
Introduction
Breast cancer is a leading cause of cancer-related death in women. This disease is diagnosed in nearly 1.4 million women and is responsible for more than 450,000 death every year [1]. According to the WHO release, there has been a 20% increase in the number of reported worldwide breast cancer patients which resulted in 522,000 deaths since 2008. According to the US National Cancer Institute, approximately 232,000 occur resulting in about 40,000 deaths in the USA each year. Breast cancer is not gender specific [2,3]. The frequency of breast cancer in men is approximately 100-fold lower than in women as approximately 2,200 males will be diagnosed with breast cancer each year in the USA.
Breast Cancer Genetics.
A prominent risk factor for the onset of breast cancer is age; however factors linked to lifestyle and diet also play important roles in breast cancer. Mutations at or deregulation of certain genes (BRCA1, BRCA2, HER2, PIK3CA) and others play important roles in breast cancer [4-18]. Mutations or aberrant or deregulated expression of TP53, MDM2 and RB also can play roles in the therapeutic responses of breast cancer [19-26]. Restoration of functional TP53 activity can increase the sensitivity of some TP53 mutant cells to certain anticancer drugs [27].
BRCA Genes and Other Genes Involved in DNA Repair Are Implicated in Breast Cancer.
Breast cancer occurrence is attributed to both genetic and environmental factors. Some breast cancers are due to hereditary mutations, namely those involving BRCA1 and BRCA2. BRCA1 encodes breast cancer type 1 susceptibility protein which is involved in DNA repair and is considered a caretaker gene. The BRCA1 protein interacts with RNA polymerase II and also with histone deacetylase complexes [28]. BRCA1 plays key roles in transcription, repair of breaks in double stranded DNA as well as ubiquitination. The BRCA1 protein also combines with other proteins which detect DNA damage and other cell signals and forms a multi-subunit protein complex known as the BRCA1-associated genome surveillance complex (BASC) [29]. Components of this complex may be mutated in certain cancers.
BRCA2 is also involved in the repair of DNA double strand breaks [30]. BRCA2 binds single stranded DNA. BRAC2 interacts with the RAD51 recombinase to stimulate strand invasion which is a critical step in homologous recombination. For RAD51 to bind the DNA double-strand breaks, a complex of BRCA1/partner and localizer of BRCA2 (PALB2)/BRCA2 is required [31].
The risk of developing breast or ovarian cancer in individuals with certain cancer-associated BRCA1/BRCA2 alleles is 60-80% for breast cancer and 20-40% for ovarian cancer. These individuals also develop cancer at an earlier age. In addition, other genes involved in DNA repair and signaling are implicated in breast cancer including: Fanconi anemia (FA) genes (FANCD2, FANCA and FANCC), mismatch repair genes (MutL homolog 1 [MLH1], MutS protein homolog 2 [MSH2], PMS1 protein homolog 1 [PMS1], mutS homolog 6 [MSH6]), mismatch repair endonuclease PMS2 [PMS2] and DNA repair genes (Ataxia telangiectasia mutated [ATM], Ataxia telangiectasia and Rad3 related [ATR] and serine/threonine-protein kinases Chk/2 (CHK1/2), and the tumor suppressor genes (TP53, Serine/threonine kinase 11 [STK11] also known as liver kinase B1[LKB1], phosphatase and tensin homolog [PTEN]) and protein phosphatase 6 (PP6) [32-44].
In an important study with triple negative breast cancer (TNBC) patients, the frequency of BRCA1 and BRCA2 mutations and survival was examined [45]. DNA was isolated from tumor samples as well as normal tissues from 77 TNBC patients and the genetic sequence of the BRCA1/2 exons and flanking regions determined. 19.5% of the TNBC patients had BRCA mutations, 15.6% were mutant at BRCA1, and 3.9% were mutant at BRCA2. It turns out that the patients with BRCA mutations were younger than the patients with WT BRCA genes. In this study which followed the patients for up to 214 months, there were 42.9% recurrences and 45.5% deaths. Interestingly, the five-year recurrence-free survival estimates were associated with the genetic status of the BRCA genes. As the five-year recurrence-free survival rates were 51.7% for patients with WT BRCA genes whereas they were 86.2% for patients with BRCA mutations.
BRCA1 and BRCA2 are also mutated in patients with ovarian cancer [46]. BRCA1/2 mutations are present in approximately 11 to 15% of unselected ovarian cancer patients. BRCA1 mutations were positively associated with TP53 mutations. The presence of BRCA1/2 mutations after platinum chemotherapy were associated with improved progression free survival.
Hereditary and Sporadic Breast and Ovarian Cancer.
Many spontaneous breast cancers are associated with environmental exposures to carcinogens [47-61]. These include: air pollution [52], exposure to polychlorinated biphenyl congeners [53]. Pesticides [54,58], electromagnetic radiation [55], cadmium and nickel [56], radiation from medical imaging [59], acrylamide [61] and other toxins.
Deregulation of BRCA1 expression has been implicated in sporadic breast cancer. The trinucleotide-repeat-containing 9 (TNRC9) gene has been shown to downregulate BRCA1 expression which results in breast cancer aggressiveness. TNRC9 is amplified in certain breast cancer patients and is associated with a poor prognosis [62]. This group also determined that ectopic expression of TNRC9 affected breast cancer cell survival. TNRC9 and BRCA1 protein expression were inversely correlated in large data sets of breast and ovarian cancer samples. Interesting this group determined that TNRC9 bound to both the BRCA1 promoter and the cAMP-responsive element-binding protein (CREB) complex. CREB is a regulator of BRCA1 transcription. Finally TNRC9 expression suppressed BRCA1 expression by altering the methylation status of the BRCA1 promoter region.
BRCA mutations have also been detected in familial and sporadic ovarian cancer patients. Germline mutations in BRCA1 or BRCA2 are present in approximately 18% of hereditary ovarian cancers. These mutations confer an estimated risk from 15 to 50% in the ovarian cancer patients [63].
In this study, the prevalence of BRCA1 mutations in 106 familial Greek ovarian cancer patients who had a strong family history of ovarian cancer or metachronous breast cancer. Metachronous breast cancer refers to a breast cancer patient which has two different breast cancers which occur at two different times, the two cancers can occur in the same breast. In addition, the prevalence of BRCA1 mutations were examined in 592 sporadic Greek ovarian cancer patients. In Greece, it had been previously determined that there were 6 types of BRCA1 mutations that accounted for 63% of all the mutations in the BRCA1 and BRCA2 genes. Deleterious BRCA1 mutations were observed in 40.6% of familial ovarian cancer cases and 4.6% of sporadic ovarian cases. This study determined that 71.2% of the BRCA1 carriers presented a high-grade serous phenotype. These studies document the importance of identifying BRCA mutations in breast and/or ovarian cancer families. The authors have stated that all serous ovarian cancer patients should consider genetic testing.
Hereditary breast cancer often results from disruption of the normal functions of BRCA1 and BRCA2. In contrast BRCA1/BRCA2 are not necessarily mutated in sporadic breast cancer, but there may be mutations in TP53 and epigenetic alterations which change the expression of other genes. These changes result in breast cancer cells which may be wild WT at BRCA1 and BRCA2 but have defects in DNA repair. In addition certain sporadic breast cancers may have hypermethylation of the BRCA1, estrogen receptor (ER), progesterone receptor (PR) and other genes which prevents or lowers their expression. These changes may results in the breast cancer cells having a mutant BRCA phenotype which is referred to as “BRCAness”. These breast cancers arise at early age and are aggressive [64].
In one study, the frequencies of methylation in the BRCA1 promoter region were examined in 96 sporadic invasive breast carcinomas and 43 sporadic ovarian carcinomas. Methylation of the BRCA promoter region was detected in 11% and 5% of the breast and ovarian carcinomas respectively. Methylation of the BRCA1 promoter was linked with lack of ER and PR expression in these tumors [65].
Alterations of Genes Involved in DNA Repair in Breast Cancer.
Sporadic breast cancers accounts for approximately 70% of breast cancers, while familiar breast cancers account for the remaining 30% which are due to the present of mutations in breast cancer families (familiar breast cancers) [66]. Familial breast cancer families have a higher incidence of breast cancers. There exist different susceptibility genes which are high-, moderate-, and low-penetrance susceptibility genes. High-penetrance genes include: BRCA1, BRCA2, PTEN and TP53, which in many cases are responsible at least in part for the familiar breast cancer. Other genes have been associated with moderate penetrance and risk. These include genes involved in DNA repair, such as CHEK2, ATM, BRIP1 (FANCJ), PALB2 (FANCN) and RAD51C (FANCO). In addition, genes involved in low penetrance and risk are being identified. The presence of mutations at these different types of breast cancer susceptibility genes may be examined in breast cancer screening in the future.
The genetic structures of the TP53, BRCA1, ATM, and PIK3CA genes were examined in 145 Bulgarian patients with sporadic breast cancer. The expression of HER2 was examined by immunohistochemistry and chromogenic hybridization in situ (CISH) [67]. In this study, mutations were observed: at TP53 in 22.07% of the patients, at BRCA1 in 0.69% of the patients, at ATM in 7.59% of the patients, and in 31.25% of the patients at PIK3CA. Overexpression of HER2 was observed in 21.21% of the patients. Mutations at TP53 were associated with both tumor size and grade of malignancy. Mutations at ATM were associated with grade of malignancy. Mutations at PIK3CA were associated with PR+ tumors. HER2 overexpression was correlated with the age of the patient when diagnosed with breast cancer, tumor size and ER+. This important study documented that TP53 mutations were an indicator for poorer outcome. Importantly, in this study the presence of two genetic mutations did not correlate with either a more aggressive carcinoma or a poorer overall survival.
Chk2 is an important kinase which is activated in response to DNA damage. Chk2 is involved both in TP53 and cell-cycle pathways. In some Li-Fraumeni syndrome families, which have germline mutations at TP53, mutations at CHK2 have also be observed. The presence of loss of heterozygosity (LOH) at the CHK2 gene was examined in 139 breast cancer tumors [68]. 139 breast tumors were screened for loss of heterozygosity (LOH) at chromosome 22q (where the CHK2 gene is located), using seven microsatellite markers, LOH was detected in 53% of the breast tumors. Further studies examined the mutational status of the CHK2 gene and a germ line variant (T59K) in the first coding exon was detected. Upon screening 1172 cancer patients with different types of cancer for the T59K sequence variant, it was detected in four breast cancer patients. This study concluded that CHK2 mutations were rare in breast cancer but the CHK2 gene product may perform a tumor suppressor function.
Effects of c-Myc Overexpression in BRCA1-Deficient Breast Cancer Patients.
Elevated c-Myc expression leads to a poor prognosis in sporadic breast cancer patients that are BRCA1-deficient [69]. In this important study, the presence of BRCA1 gene expression was examined in 374 sporadic breast cancer patients. BRCA1 expression was lacking in 60.4% of the breast cancer patients. Patients that lost BRCA1 expression often had more advanced breast cancer and were tumor node metastasis stage III positive, lymph node positive and overexpressed c-Myc. The presence of hypermethylation of the BRCA1 promoter region was examined. BRCA1 promoter hypermethylation was observed in 16.4% of the breast cancer patients examined and was associated with BRCA1-, ER- , c-Myc overexpression, and the triple-negative phenotype. Thus loss of functional BRCA1 expression combined with increased expression of c-Myc was associated with a poor prognosis.
Alteration of TP53 in Breast Cancer.
Dysregulation of the TP53 gene also occurs in sporadic breast cancer [70]. The genetic structure of the TP53 gene was examined in 136 unselected sporadic breast cancer patients’ tumors. Approximately 40% of the tumor samples had TP53 mutations. Moreover, this group postulated that these mutations were due to exposure of the breast cancer patients to environmental mutagens based on the frequency of G-T transversions and the incidence of guanosine mutations on the non-transcribed DNA strand of the TP53 gene.
Polymorphisms of the TP53 gene have been observed to have prognostic and predictive values in cancer therapy. The presence of two TP53 gene polymorphisms, Arg72Pro and PIN3 (+16 bp) was observed in ninety-four women with sporadic breast cancer who were followed for a mean of 67.9 months after therapy [71]. This study determined that the different genotypes of the Arg72Pro and PIN3 (+16 bp) polymorphisms had no significant impact on survival in the sporadic breast cancer patients. However, this study observed that the patients which were treated with chemotherapy regmins without an anthracycline, that had the A2A2 genotype of the PIN3 (+16 bp) polymorphism, had a poorer overall survival than other genotypes. Although some controversity exists on use of anthracyclines in breast cancer patients, those with the A2A2 genotype of PIN3 (+16bp) polymorphism may benefit. These important studies document the importance of genetics in personalized medicine.
Epigenetic Modification of the ER genes in Breast Cancer.
Methylation of the promoter region of the ER and other genes has been associated with their decreased expression. The methylation status of the ER-alpha promoter region was examined in 138 sporadic breast cancers. The ER-alpha promoter region was observed to be methylated in 60.1% (83/138) tumors, including 57 of 69 of the tumors which did not express ER-alpha. This study determined that the probability of ER-alpha promoter methylation was increased in those cases that were ER-alpha- and PR- [72].
In a study of 100 sporadic primary breast cancers of which 51 were ER-alpha- and 49 ER-alpha+, ER methylation was observed in 98% of ER- and 65% of ER+ tumor samples. ER- promoter region methylation was also associated with lack of PR expression and double receptor negative expression status of the breast cancer specimens [73].
The methylation of the ER-beta promoter region was examined in 178 sporadic breast cancer patients. ER-beta promoter methylation was observed in 44.9% of breast tumor samples. In contrast ER-beta promoter hypermethylation was detected in only 14.3% of patients with benign breast hyperplasia. 58% of the ER-beta- tumors exhibited ER-beta promoter region methylation whereas 36.7% of the ER-beta+-positive cases exhibited methylation at the ER-beta promoter region. As the levels of ER-beta promoter methylation increased- the levels of ER-beta protein detected decreased in the tumor samples. A strong correlation between ER-alpha promoter methylation and ER-beta promoter methylation was observed [74].
Expression of EGFR Family Members in Breast Cancer Patients.
The EGFR family consists of four members. For the sake clarity in this review, we will refer to them as EGFR1 (a.k.a., EGFR, HER1, c-erbB1), HER2 (a.k.a., EGFR2, c-erbB2), EGFR3 (a.k.a., c-erbB3, HER3) and EGFR4 (a.k.a., c-erbB4, HER4). The expression of the EGFR1, HER2, EGFR3 and EGFR4 were examined by immunohistochemistry in 220 breast cancer carcinomas [75]. Increased expression of EGFR1 was detected in 16.4% of the tumors, increased expression of HER2 was observed in 22.8% of the tumors, increased expression of EGFR3 was detected in 17.5% of the tumors, and increased expression of EGFR4 was observed in 11.9% of tumors. Breast cancer patients with tumors that overexpressed EGFR1, HER2 or EGFR3 had reduced survival. In contrast, those breast cancer patients whose tumors displayed elevated levels EGFR4 had better survival than the breast cancer patient that expressed EGFR1, HER2 or EGFR3. Thus overexpression of EGFR1, HER2 and EGFR3 was association with a poor prognosis in breast cancer patients. In contrast, overexpression of EGFR4 is associated with a good prognosis. In addition, this group investigated the association of ER expression with the different EGFR molecules. Expression of EGFR1, HER2 and EGFR3 was associated with ER negativity in the breast cancer patients. Moreover those breast cancer patients that were ER+ and also EGFR1+, HER2+ or EGFR3+ had poorer survival than those breast cancer patients which were either ER+ and HER2+ or EGFR4+.
Additional studies were performed by the same group on the proliferative potential of the different types of EGFR-expressing cells [76]. These important studies suggest that EGFR1, HER2 and EGFR3 are linked with tumor proliferation, while EGFR4 did not appear to drive proliferation but may even play protective roles.
In another study, the expression of the EGFR1 and EGFR3 proteins were examined in 104 primary breast carcinomas comprising nine comedo ductal carcinoma in situ (DCIS), 91 invasive ductal carcinomas and four invasive lobular carcinoma by histochemistry. Increased expression of EGFR3 was observed in 67% of comedo DCIS, 52% of invasive ductal carcinomas, 71% of carcinomas containing both the in situ and invasive lesions and 25% of invasive lobular carcinomas [77]. 59% of ER- tumors, 63% of lymph node+ tumors and 63% of HER2 tumors were positive for EGFR3 expression. 67% of EGFR1+, 67% of HER2+ (67%), 75% TP53+ and 60% of cathepsin-D+ DCIS were positive for EGFR3 expression.
The expression of EGFR1, HER2, EGFR3 and EGFR4 was examined in 100 breast cancer patients [78]. By using immunohistochemistry techniques, 36% of the breast cancer samples were positive for EGFR1, 27%, of the breast cancer samples were positive of HER2, 26% of the breast cancer samples were positive for EGFR3 and 82% of the breast cancer samples were positive for EGFR4. The expression of these genes was also examined by RT-PCR and the similar results were observed. An association between decreased disease-free survival and expression of HER2 was observed. Co-expression of EGFR1 and HER2 was associated with a worse prognosis. In contrast, expression of EGFR4 was associated with a better outcome. EGFR4 expression appeared to antagonize the effects on HER2 on clinical outcome in the breast cancer patients which expressed both.
The link between ER-alpha and EGFR4 expression was examined in 103 breast cancer samples by both immunohistochemistry and RT-PCR [79]. In 25% of the breast cancer samples ER-alpha were not expressed. In addition, approximately 25% of them did not express EGFR4. About one-half of the ER-alpha- tumors did not express EGFR4 at both mRNA and protein levels. The luminal breast cancer cell lines MCF-7 and T47D cells expressed both ER-alpha and EGFR4. While the basal breast cancer line MDA-MB-231 and the HER2-overexpressing SK-BR-3 line expressed neither. These results have suggested roles for EGFR4 in ER-alpha mediated signal transduction in breast cancer.
The expression patterns of EGFR1, HER2, EGFR3 and EGFR4 were examined by real-time RT-PCR in 365 unselected primary breast cancers [80]. EGFR1 and HER2 were negatively associated with ER+ and PR+ breast cancers. In contrast, EGFR3 and EGFR4 were positively associated with ER+ and PR+ breast cancers. In a subsequent study with the ER-alpha+ breast cancer cell line MCF-7, beta-estradiol down regulated the expression of all four EGFR family member receptors as determined by RT-PCR. In contrast, the ER antagonist 4-hydroxy tamoxifen (4HT) inhibited the downregulation induced by beta-estradiol [81].
Gatekeeper Mutations in HER2.
The T798M mutation in HER2 is considered a gatekeeper mutation as it affects the ability of small molecule inhibitors such as lapatinib to bind the ATP-binding pocket and the mutation confers resistance to the inhibitor [82]. In studies by a different research group which were aimed at determining the mechanism of resistance of breast cancer cells containing the HER2 T798M mutation to lapatinib, it was shown that the cells overexpressed EGFR ligands [83]. In BT474 and MCF10A breast cancer cells transfected with the construct encoding HER2-T798M mutation, elevated HER2 kinase activity was detected and lapatinib did not block phosphorylation of HER2, EGFR3 or downstream Akt and ERK1/2. Increased levels of EGFR2 associated with PI3Kp85 were detected in the transfected cells and the BT474/HER2-T798M cells were also resistant to Herceptin. However these cells were sensitive to the pan-phosphoinositol-3-kinase (PI3K) inhibitors BKM120 and XL147 but not the mitogen-activated protein kinase kinase1/2 (MEK1/2) inhibitor CI-1040. The BT474/HER2-T798M transfected cells expressed elevated levels of the EGFR ligands EGF, TGF-alpha, amphiregulin (AR), and heparin binding EGF (HB-EGF). Thus targeting breast cancers with both HER2 inhibitors and PI3K inhibitors may be an appropriate technique to treat those breast cancer patients with HER2-T798M mutations [84]. Other growth factors also bind the EGFR family of receptors including: EGF, schwannoma-derived growth factor (SDGF), vaccinia growth factor (VGF), Neu differentiation factor (NDF) or neuregulins, and heregulin. A diagram of EGFR family members is presented in Figure 1.
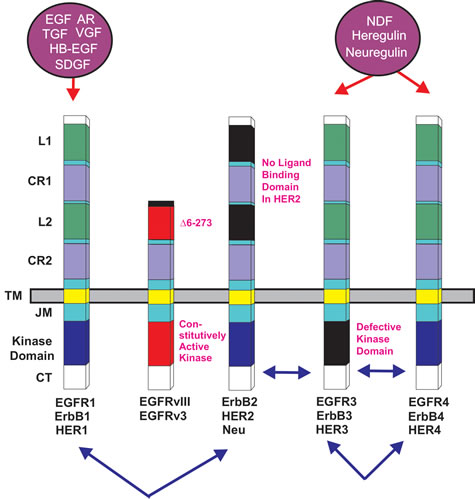
Figure 1: Epidermal Growth Factor Receptor Family. Conserved domains of the four different EGFR family members are indicated by similar shading. L = ligand binding domain, CR = cysteine-rich domains. TM = transmembrane domain. CT = C-terminal domain which contains the phosphorylation sites. JM = juxta membrane domain. HER2 does not bind a ligand. The kinase domain in ERB3 is defective. Alternative names for each receptor are written underneath each receptor. Ligands which bind the receptors are indicated in purple circles above the receptors. Epidermal growth factor (EGF), vaccinia growth factor (VGF), amphiregulin (AR), heparin binding-EGF (HB-EGF), schwannoma-derived growth factor (SDGF), Neu differentiation factor (NDF), heregulin, neuregulin. Arrows between receptors indicate possible heterodimer formation between various EGFR family receptors.
