
INTRODUCTION
Obstructive sleep apnea (OSA) is a prevalent sleep disorder that causes repetitive collapse of the upper airway, asphyxia, oxygen desaturation and chronic intermittent hypoxia (CIH) [1]. OSA is an independent risk for cardiovascular diseases, e.g. systemic arterial hypertension, ischemic heart disease, cardiac arrhythmias, metabolic disorders, and cognitive dysfunction, likely owing to its associated CIH and re-oxygenation processes [2].
The pathogenesis of OSA-induced cardiovascular diseases has been found to involve endothelial dysfunction secondary to oxidative stress and activation of inflammation [3]. The most important feature of endothelial dysfunctions are endothelial cell damage and impaired stimulated vasodilatation, as a precursor of atherosclerosis in human [2].
Upon injury, autophagic machinery is activated to endothelial cell to contradict the apoptotic progressive cell death [4]. The failure or inhibition of autophagy results in augmentation of endothelial cell apoptosis, leading to the break-down of the integrity of endothelium to facilitate endothelial dysfunction [5]. During autophagy, cytoplasmic constituents are sequestered and targeted for lysosomal degradation and re-cycling by autophagosomes, in which a cytosolic form of microtubule-associated protein 1A/1B-light chain 3 (LC3), LC3-I, is transformed and exhibits as LC3-phosphatidylethanolamine conjugate (LC3-II) [5]. Since the ratio of LC3-II to LC3-I is strictly consistent with the cellular autophagic activity, it is thus used as an indicator of autophagy level in cells [6]. Many proteins coordinate the occurrence and progression of autophagy [6]. Some of these proteins are called autophagy-associated protein, among which Beclin-1 (or ATG6) is a critical one [6]. Previous studies have shown that Beclin-1 controls the initiation of autophagy. Moreover, apoptosis-related caspases were recently shown to cleave Beclin-1 to terminate its pro-autophagic activity [7, 8]. On the other hand, the C-terminal fragment of Beclin-1 cleaved by caspases could amplify mitochondrion-mediated apoptosis, which appeared to be a feedback for caspase activity on Beclin-1 [7, 8]. These molecular crosstalk between Beclin-1 and caspases appears to be a link for interaction between autophagy and apoptosis, which highlights the important of Beclin-1 in control of cell death and survival.
MicroRNAs (miRNAs) are small non-coding RNAs that repress protein translation through their interaction with the 3′-untranslated region (3′-UTR) of the target mRNAs [9]. Interestingly, miRNAs were found to play very critical roles in autophagy [9], and in control of the expression of Beclin-1 [10–15]. Specifically, miR-30 has been shown as a Beclin-1-targeting miRNA [16–22]. Nevertheless, a role of miR-30 on endothelial cell autophagy in the regulation of CIH or OSA has not been studied.
Here, we addressed this question. In a mouse model of CIH, we detected significant upregulation of miR-30a, which subsequently attenuated the endothelial cell autophagy against cell death through Beclin-1 suppression. Suppression of miR-30a by expression of antisense of miR-30a significantly increased Beclin-1 levels to enhance endothelial cell autophagy in vitro and in vivo, which improved endothelial cell survival against CIH.
RESULTS
CIH causes increases in apoptosis but no changes in autophagy in mouse aortic endothelial cells
The exact molecular control of endothelial cell autophagy in CIH has not been determined. Here, we addressed this question. First, we performed a well-established mouse model of CIH. After 8 weeks, we found that the CIH mice exhibited echocardiographic defects, including significantly increased end-diastolic volume (EDV, Figure 1A), significantly increased end-systolic volume (ESV, Figure 1B), significantly reduced percentage of ejection fraction (EF%, Figure 1C), and significantly reduced percentage of fractional shortening (FS%, Figure 1D), compared to the control mice (CTL). Moreover, in the dissected mouse aorta, significantly higher levels of pro-inflammatory factors tumor necrosis factor alpha (TNFa), interferon gamma (INFr), hypoxia-inducible factor 1 alpha (HIF1a) and interleukin (IL-6) were detected (Figure 1E) were detected in CIH mice, compared to CTL. These data confirmed the successful establishment of the CIH model and CIH-induced cardiac dysfunction in mice.
