, Feng Ai, Dan-qing Huang and Juan Zhang.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This study was supported by National Natural Science Foundation of China (81401419), Program of Science and Technology of Henan Province (172102310003) and Program of Advanced Study to go Abroad of Henan Provincial Health System (2016047).
REFERENCES
1. Bédard E, McCarthy KP, Dimopoulos K, Giannakoulas G, Gatzoulis MA, Ho SY. Structural abnormalities of the pulmonary trunk in tetralogy of Fallot and potential clinical implications: a morphological study. J Am Coll Cardiol. 2009; 54:1883–90.
2. Beauchesne LM, Warnes CA, Connolly HM, Ammash NM, Grogan M, Jalal SM, Michels VV. Prevalence and clinical manifestations of 22q11.2 microdeletion in adults with selected conotruncal anomalies. J Am Coll Cardiol. 2005; 45:595–98.
3. Goldmuntz E, Geiger E, Benson DW. NKX2.5 mutations in patients with tetralogy of Fallot. Circulation. 2001; 104:2565–68.
4. Salazar M, Consoli F, Villegas V, Caicedo V, Maddaloni V, Daniele P, Caianiello G, Pachón S, Nuñez F, Limongelli G, Pacileo G, Marino B, Bernal JE, et al. Search of somatic GATA4 and NKX2.5 gene mutations in sporadic septal heart defects. Eur J Med Genet. 2011; 54:306–09.
5. Eldadah ZA, Hamosh A, Biery NJ, Montgomery RA, Duke M, Elkins R, Dietz HC. Familial Tetralogy of Fallot caused by mutation in the jagged1 gene. Hum Mol Genet. 2001; 10:163–69.
6. Bauer RC, Laney AO, Smith R, Gerfen J, Morrissette JJ, Woyciechowski S, Garbarini J, Loomes KM, Krantz ID, Urban Z, Gelb BD, Goldmuntz E, Spinner NB. Jagged1 (JAG1) mutations in patients with tetralogy of Fallot or pulmonic stenosis. Hum Mutat. 2010; 31:594–601.
7. Lu JH, Chung MY, Betau H, Chien HP, Lu JK. Molecular characterization of tetralogy of Fallot within Digeorge critical region of the chromosome 22. Pediatr Cardiol. 2001; 22:279–84.
8. Stoll C, Dott B, Alembik Y, Roth MP. Associated congenital anomalies among cases with Down syndrome. Eur J Med Genet. 2015; 58:674–80.
9. Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, Bamshad M, Nickerson DA, Shendure J. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009; 461:272–76.
10. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010; 26:589–95.
11. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011; 43:491–98.
12. Cuypers JA, Menting ME, Konings EE, Opić P, Utens EM, Helbing WA, Witsenburg M, van den Bosch AE, Ouhlous M, van Domburg RT, Rizopoulos D, Meijboom FJ, Boersma E, et al. Unnatural history of tetralogy of Fallot: prospective follow-up of 40 years after surgical correction. Circulation. 2014; 130:1944–53.
13. Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, Ergul E, Conta JH, Korn JM, McCarroll SA, Gorham JM, Gabriel S, Altshuler DM, et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. 2009; 41:931–35.
14. Bittel DC, Butler MG, Kibiryeva N, Marshall JA, Chen J, Lofland GK, O’Brien JE Jr. Gene expression in cardiac tissues from infants with idiopathic conotruncal defects. BMC Med Genomics. 2011; 4:1–10.
15. Sharma HS, Peters TH, Moorhouse MJ, van der Spek PJ, Bogers AJ. DNA microarray analysis for human congenital heart disease. Cell Biochem Biophys. 2006; 44:1–9.
16. Peters TH, Sharma V, Yilmaz E, Mooi WJ, Bogers AJ, Sharma HS. DNA microarray and quantitative analysis reveal enhanced myocardial VEGF expression with stunted angiogenesis in human tetralogy of Fallot. Cell Biochem Biophys. 2013; 67:305–16.
17. Feiner L, Webber AL, Brown CB, Lu MM, Jia L, Feinstein P, Mombaerts P, Epstein JA, Raper JA. Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development. 2001; 128:3061–70.
18. Brown CB, Feiner L, Lu MM, Li J, Ma X, Webber AL, Jia L, Raper JA, Epstein JA. PlexinA2 and semaphorin signaling during cardiac neural crest development. Development. 2001; 128:3071–80.
19. Amarillo IE, O’Connor S, Lee CK, Willing M, Wambach JA. De novo 9q gain in an infant with tetralogy of Fallot with absent pulmonary valve: patient report and review of congenital heart disease in 9q duplication syndrome. Am J Med Genet A. 2015; 167A:2966–74.
20. Grunert M, Dorn C, Schueler M, Dunkel I, Schlesinger J, Mebus S, Alexi-Meskishvili V, Perrot A, Wassilew K, Timmermann B, Hetzer R, Berger F, Sperling SR. Rare and private variations in neural crest, apoptosis and sarcomere genes define the polygenic background of isolated Tetralogy of Fallot. Hum Mol Genet. 2014; 23:3115–28.
21. Huffnagel IC, Clur SA, Bams-Mengerink AM, Blom NA, Wanders RJ, Waterham HR, Poll-The BT. Rhizomelic chondrodysplasia punctata and cardiac pathology. J Med Genet. 2013; 50:419–24.
22. Alazami AM, Patel N, Shamseldin HE, Anazi S, Al-Dosari MS, Alzahrani F, Hijazi H, Alshammari M, Aldahmesh MA, Salih MA, Faqeih E, Alhashem A, Bashiri FA, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Reports. 2015; 10:148–61.