Roles of Aberrant mRNA Splicing in Sensitivity to Herceptin.
The splice variant delete16HER2, which results from exon 16 skipping, has been shown to increase the transformation frequency of cancer cells. This mis-splicing results in resistance to Herceptin. In contrast, retention of intron 8 of the HER2 gene after mRNA splicing results in the creation of herstatin which inhibits tumor cell proliferation. Likewise retention of intron 15 during splicing of HER2 results in the p100 protein which also suppresses tumor cell proliferation [85].
Mutations at the EGFR Gene Family in Breast Cancer.
HER2 is amplified in 20-25% of breast cancers. However, the roles of mutations/amplifications of other EGFR family members are not so clear. Mutations and amplifications of EGFR1 have been detected in breast cancer [86, 87]. In a study consisting of 70 TNBC patient samples, mutations in the kinase domain were detected in 11% of the samples. This study amplified the region spanning exons 18 to 21 of the EGFR1 gene. The investigators detected deletions in exon 19, which encodes part of the kinase domain. However, the authors indicated that these mutations appeared to be independent of the expression levels of the EGFR1 protein that were detected by immunohistochemistry. A diagram illustrating some of the effects of mutations/amplifications in EGFR1/HER2 and other genes in breast and other cancers on signal transduction pathways is presented in Figure 2
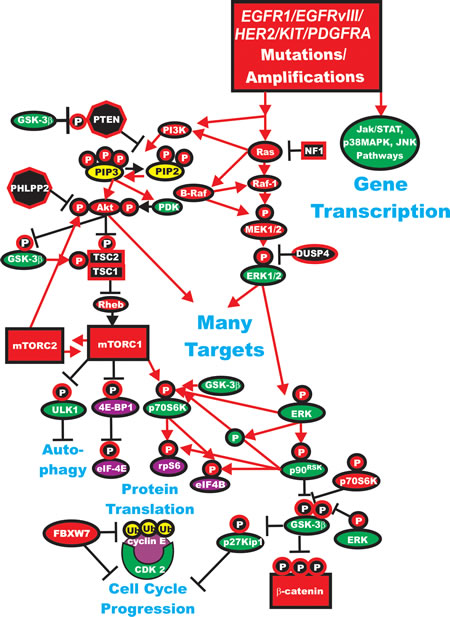
Figure 2: Dysregulated Expression of Upstream Receptors and Kinases Can Result in Activation of the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR and Other Signaling Pathways and Contribute to Malignant Transformation. Sometimes dysregulated expression of growth factor receptors occurs by increased expression, genetic translocations or genomic amplifications which can lead to activation of the Ras/Raf/MEK/ERK, Ras/PI3K/PTEN/Akt/mTOR and other signaling pathways. Alternatively chromosomal translocations can occur in non-receptor kinases and other genes which result in activation of these pathways. Genes in the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR pathways that have activating mutations detected in human cancer and proliferative diseases are indicated in red ovals and squares. Tumor suppressor genes inactivated in certain cancer are indicated in black squares or octagons. Other key genes are indicated in green ovals. Red arrows indicate activating events in pathways. Blocked black arrows indicating inactivating events in pathways.
In addition, amplification of the EGFR1 gene has been detected in certain breast cancers [88]. The EGFR1 gene was amplified in approximately 6% of the breast cancer patients they examined (n =175) and the samples displayed increased EGFR1 expression. They also examined patient samples which had amplified the EGFR1 gene for hot-spot mutations in the EGFR1 gene. No mutations were detected in exons 19 and 21.
The presence of EGFR1 or HER2 gene amplification or overexpression was investigated in a series of metaplastic breast carcinomas (MBC) [89]. MBC are basal like tumors that account for less than 1% of all invasive mammary carcinomas. Nineteen of the 25 (76%) MBC examined overexpressed EGFR1. EGFR1 gene amplifications were detected in 37% of the tumors which overexpressed EGFR1. In contrast, only one case exhibited HER2 overexpression but HER2 gene amplification was not detected in this study of MBC. The authors pointed out that some of the tumors that overexpressed EGFR1, but did not have EGFR1 gene amplification, the expression of EGFR1 could have resulted from activating mutations in EGFR1, which resulted in its expression.
In a subsequent study by the same group, the presence of EGFR1 amplification and activating mutations was examined in 47 MBC [90]. 32% of the samples exhibited overexpression of EGFR1. Within the subset that had EGFR overexpression, 34% had EGFR1 gene amplification. This group examined these MBC samples for the presence of activating mutations in exons 18, 19, 20, and 21 of the EGFR1 gene. No activating mutations of EGFR1 in exons 18, 19, 20 and 21 were detected in these MBC patient samples.
The epidermal growth factor receptor variant III (EGFRvIII) is a genetic truncation of the EGFR1 gene. It encodes a constitutively-active truncated EGFR1 protein which has been implication in many types of cancers, (e.g., brain, breast, prostate and others) [91-103].
Introduction of a construct encoding EGFRvIII into MCF-7 breast cancer cells resulted in HER2 phosphorylation, which the authors suggested occurred through heterodimerization and cross-talk [91]. The MCF-7/EGFRvIII transfectants had approximately a 3-fold increase in colony formation. MCF-7/EGFRvIII were more tumorigenic that MCF-7 cells in athymic nude
The EGFRvIII protein is detected in various human cancers, but not in normal tissues. The presence of EGFRvIII mRNA was examined in primary invasive breast cancer by utilizing laser capture microdissection (LCM)/RT-PCR. A high incidence (67.8%) of EGFRvIII transcripts was observed in pure breast cancer cells [92]. Furthermore, 57.1% of the infiltrating breast carcinomas expressed both EGFR1 WT and EGFRvIII mRNA in the same tumor. In contrast, no detectable EGFRvIII mRNA was seen in samples of normal breast tissue. These expression results were further confirmed by immunohistochemical analysis. Thus there was co-expression of EGFRvIII and EGFR WT in some human invasive breast cancer tissue but not in normal breast samples. It must be pointed out that other studies consisting of 55 breast cancer cell lines and 170 primary breast cancer did not observe similar results and the authors concluded that expression of EGFRvIII is extremely rare in breast cancer [93]. However, in a study by a different research group, the expression of EGFRvIII mRNA in women with breast cancer was examined by an RT-nested PCR. EGFRvIII mRNA was detected in the peripheral blood of 30% of 33 low risk, early stage patients, in 56% of 18 patients selected for neoadjuvant chemotherapy, in 63.6% of 11 patients with disseminated disease but not in any of 40 control women [94]. Interesting in the low risk, early stage patients, EGFRvIII expression was associated with ER- or HER2+. In another study of 225 breast cancer patients, the expression of EGFR1, phosphorylated EGFR1, and EGFRvIII, was examined by immunohistochemistry and the patient outcomes were also followed [95]. 48% of the patients displayed EGFR1, 54% of the patients were positive for phospho-EGFR, and 4% of the patients were positive for EGFRvIII. EGFR1 expression correlated with negative hormone receptor status, worse relapse-free survival and overall survival than those patients with did not express detectable EGFR1 [95]. Interestingly there did not appear to be any association between expression of phospho-EGFR or EGFRvIII expression with clinical outcome. This study indicated that prognostic value of EGFR1 expression was most important in the HER2+ and the ER-/PR- subgroups.
EGFRvIII expression may down regulate PR expression in certain luminal B tumors [96]. These tumors are ER+ but are characterized as having an aggressive behavior which is 4HT resistant. This subset of breast cancers displays increased EGFR1, HER2 and downstream PI3K/PTEN/Akt/mTORC1 pathway activation [96,97].
EGFRvIII interacts with HER2 [98] and chemokine (C-X-C motif) receptor 4 (CXCR4) [99,100] to activate signaling pathways important in migration, invasion and tumorigenesis. These interactions between EGFRvIII and HER2 and CXCR4 may be prolonged in comparison to interactions between EGFR1 and HER2 and CXCR4 as it is more difficult to down regulate the constitutive nature of EGFRvIII. CXCR4 is highly expressed in breast cancers and implicated in metastasis [101] and cancer initiating cells (CICs) [102].
Recently EGFRvIII has been implicated in primary breast cancers and breast CICs. Its expression has been associated with the Wnt/beta-catenin pathway and downstream beta-catenin target gene expression and the expression of genes associated with self-renewal. Its expression has been linked with increased in vitro mammosphere formation and tumor formation [103].
HER2 normally has to heterodimerize with another EGFR family member for activity. However, if the HER2 gene is amplified, HER2 activity is induced and the abnormal breast tumor growth is dependent on HER2 activity for growth [104]. An activating mutation has been detected in HER2+ lung cancer patients in the germline from a Japanese cancer patient with a familiar history of lung cancer as well as sporadic cancer patients [105]. The mutation occurred in the transmembrane region and may be responsible for increased stability and dimerization. This mutation activated downstream HER2 signaling molecules including Akt and p38MAPK.
Somatic mutations in the kinase domain of the HER2 gene, which result in its activation have been detected in lung cancer patients [106]. In a study involving non small cell lung cancers (NSCLC), HER2 mutations were detected in 1.6% (11 of 671) of NSCLC cancer specimens examined but were absent in other types of cancers. The HER2 mutations were in-frame insertions in exon 20 corresponding to a similar region as in the EGFR gene where insertions were detected in NSCLC patients. Interestingly the HER2 mutations were more frequent in the lung cancer patients that never smoked. HER2 mutations were detected more frequently in patients of Oriental ethnicity than other ethnicities. The mutations were also more frequently detected in females.
Recently mutations in HER2 have been detected in breast cancer patient samples which lack HER2 gene amplification. Thirteen HER2 mutations were characterized from twenty-five patient samples which had HER2 mutations but lacked HER2 gene amplification. 7 mutations were activating and resulted from point mutations and in-frame deletions. Some mutations (L755S) resulted in lapatinib resistance; however this was not an activating mutation. All of the cells containing the HER2 mutations were sensitive to the irreversible HER2 kinase inhibitor, neratinib [107].
Loss of HER2 activity also results in loss of phosphorylated EGFR3. It has been shown that HER2 can dimerize with EGFR3 to drive breast cancer proliferation [104]. Loss of EGFR3 activity in HER2+ breast cancer cells inhibited their growth. This growth inhibition mediated by loss of HER2 or EGFR3 and could be overcome by introduction of a construct encoding activated Akt. Importantly this group demonstrated that a key function of EGFR3 was to couple the response of HER2 to the PI3K/PTEN/Akt/mTORC pathway.
Less is known about the roles of EGFR3 and EGFR4 in breast cancer [108]. EGFR3 is naturally kinase-inactive. However it interacts with the EGFR family members and serves to transduce signals to the PI3K/PTEN/Akt/mTORC1 pathway. Since EGFR3 is kinase-inactive, it will be more difficult to isolate inhibitors specific to it. However, it is still important and it can serve in pathways responsible for drug resistance [109].
The functions of EGFR4 in breast and other cancers have been investigated and recently summarized [110]. Similar to the EGFR1 gene, EGFR4 encodes a protein contains an extracellular ligand-binding domain, a hydrophobic transmembrane domain, an intracellular tyrosine kinase domain, and carboxyl-terminal tyrosine residues [110,111]. These carboxyl-terminal tyrosine residues are phosphorylated after ligand binding and allows coupling of the receptor with other signaling molecules [111]. Ligand binding to EGFR4 stimulates either homodimerization or heterodimerization of EGFR4 with another EGFR family member. Activation of EGFR4 signaling results in cleavage and release of the EGFR4 cytoplasmic domain from the membrane, which then may traffic to the nucleus and mitochondria to exert additional biological effects [110,112,113].
The roles of EGFR4 in cancer are complex. In some cancers (larynx, pancreatic and prostate) EGFR4 may function as a tumor suppressor [110,114-116]. EGFR4 expression in breast, cervical and ovarian cancers is associated with a favorable prognosis [110, 113,117,118]. The EGFR4 Q646C mutant results in an EGFR4 protein which undergoes ligand-independent homodimerization and tyrosine phosphorylation which interestingly suppresses colony formation of breast, pancreatic and prostate cell lines [110, 119-121]. Introduction of the constitutively active EGFR4 I658Q mutant into breast, ovarian and prostate cell lines induces apoptosis [122].
However, in some cell types and biological situations EGFR4 has oncogenic activities [120]. EGFR4 is overexpressed in certain cancers including ependymomas and medulloblastomas [111,116,123]. Overexpression of EGFR4 in lung carcinomas results in increased proliferation [116.123]. Furthermore, overexpression of HER2, EGFR4, and the EGFR4 ligand NRG1-beta in medulloblastoma results in increased metastasis [124]. Likewise over-expression of EGFR4 in conjunction with EGFR1 and HER2 results in breast cancers correlates with poor prognosis [125]. In contrast overexpression of EGFR4 by itself results in a more favorable outcome.
Silencing endogenous EGFR4 expression in ER+ MCF7 and T47D breast tumor cell lines reduces anchorage-independent proliferation stimulated by an EGFR4 ligand such as neuregulin-2beta [110, 126]. HER2 tyrosine kinase activity, rather than EGFR4 tyrosine kinase activity was required for neuregulin-2beta to stimulate cell proliferation [110]. Interesting the sites of EGFR4 tyrosine phosphorylation, but not the sites of HER2 phosphorylation, were required for neuregulin-2beta to regulate cell proliferation [110]. Thus the roles of EGFR4 in cancer remain complex and are influenced by the expression of other EGFR family members.
Neurofibromin 1 and GTPase Activating Proteins (GAP).
Neurofibromin 1 (NF1) is a tumor suppressor gene. It encodes a GTPase which normally serves to regulate Ras signaling. It is mutated in neurofibromatosis patients. These patients have elevated (constitutive) Ras activation. The tumor suppressor NF1 has been implicated in sporadic breast cancer. Recent studies have suggested that NF1 is a breast cancer driver gene. NF1 is deleted or mutated in 27.7% of all breast carcinomas [127]. In a study of inbred Chaos3 mice, investigators noted high levels of genetic instability which can lead to mammary tumors. The genomically-characterized mammary adenocarcinomas from these mice displayed deletions of certain genes, and the NF1 gene was deleted in the vast majority of the mouse mammary adenocarcinomas. These mammary adenocarcinomas exhibited constitutive Ras hyperactivation and sensitivity to Ras pathway inhibitors.
Also alternative mRNA splicing events can give rise to different NF1 isoforms. NF1 and Ras expression were examined in 22 sporadic breast cancers, 18 benign lesions and 6 normal breast tissues by tissue microarrays. NF1 and CELF3-6 RNA expression was examined by RT-PCR in the breast samples. NF1 and Ras expression displayed no difference in expression when examined by immunohistochemistry assays. In contrast, NF1 isoforms were determined to shift from the type II mRNA isoform in normal breast in normal breast cancer to the type I mRNA isoform in breast carcinoma. The authors did not detect a shift in CELF mRNA cofactor expression that was related to the shift in NF1 mRNA isoforms. The authors suggest that there is a NF1 isoform shift in expression from type II to type I which could be important in the development and progression of sporadic breast cancer [128].
NF1 normally serves to regulate Ras and thus is implicated in the regulation of both the PI3K/PTEN/Akt/mTORC1 and Raf/MEK/ERK pathways. Successful targeting of Ras may improve the therapy of patients with NF1 mutations. Alternatively, these patients may be sensitive to combined treatment with MEK and PI3K pathway inhibitors.
Genes that serve to regulate the Ras gene may be tumor suppressors as when they are mutated the Ras pathway is turned on. The RasGAP2 gene (RASAL2) is one such gene, it is a tumor and metastasis suppressor which is mutated or suppressed in breast cancer. Inactivation of RASAL2 was shown to be associated with tumor growth, progression and metastasis in animal models. In human RASAL2 loss was associated with metastatic disease and decreased expression of RASAL2 was associated with the recurrence of luminal B breast tumors [129].
The methylation state of the Ras association domain-containing protein 1 (RASSF1A) gene was examined in 36 breast cancer patients in breast cancer tissue as well as their adjacent normal tissues [130]. RASSF1A may function as a tumor suppressor gene. Methylation of the RASSF1A gene was detected in 61.1% of the breast tissues but not in their normal adjacent tissues. The methylation of the RASSF1A gene resulted in deceases in mRNA and protein levels of 33.3 and 44.4% respectively. In contrast, the RASSF1A protein was detected in normal tissues. Methylation of the RASSF1A gene did not appear to be associated with clinical parameters, such as age, histological types, TNM stages and lymph node metastases.
RAS Gene Family Mutations/Alterations in Breast Cancer.
In a genetic study examining the mutational status of the Ras and PI3K pathway genes in 40 breast cancer cell lines, mutations were detected at approximately 25% in Ras pathway members (KRAS, HRAS, NRAS, and BRAF) and 54% of PI3K pathway members (PTEN, PIK3CA). However, unlike the mutational status of these two families in colo-rectal cancer, this study did not detect mutation in both pathway family genes in single cell lines very frequently [131]. In subsequent studies by the same group with 41 breast cancer cell lines, this group found 146 mutations among twenty-seven cancer causing genes, which resulted in an average of 3.6 mutations per cell line. Mutations in TP53, RB and PI3K pathways were frequently detected in the breast cancer cell lines. Importantly these investigators could identify mutational profiles that were associated with luminal-type and basal-type breast cancer cell lines. The luminal mutational profile included E-cadherin (CDH1) and mitogen-activated protein kinase kinase 4 (MAP2K4 a.k.a. MEK4) genes and amplifications of the cyclin D1, (CCND1), HER2 and mouse double minute 2 homolog (MDM2), while the basal mutational profile included: BRCA1, RB1, RAS and BRAF gene mutations and deletions of tumor suppressors p16 (CDKN2A) and p14ARF (CDKN2A a.k.a. INK4A) [132].
Activation of the Ras pathway is often detected in breast cancer in the absence of mutations of the RAS genes [133]. Gene expression signature analysis has revealed that Ras pathway dependence can predict the sensitivity to inhibitors targeting the Raf/MEK/ERK and PI3K/PTEN/Akt/mTORC1 pathways. The Ras pathway activation is associated with sensitivity to MEK inhibitors but resistance to Akt inhibitors in breast and lung tumors. The Ras pathway signature is a better indicator of Ras pathway dependency than mutations at KRAS, as there can be many genetic mutations which can result in Ras pathway dependency (e.g., upstream receptors, EGFR1 and others as well as mutations in BRAF and other downstream signaling molecules). The Ras pathway signature was determined to be activated in breast cancers which were sensitive to MEK inhibitors but resistant to Akt inhibitors. The Ras pathway signature was shown to be increased in ER- breast cancers and lung adenocarcinomas. The Ras pathway signature also predicts resistant to the EGFR1-targeting agent cetuximab (Erbitux) in metastatic colorectal cancer [134].
Upon novel global gene expression profiling on 47,293 gene transcripts in 128 invasive breast cancers, the Ras-like, estrogen-regulated, growth-inhibitor (RERG) gene was determined to be a key marker of the luminal BC class and could be used to separate distinct prognostic subgroups [135]. These observations were further explored by performing immunohistochemistry on tissue microarrays containing 1,140 invasive breast cancers [135]. These results showed that the RERG gene is one of the highest ranked genes to differentiate between ER+ luminal and ER- non-luminal cancers. RERG expression was positively associated with the following markers of luminal differentiation: ER+, the cytokeratins (CK7/8, CK18 and CK19) and FOXOA1. RERG expression was also associated with other markers of good prognosis namely, small size, lower histologic grade an d AR, BRCA1, fragile histidine triad protein (FHIT, Bis(5’-adenosyl)-triphosphatase), and p21Waf-1 and p27Kip-1 and inversely associated with the proliferation markers MIB1 (a monoclonal antibody which is directed to a different epitope on the Ki67 protein than the original Ki67 antibody) and TP53. Importantly RERG expression was associated with longer survival.
The Rab-coupling protein RCP (a.k.a RAB11FIP1), is located at a chromosomal region frequently amplified in breast cancer. Introduction of constructs encoding RCP into normal human mammary epithelial cells MCF-10A cells lead to cells with tumorigenic properties. Likewise knock-down of RCP in breast cancer cell lines decreased the tumorigenic properties of the cells. Therefore RCP is a frequently amplified gene in breast cancer and suggest roles for the Rab family in carcinogenesis [136].
Interactions between Ras and Bmi-1 in Breast Cancer.