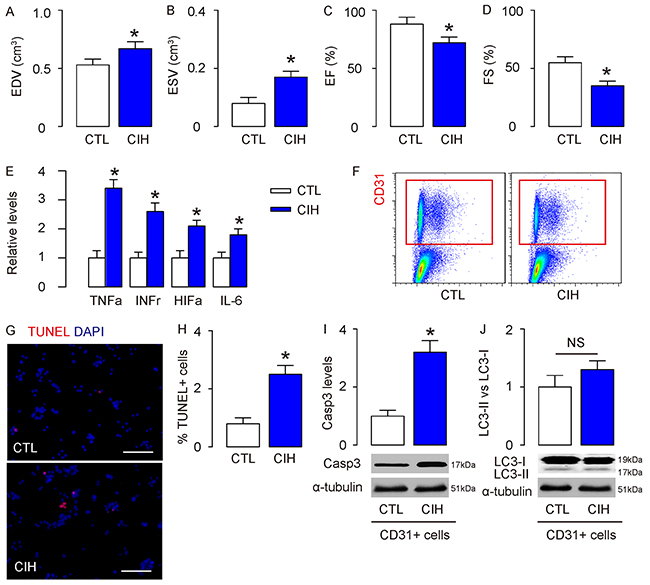
Figure 1: CIH causes increases in apoptosis but no changes in autophagy in mouse aortic endothelial cells. A well-established mouse model of CIH was performed. (A-D) After 8 weeks, echocardiography was done on mice in CIH and control (CTL) mice. (A) End-diastolic volume (EDV), (B) End-systolic volume (ESV), (C) Percentage of ejection fraction (EF%), (D) Percentage of fractional shortening (FS%). (E) The aortas were dissociated and purified for endothelial cells based on CD31 labeling by flow cytometry. (F) Levels of pro-inflammatory factors [tumor necrosis factor alpha (TNFa), interferon gamma (IFNr), hypoxia-inducible factor 1 alpha (HIF1a) and interleukin (IL-6)] in mouse aorta by ELISA. (G-H) TUNEL staining for purified CD31+ endothelial cells, shown by representative images (G), and by quantification (H). (I-J) Western blotting for cleaved caspase 3 (Casp3, I) and LC3 (J). *p<0.05. NS: non-significant. N=10.
In order to study CIH-associated endothelial cell autophagy, we isolated CD31+ mouse aortic endothelial cells by flow cytometry in CIH and CTL mice (Figure 1F), and found that the endothelia cells from CIH mice contained more apoptotic cells, in an TUNEL assay, shown by representative images (Figure 1G), and by quantification (Figure 1H). Next, we examined the levels of an apoptosis marker, cleaved caspase 3 (Casp3), and an autophagy marker, LC3, by Western blotting. We found that the Casp3 levels significantly increased in endothelial cells from CIH mice, compared to CTL (Figure 1I), confirming the increases in apoptosis by CIH. On the other hand, the LC3 II vs I levels were not significantly increased in endothelial cells from CIH mice, compared to CTL (Figure 1J). These are rather unexpected results, since hypoxia insult often cause increases in autophagy, as a resistance of the cells to apoptosis. Hence, we hypothesized presence of inhibitory mechanism of autophagy in CIH.
Autophagy is compensated by miR-30a-suppression of Beclin-1 in endothelial cells at CIH
The aim of this study was to figure out the miRNA-control of the endothelial cell autophagy at CIH. Hence, we examined the expression of published miRNAs that are related to autophagy in endothelial cells from CIH and CTL mice (Table 1, Figure 2A). Among these candidates, we specifically found that miR-30a, a Belcin-1 suppressor, was significantly upregulated in endothelial cells in CIH (Figure 2A). Bioinformatics showed that miR-30a targets 3′-UTR of Beclin-1 mRNA at one binding site of base pair 94th to 101th (Figure 2B). Hence, we prepared miR-30-overexpressing plasmids (miR-30a), miR-30a-suppressing plasmids (antisense of miR-30a; as-miR-30a), and null plasmids as a control. Transfection of mouse endothelial cells by these plasmids confirmed their potential in modifying miR-30a levels in mouse endothelial cells (Figure 2C). The functional binding of miR-30a to Beclin-1 mRNA was then examined in a dual luciferase reporter assay. The intact 3′-UTR of wildtype Beclin-1 mRNA (Beclin-1 3′-UTR) and the 3′-UTR of Beclin-1 mRNA with a mutant at miR-30a-binding site (Beclin-1 3′-UTR mut) were respectively cloned into luciferase reporter plasmids. Mouse endothelial cells were then co-transfected with one plasmid from miR-30a/as-miR-30a/null plasmids and one plasmid from either Beclin-1 3′-UTR or Beclin-1 3′-UTR mut, and subsequently subjected to a dual luciferase reporter assay. We found that depletion of miR-30a increased luciferase activity of Beclin-1 3′-UTR, while overexpression of miR-30a reduced luciferase activity of Beclin-1 3′-UTR but had no effects on Beclin-1 3′-UTR mut. These results suggest that miR-30a specifically targets 3′-UTR of Beclin-1 mRNA to inhibit its translation in endothelial cells (Figure 2D).