The B-lymphoma Moloney murine leukemia virus insertion region-1 (Bmi-1) gene functions in stem cell maintenance. It is a member of the polycomb group of transcription repressors. Bmi-1 exerts it effects by suppression of the p16Ink4A/ARF tumor suppressor. Increased expression of Bmi-1 is detected in many cancers. The effects on Bmi-1 overexpression were examined on MCF-10A cells. While Bmi-1 overexpression by itself did not result in the oncogenic transformation of MCF-10A cell, co-expression of activated H-Ras (RasG12) resulted in the oncogenic transformation of MCF-10A cells. The Bmi-1/H-Ras transformed cells exhibited properties of cells that had undergone the epithelial to mesenchymal transition (EMT). Bmi-1 inhibited senescence and allowed the proliferation of cells expressing high levels of activated H-Ras [137]. Subsequent studies by the same group have indicated that knock-down of Bmi-1 in breast cancer cell lines decreased their aggressive nature in vivo in tumor transplant studies [138].
Mutations in Components of the PI3K/PTEN/Akt mTOR Pathway in Breast Cancer.
In a study which examined MBC, PIK3CA mutations were detected in 47.4% of the cancers which were aggressive and also chemoresistant. In contrast, PI3KCA mutations were detected in 34.5% of hormone receptor-positive cancers, 22.7% of 75 HER2-positive cancers, 8.3% of basal-like cancers and none of claudin-low tumors examined in this study. MBCs and claudin-low breast cancer subsets displayed enrichment for markers linked to stem cell function and EMT. It was postulated that MBCs and claudin-low tumors are enriched with CICs and may arise from an earlier, more chemoresistant breast epithelial precursor than either basal-like or luminal cancers. However in this study, no mutations at PIK3CA were detected in the claudin-low cancers, so the roles of PIK3CA mutations in these cancers are not clear [139].
In a study with 547 human breast cancer patient samples and 41 established cell lines, the mutational status of PIK3CA, AKT and PTEN were analyzed as well as the effects of pathway mutations on the sensitivity to PI3K inhibitor LY294002 [140]. PIK3CA mutations were determined to be more frequent in ER+ (34.5%) and HER2+ (22.7%) than in basal-like tumors (8.3%). AKT1 (1.4%) and PTEN (2.3%) mutations were determined to be restricted to ER+ breast cancers. Interestingly cells with PIK3CA mutations were less sensitive to the PI3K inhibitor LY294002 inhibitor than breast cancer cells which had loss of PTEN activity. Thus, PI3K pathway aberrations likely play a distinct role in the pathogenesis of different breast cancer subtypes. The specific aberration present may have implications for the selection of PI3K-targeted therapies in hormone receptor-positive breast cancer.
In a study which examined the genetic structure of the PIK3CA gene in 452 breast cancer patients, PIK3CA mutations were observed in 33.4% of the breast cancer patients [141]. PIK3CA mutations were more frequently detected in ERalpha+ and PR+ breast cancers (41.1%), than in TNBCs (ER-,PR-, HER2-) (12.5%.). Patients which had PIK3CA mutations had a longer metastasis-free survival period than the overall population.
The mutational status of the PIK3CA gene was examined in eighty HER2+ breast cancer patients as well as clinical outcome of the patients after Herceptin treatment [142]. The PIK3CA gene was determined to be mutated in 21.3% of HER2+ breast cancer patients that had been treated with herceptin for one year. Improved disease free survival was observed in those patients with WT PIK3CA as opposed to those patients with mutant PIK3CA. The PIK3CA gene is also mutated in some ovarian cancer patients [143].
PI3K-p110 (PIK3CA) protein expression was examined in 1,394 early stage breast cancer samples. Elevated PI3K-p110 was associated with the basal-like breast cancers, HER2+ breast cancer, and triple negative non-basal breast cancers. In contrast, the luminal class of breast cancers had reduced levels of PI3K-p110 in comparison to the other classes. PI3K-p110+ breast cancer patients had shorter disease free survival. Thus these studies demonstrated that PI3K-p110 expression is a biomarker associated with poor prognosis in breast cancer [144].
Interactions between PIK3CA Mutations and HER2 Amplification in Breast Cancer.
Introduction of genetic constructs containing PIK3CA mutations E545K and H1047R into MCF-10A human mammary epithelial cells that also overexpress HER2 conferred enhanced growth properties to MCF10A/HER2 cells in comparison to cells which lacked the introduced mutant PIK3CA gene [145]. Upon introduction of mutant PIK3CA (H1047) into MCF-10A cells which overexpress HER2, the expression of EGFR3/EGFR4 ligand heregulin (HRG) was detected. In contrast, introduction of mutant PIK3CA (E545K) gene into the MCF-10A cells which overexpressed HER2 did not result in the expression of HRG. Silencing HRG with siRNA inhibited the growth of MCF-10A/HER2/PIK3CA-H1047R cells but not MCF-10A/HER2/PIK3CA-E545K-expressing cells. The HRG siRNA synergized with the HER2 inhibitors herceptin (trastuzumab) and lapatinib. Treatment of the cells with a PI3K inhibitor (BEZ235) suppressed HRG and P-AKT levels. When the cells were cotreated with BEZ235 and lapatinib, a complete suppression of growth was observed. These important result document interactions between HER2 and certain PIK3CA mutations and point to the possible co-targeting of HER2 and PI3K in certain breast cancers.
Deregulation of the PI3K/PTEN/Akt/mTORC1 pathway by gene mutations has been estimated to occur in >70% of breast cancers [146]. In ER+ breast cancers, activation of the PI3K/PTEN/Akt/mTORC1 pathway can result in both estrogen-dependent and estrogen-independent ER activity and contribute to estrogen-independence and potentially loss of sensitivity to hormonal based therapies. Activation of the PI3K/PTEN/Akt/mTORC1 pathway can also result in resistance to HER2 inhibitors in HER2+ cells. Inhibition of the PI3K/PTEN/Akt/mTORC1 pathway can overcome resistant to hormonal and anti-HER2 targeted therapies [146]. Encouraging results have been obtained in breast cancer clinical trials with combinations of various inhibitors. These results predict that combinations of HER2, PI3K, mTORC2 inhibitors and hormonal based therapeutics may be appropriate for the treatment of certain breast cancer patients which are resistant to current therapies.
The transforming activity of mutant PI3K has been linked to its ability to bind phosphorylated YXXM motifs present in activated receptor tyrosine kinases (RTK) or adaptor molecules, potentially in association with the PI3K-regulatory subunit, p85. EGFR3 is a potent activator of the PI3K/PTEN/Akt/mTORC1 pathway. The requirement for EGFR3 in PI3K-mediated transformation of mammary epithelium was investigated [147]. Conditional loss of EGFR3 in mammary epithelium lead to a delay in PI3K-H1047R-mediated mammary hyperplasia. However, these studies demonstrated that tumor latency and PI3K signaling were not perturbed. In the EGFR3-deficient mammary tumors, the PI3K-H1407R protein was determined to be associated with several tyrosyl phosphoproteins. These studies also demonstrated that inhibition of other EGFR family members with lapatinib did not inhibit the mutant PI3K-mediated signaling. However, co-inhibition of PI3K with the PI3K-alpha specific inhibitor BYL719 and EGFR-mediated signaling with lapatinib was more effective in suppressing growth than treatment with either the PI3K-alpha inhibitor or lapatinib alone. Additional studies by this group determined that co-inhibition of PI3K and EGFR suppressed growth and PI3K signaling in human breast cancer cells containing the mutant PIK3CA-H1047R gene. These studies point to the possibility of co-targeting of PI3K and EGFR in certain breast cancers.
Mutations at PIK3CA and PTEN Can Confer Resistance to Herceptin in HER2+ Breast Cancer.
An RNA interference screen was performed to identify some of the genes involved in resistance to herceptin. PTEN was identified as a modulator of resistance to Herceptin in cell cultures experiments. Likewise oncogenic PIK3CA mutations would confer resistance to Herceptin in cell cultures [148]. In a screening of samples from 55 breast cancer patients, PIK3CA mutations or low expression of PTEN was associated with resistance to Herceptin.
Deregulation of PI3K and PTEN in Metastatic Breast Cancer.
The mutational status of the PIK3CA gene and expression of PTEN was examined in breast cancer specimens that differed in the state of malignancy (e.g., primary vs. metastatic breast cancer from the same cancer patient) [149]. The PIK3CA gene was determined to be mutated in 19 (40%) of primary tumors and 21 (42%) of metastatic cancers. PTEN expression was examined by immunohistochemistry and PTEN expression was lost in 14 (30%) primary tumors and 13 (25%) metastases. Thus the PI3KCA gene is frequently mutated and PTEN expression is often lost in breast cancer.
Epigenetic Regulation of Additional and Novel Genes Associated with Breast Cancer.
Epigenetic profiling has recently resulted in the identification of genes associated with breast cancer tumorigenicity that were methylated [150]. This study identified 264 hypermethylated loci in genomic CpG islands. Hierarchical clustering in terms of the levels of methylation at the loci resulted in generation of at least three distinct groups of breast cancer patients. Namely the methylation levels were distributed into three different groups, ER+/PR+ breast cancer patients, time to tumor relapse and lymph note metastasis. Methylation of six genes (RECK, SFRP2, UAP1L1, ACADL, ITR, and UGT3A1) was associated with decreased relapse free survival. Reversion-inducing-cysteine-rich protein with kazal motifs (RECK) is thought to be a metastasis-suppressor gene and may interact negatively with matrix metalloproteinase (MMP9). Secreted frizzled-related protein 2 (SFRP2) is a soluble modulator of Wnt signaling. Methylation of this gene has been association with breast and other cancers [151]. UAP1L1 encodes a UDP-N- acteylglucosamine pyrophosphorylase 1-like 1 protein. ACADL is a member of the acyl-CoA dehydrogenase family. This is a family of mitochondrial flavoenzymes involved in fatty acid and branched chain amino-acid metabolism. The ACADL gene product is associated with long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. The ITR gene (ITR/GPR180) is a G protein-coupled receptor). The UGT3A1 gene is a member of the UDP glycosyltransferase 3 family.
Drug Resistance and CICs.
Drug resistance breast cancer cells are enriched in populations of cells which have characteristics of cancer stem cells. These cancer cells with stem like characteristics are referred to cancer initiating cells (CICs) [152-175]. In the study by Britton and colleagues with both clinical fine needle aspirates obtained from breast cancer patients as well as established breast cancer cell lines, the expression of ATP-binding cassette sub-family G member 2 (ABCG2=BCRP1) was monitored [175]. In the studies with the side populations from MCF-7 and MDA-MB-231 cells, increased ABCG2 was observed in the side populations as well as elevated resistance to the chemotherapeutic drug mitoxantrone. The increase in the side populations may be due to increased drug transporter expression in the cells with the CIC phenotype as the drug transporter would exclude the drugs from the cells. The presence of the side populations in the fine needle aspirates was associated with ER-negative breast cancers and TNBCs which also had elevated ABCG2 protein expression. Breast CICs can be characterized by increased expression of CD44 and decreased expression of CD24 (CD44↑/CD24↓) compared to the remaining cancer cells which are referred to as the bulk cancer cells (BCs). The CICs and BCs differ in their gene expression patterns which may have resulted from epigenetic and other mechanisms [176]. Thus the CICs and BCs will likely have different signaling pathways activated/suppressed which will require different therapeutic approaches to eliminate both populations of cancer cells.
Involvement of HER2 in Breast CICs.
HER2 is expressed in the CIC population [177, 178]. The expression of HER2 is modulated by the tumor microenvironment. Targeting of HER2 in these CICs may be an appropriate therapeutic approach. Interestingly herceptin suppressed tumor growth of the CIC in mouse xenograft models but in not established breast tumors [176].
Initially was thought that herceptin would only be effective in breast cancer patients which overexpressed HER2, some clinical studies have shown that herceptin may target breast cells which did not overexpress HER2 [179]. These results have led to the hypothesis that HER2 is an important molecule expressed on breast CICs and further studies have suggested that HER2 expression may be induced by signals in the microenvironment in breast CICs which lack HER2 gene amplification.The effectiveness of herceptin is thought to be due to its ability to target the breast CIC population as well as the PI3K/PTEN/Akt/mTORC and other signaling pathways [176, 177, 180].
Herceptin may also be effective in the treatment of breast cancer patients which lack amplification of HER2, documenting a role for HER2 in the growth of these normally HER2-negative cancers [181]. These authors demonstrated that HER2 was expressed in ER+, HER2- luminal breast cancers and regulates the self renewal of the CIC sub-population. HER2 expression was not due to gene amplification but was determined to result from receptor activation of NF-kappaB (RANK)-ligand in the bone microenvironment.
PI3K Pathway in Breast CICs.
The PIK3CA gene may be mutated in some breast CICs [182]. The PI3K/PTEN/Akt/mTORC pathway has been reported to be important in breast CICs. Side-population positive MCF-7 breast cancer cells [MCF-7/(CIC)] were isolated from parent MCF-7 bulk cancer [MCF-7/(BC)] [183]. The MCF-7/(CIC) cells displayed increased drug transporter activity and enhanced colony-formation ability in vitro and greater tumorigenicity in vivo than the MCF-7/(BC). The expression of critical pathways were compared between MCF-7/(CIC) and MCF-7/(BC). The PI3K/PTEN/Akt/mTORC1 and STAT3 pathways were shown to be differentially expressed in the MCF-7/(CIC) population and responsible in part for their enhanced survival [183].
Deregulation of Akt in Breast Cancer.
Mutations of Akt are relatively rare in breast cancer, however, aberrant activation of either upstream PIK3CA or polymorphism of the PH domain and leucine rich repeat protein phosphatase 2 (PHLPP2) gene can result in Akt expression. In a study which examined the mutational status and polymorphism of genes in the PI3K/PTEN/Akt/mTORC1 pathway, DNA was isolated from fine needle aspirations of 267 stage I-III breast cancers [184]. In this study, 28 genes were examined for 163 known cancer-related DNA sequence variations by Sequenom technology. The PI3K pathway was determined to be frequently altered in breast cancers as at least one mutation in 38 alleles corresponding to 15 genes in 108 (40%) of the breast cancer samples. The PIK3CA gene was determined to be the most frequently mutated (16.1% of all samples), the F-box and WD repeat domain containing 7, E3 ubiquitin protein ligase (FBXW7) gene, the second (8%), the BRAF gene, the third (3.0%), the EGFR1 gene, the fourth (2.6%), the AKT1 and CTNNB1 genes (beta-catenin) the fifth and sixth (1.9% each), the KIT and KRAS genes (1.5% each), and the PDGFRA gene, the seventh (1.1%). Polymorphism at the PHLPP2 phosphatase which activates Akt was observed in 13.5% of the patient samples. PIK3CA mutations were observed more frequently in ER+ cancers compared to TNBC (19 vs. 8%). Interesting a high frequency of PIK3CA mutations (28%) was observed in HER2+ breast tumors. In TNBC, FBXW7 mutations were significantly more frequent compared to ER+ tumors (13 vs. 5%). FBXWZ is a component of ubiquitin ligase (SKP-cullin-F-box) and may bind cyclin E and target it for ubiquitin-mediated degradation (Figure 2).
Some investigators have suggested that other molecules besides Akt are important in breast cancer and have proposed Akt-independent signaling mechanisms [185]. This group demonstrated that in some breast tumors cultured in an anchorage-independent fashion displayed minimal Akt activation and decreased reliance on Akt for growth. In contrast, these cells had strong PDK1 activation and membrane localization and were dependent on the activated PDK1 substrate serum/glucocorticoid regulated kinase family, member 3 (SGK3), which is related in structure to Akt. SGK3 has been shown in additional studies to be linked with breast cancer and is induced by estrogen in breast cancer cells and is associated with ER expression [186]. Like Akt, SGK3 can phosphorylate GSK-3beta and TSC-2 which results in their inactivation and activation of mTORC1 and stimulation of protein translation [187, 188]. Knowledge of the particular kinase or other type of protein, responsible for the malignant potential of breast cancer cells could aid therapy by the use of more effective inhibitors which target the particular enzyme affected and responsible for the abnormal growth.
Activated Akt as a Marker for Sensitivity to Drug Therapy.
Phosphorylation (activation) of Akt has been shown to predict the effectiveness of paclitaxel chemotherapy in node-positive breast cancer patients [189]. In the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-28 trial, the effectiveness of adding paclitaxel to doxorubicin (a.k.a Adriamycin) plus cyclophosphamide (AC) was examined in breast cancer patients (median follow up 9.1 years). Enhanced effectiveness of adding paclitaxel to AC was observed in those breast cancer patients who expressed elevated P-Akt. In contrast, no enhanced effectiveness of adding paclitaxel to AC was detected in the breast cancer patients who did not express activated P-Akt. Thus addition of paclitaxel to breast cancer patients which are P-Akt- does not appear to increase the effect of AC therapy, while addition of paclitaxel to those breast cancer patients which are P-Akt+ does appear to improve therapy.
PTEN and Cytokines and their Involvement in HER2-Resistance and CIC Survival.
PTEN is important in breast CIC survival. Knockdown of PTEN expression was shown to result in increases in normal and malignant human mammary stem/progenitor cells both in vitro and in vivo. This increase in progenitor cells was mediated by increased Akt activation which resulted in the phosphorylation of GSK-3beta which in turn led to activation of the Wnt/beta-catenin pathway. The increases in progenitor cells could be suppressed by the Akt inhibitor perifosine [190]. PTEN is also important in the resistance of breast cancers to herceptin and other therapeutic approaches [191].
Interleukin-6 (IL-6) is cytokine which is an important immune-regulator. IL-6 is also important in HER2-resistance as it can expand the CIC population [192-194]. Decreases in PTEN expression has been implicated in herceptin-resistance. In HER2+ cell lines generated by knocking down PTEN, which were resistant to herceptin, the resistance was shown to be due to activation of an IL-6 inflammatory feed back loop. This IL-6 inflammatory loop resulted in the expansion of breast CIC which have an EMT phenotype and secrete 100-fold more IL-6 than in the parental cells which did not have PTEN knocked down. The authors of this important study also determined than treatment with an IL-6R Ab inhibited this IL-6 regulatory loop and reduced the CIC population and importantly reduced tumor growth and metastasis in mouse xenographs. A figure depicting the effects of PTEN on certain CICs as well as ER-signaling is present in Figure 3.
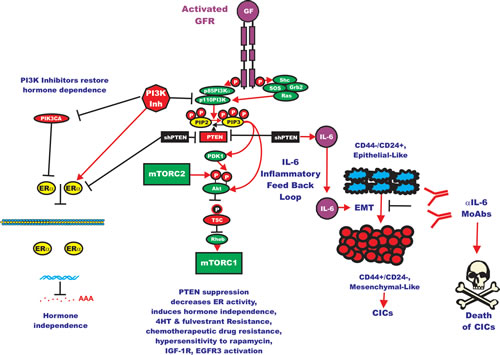
Figure 3: Effects of Targeting PI3K/PTEN/Akt/mTORC1 and IL-6 on Breast Cancer Hormonal Dependency, EMT and CICs. Mutations at PIK3CA can alter hormonal dependency. Silencing PI3K with PI3K inhibitors can restore hormonal dependency. Silencing PTEN can also result in hormonal-independenc, 4HT and fulvestrant resistance. Silencing PTEN can in some cases also lead in some cases to IL-6 production and an IL-6 inflammatory feed back loop which results in EMT and CIC formation. Silencing of IL-6 with monoclonal antibodies (MoAb) can prevent this loop and result in the death of the breast CICs.
The tumor microenvironment is important in CICs. Interactions between the interleukin-8 receptor (IL-8R) and HER2 have been determined to be important in the survival of breast CICs [195]. NF-kappaB has been shown to be important in breast cancer CIC survival and HER2-dependent tumorigenesis. NF-kappaB activity can be regulated by the PI3K/PTEN/Akt/mTORC1 pathway [193].
Targeting PI3K Pathway to Inhibit Breast Cancer Resistance to Therapy.
Treatment of breast cancer patients with the aromatase inhibitor (AI) letrozole has been shown to result in suppression of the PI3K/PTEN/Akt/mTORC1 pathway [194]. This clinical study examined the expression of PI3K (p110), P-Akt, and P-mTOR by immunohistochemistry on breast cancer samples from 113 patients. The patients had been enrolled in a phase II study of letrozole or letrozole and cyclphosphamide. Either letrozole or letrozole plus cyclophosphamide-treated patients displayed a reduction in PI3K and P-mTOR expression. In contrast, expression of P-Akt did not change in the letrozole-treated patients whereas it decreased in the letrozole and cyclophosphamide-treated patients. The reduction of P-Akt expression was associated with a better response rate and reduction in Ki67 staining. The reduction in P-mTOR expression was associated with a longer disease-free survival. Thus the AI letrozole targets key components of the PI3K/PTEN/Akt/mTORC1 pathway which may be important in the successful treatment of certain breast cancers.
This same group developed some letrozole-resistant MCF-7 cells by culturing the cells for prolonged periods of time in letrozole. The letrozole-resistant cells displayed elevated expression of key components of the PI3K/PTEN/Akt/mTORC1 pathway. They observed that suppression of the PI3K/PTEN/Akt/mTORC1 pathways with PI3K or mTORC1 inhibitors reversed the acquired letrozole- resistance [195]. A diagram of the effects of letrozole on ER and PI3K/PTEN/Akt/mTORC signaling and letrozole-resistance is presented in Figure 4.
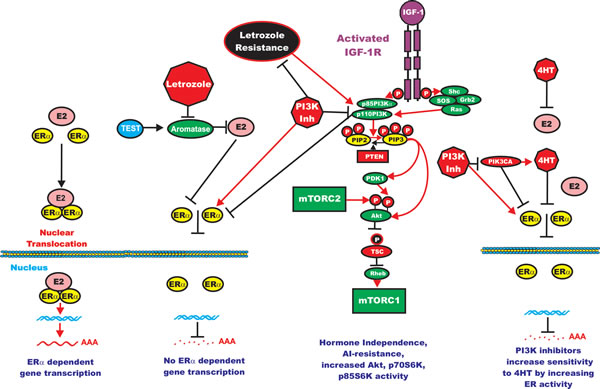
Figure 4: Effects of PI3K/PTEN/Akt/mTORC1 on Aromatase Resistance and Sensitivity to Tamoxifen (4HT). The aromatase inhibitor (AI) letrozole prevents the conversion of testosterone (TEST) into estrogen (ER) and hence there is no ERalpha mediated gene transcription. Letrozole alters the expression of the PI3K/PTEN/Akt/mTORC1 pathway. PI3K inhibitors will restore ERalpha mediated gene expression in Letrozole resistant cells and the cells revert to hormonal sensitivity. Likewise PI3K inhibitors will restore the sensitivity of 4HT-resistant cells to 4HT. Breast cells with mutations at PIK3CA may develop resistance to 4HT and PI3K inhibitors may restore sensitivity to 4HT.
Effects of PI3K/PTEN/Akt/mTORC1 Pathway Activation and ER Expression on Breast Cancer.
PIK3CA mutations in ER+/HER2- luminal breast cancer actually result in low levels of mTORC1 expression and these breast cancers have some of the better treatment successes after 4HT therapy [196]. These PIK3CA mutations are predicted to render these breast cancers sensitive to targeted therapy. The authors of this study have devised a PIK3CA gene expression pattern. This pattern was associated with prognosis in those breast cancers with PIK3CA mutations that remained ER+/HER2-, it was not associated with prognosis in breast cancers which were either HER2+ or ER-.
An inverse relationship between PI3K pathway activation and ER expression was observed in ER+ breast cancers. When the PI3K pathway was activated, lower levels of ER were detected, indicating a negative correlation. Treatment of breast cancer cells with insulin like growth factor-1 (IGF-1), which activated the PI3K pathway resulted in decreased ER expression. Likewise treatment of breast cancers with the PI3K inhibitor BEZ-235 resulted in increased ER activity as well as ER-regulated gene expression [197]. The PI3K pathway activity was increased in ER+ tumors and cell lines of the more aggressive luminal B subtype versus those of the less aggressive luminal A subtype. The PI3K inhibitor would increase the effects of 4HT on the more aggressive luminal B breast cancer, potentially by increasing ER expression and restoring sensitivity to hormone based therapies. This study suggests a potential therapeutic approach by combining PI3K inhibitors and 4HT.
The PI3K/PTEN/Akt/mTORC1 pathway is also important in the growth of breast cancers which have become hormone-independent. In four hormone-independent breast cancer cell lines created after long term estrogen deprivation, increased phosphorylation of p70S6K, p85S6K and Akt was observed [198]. Inhibition of the PI3K pathway in these hormone-independent cells resulted in apoptosis. These results indicate that some hormone-independent breast cancers may be sensitive to the combination of ER and PI3K pathway inhibitors.
Inhibition of PTEN activity has been associated with resistance to chemotherapeutic drugs, hypersensitivity to the mTORC1 inhibitor rapamycin as well as hormonal based therapies [199,200] Inhibition of PTEN expression by shRNA resulted in three ERalpha+ breast cancer cell lines that were able to grow in the absence of hormone. Furthermore the cells were resistant to 4HT and fulvestrant. Fulvestrant is an ER antagonist that functions by down regulating the ER. Knock down of PTEN also increased ERalpha transcriptional activity in MCF-7 cells, but decreased ER protein levels and transcriptional activity in T47D and MDA-361 cells. Inhibition of PTEN by shRNA increased basal and ligand-induced activation of IGF-1R and EGFR-3, documenting the effects that PTEN can have on the regulation of these upstream tyrosine kinases. Inhibition of IGF-1R or EGFR-3 restored hormonal dependency and the effects of hormonal therapy on the breast cancer cells with PTEN-knocked down. These studies indicate a possible treatment strategy for breast cancers which are either PTEN-negative or have decreased PTEN expression. These results document the complex interactions between hormonal- and growth factor-dependent signaling.
Association between PIK3CA Mutations and AR Expression in Breast Cancer.
An association between AR expression and PIK3CA mutations was observed in a study which examined AR and ER expression and PIK3CA mutational status in 347 breast cancer patients [201]. AR expression was higher in those breast cancers which also expressed ER and PR. In those samples that expressed AR, mutations in the PIK3CA kinase domain were more frequently detected than mutations in the helical domain or those lacking PIK3CA mutations. High AR expression was linked with an improved recurrence-free survival in 207 patients with early-stage ER+/PR+ positive tumors after hormonal-based therapy. Higher AR expression was also associated with PIK3CA mutations and not with PIK3CA WT or TNBCs. These studies indicate that AR and PIK3CA (as well as ER and PR) may be prognostic markers for breast cancer [201].
Dysregulation of the PI3K/PTEN/Akt/mTORC1 Pathway in Endometrial Cancer.
Endometrial cancer is one of the most frequent gynecological malignancies [202]. Many signaling pathways have been implicated in endometriod cancer [203-206]. Greater than 90% of endometriod cancers suffer from some type of mutation in the PI3K/PTEN/Akt/mTORC1 pathway. Thus, this pathway is a key therapeutic target in endometriod cancer [207,208]. Endometrioid endometrial cancers (EEC) frequently have multiple mutations at PTEN, PIK3CA, PIK3R1 and KRAS. The effects of the pan-class I PI3K inhibitor GDC-0941 and the mTORC1 inhibitor temsirolimus were examined on EEC cells with concurrent PIK3CA and PTEN mutations [209]. ECC with PIK3CA mutations were sensitive to GDC-0941, while ECC with PTEN mutations were sensitive to the mTORC1 blocker. Only 2 of 6 EEC cells with KRAS mutations were sensitive to MEK inhibitors. The PI3K p110alpha selective inhibitor A66 was more effective than the PI3K p110beta inhibitors GSK2636771 and AZD6482 in suppressing the growth of the PTEN-mutant EEC cell lines.
Expression of the PI3K-p110 alpha and beta Subunits in Breast Carcinomas and EEC.
A study was performed on 315 invasive breast carcinomas to compare the expression of the PI3K-p110 alpha and beta subunits in these breast cancer patients. Then the expression results were correlated with clinical outcomes [210]. This immunohistochemistry study determined that overall the p110 subunits were expressed in 23.8% of invasive breast carcinomas. PI3K-p110-alpha was expressed in 11.8% and 15.2% expressed PI3K-p110-beta. This study observed that PI3K-p110-alpha expression was associated with hormone receptor expression but was not associated with overall survival. PI3K-p110-beta expression was linked to HER2 overexpression and lack of hormone receptor expression. PI3K-p110-beta+ breast cancer patients had lower age of onset, lymph node involvement and distant metastasis. Those breast cancer patients that expressed membrane PI3K-p110-beta had a worse prognosis and overall survival. These important clinical studies point to the possibility of co-targeting of HER2 and PI3K-p110-beta in certain breast cancer patients.
Recently it was determined that the PIK3R1 (p85alpha) and PIK3R2 (p85beta) regulatory subunits are mutated in EEC [211]. PIK3R1 mutations were reported to occur at a higher rate in EEC than any other cancer type. Also this study demonstrated that the PIK3R2 gene is mutated in EEC, which was previously not thought to be a cancer gene. Many PIK3R1 and PIK3R2 mutations are gain of function mutations. Some PIK3R1 mutations bind and stabilize PTEN. KRAS mutations are also common in EECs. PI3K pathway mutations can occur in the presence of WT PTEN and they phenocopy PTEN loss as the pathway is activated.
Deregulation of Downstream Components of the PI3K/PTEN/Akt/mTORC1 Pathway Involved in the Regulation of mRNA Translation in Breast Cancer.
The PI3K/PTEN/Akt/mTORC1 pathway serves to regulate the translation of certain mRNAs which are considered difficult to translate due to their structures. The PI3K/PTEN/Akt/mTORC1 pathway can regulate the activity of key components of the translational apparatus such as eIF4E, eIF4G, 4E-BP1, rpS6, programmed cell death protein 4 (pdcd4), eEF2 and eEF2K. The expression of eIF4E, eIF4G, 4E-BP1, p4E-BP1 (T37/46), p4E-BP1 (S65), p4E-BP1 (T70), S6, pS6 (S235/236), pS6 (S240/244), pdcd4, eEF2 and eEF2K was examined in 190 hormone receptor-positive breast cancer patients [212]. This study followed the course of the breast cancer patients for 96 months. Elevated eEF2K, rpS6, and p4E-BP and decreased pdcd4 were associated with poor prognosis in hormone receptor+ breast cancer. These molecules may be prognostic markers and therapeutic targets for certain classes of breast cancer (e.g., hormone-responsive breast cancers).
Involvement of GSK-3 in Breast Cancer.
Phosphorylation by Akt also inhibits the activity of many key molecules involved in signaling and apoptosis. Activated Akt can stimulate carcinogenesis by inactivating proteins that normally function to limit cell growth and regulate apoptosis [213]. GSK-3beta is a down stream target of Akt. Introduction of kinase-dead GSK-3beta [GSK-3beta(KD)] into epithelial cells promoted tumorigenesis of breast and skin tumors [214]. Overexpression of constitutively-active GSK-3beta altered chemosensitivity, cell cycle arrest and tumorigenicity of breast cancers [215-218]. Inhibition of GSK-3beta, by small molecule inhibitors induced epithelial mesenchymal transition (EMT) and invasion in breast cancer [219].
The localization of GSK-3beta was altered in a study of invasive ductal carcinomas (IDC). A reduction or loss of cytoplasmic GSK-3beta in was observed in 53% of IDC examined [220]. Nuclear accumulation of GSK-3beta was detected in 35% of the IDC samples examined. This nuclear accumulation of GSK-3beta was associated positively with tumor grade [220]. A downstream target of GSK-3 is p27Kip-1 which is also implicated in breast cancer [221]. In addition, p70S6K can be regulated by GSK-3 and is involved in breast cancer [222]. Figure 2 depicts some of the interactions with GSK-3 and these and other signaling molecules.
GSK-3beta and beta-catenin can regulate cadherin-11 post-transcriptionally in breast and prostate cancer cells. Inactivation of GSK-3beta lead to repression of cadherin-11 mRNA and protein levels [223]. Loss of cytoplasmic GSK-3beta may promote EMT in breast cancer [223]. GSK-3 can regulate c-Myb which is important in EMT in breast cancer [224-227]. A studyl performed on breast cancer biopsies observed that inactivation of GSK-3beta was associated with elevated levels of the prolactin receptor, which is implicated in tumorigenesis [228]. The breast cancer resistance protein (BCRP) was determined to be downregulated in breast cancer cells when GSK-3beta was active, documenting that GSK-3beta can suppress active drug efflux [229]. Suppression of GSK-3beta activity by Akt phosphorylation enriched for mammary stem cells in both normal and breast cancer cells through activation of beta-catenin [230]. Thus at least with regards to mammary epithelial cells GSK-3beta activity appears to limit proliferation and suppress the stem-like cell population.
Often GSK-3beta is thought to have roles in tumor suppression. However, this is not always the case. In certain cancer types (e.g., pancreatic cancer) GSK-3beta was shown to participate in pro-inflammatory and anti-apoptotic processes by positively regulating NF-kappaB activity in the nucleus [231-233]. The roles of GSK-3beta in cancer progression remain controversial and extremely complex and may depend on the cancer type. The cellular localization of GSK-3beta is an important factor in controlling GSK-3beta ability to provide growth-limiting and survival-promoting activities. Aberrant nuclear accumulation of GSK-3beta may be important in many cancers.
The biochemical events leading to loss of functional GSK-3beta activity may also enrich for a subpopulation of CICs that demonstrate enhanced motility, clonogenicity, and drug resistance. However, the production of CICs may depend on the cancer type as well as stage of differentiation. Micro RNAs (miRs) also play important roles in CICs. TNBCs often have a high percentage of CICs [234]. Different miRs are expressed in TNBC than other breast cancer. Breast CICs often express high levels of CD44 and lower levels of CD24 (CD44↑CD24↓) than the non-CIC population which is referred frequently to as the bulk cancer. Autophagy is important in the regulation of the CIC population [235]. GSK-3 plays important roles in the regulation of autophagy and other age-related diseases [236]. Pharmacological inhibition of GSK-3 by small molecule inhibitors or deletion of GSK-3alpha leads to the prevention of autophagy.
We determined recently that GSK-3beta play important a role in MCF-7 breast cancer clonogenicity, drug resistance, and cell signaling [218]. Introduction of GSK-3beta(KD) into MCF-7 cells resulted in increases in both anchorage-dependent and anchorage-independent clonogenicity compared to cells transfected with the GSK-3beta(WT) construct. This increase in clonogenicity was observed when doxorubicin was absent or present. More colonies were observed in MCF-7/GSK-3beta(KD) cells than MCF-7/GSK-3beta(WT) or MCF-7/GSK-3beta(A9) cells when the cells were treated with doxorubicin. MCF-7/GSK-3beta(A9) cells with constitutive GSK-3beta kinase activity displayed higher anchorage-dependent, but not anchorage-independent, clonogenicity than MCF-7/GSK-3beta(WT) cells. Anchorage-independent growth is one measurement of transformed cells. In essence, it measures the ability of the cells to grow without adherence to a tissue culture plate surface. Cells which are more transformed will form more colonies in soft agar than cells which are either “not”-tranformed or “less”-transformed. GSK-3beta may play multiple roles by both limiting proliferation under certain conditions and allowing cell growth in others. In some cases GSK-3beta functions as a tumor promoter by phosphorylation of Axin which leads to beta-catenin stabilization and potentially cancer. In contrast, other cases GSK-3beta acts as a tumor suppressor and induces beta-catenin phosphorylation and proteasomal degradation.
When the cells were plated n doxorubicin, the anchorage-independent colony forming ability of MCF-7/GSK-3beta(KD) cells in doxorubicin was enhanced five-fold in comparison to MCF-7/GSK-3beta(WT) cells. Our results indicated that GSK-3beta activity influences breast cancer proliferation, motility and response to chemotherapy. Loss of GSK-3beta kinase activity may confer survival advantages by upregulating factors involved in cell cycle progression, prevention of apoptosis and anchorage-independence.
Resistance to doxorubicin and 4HT was increased in MCF-7/GSK-3beta(KD) cells in comparison to MCF-7/GSK-3beta(WT) cells. However, drug resistant MCF-7/GSK-3beta(KD) cells responded to mTOR inhibition by treatment with rapamycin. Additionally, a combination treatment of consisting of a MEK inhibitor and doxorubicin or 4HT was determined to have a synergistic effect that eliminated drug resistance in MCF-7/GSK-3beta(KD) cells. Targeting signaling molecules involved with the PI3K/PTEN/Akt/mTORC1 and Raf/MEK/ERK pathways may be appropriate to overcome resistance to chemo- and hormonal therapy. Genetic alterations which result in abnormal GSK-3beta activity should be taken into considered when designing a course of breast cancer therapy. Combination drug/inhibitor treatments could be advantageous by lowering the concentration of the chemotherapeutic drug and reduce therapy-related side effects.
There are alternate routes of GSK-3beta activation that are independent of Akt. To add to the complexity, GSK-3beta plays roles in the Wnt/beta-catenin pathway by being a component of the beta-catenin destruction complex as well as in the phosphorylation of Axin which results in the stabilization of Axin. This interaction of GSK-3 with the Axin protein complex prevents Akt from accessing and phosphorylating GSK-3beta. There are likely different cellular pools of GSK-3beta that are under separate regulation by Wnt and Akt, in which the activation of one does not affect the other. Simultaneous Akt and GSK-3beta activity has been observed in pancreatic and colon cancer cell lines, indicating that increased Akt expression does not always result with decreased GSK-3beta activity [231]. Since certain pools of GSK-3beta may remain active in cancer, the precise cellular localization of these pools may also be an important factor involved in their regulation. GSK-3beta is thought to be active predominantly in the cytoplasm, but it can also translocate to the nucleus. The nuclear activities of GSK-3beta may appear to conflict with its cytosolic roles. Aberrant nuclear accumulation of GSK-3beta has been observed in certain cancers [231]. A nuclear localization signal (NLS) is present in the basic domain of GSK-3beta [237]. Increased translocation of GSK-3beta to the nucleus may be involved with cancer progression.
Involvement of the Wnt Signaling Pathway in ERalpha+ Breast Cancers with PIK3CA Mutations.
The PIK3CA gene is mutated in 30-40% of ERalpha+ breast cancers. Gene expression profiling of 249 ER-alpha+ breast tumors revealed that nineteen genes were differently expressed in PIK3CA-mutated tumors in comparison to samples lacking PIK3CA mutations. Interesting PIK3CA mutations were associated with over-expression of several genes critical for the Wnt signaling pathway [WNT5A, transcription factor 7-like 2 (T-cell specific, HMG-box (TCF7L2), msh homeobox 2 (MSX2), tumor necrosis factor receptor superfamily, member 11b (TNFRSF11B)], as well as genes important in the regulation of gene transcription [SEC14-like 2 (SEC14L2), transcription factor AP-2 beta (activating enhancer binding protein 2 beta) (TFAP2B), nuclear receptor interacting protein 3 (NRIP3)] and metal ion binding [cytochrome P450, family 4, subfamily Z, polypeptide 1 (CYP4Z1), cytochrome P450, family 4, subfamily Z, polypeptide 2, pseudogene (CYP4Z2P), solute carrier family 40 (iron-regulated transporter), member 1 (SLC40A1), lactotransferrin (LTF), LIM and calponin homology domains 1 (LIMCH1) [238]. These studies suggest that targeting the Wnt signaling pathway may be an appropriate approach to treat certain ERalpha+ breast cancers containing PIK3CA mutations.
Interactions between PI3K/PTEN/Akt/mTORC1 and Raf/MEK/ERK Pathways in Breast Cancer and Drug Resistance.
The Raf/MEK/ERK pathway is also important in breast cancer drug resistance [239,240]. The Raf/MEK/ERK pathway often interacts with the PI3K/PTEN/Akt/mTORC1 pathway and the two pathways often co regulate many signaling molecules such as p70S6K, eIF-4B, eIF-4E, rpS6 and others. A figure illustrating some of the sites of targeting these pathways is present in Figure 5. A recent component which has been shown to be involved in breast cancer drug resistance is the phosphatase DUSP4 which removes the phosphate from active ERK1,2. Decreased expression of DUSP4 is associated with breast cancer drug resistance and increased expression of active ERK1,2 [241].
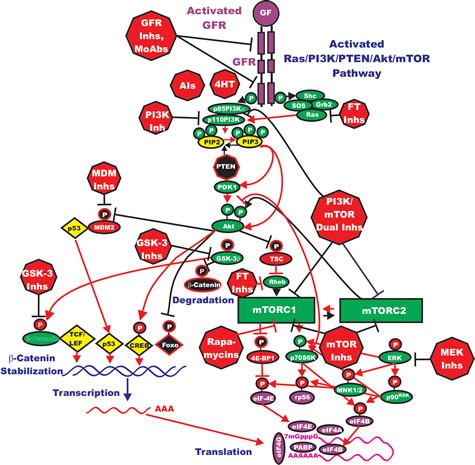
Figure 5: Sites of Targeting the EGFR/PI3K/PTEN/Akt/mTORC Pathway with Small Molecule Membrane-Permeable Inhibitors and Monoclonal Antibodies (MoAbs). The PI3K/PTEN/Akt/mTORC1 pathway is regulated by Ras (indicated in green ovals), as well as various upstream growth factor receptors (indicated in purple) and PTEN indicated in a black rectangle. Sites where various small molecule inhibitors suppress this pathway are indicated by red octagons. The downstream transcription factors regulated by this pathway are indicated in yellow diamond shaped outlines. The Ras/Raf/MEK/ERK pathway also interacts with key proteins involved in protein translation (indicated in green ovals). The two pathways aid in the assembly of the protein translation complex (indicated in purple ovals) responsible for the translation of “weak” mRNAs (indicated in a red line folding over on itself) important in the prevention of apoptosis. Other key proteins inactivated by PI3K/PTEN/Akt/mTORC1 pathway (e.g., TSC, PTEN, beta-catenin, Foxo and 4E-BP1 are indicated by black ovals, diamonds and rectangles. Red arrows indicate activating events in pathways. Blocked black arrows indicating inactivating events in pathways. GF = growth factor, GFR = growth factor receptor.