Table 1: Published miRNAs associated with autophagy
miRNA |
Target autophagy regulatory proteins |
|---|---|
miR-7 |
EGFR |
miR-31 |
HIF-1a |
miR-34 |
HIF-1a |
miR-30a |
Beclin-1 |
miR-376b |
Beclin-1/ATG4 |
miR-519a |
Beclin-1 |
miR-199a-5p |
ATG7 |
miR-375 |
ATG7 |
miR-519a |
ATG10/ATG16 |
miR-181a |
ATG5/ATG12 |
miR-374a |
ATG5/ATG12 |
miR-101 |
ATG4/Rab5A |
miR-502 |
P53/Rab1B |
miR-130a |
ATG2B/DICER1 |
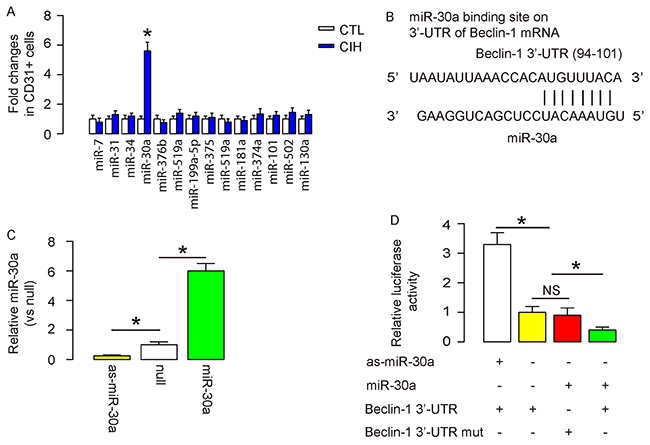
Figure 2: Autophagy is compensated by miR-30a-suppression of Beclin-1 in endothelial cells at CIH. (A) RT-qPCR for expression levels of autophagy regulatory miRNAs in purified endothelia cells from CIH and CTL mice. (B) Bioinformatics showed that miR-30a targets 3′-UTR of Beclin-1 mRNA at one binding site of base pair 94th to 101th. (C) RT-qPCR for the transfected mouse endothelial cells by miR-30-overexpressing plasmids (miR-30a), miR-30a-suppressing plasmids (antisense of miR-30a; as-miR-30a), and null plasmids as a control. (D) The functional binding of miR-30a to Beclin-1 mRNA was then examined in a dual luciferase reporter assay. The intact 3′-UTR of wildtype Beclin-1 mRNA (Beclin-1 3′-UTR) and the 3′-UTR of Beclin-1 mRNA with a mutant at miR-30a-binding site (Beclin-1 3′-UTR mut) were respectively cloned into luciferase reporter plasmids. Mouse endothelial cells were then co-transfected with one plasmid from miR-30a/as-miR-30a/null plasmids and one plasmid from either Beclin-1 3′-UTR or Beclin-1 3′-UTR mut, and subsequently subjected to a dual luciferase reporter assay. The luciferase activity was determined. *p<0.05. NS: non-significant. N=10.
Suppression of miR-30a by as-miR-30a significantly increases CIH-associated endothelial cell autophagy in vitro
Next, mouse endothelial cells expressing as-miR-30a (EC) or null were subjected to an in vitro CIH model. In group 1, null-transfected cells were exposed to normal oxygen (EC/CTL); in group 2, null-transfected cells were exposed to intermittent hypoxic (EC/IH); in group 3, as-miR-30a-transfected cells were exposed to intermittent hypoxic (EC-as-miR-30a/IH). After treatment, apoptosis of the cells was determined in TUNEL assay. We found that IH significantly increased apoptotic cells, which was significantly attenuated by suppression of miR-30a in endothelia cells, shown by representative images (Figure 3A), and by quantification (Figure 3B). Next, we examined the levels of Casp3 by Western blotting. We found that the Casp3 levels significantly increased in endothelial cells by IH, which was significantly attenuated by suppression of miR-30a in endothelia cells (Figure 3C), suggesting that the increases in apoptosis by CIH are reduced by miR-30a suppression. In an CCK-8 assay, the viable cell number significantly decreased in endothelial cells by IH, which was significantly attenuated by suppression of miR-30a in endothelia cells (Figure 3D). On the other hand, when we examined LC3 for determining autophagy activity, we found that although IH did not significantly altered the LC3 II vs I levels in endothelial cells, suppression of miR-30a significantly increased this ratio (Figure 3E). Thus, suppression of miR-30a in endothelial cells enhanced cell autophagy at IH. The levels of Beclin-1 protein in endothelial cells were not altered by IH (Figure 3F), but the levels of Beclin-1 mRNA were (Figure 3G). Furthermore, suppression of miR-30a in endothelial cells significantly increased Beclin-1 protein in IH (Figure 3F), without altering Beclin-1 mRNA (Figure 3G), consistent with the abovementioned presence of miR-30a suppression of Belcin-1 protein translation (Figure 2). Thus, suppression of miR-30a by as-miR-30a appeared to enhance CIH-associated endothelial cell autophagy in vitro through increasing Beclin-1.