Other studies have indicated that increased MAPK signaling is a predicator of resistance to successful endocrine therapy of breast cancer patients [242]. This study included 114 women who were ER-alpha+ that were randomly assigned to neoadjuvant letrozole or letrozole plus metronomic cyclophosphamide (frequent low doses of cyclophosphamide). The expression of twenty-four proteins involved in apoptosis, cell survival, hypoxia, angiogenesis and signaling were examined by immunohistochemistry in pretreatment samples. The ages, sizes of the tumors, the nodal status of the tumor, tumor grade, histological type and treatment were also followed. 81% of the patients had a disease response, 43% achieved a complete clinical response and 19% did not respond. This study determined that increased phosphorylated ER-alpha and decreased phosphorylated ERK1,2 were factors associated with complete response to therapy. Phosphorylated and activated ER-alpha was an independent factor for sensitivity to chemoendocrine therapy. In contrast, HIF-alpha and phosphorylated ERK1/2 were independent factors associated with resistance to therapy. These and other results indicating that targeting ERK (e.g., with MEK inhibitors) may be effective in reducing the drug resistance of certain breast cancers.
Interactions Between Raf-1 and Aurora Kinase Signaling in ER+ Breast Cancer.
Recently, it has been shown that there is a Raf-1-mediated involvement of Aurora-A kinase signaling which is important in regulating the balance between EMT and MET in ER+ breast cancer cells. This interaction is also important in chemoresistance and regulating the expression of many genes involved in EMT and MET. The interactions between Raf-1 and Aurora-1 kinase signaling altered the metastatic potential of the cells. Constitutive activation of Raf-1 oncogenic signaling induced HER-2 overexpression and resulted in the development of distant metastases in ERα+ MCF-7/∆Raf-1 breast cancer xenografts. These distant metastases in xenograft models were associated with activation of MET as characterized by reduced expression of EMT-inducer genes (TGFB2, TWIST1 and FOXC1) while overexpression of the BMB7, CXCR7 and EGR genes. Constitutive activation of Raf/MEK/MAPK oncogenic signaling during tumor growth promoted the development of metastatic lesions from primary tumors by activating MET [174].
Further studies by this same group demonstrated that constitutive activation of Raf-1 oncogenic signaling induced stabilization and accumulation of Aurora-A mitotic kinase. This promoted EMT and stemness in ER+ MCF-7/∆Raf-1 cells. The EMT transition was associated by reduced expression of CD24 and ERalpha, while HER2 and CD44 were upregulated. The upregulation of the stemness gene SOX2 was linked to acquisition of multiple stem cell-like properties. Namely the cells displayed an enhanced ability to form mammospheres in vitro and self-renewal in vivo. The aberrant Aurora-A kinase activity induced phosphorylation and nuclear translocation of SMAD5, indicating a novel interplay between Aurora-A and SMAD5 signaling pathways in the development of EMT, stemness and ultimately tumor progression. Pharmacological and molecular inhibition of Aurora-A kinase activity restored the CD24↑ epithelial phenotype that was coupled with ERalpha expression, downregulation of HER2, inhibition of EMT and impaired self-renewal ability and suppression of distant metastases. These findings demonstrated the importance of Aurora-A kinase in the activation of EMT pathway responsible for the development of distant metastases in ERalpha+ breast cancer cells. This study has translational implications because it highlighted the mitotic kinase Aurora-A as a novel, promising therapeutic target to eliminate invasive breast cancer cells and improve the disease-free and overall survival of ERalpha+ breast cancer patients resistant to conventional endocrine therapy [164].
Further studies indicated that cross-talk between ERalpha+ and Raf/MEK/ERK signaling pathways is a key oncogenic axis which is responsible in part for the development of ER-independent growth of breast cancers which were initially ERalpha+ and hormone sensitive. In a metastatic breast cancer xenograft model harboring constitutive activation of Raf-1, the link between aberrant Raf/MEK/ERK signaling and development of endocrine resistance through abrogation of the ERα signaling axis was elucidated. The Aurora-A mitotic kinase was demonstrated to be important in the development of endocrine resistance. Activation of SMAD5 nuclear signaling was detected as well as down-regulation of ERalpha. These results suggest that the development of novel molecular therapies targeting the Aurora-A/SMAD5 oncogenic axis may be beneficial for the selective eradication of endocrine resistant ERalpha- cancer cells from the bulk tumor with benefits for breast cancer patients [243].
Relationship between Raf-1/p53/Aurora Kinase and Centrosome Amplification in Breast Cancer.
Centrosome amplification has been shown to play key roles in the origin of chromosomal instability (CIN) that affects cancer development and progression. The relationships between induction of genotoxic stress, activation of cyclin-A/Cdk2 and Aurora-A and development of centrosome amplification were investigated in MCF-7 breast cancer cell lines harboring a dominant negative (DN) p53 mutation (vMCF-7DNp53). Genotoxic stress was induced in the MCF-7 cells harboring the DN p53 by treatment with hydroxyurea (HU) which induced centrosome amplification. Aurora-A kinase activity was linked with centrosome amplication. The Aurora-A kinase-induced centrosome amplification was determined to be mediated by Cdk2 kinase. Molecular inhibition of Cdk2 activity by SU9516 suppressed Aurora-A centrosomal localization and consequent centrosome amplification. MCF-7 cells harboring constitutively-activated Raf-1 displayed high levels of endogenous cyclin-A. Targeting of Aurora-A by the Aurora kinase inhibitor Alisertib reduced cyclin-A expression. Thus there is a positive feed-back loop between cyclin-A/Cdk2 and Aurora-A pathways in the development of centrosome amplification in breast cancer cells which may be which may provide additional approaches to target drug resistant breast cancer patients [244]. These studies document the key interactions between the Raf/MEK/ERK and Aurora kinase pathways which may be important in regulating breast cancer progression and serve as targets for therapeutic intervention.
Breast Cancer Therapy.
The most commonly utilized treatment for breast cancer is surgical resection with adjuvant chemotherapy, hormone therapy or radiation (National Cancer Institute, 2013). Radiation and chemotherapy are effective in killing or limiting the growth of actively dividing cancer cells through various mechanisms including the production of oxygen free radicals, DNA damage, and subsequent apoptosis [245-251]. Common chemotherapeutic drugs used to treat breast cancer include the anthracyclines, taxanes, 5-fluorouracil, cyclophosphamide, and methotrexate [252]. Doxorubicin, also known as adriamycin, is in the anthracycline class of antibiotic chemotherapeutic drugs. These compounds work by intercalating between adjacent DNA base pairs and inhibiting topoisomerase II, thereby interfering with DNA, RNA, and protein synthesis.
The effects of combining chemotherapeutic drugs have been analyzed by clinical trials.
A phase III, clinical trial with 1491 patients with node-positive, early breast cancer were randomly assigned to adjuvant treatment with docetaxel, doxorubicin, and cyclophosphamide (ACT) or fluorouracil, doxorubicin, and cyclophosphamide (FAC) every 3 weeks for six cycles. The 10 year follow-up analysis of disease-free survival, overall survival, and long-term safety in the clinical trial was reported [253]. In general, breast cancer patients in ACT treatment group had better disease-free survival relative to breast cancer patients in the FAC treatment group. In this study, hormone receptor, and HER2 status and nodal involvement status did not appear to change the disease-free survival between the enhanced survival in ACT treatment group vs. the FAC treatment group. This study also documented that there were a significant percentage of patients who had a decrease in ventricular ejections secondary to anthracycline therapy.
An additional study evaluated the effects of ACT and FAC treatments on node negative breast cancer patients over a 5 year observation period. This study was performed as it was known previously that ACT is superior to FAC when used as adjuvant therapy in women with node-positive breast cancer but the effects of taxanes on breast cancer patients with node cancer were not known [254]. The breast cancer patients in the two treatment groups were examined for 77 months. The disease-free survival was higher in the ACT group (87.8%) than the FAC group (81.8%). Thus the ACT treatment may be appropriate for some high risk, lymph node negative breast cancer patients.
Hormonal Therapy Combined with mTORC1 Blockage.
Another common treatment option for breast cancer consists of a hormonal-based approach [255]. Many early stage breast cancers overexpress various isoforms of the ER, making their growth dependent on estrogen. 4HT is a selective ER modulator. 4HT can block estrogen signaling by competitively binding the ER and antagonizing its proliferative effects [256]. Once a breast cancer has undergone additional molecular changes allowing it to overcome estrogen-dependence, hormonal therapy is no longer effective, however, certain inhibitors can reverse the estrogen-independence (see Figures 3 and 4) [257-261].
Exemestane is an AI and prevents conversion of testosterone into estrogen. The quality of life was observed to be better in hormone-responsive metastatic breast cancer patients treated with exemestane and the mTORC1 blocker everolimus than patients treated with exemestane by itself. In the BOLERO-3 study with herceptin-resistant metastatic breast cancer patients, treatment with everolimus, herceptin and vinorelbine was more effective than treatment of with herceptin and vinorelbine [262]. Vinorelbine (Navelbine) is anti-mitotic drug [261]. It is used to treat non-small cell lung cancer and metastatic breast cancer. Thus suppressing mTORC1 activity could enhance the effectiveness of exemestane and Herceptin in different types of breast cancer patients.
Previously it was thought that hormonal therapy would only be effective in treatment of breast cancer cells which express ER-alpha. Triple negative breast and HER2+ cancers often do not expresses ER-alpha, but some express ER-beta and may be sensitive to hormonal therapy [262]. The expression of hormonal receptors (ER, PR) and HER2 may change with the progression of breast cancer into metastatic cancer. In addition, the combination of hormonal based therapy can be enhanced by the addition of herceptin and mTORC1 blockers in some cases.
Antibody Therapy of Breast Cancer.
The genetically engineered antibody herceptin (trastuzumab) is used to treat HER2+ breast cancers. Combining herceptin with chemotherapy increases survival and response rates [263]. However a significant problem with chemotherapy is cardiotoxicity. Herceptin therapy is also expensive and not always covered by insurance companies [264]. A diagram depicting the targeting of HER2 and other signaling molecules and sites where herceptin-resistance occurs is presented in Figure 6.
Figure 6: Sites of Resistance in the HER2/PI3K/PTEN/Akt/mTORC Pathway and Potential Sites for Intervention with Small Molecule Membrane-Permeable Inhibitors and Monoclonal Antibodies (MoAbs). The HER2/EGFR receptor is indicated in a blue figure. The downstream PI3K/PTEN/Akt/mTORC1 pathway is regulated by Ras (indicated in green ovals), PTEN indicated in a black octagon, activated Src is indicated by a red oval, IRS1 is indicated by an orange oval. Sites where various small molecule inhibitors suppress this pathway are indicated by red octagons. Sites which stimulate proteins involved in autophagy are indicated by green octagons and ovals. The Serine/threonine-protein kinase ULK1 (ULK1) which is regulated by mTORC1 is indicated in a black oval. The downstream transcription factors regulated by this pathway are indicated in yellow diamond shaped outlines. The Ras/Raf/MEK/ERK pathway also interacts with key proteins involved in protein translation (indicated in green ovals). The two pathways aid in the assembly of the protein translation complex (indicated in purple ovals) responsible for the translation of “weak” mRNAs (indicated in a red line folding over on itself) important in the prevention of apoptosis. Other key proteins inactivated by PI3K/PTEN/Akt/mTORC1 pathway (e.g., TSC, PTEN, GSK-3beta, beta-catenin, Foxo and 4E-BP1 are indicated by black ovals, diamonds and rectangles. Red arrows indicate activating events in pathways. Blocked black arrows indicating inactivating events in pathways.
Pertuzumab (Perjeta(®) is a newer antibody developed by Genentech/Roche. Pertuzumab and herceptin binding to different sites on HER2 and the combined administration of pertuzumab and herceptin had synergistic inhibitory effects on HER2+ breast cancers [265].
Pertuzumab prevents the dimerization of HER2 with other EGFR family members (EGFR1, EGFR3, EGFR4) [266]. Pertuzumab prevents the interaction of HER2 with EGFR3 and subsequent activation of the PI3K/PTEN/Akt/mTOR pathway. This has been proposed to be in part responsible to the anti-cancer effects of pertuzumab [267].
Initial studies with just pertuzumab did not reveal impressive results in suppressing cancer growth. However, in the more recent phase III CLEOPATA trial have revealed that combination of pertuzumab with herceptin and docetaxel were promising and significantly improved prolonged progression-free survival for a first-line treatment of HER2+ metastatic breast cancer and importantly no increase in cardio-toxic effects were observed [268]. Pertuzumab received approval by the FDA to treat HER2+ metastatic breast cancer patients in 2012. Pertuzumab in combination with herceptin and a taxol has been approved by the FDA and is being evaluated as a first line treatment option for HER2+ metastatic breast cancer patients who have not been previously treated with anti-HER2 therapy or chemotherapy [269].
Pertuzumab is being further evaluated in the following clinical trials: MARIANNE, for advanced breast cancer, NEOSPHERE, for early breast cancer, TRYPAHENA, for HER2+ stage II/III breast cancer and APHINITY for HER2+ nonmetastatic breast cancer [270].
Trastuzumab emtansine is an antibody-drug modification of herceptin which is now linked with the cytotoxic agent mertansine (DM1). It is abbreviated T-DM1 (trastuzumab-DM1) and also called Kadcyla, ado-trastuzumab emtansine and PRO132365. Mertansine inhibits cell growth by binding to tubulin [271]. Herceptin targets the antibody-drug conjugate to HER+ cells [272]. T-DM1 is more effective than Herceptin in the treatment of HER+ advanced metastatic breast cancer patients. It was evaluated in the EMILIA phase III clinical trial. This trial consisted of 991 cancer patients with unrectable, locally advanced or metastatic HER+ breast cancer which had been previously treated with herceptin and taxanes with breast cancer patients treated with capecitabine (Xeloda) plus lapatinib (Tykerb). Progression free survival was enhanced in breast cancer patients treated with T-DM1. It has been approved by the FDA for the treatment of HER2+ advanced metastatic breast cancer patients. T-DM1 has also been evaluated in other clinical trials such as MARIANNE, which compares the effectiveness of the taxanes (docetaxel or paclitaxel) combined with Herceptin vs T-DM1 by itself vs T-DM1 plus pertuzumab as a first line therapy for HER2+ breast cancers. Patients in this study are HER2+, which have either unresectable locally advanced or metastatic breast cancer. T-DM1 is also being investigated in the TH3RESA clinical trial which is evaluating the effectiveness T-DM1 in HER2+ metastatic breast cancer patients that were previously treated with Herceptin and the EGFR2 kinase inhibitor lapatinib. T-DM1 is also being evaluated in gastric cancers in clinical trials. Tables 1-3 and Supplementary Tables 1-3 list the various clinical trials in breast cancer which are listed on the ClinicalTrial.gov website.
EGFR, PI3K, and mTORC1 inhibitors are being evaluated to treat breast cancer patients. Drug targeting the ER have been developed and many have been evaluated in clinical trials [273]. The ER modulators: 4HT, raloxifene, and lasofoxifene) and AIs (anastrozole, letrozole, and exemestane) have been evaluated extensively [273]. An important pitfall with these drugs is they were thought to be only effective in ER+ cancers. Drugs such as herceptin are more effective on HER2+ breast cancers.
Lapatinib is a small molecule HER2/EGFR1 dual kinase inhibitor that has also shown be effective in inhibiting the growth of HER2+ breast cancers. TNBCs do not express either ER or HER2, thus novel therapies need to be developed for this class of breast cancer. Drugs being evaluated to treat TNBC include poly ADP-ribose polymerase (PARP) inhibitors, vitamin D, and rexinoids, which activate the vitamin D and retinoid X receptors.
Mechanisms of Lapatinib-Resistance in HER2+ Cells: Activation of PI3K and Src.
The PI3K pathway and Src activation may be a mechanism by which some HER2+ cells grow in response to HER2 inhibition by treatment with small molecule HER2 inhibitors such as lapatinib. Lapatinib-resistant HER2+ breast cancer cell lines were generated by culturing the cells in the presence of lapatinib for prolonged periods of time. These cells did not express activated HER2, but they did express the PI3K/PTEN/Akt/mTORC and Raf/MEK/ERK pathways which is believed to be due to activated Src family members. Treatment of the lapatinib-resistant HER2+ cells with Src inhibitors suppressed the PI3K/PTEN/Akt/mTOR pathway as well as growth and restored sensitivity to lapatinib. Treatment of primary HER2+ tumors with lapatinib actually resulted in the expression of Src-family kinases as detected by mRNA analysis. Treatment of HER2+ BT-474 cells with lapatinib and Src inhibitors was more effective in suppressing the growth of these cells in xenograft models than treatment with lapatinib alone. These studies provide a rationale for the treatment of certain HER2+ cells with the combination of lapatinib and Src inhibitors as a means to prevent drug resistance of HER2+ cells [274]. Thus activation of Src family kinases may be a mechanism by which some HER2+ cells become resistant to HER2 inhibitors such as lapatinib.
Cotargeting of mTORC1 and Other Signaling Pathways in Breast Cancer Therapy.
The possibility of targeting mTORC1 and other signaling pathways such as Raf/MEK/ERK, PI3K, IGF-1R to treat breast cancer is also being examined [275]. In some cases, treatment with rapalogs induces the IGF-1R or PDGFR pathways which in turn will activate the Raf/MEK/ERK pathway. This type of approach may also be appropriate to over come the rapamycin-resistance that certain breast cancers have developed. There are clinical trials in progress evaluating the effectiveness of co targeting mTORC1 and pathways such as IGF-1R, PI3K and Raf/MEK/ERK (find out how many trials).
Combinations of Herceptin with pertuzumab, or T-DM1 and mTOR inhibitors added to an aromatase inhibitor are new therapeutic approaches for the treatment of HER2+ breast cancers [276].
Inhibition of mTORC1 in combination with endocrine therapy may be an approach for treatment of metastatic breast cancer patients which are resistant to aromatase in inhibitors. Two clinical trials (TAMRAD and BOLERO-2) have revealed significant effects [277].
Combining Herceptin with PI3K, mTORC1 Inhibitors or Chemotherapy to Improve Breast Cancer Therapy.
Recently it was demonstrated that combining a pan PI3K inhibitor (XL 147) with Herceptin may overcome herceptin-resistance in breast cancer by suppressing HER2/PI3K/FOXO/survivin signaling [278].
Clinical trials have been performed examining the ability to combine lapatinib, herceptin with paclitaxel in first line HER2+ positive breast cancer patients. The dose limiting toxicity observed in the study was diarrhea [279]. Clinical studies examining the combination of the mTORC1 blocker everolimus and either 4HT or exemestane (Aromasin) have been reviewed [280]. Previous clinical studies combining endocrine therapy with rapalogs to treat metastatic hormone receptor+ breast cancer patients yielded variable results. However, two recent independent trials which selected patients refractory to endocrine therapy, demonstrated that combining the rapalog everolimus (Afinitor) with 4HT or combining everolimus with exemestane (Aromasin) was more effective than either endocrine agent alone. The rapalogs may sensitize the normally endocrine therapy resistant patients to endocrine therapy. Likewise additional clinical trials with HER2+ breast cancer patients and the inclusion of PI3K/mTOR inhibitors may improve therapy of the HER2 inhibitors.
Novel Approaches to Treat TNBC Patients.
While therapies for ER+ and HER+ breast cancer patients have improved over the years, the treatment possibilities for TNBC are more limited. A recent review has summarized the clinical results with more novel approaches to treat TNBCs which include inhibitors of enzymes such as PARP and HDAC, kinases such as Jak2 and Src and receptor kinases as well as biological processes such as angiogenesis [281].
Resistance to Therapy.
Rb is a critical protein involved in the regulation of cell cycle progression and is a tumor suppressor. The Rb pathway is involved in sensitivity to tamoxifen. Loss of functional Rb activity is associated with resistance to tamoxifen [282,284]. Thus oncogenes and tumor suppressor genes often are important in the sensitivity to therapy [168,285]. A diagram depicting some of the sites where mutations result in resistance to therapy is presented in Figure 7. Breast cancers can be inherently drug resistance or develop an acquired resistance after exposure to the particular drug [213,252]. Resistance can develop in patients and breast cancer cells when treated with anthracycline chemotherapy drugs, such as doxorubicin, daunorubicin, and epirubicin as well as hormonal based therapies [199,285,286]. Therefore, cancer cells are capable of undergoing calculated changes that confer survival advantages in otherwise nutrient-restricted or toxic environments [287,288]. Thus novel targets and approaches to treat breast cancer are being evaluated [167,289-308]. TGF-beta and Smads are also important targets in breast cancer [309-312]. The anti-diabetes drug metformin is showing some successes in treating breast cancer including Herceptin-resistant breast cancers [313-317] A target of metformin is AMPK which is involved in regulation of components of the PI3K/Akt/mTORC1 and GSK-3 pathways. An additional target of metformin is Stat3 and it has been shown that metformin can inhibit the growth of TNBC [318]. Additional Stats may be important in the progression of breast cancer [319].