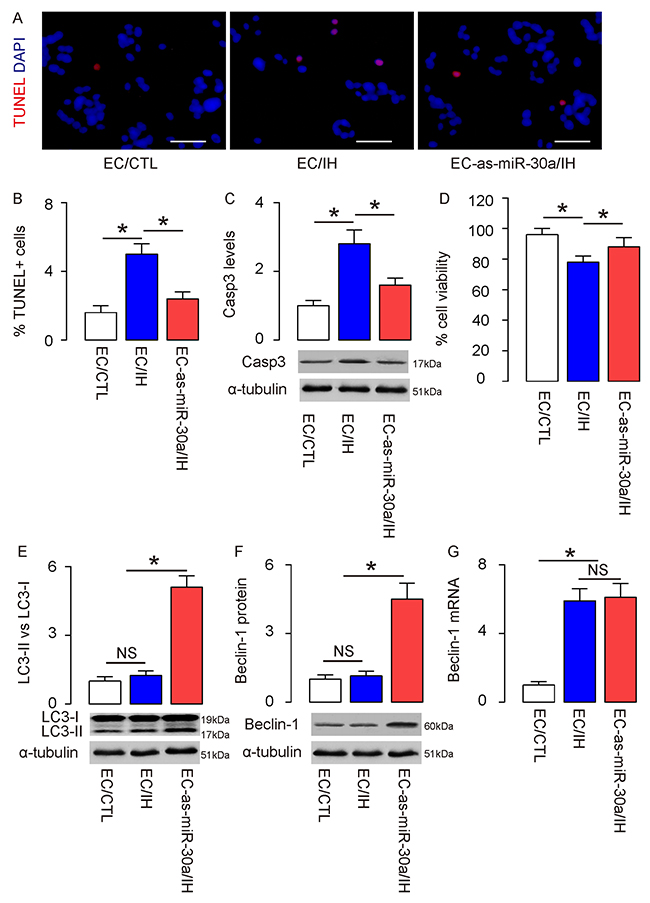
Figure 3: Suppression of miR-30a by as-miR-30a significantly increases CIH-associated endothelial cell autophagy in vitro. Mouse endothelial cells expressing as-miR-30a (EC) or null were subjected to an in vitro CIH model. In group 1, null-transfected cells were exposed to normal oxygen (EC/CTL); in group 2, null-transfected cells were exposed to intermittent hypoxic (EC/IH); in group 3, as-miR-30a-transfected cells were exposed to intermittent hypoxic (EC-as-miR-30a/IH). (A-B) After treatment, apoptosis of the cells was determined in TUNEL assay, shown by representative images (A), and by quantification (B). (C) Western blotting for Casp3. (D) An CCK-8 assay for these cells. (E) Western blotting for LC3. (F) Western blotting for Beclin-1. (G) RT-qPCR for Beclin-1. *p<0.05. NS: non-significant. N=5.
Suppression of miR-30a by as-miR-30a significantly increases CIH-associated endothelial cell autophagy and attenuates cardiac dysfunction in vivo
Finally, we examined this model in vivo. Suppression of miR-30a in mouse endothelial cells in vivo was realized through i.v. infusion of a lentivirus carrying as-miR-30a or null under control of a cytomegalovirus (CMV) promoter immediately before CIH treatment. In group 1, mice did not receive CIH but null virus (CTL); in group 2, mice received CIH and null virus (CIH); in group 3, mice received CIH and as-miR-30a virus (CIH+as-miR-30a). At analysis, we isolated CD31+ mouse aortic endothelial cells by flow cytometry in these mice (Figure 4A), and found that the increased endothelia cell apoptosis in CIH mice was significantly attenuated by suppression of miR-30a, in an TUNEL assay (Figure 4B–4C), and by Casp3 Western blotting (Figure 4D). On the other hand, the unchanged endothelial cell autophagy by CIH was significantly enhanced by miR-30a suppression, evident by LC3 II vs I ratio (Figure 4E) and Beclin-1 protein (Figure 4F). Thus, suppression of miR-30a by as-miR-30a appeared to enhance CIH-associated endothelial cell autophagy in vivo through increasing Beclin-1. Together, our data suggest that endothelial cell autophagy in CIH may be attenuated by miR-30a-mediated translational control of Beclin-1, as an important cause of endothelial cell dysfunction and damage (Figure 5).
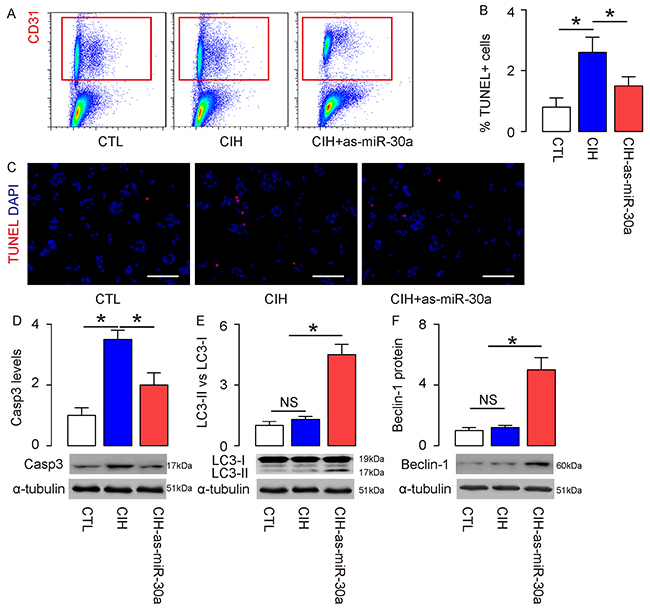
Figure 4: Suppression of miR-30a by as-miR-30a significantly increases CIH-associated endothelial cell autophagy and attenuates cardiac dysfunction in vivo. Suppression of miR-30a in mouse endothelial cells in vivo was realized through i.v. infusion of a lentivirus carrying as-miR-30a or null under control of a cytomegalovirus (CMV) promoter immediately before CIH treatment. In group 1, mice did not receive CIH but null virus (CTL); in group 2, mice received CIH and null virus (CIH); in group 3, mice received CIH and as-miR-30a virus (CIH+as-miR-30a). (A) At analysis, CD31+ mouse aortic endothelial cells were isolated by flow cytometry in these mice. (B-C) TUNEL staining for purified CD31+ endothelial cells, shown by quantification (B), and by representative images (C). (D-F) Western blotting for Casp3 (D), LC3 (E) and Beclin-1 (F). *p<0.05. NS: non-significant. N=10.
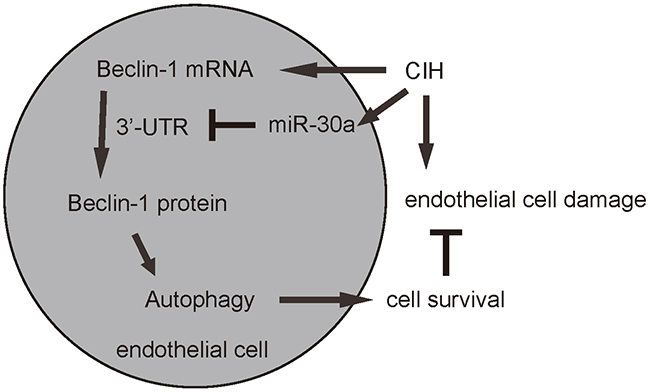
Figure 5: Schematic of the model. CIH induces damages of endothelial cells partially through upregulation of miR-30a, which inhibits the Beclin-1 through 3′-UTR binding, resulting in abolished increases in autophagy to antagonize CIH-induced apoptosis of endothelial cells. Endothelial cell autophagy in CIH attenuated by miR-30a-mediated translational control of Beclin-1 is thus an important cause of endothelial cell dysfunction and damage.
DISCUSSION
OSA is recognized as a significant and highly prevalent health problem with cardiovascular and metabolic morbidity, which is primarily attributable to CIH-associated neurobehavioral impairments, increased cell apoptosis, oxidant stress and inflammation [2]. A previous study has shown the role of miRNAs in CIH as well as the relationship between endothelial cell apoptosis and endothelial dysfunction [23], but the molecular mechanisms remain undetermined. Very recently, CIH was shown to cause body weight reductions and insulin resistance, seemingly via control of ATG7 [24]. However, this study revealed the involvement of autophagy in the CIH-mediated endothelial cell damage and dysfunction. These previous studies encouraged us to study the association of miss-expressed miRNAs and endothelia cell autophagy in the pathogenesis of CIH.
In a mouse model of CIH, we detected significant increases in endothelial cell apoptosis. However, the endothelial cell autophagy was not altered. Since hypoxia insult often causes increases in autophagy to contradict the enhanced apoptosis, this result was rather surprising and may indicate that the autophagy activation here may be suppressed by some factors associated with CIH. Indeed, in an in vitro model for CIH, uncoupled alteration of Beclin-1 protein and mRNA indicated presence of post-transcriptional control of this key regulator of autophagy. Although post-transcriptional control of a protein involves protein degradation and translation by miRNAs, we focused on the later possibility based on the aim of this study.