Figure 7: Induction of the Ras/Raf/MEK/ERK Pathway after Leukemia Therapy and Subsequent Effects on Cell Cycle Progression, Survival Pathways and Protein Translation. After chemotherapy or radiotherapy there can be activation of signaling pathways which can actually promote cell survival and may lead to therapy resistance. Chemotherapeutic drug treatment (shown in irregular black oval) frequently results in the induction of reactive oxygen species (ROS) (shown in black square). ROS can induce the calcium calmodulin kinase (CaM-K) cascade which can induce Ras which can subsequently activate both the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades (most components of the two cascades which promote signaling are show in green ovals, transcription factors activated by events are shown in yellow diamonds, transcription factors inactivated by events shown in black diamonds). Induction of the Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways can result in the activation of many survival pathways, and regulate both cell cycle progression as well as protein translation. Some of the phosphorylation events mediated by Akt actually serve to inhibit the activities of key proteins such as the Foxo transcription factors and the murine double minute (MDM2) ubiquitin ligase (depicted in black ovals). MDM2 serves to regulate p53 protein stability by ubiquitination, however when it is phosphorylated by Akt it is inactivated. Moreover chemotherapeutic drugs and radiotherapy can induce the ataxia telangiectasia mutated (ATM) protein shown in a black oval, which can in turn phosphorylate and regulate p53. p53 can have complex positive and negative effects on cell growth (depicted in yellow diamond), it can regulate the expression of p21 cyclin dependent kinase inhibitory protein-1 (p21Cip1) which controls cell cycle progression. p53 can also control the transcription of genes such as Puma, Noxa and Bax which are involved in apoptosis (all of these molecules are shown in black ovals, as they tend to inhibit cell cycle progression or promote apoptosis). Both Akt and ERK can phosphorylate p21Cip1 which alters its activity and ability to inhibit cell growth (shown as black phosphorylation sites) and subsequently influence cell growth and therapy resistance. p27Kip1 can also be phosphorylated by both Akt and ERK, however the effects of these phosphorylation events are unclear. Akt phosphorylation of p27Kip1 may result in its cytoplasmic localization, while ERK phosphorylation of p27Kip1 may result in elevated levels of the protein. Hence phosphorylation of proteins by ERK and Akt can have dramatic effects on cell proliferation and contribute to the therapy resistance. Chemo- and radiotherapy also induce breaks in the DNA. In the presence of functional BRCA1 and BRCA2 (indicated in yellow ovals) these breaks may be repaired and normal gene transcription can occur. However when BRCA1 or BRCA2 are mutated, the repair of these genes may not occur and proper gene transcription might not occur. This figure serves as an introduction as to how activation of these pathways by chemotherapy and radiotherapy may contribute to therapeutic resistance.
Drug resistance may involve multiple approaches such as pumping the compound out of the cell, modifying or detoxifying the drug, or activating survival signaling pathways that prevent drug-induced apoptosis [287]. ATP-binding cassette transporters such as multidrug resistance protein (MRP1) and the MDR1 product, P-glycoprotein (Pgp), actively expel chemotherapeutic drugs from the cell [239,240]. Drug transporters pumps have been detected in a large number of untreated breast cancers, and their expression increases upon chemotherapy exposure [320]. Certain (e.g., EGFR inhibitors) targeted therapeutics antagonize drug transporter activity [321]. Certain CDK4 inhibitors antagonize the response of breast cancer cells to antracyclines [322]. Activation of cell survival pathways such as PI3K/PTEN/Akt/mTORC1 pathway can prevent apoptosis in the presence of a drug by altering mitochondrial bioenergetics and inhibiting the release of cytochrome c and result in resistance to autophagy [323-329]. The tumor microenvironment and metabolism may influence the response to targeted therapy as well as drug resistance of breast cancer [330]. Genes and biotargets are being identified which can confer resistance or sensitivity to other targeted therapies such as PARP inhibitors [331,332].
Effects of Metformin on HER2+ Breast CICs which are Resistant to Herceptin.
JIMT-1 is a human breast cancer cell line that was isolated from a pleural metastasis of a patient who was resistant to herceptin. CICs were isolated from this cell line JIMT-1(CIC). JIMT-1(CICs) were shown to be preferentially sensitive to the anti-diabetes drug metformin compared to the non-CIC JIMT(BC) population. Furthermore, inclusion of metformin in tumor xenograft studies increased the ability of Herceptin to suppress the growth of JIMT xenografts. These important studies document the potential usefulness of metformin in the treatment of HER2+ breast cancer patients [298].
Similar studies were performed with a herceptin-sensitive (SKBR3-TzbS) and a derivative Herceptin-resistant (SKBR3-TzbR) breast cancer cell lines. While herceptin inhibited the formation of mammospheres from herceptin-sensitive (SKBR3-TzbS), it did not suppress the formation of mammospheres from herceptin-resistant (SKBR3-TzbR) cells. Metformin would reduce the formation of mamospheres from both cell types but the SKBR3-TzbR cells were more sensitive to metformin. Metformin could be combined with herceptin and synergistically reduced mammosphere formation ability in SKBR-TzbR cells [333].
Induction of EMT can lead to Herceptin Resistance.
Induction of EMT in breast cancer is also another mechanism of resistance to such drugs as herceptin [334]. HER2 is also associated with breast cancer stem cells and their aggressiveness [335]. Inherent herceptin resistance in HER2+ breast cancers is a significant problem and it has been reported to be as high as 70% [334]. In the above study, basal HER2+ breast cancer cells resistant to herceptin were infected with lentiviruses containing small hairpin (sh) RNAs specific for EMT-specific transcription factors. The authors had determined appropriate genes to target by analysis of the public Lawrence Berkeley Laboratory (LBL) Breast Cancer Collection database.
These authors demonstrated that herceptin sensitivity was restricted to the SLUG/SNAIL2-negative subset of luminal/HER2+ cell lines. In contrast, breast cancer lines which expressed SLUG/SNAIL2 were inherently resistance to herceptin. Knockdown of SLUG/SNAIL2 suppressed the CIC phenotype by upregulating the expression of the luminal epithelial marker CD24 in basal/HER2+ cells and these cells were also sensitive to herceptin and underwent the mesenchymal to epithelial transition (MET). This important study also documented a reduction in tumor growth and sensitivity to herceptin when SLUG and SNAIL2 were knocked-down in HER+ cells in tumor xenograft studies. This group has also postulated that other mechanisms may be involved in the induction of herceptin-refractory CICs from more differentiated cells via the activation of intrinsic or microenvironmental paths-to-stemness, also involving EMT [336].
Table 1: Clinical Trials with Single Targeted Agent Treatments (Monotherapy)
Official Trial Name |
Clinical Trial # |
Phase of Trial |
Type of Cancer Patient in Trial |
Status of Trial |
Intervention |
Publications |
Phase II Trial of Akt Inhibitor MK2206 in Patients With Advanced Breast Cancer Who Have Tumors With a PIK3CA Mutation, or an AKT Mutation, and/or PTEN Loss/PTEN Mutation |
NCT01277757 |
II |
Male Breast Cancer (BC), recurrent BC, stage IIIB BC, stage IIIC BC, stage IV BC |
Recruiting |
MK2206 Akt inhibitor |
Not Provided |
Pre-surgical Evaluation of MK-2206 in Patients With Operable Invasive Breast Cancer |
NCT01319539 |
II |
ER- BC, ER+ BC, HER2- BC, HER2+ BC, PR- BC, PR+ BC, stage IB BC, stage II BC, stage IIIA BC, stage IIIB BC, stage IIIC BC, TNBC |
Ongoing, but not recruiting participants |
MK2206 |
Not provided |
A Phase I, Open-Label, Two-Stage Study to Investigate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of the Oral AKT Inhibitor GSK2141795 in Subjects With Solid Tumors or Lymphomas |
NCT00920257 |
I |
Cancer |
Completed |
GSK2141795 Akt Inhibitor |
Not provided |
A Phase II Trial of BKM120 (a PI3K Inhibitor) in Patients With Triple Negative Metastatic Breast Cancer |
NCT01629615 |
II |
BC |
Currently recruiting participants |
BKM120 PI3K Inhibitor |
Not provided |
A Phase I/II, Multi-center, Open-label Study of BEZ235, Administered Orally on a Continuous Daily Dosing Schedule in Adult Patients With Advanced Solid Malignancies Including Patients With Advanced Breast Cancer |
NCT00620594 |
I |
BC, Advanced Solid Tumors, Cowden Syndrome |
Completed |
BEZ235 PI3K/mTOR Inhibitor |
Not provided |
A Phase II Trial of Short-Term Everolimus (RAD001) to Predict Response in Women With Operable Breast Cancer |
NCT00855114 |
II |
BC |
Withdrawn |
Everolimus (mTORC1 blocker) |
Not provided |
Phase II Trial of CCI-779 (Temsirolimus) in Patients With Locally Advanced or Metastatic Breast Cancer |
NCT00376688 |
II |
Male BC, Recurrent BC, Stage IIIA, Stage IIIB, Stage IIIC, Stage IV BC |
Ongoing but not recruiting |
Temsirolimus (mTORC1 blocker) |
Not provided |
A Phase Ib/II Study Investigating the Combination of Everolimus With Trastuzumab and Paclitaxel in Patients With HER2-overexpressing Metastatic Breast Cancer |
NCT00426556 |
I |
Metastatic BC |
Completed |
Everolimus |
Not provided |
A Phase II Trial of RAD001 in Triple Negative Metastatic Breast Cancer |
NCT00827567 |
II |
Breast cancer |
Terminated |
RAD 001 (everolimus) |
|
A Phase Ib Study Investigating the Combination of Everolimus With Trastuzumab and Vinorelbine in Patients With HER2-overexpressing Metastatic Breast Cancer |
NCT00426530 |
I |
Breast Neoplasms, Neoplasm Metastasis |
Completed |
Everolimus |
[340] |
Neoadjuvant Phase II Study Of Everolimus Plus Cisplatin In Triple Negative Breast Cancer Patients With Residual Disease After Standard Chemotherapy |
NCT01931163 |
II |
BC, TNBC |
Recruiting |
Everolimus |
Not provided |
Randomized, Double Blind, Multicentric Phase III Trial Evaluating the Safety and Benefit of Adding Everolimus to Adjuvant Hormone Therapy in Women With Poor Prognosis, ER+ and HER2- Primary Breast Cancer Who Remain Free of Disease After Receiving 3 Years of Adjuvant Hormone Therapy |
NCT01805271 |
III |
ER+, HER2- BC |
Recruiting |
Everolimus |
Not provided |
RADAR: A Randomized Discontinuation Phase II Study to Determine the Efficacy of RAD001 in Breast Cancer Patients With Bone Metastases |
NCT00466102 |
II |
BC |
Ongoing, but not recruiting |
RAD001 |
Not provided |
Phase II Trial of RAD001 Plus Carboplatin in Patients With Triple-Negative Metastatic Breast Cancer |
NCT01127763 |
II |
BC |
Ongoing, but not recruiting |
RAD001 |
Not provided |
A Randomized Phase II Study of Two Different Schedules of RAD001C in Patients With Recurrent/Metastatic Breast Cancer |
NCT00255788 |
II |
BC |
Completed |
Everolimus |
Not provided |
A Randomized Study of mTOR Inhibition by RAD001 (Everolimus) in Invasive Breast Cancer Patients After Pre-operative Use of Anthracycline and/or Taxane-based Chemotherapy |
NCT01088893 |
II |
BC |
Unknown because information has not been verified recently |
Everolimus |
Not provided |
A Multicenter Randomized, Double Blind, Placebo- Controlled, Phase II Study to Compare Endocrine Treatment Alone Versus Endocrine Treatment With Everolimus in Patients With HR+/HER2- Metastatic Breast Cancer and Progression After Previous Treatment With Exemestane and Everolimus |
NCT01773460 |
III |
Metastatic BC |
Recruiting |
Everolimus |
Not provided |
Influence of Exceptional Patient Characteristics on Everolimus Exposure |
NCT01948960 |
IV |
BC |
Recruiting |
Everolimus |
Not provided |
A Phase Ib Study Administering Rapamycin (Sirolimus) With Grapefruit Juice in Patients With Advanced Malignancies |
NCT00375245 |
I |
Tumors, Neoplasm Metastasis |
Completed |
Rapamycin (sirolimus) |
Not provided |
A Phase Ib, Open-label Study to Evaluate RAD001 as Monotherapy Treatment in Chinese Patients With Advanced Pulmonary Neuroendocrine Tumor |
NCT01175096 |
I/II |
Neuroendocrine Tumors, Carcinoid Tumor |
Recruitment unknown because information has not been verified recently |
RAD001 (everolimus, Afinitor®) |
Not provided |
A Phase 1/2, Multi-Center, Open-Label, Dose Finding Study to Assess the Safety, Tolerability, Pharmacokinetics and Preliminary Efficacy of the mTOR Kinase Inhibitor CC-223 Administered Orally to Subjects With Advanced Solid Tumors, Non-Hodgkin Lymphoma or Multiple Myeloma. |
NCT01177397 |
I/II |
Multiple Myeloma, Diffuse Large B-Cell Lymphoma, Glioblastoma Multiforme, Hepatocellular Carcinoma, Non-Small Cell Lung Cancer, Neuroendocrine Tumors of Non-Pancreatic Origin, Hormone Receptor-Positive BC |
Currently recruiting participants |
CC-223 |
Not provided |
A Randomized Study of mTOR Inhibition by RAD001 (Everolimus) in Invasive Breast Cancer Patients After Pre-operative Use of Anthracycline and/or Taxane-based Chemotherapy |
NCT01088893 |
II |
BC |
Recruitment unknown because the information has not been verified recently |
Everolimus |
Not provided |
Clinical Trials with Breast Cancer Patients Regarding Key Pathways Discussed.
In Tables 1-3 and Supplementary Tables 1-3, we present the listing of clinical trials with breast cancer patients derived from the ClinicalTrials.gov data base. We have focused our searches to the key pathways discussed in this review. Table 1 presents the clinical trials with single inhibitors (mono therapy) such as PI3K, PI3K/mTOR, Akt, mTOR inhibitors and mTORC1 blockers (Everolimus, Temsirolimus, Rapamycin). Some trials have presented publication regarding their clinical trials, either in the form of abstracts or scientific manuscripts [333-340]. In Table 2, we list the trials which have used 2 or more agents, usually a combination of inhibitors which target two different signaling pathways. Table 3 lists clinical trials which often combine a signal transduction inhibitor with chemotherapy. Some of these clinical trails have presented publications in the database [341-343]. In Supplementary Table 1, clinical trials which combine hormonal therapy with targeted or chemotherapy are presented. Some of these clinical trails have presented publications in the database [344-350]. Clinical trials using metformin as a single agent are presented in Supplementary Table 2. Finally clinical trails combining metformin and AIs and other drugs are presented in Supplementary Table 3 as well as a publication from one study [351].