We examined the expression levels of published autophagy regulatory miRNAs in endothelial cells from CIH and CTL mice, and found exclusive upregulation of miR-30a by CIH. Interestingly, miR-30a is a well known miRNA that targets 3′-UTR of Beclin-1 mRNA to suppress the protein translation. For example, Lai et al. showed that angiotensin-converting enzyme 2 suppressed harmful action of Doxorubicin and caused a significant improvement of left ventricular contractility function, through upregulation in miR-30, which inhibited Beclin-1 and then reduced LC3-II/I ratio and autophagy in cardiomyocytes [25]. Zhang et al. showed that high-fat-diet-treated mice had significantly lower levels of endothelial cell autophagy, resulting from miR-30 suppression on Beclin-1. Upregulation of miR-30 in endothelial cells by high-fat-diet impair the protective effects of endothelial cell autophagy against development of atherosclerosis [16]. In the current study, this regulatory axis apparently attenuated the endothelial cell autophagy against cell death. Indeed, apoptosis-related caspases were recently shown to cleave Beclin-1 to terminate its pro-autophagic activity, while cleaved C-terminal fragment of Beclin-1 strengthened mitochondrion-mediated apoptosis [7, 8]. Thus, the control of Beclin-1 here appeared to the key regulator for the balance between cellular apoptosis and autophagy within endothelia cells in responsive to the CIH insult. These findings were further strengthened by in vitro and in vivo loss-of-function experiments for miR-30a, in which depletion of miR-30a by expression of as-miR-30a abolished the suppressive effects on endothelial cell autophagy, resulting in improved cardiac function in vivo.
Together, these data suggest that endothelial cell autophagy in CIH may be attenuated by miR-30a-mediated translational control of Beclin-1, as an important cause of endothelial cell dysfunction and damage.
MATERIALS AND METHODS
Ethics statement
The animal study was approved by the Animal Care and Use Committee of the First Affiliated Hospital of Fujian Medical University. All the animal and experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health.
Animal model for CIH
Male C57BL/6 mice of 8–10 weeks old were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and maintained under sterile conditions and standard animal room conditions (temperature, 22°C; humidity, 55%) with a 12-h: 12-h dark/light cycle and free access to water and food. The CIH protocol has been previously described [26]. Mice were housed in customized cages suppled with either an intermittent hypoxic stimulus or intermittent room air, controlled by a gas delivery port for flow of room air, N2, and O2 into the customized cages. During the 12-h light cycle, O2 fraction levels was reduced from 21% to 5% over a 30-s period and rapidly re-oxygenated to room air levels using a burst of 100% O2 from a medical air compressor during the following 30-s period. Room air was delivered to the cages throughout the 12-h dark cycle. Mice were exposed for 8 weeks in either CIH or intermittent air as a control (CTL).
Flow cytometry based isolation of endothelial cells
For analyses and isolation of endothelial cells, the mouse aorta was dissociated by 20 μg/ml trypsin (Sigma-Aldrich, St. Louis, MO, USA) and 10 μg/ml DNase (Roche, Nutley, NJ, USA) for 40 minutes. Single cell fraction was obtained through a 30μm-filter. Single cell fraction was then incubated with APC-conjugated-anti-CD31 antibody (Becton-Dickinson Biosciences, San Jose, CA, USA) and then sorted on a FACSAria (Becton-Dickinson Biosciences). Data were presented using Flowjo software (Flowjo LLC, Ashland, OR, USA).
Endothelial cell transfection and transduction
Isolated CD31+ mouse endothelial cells were cultured in Endothelial Cell Media supplemented with endothelial cell growth factors, 10% fetal bovine serum (FBS, Invitrogen, CA, Carlsbad, USA) and 1% penicillin/streptomycin (Invitrogen) at 37°C with 5% CO2. Mouse endothelial cells were transfected with miR-30a mimics, antisense for miR-30a (as-miR-30a) or null as a control (RiboBio Co., Ltd., shanghai, China), using Lipofectamine 3000 (Invitrogen). The transfection efficiency was about 90%. For suppression of miR-30a in vivo, we generated lentiviruses carrying as-miR-30a or null under control of a cytomegalovirus (CMV) promoter. The plasmids for generating these 2 viruses were prepared from a pcDNA3.1-CMV-GFP plasmid (Clontech, Mountain View, CA, USA), using molecular cloning. For lentiviral production, seeded HEK293T cells (American Type Culture Collection, ATCC, Manassas, VA, USA) were co-transfected with 5 μg of the prepared plasmids and 5 μg each of 3 packaging plasmids (REV, pMDL and VSV-G) by Lipofectamine-3000 (Invitrogen). The supernatant was removed 48 hours after transfection and filtered through the 0.45 μm syringe filter, after which the lentivirus in supernatant was isolated for titration. For in vivo delivery, 107 viruses were injected into the mouse through tail vein.
In vitro IH treatment of the cells
Purified mouse endothelial cells were treated in serum-free medium for 12 h till synchronization, and then cultured in serum-free medium to prepare for in vitro intermittent hypoxic (IH) treatment. Serum-free culture under normal oxygen conditions was used as the control. In the IH group, the medium was placed in the hypoxia cabin by filling the cabin circulation with 5% CO2 for 15 s and 1% O2 for 25 s, totaling 60 cycles. After hypoxic treatment, the medium was washed off, and the mouse aortic endothelial cells were returned to the maintenance medium in the normal incubator for 60-min recovery.
TUNEL assay and immunohistochemistry
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay was designed to detect apoptotic cells at the late stages of apoptosis. The method is based on the ability of TdT to label blunt ends of double-stranded DNA breaks independent of a template. The assay was performed using a TUNEL Assay kit (R&D Biosystems, Shanghai, China). Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole, Sigma-Aldrich).
Cell viability by cell counting kit-8 (CCK-8) assay
A CCK-8 detection kit (Sigma-Aldrich) was used to measure cell viability. Briefly, cells were seeded in a 96-well microplate at a density of 5000/well. After treatment, CCK-8 solution (20ml/well) was added and the plate was incubated at 37°C for 2 hours. The viable cells were counted at a wavelength of 450 nm.
Real-time quantitative PCR
For real-time quantitative PCR (RT-qPCR), total RNA was extracted from mouse endothelial cells with miRNeasy mini kit (Qiagen, Hilden, Germany). After reverse transcription was done to generate cDNA, RT-qPCR was performed in triplicate with QuantiTect SYBR Green PCR Kit (Qiagen). All primers were purchased from Qiagen. Data were collected and analyzed using 2-ΔΔCt method for quantification of the relative mRNA expression levels. Values of genes were first normalized against α-tubulin, and then compared to the experimental controls.
Western blotting and ELISA
The extracted protein was mixed with the buffer solution, boiled, electrophoresed by 10% SDS-PAGE, and transferred to the membrane using the semi-dry transfer method. Primary antibodies are rabbit-anti-caspase3 (Abcam, Shanghai, China), rat anti-CD31 (Becton-Dickinson Biosciences), rabbit anti-LC3, rabbit-anti-Beclin-1, and rabbit-anti-α-tubulin (Cell Signaling Technology, Danvers, MA, USA). Secondary antibodies are HRP-conjugated against rat or rabbit (Jackson ImmunoResearch Labs, West Grove, PA, USA). Images were collected on the AlphaImager HP Fluorescent and Visible Light Gel Imaging System. The protein levels were first normalized to α-tubulin, and then normalized to the experimental controls. Densitometry of Western blots was quantified with NIH ImageJ software. For ELISA, total protein was isolated and TNFa, IFNr, HIF-1a and IL-6levels were determined by ELISA kits (R&D System, Los Angeles, CA, USA).
MiRNA target prediction and 3′-UTR luciferase-reporter assay
MiRNA target prediction was done using the algorithms TargetSan. The Beclin-1 3′-UTR reporter plasmid (pRL-Beclin-1) and mutants were purchased from Creative Biogene (Shirley, NY, USA). Co-transfection was done by Lipofectamine 3000. Cells were collected 48 hours after transfection and subjected to a dual-luciferase reporter assay (Promega, Madison, WI, USA), according to the manufacturer’s instructions.
Statistical analyses
The statistical analysis was performed with the GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA), using one-way analysis of variance (ANOVA) test followed by a Turkey multiple comparison post-hoc analysis. The values were shown as mean ± standard deviation (SD). A value of p<0.05 was considered as significant after Bonferroni correction.
CONFLICTS OF INTEREST
The authors have declared that no competing interests exist.
FUNDING
This work was supported by National Natural Science foundation of China (NO: 81370182).
REFERENCES
1. Ma L, Zhang J, Liu Y. Roles and Mechanisms of Obstructive Sleep Apnea-Hypopnea Syndrome and Chronic Intermittent Hypoxia in Atherosclerosis: evidence and Prospective. Oxid Med Cell Longev. 2016; 2016:8215082.
2. Sforza E, Roche F. Chronic intermittent hypoxia and obstructive sleep apnea: an experimental and clinical approach. Hypoxia (Auckl). 2016; 4:99–108.
3. Ozeke O, Ozer C, Gungor M, Celenk MK, Dincer H, Ilicin G. Chronic intermittent hypoxia caused by obstructive sleep apnea may play an important role in explaining the morbidity-mortality paradox of obesity. Med Hypotheses. 2011; 76:61–63.
4. Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014; 157:65–75.
5. Jiang F. Autophagy in vascular endothelial cells. Clin Exp Pharmacol Physiol. 2016; 43:1021–28.
6. Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015; 25:354–63.
7. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011; 18:571–80.
8. Djavaheri-Mergny M, Maiuri MC, Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene. 2010; 29:1717–19.
9. Zhai H, Fesler A, Ju J. MicroRNA: a third dimension in autophagy. Cell Cycle. 2013; 12:246–50.
10. Chen X, Zhang Y, Shi Y, Lian H, Tu H, Han S, Yin J, Peng B, Zhou B, He X, Liu W. MiR-129 triggers autophagic flux by regulating a novel Notch-1/ E2F7/Beclin-1 axis to impair the viability of human malignant glioma cells. Oncotarget. 2016; 7:9222–35. https://doi.org/10.18632/oncotarget.7003.