Table 2: Combined Targeted Agent Treatments
Official Trial Name |
Clinical Trial # |
Phase of Trial |
Type of Cancer Patient in Trial |
Status of Trial |
Intervention |
Publications |
An Open-Label, Two Part, Phase I/II Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics, and Clinical Activity of the MEK Inhibitor GSK1120212 in Combination With the AKT Inhibitor GSK2110183 in Subjects With Solid Tumors and Multiple Myeloma |
NCT01476137 |
I |
Cancer |
Completed |
GSK1120212, GSK2110183 |
Not provided |
A Phase Ib, Open-label, Multi-center, Dose-escalation and Expansion Study of an Orally Administered Combination of BKM120 Plus MEK162 in Adult Patients With Selected Advanced Solid Tumors |
NCT01363232 |
I |
Advanced Solid Tumors, Selected Solid Tumors |
Ongoing, but not participating |
BKM120 (a pan-class I PI3K inhibitor)+ MEK162 (MEK inhibitor) |
Not provided |
A Phase Ib, Open-label, Multi-center, Dose-escalation Study of Oral BKM120 in Combination With Oral GSK1120212 in Adult Patients With Selected Advanced Solid Tumors. |
NCT01155453 |
I |
Advanced Solid Tumors, Selected Solid Tumors |
Ongoing, but not recruiting |
BKM120, GSK1120212 |
Not provided |
A Phase I/II Trial of an Oral MTOR Protein Kinase Inhibitor (Everolimus, RAD001) in Combination With an Oral EGFR Tyrosine Kinase Inhibitor (Erlotinib, Tarceva™) In Patients With Metastatic Breast Cancer |
NCT00574366 |
I |
BC |
Completed |
Erlotinib (EGFR1 inhibitor), Everolimus (RAD001) |
Not provided |
An Open-label, Multi-center Phase I Dose-finding Study of RAD001 (Everolimus, Afinitor®) in Combination With BEZ235 in Patients With Advanced Solid Tumors |
NCT01482156 |
I |
Advanced Solid Tumors, Metastatic BC, Metastatic Renal Cell Carcinoma |
Ongoing, but not recruiting |
RAD001 + BEZ235, a dual inhibitor of PI3K and mTOR |
Not provided |
Phase I/II Trial of IMC-A12 in Combination With Temsirolimus in Patients With Metastatic Breast Cancer |
NCT00699491 |
I/II |
Male BC, Recurrent BC, Stage IV BC |
Ongoing, but not recruiting |
Cixutumumab (an IGF-1R inhibitor), temsirolimus |
Not provided |
Phase I Clinical Trial of Temsirolimus and Vinorelbine in Advanced Solid Tumors. |
NCT01155258 |
I |
Extensive Stage Small Cell Lung Cancer, Hereditary Paraganglioma, Male BC, Malignant Paraganglioma, Metastatic Gastrointestinal Carcinoid Tumor, Metastatic Pheochromocytoma, Pancreatic Polypeptide Tumor, Recurrent Breast Cancer, Recurrent Cervical Cancer, Recurrent Endometrial Carcinoma, Recurrent Gastrointestinal Carcinoid Tumor, Recurrent Islet Cell Carcinoma Recurrent Neuroendocrine Carcinoma of the Skin, Recurrent Non-small Cell Lung Cancer, Recurrent Ovarian Epithelial Cancer Recurrent Ovarian Germ Cell Tumor, Recurrent Pheochromocytoma, -Recurrent Prostate Cancer, Recurrent Renal Cell Cancer, Recurrent Small Cell Lung Cancer, Recurrent Uterine Sarcoma, Regional Gastrointestinal Carcinoid Tumor, Regional Pheochromocytoma, Stage III Cervical Cancer, Stage III Endometrial Carcinoma, Stage III Neuroendocrine Carcinoma of the Skin, Stage III Ovarian Epithelial Cancer, Stage III Ovarian Germ Cell Tumor, Stage III Prostate Cancer, Stage III Renal Cell Cancer, Stage III Uterine Sarcoma, Stage IIIA BC, Stage IIIA Non-small Cell Lung Cancer, Stage IIIB BC, Stage IIIB Non-small Cell Lung Cancer, Stage IIIC BC, Stage IV BC, Stage IV Endometrial Carcinoma, Stage IV Neuroendocrine Carcinoma of the Skin, Stage IV Non-small Cell Lung Cancer, Stage IV Ovarian Epithelial Cancer, Stage IV Ovarian Germ Cell Tumor, Stage IV Prostate Cancer, Stage IV Renal Cell Cancer, Stage IV Uterine Sarcoma, Stage IVA Cervical Cancer, Stage IVB Cervical Cancer Thyroid Gland Medullary Carcinoma |
Ongoing, but not recruiting |
Temsirolimus, vinorelbine ditartrate (an anti-mitotic vinca alkaloid) |
Not provided |
A Phase I Study of Temsirolimus in Combination With Metformin in Advanced Solid Tumours |
NCT00659568 |
I |
BC, Endometrial Cancer, Kidney Cancer |
Completed |
Metformin hydrochloride, temsirolimus |
Not provided |
An Open-Label, Phase Ib Dose Escalation Trial of Oral Combination Therapy With MSC1936369B and SAR245409 in Subjects With Locally Advanced or Metastatic Solid Tumors |
NCT01390818 |
I |
Locally Advanced Solid Tumor, Metastatic Solid Tumor, BC, Non Small Cell Lung Cancer, Melanoma, Colorectal Cancer |
Ongoing, but not recruiting |
MSC1936369B (a MEK inhibitor) and SAR245409 (a dual PI3K/mTOR inhibitor) |
Not provided |
A Phase I Study of BKM120 and Everolimus in Advanced Solid Malignancies |
NCT01470209 |
I |
Solid Tumors |
Recruiting participants |
BKM120, everolimus |
Not provided |
A Phase Ib, Open-label, Multi-center, Dose-escalation and Expansion Study of an Orally Administered Combination of BEZ235 Plus MEK162 in Adult Patients With Selected Advanced Solid Tumors |
NCT01337765 |
I |
Unspecified Adult Solid Tumor, Protocol Specific Solid Tumor |
Completed |
BEZ235, MEK162 |
Not provided |
A Clinical Trial to Qualify the Growth Factor Signature (GFS) as an Intermediate Biomarker of Response for Development of PI3K-Pathway Inhibitors in Patients With Breast Cancer |
NCT01220570 |
I |
BC |
Completed |
Ridaforolimus (mTORC1 blocker), Dalotuzumab (IGF-1R inhibitor) |
Not provided |
Phase I Parallel Protocol of MK-8669 (Ridaforolimus) + MK-2206 and MK-8669 (Ridaforolimus) + MK-0752 Doublets (MK-MK) in Patients With Advanced Cancer |
NCT01295632 |
I |
Advanced Cancer |
Ongoing, but not recruiting |
Ridaforolimus, MK-0752 (gamma secretase inhibitor, a Notch signaling pathway inhibitor). MK-2206 |
Not provided |
Table 3: Targeted Agents in Combination with Cytotoxic Therapy
Official Trial Name |
Clinical Trial # |
Phase of Trial |
Type of Cancer Patient in Trial |
Status of Trial |
Intervention |
Publications |
Phase Ib Dose Escalation and Biomarker Study of MK-2206 in Combination With Standard Doses of Weekly Paclitaxel in Patients With Locally Advanced or Metastatic Solid Tumors With an Expansion in Advanced Breast Cancer |
NCT01263145 |
I |
Recurrent BC, -Stage IV BC, Unspecified Adult Solid Tumor |
Active, not recruiting |
MK2206, paclitaxel |
Not Provided |
A Phase I-II Study of Triciribine Phosphate Monohydrate (TCN-PM) Plus Sequential Weekly Paclitaxel Followed by Dose-Dense Doxorubicin and Cyclophosphamide in Patients With Metastatic and Locally Advanced Breast Cancer |
NCT01697293 |
I/ II |
Metastatic BC, Carcinoma Breast Stage IV |
Currently recruiting participants |
Triciribine (an Akt inhibitor), paclitaxel, |
Not provided |
A Phase Ib/II Trial of GDC-0941 (a PI3K Inhibitor) in Combination With Cisplatin in Patients With Androgen Receptor Negative Triple Negative Metastatic Breast Cancer |
NCT01918306 |
I/II |
ER- BC, HER- BC, TNBC, Recurrent BC, Stage IV BC |
Currently recruiting participants |
Cisplatin, GDC-0941, a PI3K inhibitor |
Not provided |
*NeoPHOEBE: PI3k Inhibition in Her2 OverExpressing Breast cancEr: A Phase II, Randomized, Parallel Cohort, Two Stage, Double-blind, Placebo-controlled Study of Neoadjuvant Trastuzumab Versus Trastuzumab + BKM120 in Combination With Weekly Paclitaxel in HER2-positive, PIK3CA Wild-type and PIK3CA Mutant Primary Breast Cancer |
NCT01816594 |
II |
HER2+, Newly Diagnosed, Primary BC Neoadjuvant Therapy, Trastuzumab |
Currently recruiting participants |
BKM120, trastuzumab (trastuzumab = herceptin, a anti-HER2 MoAb), |
Not provided |
Neoadjuvant Phase II Trial of Paclitaxel in Combination With BKM120 in Endocrine Resistant Clinical Stage II or III Estrogen Receptor-Positive and HER2 Negative Breast Cancer |
NCT01953445 |
II |
BC |
Not yet open for participant study |
Paclitaxel, BKM120 |
Not provided |
A Phase Ib Trial of Gemcitabine and Cisplatin With RAD001 in Patients With Metastatic Triple Negative Breast Cancer Proceeding to an Open Label Randomized Phase II Trial Comparing Gemcitabine/Cisplatin With or Without RAD001. |
NCT01939418 |
I/II |
Metastatic BC |
Currently recruiting patients |
RAD001 (Afinitor everolimus), gemcitabine (a nucleoside analog), cisplatin |
Not provided |
A Randomized, Double-Blind, Placebo-Controlled Phase II Trial of Weekly Paclitaxel/Bevacizumab +/- Everolimus as First-Line Chemotherapy for Patients With HER2-Negative Metastatic Breast Cancer (MBC) |
NCT00915603 |
II |
Metastatic BC |
Ongoing, but not recruiting |
Everolimus, |
Not provided |
*A Phase II Study Evaluating The Efficacy And Tolerability Of Everolimus (RAD001) In Combination With Trastuzumab And Vinorelbine In The Treatment Of Progressive HER2-Positive Breast Cancer Brain Metastases |
NCT01305941 |
II |
HER2+ BC |
Recruiting |
Everolimus, vinorelbine, trastuzumab (HER2 inhibibitor, MoAb) |
Not provided |
*Phase II Study of Everolimus in Combination With Exemestane Versus Everolimus Alone Versus Capecitabine in the Treatment of Postmenopausal Women With ER+Locally Advanced, Recurrent, or Metastatic Breast Cancer After Recurrence or Progression on Prior Letrozole or Anastrozole. |
NCT01783444 |
II |
BC |
Recruiting |
Capecitabine (a pro-drug, which is converted to 5-flurouracil (5-FU) which inhibits thymidylate synthase, exemestane, Everolimus |
Not provided |
*A Phase III Trials Program Exploring the Integration of Bevacizumab, Everolimus (RAD001), and Lapatinib Into Current Neoadjuvant Chemotherapy Regimes for Primary Breast Cancer |
NCT00567554 |
III |
BC |
Ongoing, but not recruiting |
Epirubicin , cyclophosphamide, docetaxel, bevacizumab, paclitaxel, |
|
Phase I, Open Label, Dose Escalation Study of the Safety, Tolerability, and Pharmacokinetics of the Combination RAD001 Plus Docetaxel in Patients With Metastatic Breast Cancer |
NCT00253318 |
I |
BC |
Terminated |
Docetaxel, RAD001, dexamethasone, a glucocorticoid /steroidial drugs that has anti-inflammatory, immunosuppressant properties |
Not provided |
Phase II, Open Label, Dose Escalation Study of the Safety, Tolerability, and Pharmacokinetics of the Combination RAD001 Plus Docetaxel in Patients With Metastatic Breast Cancer |
NCT01825265 |
II |
BC |
Withdrawn prior to enrollment |
Docetaxel, RAD001, Dexamethasone |
Not provided |
*Phase 1b/2 Single-arm Trial Evaluating the Combination of Lapatinib, Everolimus and Capecitabine for the Treatment of Patients With HER2-positive Metastatic Breast Cancer With CNS Progression After Trastuzumab |
NCT01783756 |
I/II |
Central Nervous System Metastases HER2+ BC, Male BC, Recurrent BC, |
Currently recruiting |
Lapatinib ditosylate, everolimus, capecitabine |
Not provided |
*A Phase Ib/II Trial of LEE011 in Combination With Everolimus (RAD001) and Exemestane in the Treatment of Postmenopausal Women With Estrogen Receptor Positive, Her2- Locally Advanced or Metastatic Breast Cancer |
NCT01857193 |
I/II |
BC |
Currently recruiting |
LEE011, a CDK4/6 inhibitor, exemestane, everolimus |
Not provided |
Phase I/II Study of Weekly Abraxane and RAD001 in Women With Locally Advanced or Metastatic Breast Cancer. A Study of the Cancer Institute of New Jersey Oncology Group (CINJOG) |
NCT00934895 |
I/II |
BC |
Currently recruiting |
Everolimus, abraxane, abraxane is paclitaxel bonded to albumin as a delivery vehicle |
Not provided |
A Phase Ib/II Study of Cisplatin, Paclitaxel, and RAD001 in Patients With Metastatic Breast Cancer |
NCT01031446 |
I/II |
BC |
Completed |
Cisplatin, everolimus, paclitaxel |
Not provided |
Everolimus (RAD001) in Combination With Intravenous Carboplatin in Taxane- and Anthracycline-pretreated Patients With Progressive Metastatic Breast Cancer |
NCT00930475 |
I/II |
BC |
Unknown |
RAD001 in combination with carboplatin. Carboplatin is related to cisplatin, but is modified. |
Not provided |
Open Label Randomized Clinical Trial of Standard Neoadjuvant Chemotherapy (Paclitaxel Followed by FEC) Versus the Combination of Paclitaxel and RAD001 Followed by FEC in Women With Triple Receptor-Negative Breast Cancer (CRAD001C24101) |
NCT00499603 |
II |
BC |
Completed |
Paclitaxel, 5-FU, |
Not provided |
A Phase I Study of Cisplatin, Paclitaxel, and RAD001 Patients With Metastatic Breast Cancer |
NCT00680758 |
I |
BC |
Completed |
Cisplatin, everolimus, paclitaxel |
Not provided |
A Phase II Neo-Adjuvant Study of Cisplatin, Paclitaxel With or Without RAD001 in Patients With Triple-negative Locally Advanced Breast Cancer. |
NCT00930930 |
II |
BC |
Ongoing, but not recruiting |
Cisplatin, everolimus, paclitaxel |
Not provided |
A Phase I Pilot Study of the Oral mTOR Inhibitor RAD001 in Combination With Capecitabine for Metastatic Breast Cancer |
NCT00473005 |
I |
BC |
Terminated due to principle investigator leaving sponsor |
Capecitabine, RAD001 |
Not provided |
Phase I Study of Combined Temosirolimus, Erlotinib and Cisplatin in Advanced Solid Tumors |
NCT00998036 |
I |
TNBC |
Completed |
Temsirolimus, cisplatin, erlotinib |
Not provided |
A Phase Ib Study of Combination of Temsirolimus (Torisel®) and Pegylated Liposomal Doxorubicin (PLD, Doxil®/ Caelyx®) in Advanced or Recurrent Breast, Endometrial and Ovarian Cancer |
NCT00982631 |
I |
Advanced/Recurrent BC, Endometrial Cancer, Ovarian Cancer |
Recruitment is unknown because the information has not been verified recently |
Temsirolimus/PLD |
Not provided |
A Phase I, Open-Label, Multi-center Study to Assess the Safety, Tolerability and Pharmacokinetics of AZD6244 (ARRY-142886) When Given in Combination With Standard Doses of Selected Chemotherapies to Patients With Advanced Solid Tumors |
NCT00600496 |
I |
BC, Colon Cancer, Lung Cancer, Melanoma, Kidney Cancer |
Ongoing, but not recruiting participants |
AZD6244 (MEK inhibitor), dacarbazine, |
[343] |
A Phase I Study of Lenalidomide in Combination With Bevacizumab, Sorafenib, Temsirolimus, or 5-fluorouracil, Leucovorin, Oxaliplatin (FOLFOX) in Patients With Advanced Cancers |
NCT01183663 |
I |
Advanced Cancers |
Ongoing, but not recruiting |
Lenalidomide (related to thalidomide), bevacizumab |
Not provided |
Phase I Study of Pegylated Liposomal Doxorubicin and Temsirolimus in Resistant Solid Malignancies |
NCT00703170 |
I |
Resistant Solid Malignancies |
Completed |
Temsirolimus, pegylated liposomal doxorubicin |
Not provided |
A Phase I Study of the mTOR Inhibitor Rapamycin (Rapamune, Sirolimus) in Combination With Abraxane (Paclitaxel Protein-bound Particles) in Advanced Solid Cancers |
NCT00337376 |
I |
Advanced Solid Cancers |
Terminated |
Rapamune, Abraxane |
Not provided |
A Dose-finding Phase Ib Study Followed by an Open-label, Randomized Phase II Study of BEZ235 Plus Paclitaxel in Patients With HER2 Negative, Inoperable Locally Advanced or Metastatic Breast Cancer |
NCT01495247 |
I/II |
Inoperable Locally Advanced Breast Cancer, Metastatic MBC |
Ongoing, but not recruiting |
BEZ235, paclitaxel |
Not provided |
Phase I Study of Docetaxel and Temsirolimus in Resistant Solid Malignancies |
NCT00703625 |
I |
Resistant Solid Malignancies |
Completed |
Temsirolimus, docetaxel |
Not provided |
Summary
HER2, EGFR1, EGFRvIII and the PI3K/PTEN/Akt/mTORC1 pathway are clearly important not only in the development of breast cancer, but also in the classification and prognosis of breast cancer patients which display mutations or changes in the expression of components of these pathways. Both HER2 and PIK3CA are aberrantly expressed or mutated in many breast cancers. These genes may be altered in breast CICs and successful targeting of them may prove to be effectively therapeutically. HER2 and PI3K control the expression of many downstream genes involved in many different biological processes including protein translation and gene transcription.
ACKNOWLEDGEMENTS
ABD was supported in part by grants from the USAMRMC BC022276, the Intramural RECDA Award and the Italian Association for Cancer Research (AIRC). LC was supported in part by MIUR-PRIN 2009 and Italian MIUR-FIRB Accordi di Programma 2010. MC and GM were supported in part by grants from the Italian “Ministero dell’Istruzione, dell’Università e della Ricerca (Ministry for Education, Universities and Research) – FIRB-MERIT (RBNE08YYBM). MC was also supported in part by a grant to the CNR from the Italian Ministry of Economy and Finance for the Project FaReBio di Qualità. ML was supported in part by a grant from the Italian Ministry of Health, Ricerca Finalizzata Stemness 2008 entitled “Molecular Determinants of Stemness and Mesenchymal Phenotype in Breast Cancer”. AMM was supported by MIUR FIRB 2011 (RBAP11ZJFA_001) grant. RM was supported in part by grants from: CRO, the Italian Association for Cancer Research, (AIRC) (RM PI grant and MCO10016), the Italian Ministry of Health, and Regione Friuli Venezia-Giulia.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013; 63: 11-30.
2. Weiss JR, Moysich KB, Swede H. Epidemiology of male breast cancer. Cancer Epidemiol Biomarkers Prev. 2005; 14: 20-26.
3. Deb S, Jene N, Kconfab Investigators, Fox SB. Genotypic and phenotypic analysis of familial male breast cancer shows under representation of the HER2 and basal subtypes in BRCA-associated carcinomas. BMC Cancer. 2012; 12: 510.
4. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, Bell R, Rosenthal J, Hussey C, Tran T, McClure M, Fry Cet al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994; 266: 66-71.
5. Easton DF, Steele L, Fields P, Ormiston W, Averill D, Daly PA, McManus R, Neuhausen SL, Ford D, Wooster R, Cannon-Albright LA, Stratton MR, Goldgar DE. Cancer risks in two large breast cancer families linked to BRCA2 on chromosome 13q12-13. Am J Hum Genet. 1997; 61: 120-128.
6. Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, Cristiano BE, Pearson RB, Phillips WA. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004; 64: 7678-7681.
7. Bartlett JM, Going JJ, Mallon EA, Watters AD, Reeves JR, Stanton P, Richmond J, Donald B, Ferrier R, Cooke TG. Evaluating HER2 amplification and overexpression in breast cancer. J Pathol. 2001; 195: 422-428.
8. Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, Shak S, Stewart SJ, Press M. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002; 20: 719-726.
9. Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, Monsey J, Goel N, Aronson AB, Li S, Ma CX, Ding L, Mardis ER, Ellis MJ. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013; 3: 224-237.
10. Harris JL, Khanna KK. BRCA1 A-complex fine tunes repair functions of BRCA1. Aging. 2011; 3: 461-563.
11. Martinez-Outschoorn UE, Balliet R, Lin Z, Whitaker-Menezes D, Birbe RC, Bombonati A, Pavlides S, Lamb R, Sneddon S, Howell A, Sotgia F, Lisanti MP. BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: implications for breast cancer prevention with antioxidant therapies. Cell Cycle. 2012; 11: 4402-4413.
12. Shimizu Y, Mullins N, Blanchard Z, Elshamy WM. CA1/p220 loss triggers BRCA1-IRIS overexpression via mRNA stabilization in breast cancer cells. Oncotarget. 2012; 3: 299-313.
13. Chang S, Sharan SK. Epigenetic control of an oncogenic microRNA, miR-155, by BRCA1. Oncotarget. 2012; 3: 5-6.
14. Adams JR, Schachter NF, Liu JC, Zacksenhaus E, Egan SE. Elevated PI3K signaling drives multiple breast cancer subtypes. Oncotarget. 2011; 2: 435-447.
15. Karanja KK, Cox SW, Duxin JP, Stewart SA, Campbell JL. DNA2 and EXO1 in replication-coupled, homology-directed repair and in the interplay between HDR and the FA/BRCA network. Cell Cycle. 2012; 11: 3983-3996.
16. Tarapore P, Hanashiro K, Fukasawa K. Analysis of centrosome localization of BRCA1 and its activity in suppressing centrosomal aster formation. Cell Cycle. 2012; 11:2931-2946.
17. Almeida MI, Reis RM, Calin GA. BRCA1, microRNAs and cancer predisposition: challenging the dogma. Cell Cycle. 2011; 10: 377.
18. Pelletier C, Speed WC, Paranjape T, Keane K, Blitzblau R, Hollestelle A, Safavi K, van den Ouweland A, Zelterman D, Slack FJ, Kidd KK, Weidhaas JB. Rare BRCA1 haplotypes including 3’UTR SNPs associated with breast cancer risk. Cell Cycle. 2011; 10: 90-99.
19. Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990; 250: 1233-1238.
20. Coles C, Condie A, Chetty U, Steel CM, Evans HJ, Prosser J. p53 mutations in breast cancer. Cancer Res. 1992; 52: 5291-5298.
21. Chrisanthar R, Knappskog S, Løkkevik E, Anker G, Ostenstad B, Lundgren S, Risberg T, Mjaaland I, Skjønsberg G, Aas T, Schlichting E, Fjösne HE, Nysted A, Lillehaug JR, Lønning PE. Predictive and prognostic impact of TP53 mutations and MDM2 promoter genotype in primary breast cancer patients treated with epirubicin or paclitaxel. PLoS One. 2011; 6: e19249. doi: 10.1371/journal.pone.0019249.
22. Wang NP, To H, Lee WH, Lee EY. Tumor suppressor activity of RB and p53 genes in human breast carcinoma cells. Oncogene. 1993; 8: 279-288.
23. Azmi AS. Banerjee S. Ali S. Wang Z. Bao B. Beck FW. Maitah M. Choi M. Shields TF. Philip PA. Sarkar FH. Mohammad RM. Network modeling of MDM2 inhibitor-oxaliplatin combination reveals biological synergy in wt-p53 solid tumors. Oncotarget. 2011; 2: 378-392.
24. Knappskog S, Lonning PE. MDM2 promoter SNP285 and SNP309; phylogeny and impact on cancer risk. Oncotarget. 2011; 2: 251-258.
25. Bazarov AV, Lee WJ, Bazarov I, Bosire M, Hines WC, Stankovich B, Chicas A, Lowe SW, Yaswen P. The specific role of pRb in p16 (INK4A) -mediated arrest of normal and malignant human breast cells. Cell Cycle. 2012; 11: 1008-1013.
26. Jiang Z, Jones R, Liu JC, Deng T, Robinson T, Chung PE, Wang S, Herschkowitz JI, Egan SE, Perou CM, Zacksenhaus E. RB1 and p53 at the crossroad of EMT and triple-negative breast cancer. Cell Cycle. 2011; 10: 1563-1570.
27. Puca R, Nardinocchi L, Porru M, Simon AJ, Rechavi G, Leonetti C, Givol D, D’Orazi G. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle. 2011; 10: 1679-1689.
28. Starita LM, Parvin JD. The multiple nuclear functions of BRCA1: transcription, ubiquitination and DNA repair. Current Opin Cell Biol. 2013; 15: 345–350.
29. Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J (April 2000). “BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures”. Genes Dev. 14 (8): 927–39.
30. Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004; 95: 866–871.
31. Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2”. Mol. Cell. 2006; 22: 719–729.
32. Kobayashi H, Ohno S, Sasaki Y, Matsuura M. Hereditary breast and ovarian cancer susceptibility genes. Oncol Rep. 2013; 30: 1019-1029.
33. Arun B. Bayraktar S. Liu DD. Gutierrez Barrera AM. Atchley D. Pusztai L. Litton JK. Valero V. Meric-Bernstam F. Hortobagyi GN. Albarracin C. Response to neoadjuvant systemic therapy for breast cancer in BRCA mutation carriers and noncarriers: a single-institution experience. J Clin Oncol. 2011; 29: 3739-3746.
34. Lee LJ, Alexander B, Schnitt SJ, Comander A, Gallagher B, Garber JE, Tung N. Clinical outcome of triple negative breast cancer in BRCA1 mutation carriers and noncarriers. Cancer. 2012; 117: 3093-3100.
35. Gonzalez-Angulo AM, Timms KM, Liu S, Chen H, Litton JK, Potter J, Lanchbury JS, Stemke-Hale K, Hennessy BT, Arun BK, Hortobagyi GN, Do KA, Mills GB, Meric-Bernstam F. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res. 2011; 17: 1082-1089.
36. Lee EH, Park SK, Park B, Kim SW, Lee MH, Ahn SH, Son BH, Moon BI, Yom CK, Lee CH, Yoon Ds, Noh DY, Choi DH, Chang ED Lee HK, Lee H et al. Effect of BRCA1/2 mutation on short-term and long-term breast cancer survival: a systematic review and meta-analysis. Breast Cancer Res & Treat. 2010; 122: 11-25.