11. Gaur S, Wen Y, Song JH, Parikh NU, Mangala LS, Blessing AM, Ivan C, Wu SY, Varkaris A, Shi Y, Lopez-Berestein G, Frigo DE, Sood AK, Gallick GE. Chitosan nanoparticle-mediated delivery of miRNA-34a decreases prostate tumor growth in the bone and its expression induces non-canonical autophagy. Oncotarget. 2015; 6:29161–77. https://doi.org/10.18632/oncotarget.4971.
12. Sun L, Zhao M, Zhang J, Lv M, Li Y, Yang X, Liu A, Wu Z. MiR-29b Downregulation Induces Phenotypic Modulation of Vascular Smooth Muscle Cells: Implication for Intracranial Aneurysm Formation and Progression to Rupture. Cell Physiol Biochem. 2017; 41:510–18.
13. Zhi Y, Pan J, Shen W, He P, Zheng J, Zhou X, Lu G, Chen Z, Zhou Z, Ginkgolide B. Ginkgolide B Inhibits Human Bladder Cancer Cell Migration and Invasion Through MicroRNA-223-3p. Cell Physiol Biochem. 2016; 39:1787–94.
14. Wu X, Feng X, Zhao X, Ma F, Liu N, Guo H, Li C, Du H, Zhang B. Role of Beclin-1-Mediated Autophagy in the Survival of Pediatric Leukemia Cells. Cell Physiol Biochem. 2016; 39:1827–36.
15. Zhou Z, You Z. Mesenchymal Stem Cells Alleviate LPS-Induced Acute Lung Injury in Mice by MiR-142a-5p-Controlled Pulmonary Endothelial Cell Autophagy. Cell Physiol Biochem. 2016; 38:258–66.
16. Zhang T, Tian F, Wang J, Jing J, Zhou SS, Chen YD. Endothelial Cell Autophagy in Atherosclerosis is Regulated by miR-30-Mediated Translational Control of ATG6. Cell Physiol Biochem. 2015; 37:1369–78.
17. Pan W, Zhong Y, Cheng C, Liu B, Wang L, Li A, Xiong L, Liu S. MiR-30-regulated autophagy mediates angiotensin II-induced myocardial hypertrophy. PLoS One. 2013; 8:e53950.
18. Zheng B, Zhu H, Gu D, Pan X, Qian L, Xue B, Yang D, Zhou J, Shan Y. MiRNA-30a-mediated autophagy inhibition sensitizes renal cell carcinoma cells to sorafenib. Biochem Biophys Res Commun. 2015; 459:234–39.
19. Xu R, Liu S, Chen H, Lao L. MicroRNA-30a downregulation contributes to chemoresistance of osteosarcoma cells through activating Beclin-1-mediated autophagy. Oncol Rep. 2016; 35:1757–63.
20. Zou Z, Wu L, Ding H, Wang Y, Zhang Y, Chen X, Chen X, Zhang CY, Zhang Q, Zen K. MicroRNA-30a sensitizes tumor cells to cis-platinum via suppressing beclin 1-mediated autophagy. J Biol Chem. 2012; 287:4148–56.
21. Chen Z, Jin T, Lu Y. AntimiR-30b Inhibits TNF-α Mediated Apoptosis and Attenuated Cartilage Degradation through Enhancing Autophagy. Cell Physiol Biochem. 2016; 40:883–94.
22. Wang J, Sun YT, Xu TH, Sun W, Tian BY, Sheng ZT, Sun L, Liu LL, Ma JF, Wang LN, Yao L. MicroRNA-30b Regulates High Phosphorus Level-Induced Autophagy in Vascular Smooth Muscle Cells by Targeting BECN1. Cell Physiol Biochem. 2017; 42:530–36.
23. Akinnusi ME, Laporta R, El-Solh AA. Lectin-like oxidized low-density lipoprotein receptor-1 modulates endothelial apoptosis in obstructive sleep apnea. Chest. 2011; 140:1503–10.
24. Gozal D, Gileles-Hillel A, Cortese R, Li Y, Almendros I, Qiao Z, Khalyfa AA, Andrade J, Khalyfa A. Visceral White Adipose Tissue after Chronic Intermittent and Sustained Hypoxia in Mice. Am J Respir Cell Mol Biol. 2017; 56:477–87.
25. Lai L, Chen J, Wang N, Zhu G, Duan X, Ling F. MiRNA-30e mediated cardioprotection of ACE2 in rats with Doxorubicin-induced heart failure through inhibiting cardiomyocytes autophagy. Life Sci. 2017; 169:69–75.
26. Tagaito Y, Polotsky VY, Campen MJ, Wilson JA, Balbir A, Smith PL, Schwartz AR, O’Donnell CP. A model of sleep-disordered breathing in the C57BL/6J mouse. J Appl Physiol (1985). 2001; 91:2758–66.