37. Cortesi L, Masini C, Cirilli C, Medici V, Marchi I, Cavazzini G, Pasini G, Turchetti D, Federico M.. Favourable ten-year overall survival in a Caucasian population with high probability of hereditary breast cancer. BMC Cancer. 2010; 10: 90.
38. Lubinski J, Huzarski T, Byrski T, Lynch HT, Cybulski C, Ghadirian P, Stawicka M, Foulkes WD, Kilar E, Kim-Sing C, Neuhausen SL, Armel S, Gilchrist D, Sweet K, Gronwald J, Eisen A, et al. Hereditary Breast Cancer Clinical Study Group. The risk of breast cancer in women with a BRCA1 mutation from North America and Poland. Int J Cancer. 2012; 131: 229-234.
39. Dever SM, Golding SE, Rosenberg E, Adams BR, Idowu MO, Quillin JM, Valerie N, Xu B, Povirk LF, Valerie K. Mutations in the BRCT binding site of BRCA1 result in hyper-recombination. Aging. 2011; 3: 515-532.
40. Dever SM, White ER, Hartman MC, Valerie K. BRCA1-directed, enhanced and aberrant homologous recombination: mechanism and potential treatment strategies. Cell Cycle. 2012; 11: 687-694.
41. Salem AF, Howell A, Sartini M, Sotgia F, Lisanti MP. Downregulation of stromal BRCA1 drives breast cancer tumor growth via upregulation of HIF-1, autophagy and ketone body production. Cell Cycle. 2012; 11: 4167-4173.
42. Menendez JA. Cufi S, Oliveras-Ferraros C, Vellon L, Joven J, Vazquez-Martin A. Gerosuppressant metformin: less is more. Aging. 2011; 3: 348-362.
43. Thompson AM. Molecular pathways: Preclinical models and clinical trials with metformin in breast cancer. Clin Cancer Res. 2014; In Press.
44. Zhong J, Liao J, Liu X, Wang P, Liu J, Hou W, Zhu B, Yao L, Wang J, Li J, Stark JM, Xie Y, Xu X. Protein phosphatase PP6 is required for homology-directed repair of DNA double-strand breaks. Cell Cycle. 2011; 10: 1411-1419.
45. Bonotto M, Gerratana L, Poletto E, Driol P, Giangreco M, Russo S, Minisini AM, Andreetta C, Mansutti M, Pisa FE, Fasola G, Puglisi F. Measures of Outcome in Metastatic Breast Cancer: Insights From a Real-World Scenario. Oncologist. 2014 In Press.
46. Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD 2nd, Abkevich V, Potter J, Pruss D, Glenn P, Li Y, Li J, Gonzalez-Angulo AM, McCune KS, Markman M, Broaddus RR, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010; 28: 3570-3576.
47. Rudel RA, Fenton SE, Ackerman JM, Euling SY, Makris SL. Environmental exposures and mammary gland development: state of the science, public health implications, and research recommendations. Envir Health Pers. 2011; 119: 1053-1061.
48. Stuckey A. Breast cancer: epidemiology and risk factors. Clin Obst & Gyn. 2011; 54: 96-102.
49. St-Hilaire S, Mandal R, Commendador A, Mannel S, Derryberry D. Estrogen receptor positive breast cancers and their association with environmental factors. Int J Health Geograp. 2011; 10: 32.
50. Schmidt CW. IOM issues report on breast cancer and the environment. Environmental Health Perspectives 2012; 120:a60-1.; Hampton T. Breast cancer symposium highlights risk, recurrence, and research trials. JAMA. 2012; 307: 348-350.
51. Fenton SE, Reed C, Newbold RR. Perinatal environmental exposures affect mammary development, function, and cancer risk in adulthood. Ann Rev Pharm & Tox 2012; 52: 455-479.
52. Wei Y, Davis J, Bina WF. Ambient air pollution is associated with the increased incidence of breast cancer in US. Int J Envir Health Res. 2012; 22: 12-21.
53. Cohn BA, Terry MB, Plumb M, Cirillo PM. Exposure to polychlorinated biphenyl (PCB) congeners measured shortly after giving birth and subsequent risk of maternal breast cancer before age 50. Breast Cancer Res & Treat. 2012; 136: 267-275.
54. Alavanja MC, Ross MK, Bonner MR. Increased cancer burden among pesticide applicators and others due to pesticide exposure. [Review] CA: a Cancer Journal for Clinicians 2013; 63:120-42.
55. Naziroglu M, Tokat S, Demirci S. Role of melatonin on electromagnetic radiation-induced oxidative stress and Ca2+ signaling molecular pathways in breast cancer. J Rec & Signal Trans Res. 2012; 32: 290-297.
56. Aquino NB, Sevigny MB, Sabangan J, Louie MC. The role of cadmium and nickel in estrogen receptor signaling and breast cancer: metalloestrogens or not?. J Envir Science & Health Part C Envir Carcino & Ecotox Rev 2012; 30: 189-224.
57. Ashley-Martin J, VanLeeuwen J, Cribb A, Andreou P, Guernsey JR. Breast cancer risk, fungicide exposure and CYP1A1*2A gene-environment interactions in a province-wide case control study in Prince Edward Island, Canada. Int J Envir Res & Public Health 2012; 9: 1846-1858.
58. Boada LD, Zumbado M, Henriquez-Hernandez LA, Almeida-Gonzalez M, Alvarez-Leon EE, Serra-Majem L, Luzardo OP. Complex organochlorine pesticide mixtures as determinant factor for breast cancer risk: a population-based case-control study in the Canary Islands (Spain). Envir Health: A Global Access Sci Source 2012; 11: 28.
59. Smith-Bindman R. Environmental causes of breast cancer and radiation from medical imaging: findings from the Institute of Medicine report. Arc Intern Med. 2012; 172: 1023-1027.
60. Reeves GK, Pirie K, Green J, Bull D, Beral V. Million Women Study Collaborators. Comparison of the effects of genetic and environmental risk factors on in situ and invasive ductal breast cancer. Int JCancer. 2012; 131: 930-937.
61. Olsen A, Christensen J, Outzen M, Olesen PT, Frandsen H, Overvad K, Halkjær J. Pre-diagnostic acrylamide exposure and survival after breast cancer among postmenopausal Danish women. Toxicology 2012; 296:67-72.
62. Shan J, Dsouza SP, Bakhru S, Al-Azwani EK, Ascierto ML, Sastry KS, Bedri S, Kizhakayil D, Aigha II, Malek J, Al-Bozom I, Gehani S, Furtado S, Mathiowitz E, Wang E, Marincola FM, et al. TNRC9 downregulates BRCA1 expression and promotes breast cancer aggressiveness. Cancer Res. 2013; 73: 2840-2849.
63. Stavropoulou AV, Fostira F, Pertesi M, Tsitlaidou M, Voutsinas GE, Triantafyllidou O, Bamias A, Dimopoulos MA, Timotheadou E, Pectasides D, Christodoulou C, Klouvas G, Papadimitriou C, Makatsoris T, et al. Prevalence of BRCA1 mutations in familial and sporadic Greek ovarian cancer cases. PLoS One. 2013; 8(3):e58182. doi: 10.1371/journal.pone.0058182.
64. Kobayashi H, Ohno S, Sasaki Y, Matsuura M. Hereditary breast and ovarian cancer susceptibility genes. Oncol Rep. 2013; 30: 1019-1029.
65. Catteau A, Harris WH, Xu CF, Solomon E. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: correlation with disease characteristics. Oncogene. 1999; 18: 1957-1965.
66. Filippini SE, Vega A. Breast cancer genes: beyond BRCA1 and BRCA2. Front Biosci. 2013; 18: 1358-1372.
67. Bozhanov SS, Angelova SG, Krasteva ME, Markov TL, Christova SL, Gavrilov IG, Georgieva EI. Alterations in p53, BRCA1, ATM, PIK3CA, and HER2 genes and their effect in modifying clinicopathological characteristics and overall survival of Bulgarian patients with breast cancer. J Cancer Res Clin Oncol. 2010; 136: 1657-1669.
68. Ingvarsson S, Sigbjornsdottir BI, Huiping C, Hafsteinsdottir SH, Ragnarsson G, Barkardottir RB, Arason A, Egilsson V, Bergthorsson JT. Mutation analysis of the CHK2 gene in breast carcinoma and other cancers. Breast Cancer Res. 2002; 4: R4.
69. Ren J, Jin F, Yu Z, Zhao L, Wang L, Bai X, Zhao H, Yao W, Mi X, Wang E, Olopade OI, Wei M. MYC overexpression and poor prognosis in sporadic breast cancer with BRCA1 deficiency. Tumour Biol. 2013; 34: 3945-3958.
70. Coles C, Condie A, Chetty U, Steel CM, Evans HJ, Prosser J. p53 mutations in breast cancer. Cancer Res. 1992; 52: 5291-5298.
71. Bišof V, Salihović MP, Narančić NS, Skarić-Jurić T, Jakić-Razumović J, Janićijević B, Rudan P. The TP53 gene polymorphisms and survival of sporadic breast cancer patients. Med Oncol. 2012; 29: 472-478.
72. Zhao L, Wang L, Jin F, Ma W, Ren J, Wen X, He M, Sun M, Tang H, Wei M. Silencing of estrogen receptor alpha (ERalpha) gene by promoter hypermethylation is a frequent event in Chinese women with sporadic breast cancer. Breast Cancer Res Treat. 2009; 117: 253-259.
73. Izadi P, Noruzinia M, Karimipoor M, Karbassian MH, Akbari MT. Promoter hypermethylation of estrogen receptor alpha gene is correlated to estrogen receptor negativity in Iranian patients with sporadic breast cancer. Cell J. 2012; 14: 102-109.
74. Zhao L, Yu Z, Li Y, Wen X, Ma W, Wang L, Ren J, Liu C, He M, Bai X, Sun M, Zheng Z, Mi X, Wang E, Olopade OI, Jin F, et al. Clinical implications of ERβ methylation on sporadic breast cancers in Chinese women. Med Oncol. 2012; 29: 1569-1575.
75. Witton CJ, Reeves JR, Going JJ, Cooke TG, Bartlett JM. Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J Pathol. 2003; 200: 290-297.
76. Tovey SM, Witton CJ, Bartlett JM, Stanton PD, Reeves JR, Cooke TG. Outcome and human epidermal growth factor receptor (HER) 1-4 status in invasive breast carcinomas with proliferation indices evaluated by bromodeoxyuridine labelling. Breast Cancer Res. 2004; 6: R246-251.
77. Naidu R, Yadav M, Nair S, Kutty MK. Expression of c-erbB3 protein in primary breast carcinomas. Br J Cancer. 1998; 78: 1385-1390.
78. Suo Z, Risberg B, Kalsson MG, Willman K, Tierens A, Skovlund E, Nesland JM. EGFR family expression in breast carcinomas. c-erbB-2 and c-erbB-4 receptors have different effects on survival. J Pathol. 2002; 196: 17-25.
79. Suo Z, Berner HS, Risberg B, Karlsson MG, Nesland JM. Estrogen receptor-alpha and C-ERBB-4 expression in breast carcinomas. Virchows Arch. 2001; 439: 62-69.
80. Pawlowski V, Révillion F, Hebbar M, Hornez L, Peyrat JP. Prognostic value of the type I growth factor receptors in a large series of human primary breast cancers quantified with a real-time reverse transcription-polymerase chain reaction assay. Clin Cancer Res. 2000; 6: 4217-4225.
81. Revillion F, Pawlowski V, Lhotellier V, Louchez MM, Peyrat JP. mRNA expression of the type I growth factor receptors in the human breast cancer cells MCF-7: regulation by estradiol and tamoxifen. Anticancer Res. 2003; 23: 1455-1460.
82. Kancha RK, von Bubnoff N, Bartosch N, Peschel C, Engh RA, Duyster J. Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PLoS One. 2011; 6: e26760. doi: 10.1371/journal.pone.0026760. Epub 2011 Oct 28.
83. Rexer BN, Ghosh R, Narasanna A, Estrada MV, Chakrabarty A, Song Y, Engelman JA, Arteaga CL. Human breast cancer cells harboring a gatekeeper T798M mutation in HER2 overexpress EGFR ligands and are sensitive to dual inhibition of EGFR and HER2. Clin Cancer Res. 2013; 19: 5390-5401.
84. Rexer BN, Arteaga CL. Optimal targeting of HER2-PI3K signaling in breast cancer: mechanistic insights and clinical implications. Cancer Res. 2013; 73: 3817-3820.
85. Jackson C, Browell D, Gautrey H, Tyson-Capper A. Clinical significance of HER-2 splice variants in breast cancer progression and drug resistance. Int J Cell Biol. 2013; 2013:973584. doi: 10.1155/2013/973584. Epub 2013 Jul 1.
86. Generali D, Leek R, Fox SB, Moore JW, Taylor C, Chambers P, Harris AL. EGFR mutations in exons 18-21 in sporadic breast cancer. Ann Oncol. 2007; 18: 203-205.
87. Teng YH, Tan WJ, Thike AA, Cheok PY, Tse GM, Wong NS, Yip GW, Bay BH, Tan PH. Mutations in the epidermal growth factor receptor (EGFR) gene in triple negative breast cancer: possible implications for targeted therapy. Breast Cancer Res. 2011; 13:R35.
88. Bhargava R, Gerald WL, Li AR, Pan Q, Lal P, Ladanyi M, Chen B., EGFR gene amplification in breast cancer: correlation with epidermal growth factor receptor mRNA and protein expression and HER-2 status and absence of EGFR-activating mutations. Mod Pathol. 2005; 18: 1027-1033.
89. Reis-Filho JS, Milanezi F, Carvalho S, Simpson PT, Steele D, Savage K, Lambros MB, Pereira EM, Nesland JM, Lakhani SR, Schmitt FC. Metaplastic breast carcinomas exhibit EGFR, but not HER2, gene amplification and overexpression: immunohistochemical and chromogenic in situ hybridization analysis. Breast Cancer Res. 2005; 7: R1028-35.
90. Reis-Filho JS, Pinheiro C, Lambros MB, Milanezi F, Carvalho S, Savage K, Simpson PT, Jones C, Swift S, Mackay A, Reis RM, Hornick JL, Pereira EM, Baltazar F, Fletcher CD, Ashworth A, et al. EGFR amplification and lack of activating mutations in metaplastic breast carcinomas. J Pathol. 2006; 209: 445-453.
91. Tang CK, Gong XQ, Moscatello DK, Wong AJ, Lippman ME. Epidermal growth factor receptor vIII enhances tumorigenicity in human breast cancer. Cancer Research. 2000; 60: 3081-3087.
92. Ge H, Gong X, Tang CK. Evidence of high incidence of EGFRvIII expression and coexpression with EGFR in human invasive breast cancer by laser capture microdissection and immunohistochemical analysis. Int J Cancer. 2002; 98: 357-361.
93. Rae JM, Scheys JO, Clark KM, Chadwick RB, Kiefer MC, Lippman ME. EGFR and EGFRvIII expression in primary breast cancer and cell lines. Breast Cancer Res & Treat. 2004; 87: 87-95.
94. Silva HA, Abraul E, Raimundo D, Dias MF, Marques C, Guerra C, de Oliveira CF, Regateiro FJ. Molecular detection of EGFRvIII-positive cells in the peripheral blood of breast cancer patients. Eur J Cancer. 2006; 42: 2617-2622.
95. Nieto Y, Nawaz F, Jones RB, Shpall EJ, Nawaz S. Prognostic significance of overexpression and phosphorylation of epidermal growth factor receptor (EGFR) and the presence of truncated EGFRvIII in locoregionally advanced breast cancer. J Clin Oncol. 2007; 25: 4405-4413.
96. Zhang Y, Su H, Rahimi M, Tochihara R, Tang C. EGFRvIII-induced estrogen-independence, tamoxifen-resistance phenotype correlates with PgR expression and modulation of apoptotic molecules in breast cancer. Int J Cancer. 2009; 125: 2021-2028.
97. Thakkar JP, Mehta DG. A review of an unfavorable subset of breast cancer: estrogen receptor positive progesterone receptor negative. Oncologist. 2011; 16: 276-285.
98. Yu H, Gong X, Luo X, Han W, Hong G, Singh B, Tang CK. Co-expression of EGFRvIII with ErbB-2 enhances tumorigenesis: EGFRvIII mediated constitutively activated and sustained signaling pathways, whereas EGF-induced a transient effect on EGFR-mediated signaling pathways. Cancer Biol & Ther. 2008; 7: 1818-1828.
99. Rahimi M, George J, Tang C. EGFR variant-mediated invasion by enhanced CXCR4 expression through transcriptional and post-translational mechanisms. Int J Cancer. 2010; 126: 1850-1860.
100. Rahimi M, Toth TA, Tang CK.CXCR4 suppression attenuates EGFRvIII-mediated invasion and induces p38 MAPK-dependent protein trafficking and degradation of EGFRvIII in breast cancer cells. Cancer Letters. 2011; 306: 43-51.
101. Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verástegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001 Mar 1;410(6824):50-6.
102. Mukherjee D, Zhao J. The role of chemokine receptor CXCR4 in breast cancer metastasis. Am J Cancer Res. 2013; 3: 46-57.
103. Del Vecchio CA, Jensen KC, Nitta RT, Shain AH, Giacomini C,. Wong AJ. Epidermal growth factor receptor variant III contributes to cancer stem cell phenotypes in invasive breast carcinoma. Cancer Research. 2012; 72: 2657-2671.
104. Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003; 100: 8933-8938.
105. Yamamoto H, Higasa K, Sakaguchi M, Shien K, Soh J, Ichimura K, Furukawa M, Hashida S, Tsukuda K, Takigawa N, Matsuo K, Kiura K, Miyoshi S, Matsuda F, Toyooka S. Novel germline mutation in the transmembrane domain of HER2 in familial lung adenocarcinomas. J Natl Cancer Inst. 2014; 106: In Press.
106. Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, Wistuba II, Fong KM, Toyooka S, Shimizu N, Fujisawa T, Minna JD, Gazdar AF. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005; 65: 1642-1646.
107. Bose R1, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, Monsey J, Goel N, Aronson AB, Li S, Ma CX, Ding L, Mardis ER, Ellis MJ. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013; 3: 224-237.
108. Milanezi F, Carvalho S, Schmitt FC. EGFR/HER2 in breast cancer: a biological approach for molecular diagnosis and therapy. Expert Rev Mol Diagn. 2008; 8: 417-434. doi: 10.1586/14737159.8.4.417.
109. Hsieh AC, Moasser MM.Targeting HER proteins in cancer therapy and the role of the non-target HER3. Br J Cancer. 2007; 97: 453-457.
110. Mill CP, Zordan MD, Rothenberg SM, Settleman J, Leary JF, Riese DJ 2nd. ErbB2 Is necessary for ErbB4 ligands to stimulate oncogenic activities in models of human breast cancer. Genes Cancer. 2011; 2:792-804.
111. Carpenter G. ErbB-4: mechanism of action and biology. Exp Cell Res. 2003; 284: 66-77.
112. Jones FE. HER4 intracellular domain (4ICD) activity in the developing mammary gland and breast cancer. J Mammary Gland Biol Neoplasia. 2008; 13:247-258.
113. Sundvall M, Iljin K, Kilpinen S, Sara H, Kallioniemi OP, Elenius K. Role of ErbB4 in breast cancer. J Mammary Gland Biol Neoplasia. 2008; 13: 259-268.
114. Thybusch-Bernhardt A, Beckmann S, Juhl H. Comparative analysis of the EGF-receptor family in pancreatic cancer: expression of HER-4 correlates with a favourable tumor stage. Int J Surg Investig. 2001;2:393-400.
115. Saglam O, Shah V, Worsham MJ. Molecular differentiation of early and late stage laryngeal squamous cell carcinoma: an exploratory analysis. Diagn Mol Pathol. 2007; 16: 218-221.
116. Uberall I, Kolar Z, Trojanec R, Berkovcova J, Hajduch M. The status and role of ErbB receptors in human cancer. Exp Mol Pathol. 2008; 84: 79-89.
117. Gilmour LM, Macleod KG, McCaig A, Gullick WJ, Smyth JF, Langdon SP. Expression of erbB-4/HER-4 growth factor receptor isoforms in ovarian cancer. Cancer Res. 2001; 61: 2169-2176.
118. Lee CM, Shrieve DC, Zempolich KA, Lee RJ, Hammond E, Handrahan DL, Gaffney DK. Correlation between human epidermal growth factor receptor family (EGFR, HER2, HER3, HER4), phosphorylated Akt (P-Akt), and clinical outcomes after radiation therapy in carcinoma of the cervix. Gynecol Oncol. 2005; 99: 415-421.
119. Pitfield SE, Bryant I, Penington DJ, Park G, Riese DJ., II. Phosphorylation of ErbB4 on tyrosine 1056 is critical for ErbB4 coupling to inhibition of colony formation by human mammary cell lines. Oncol Res. 2006;16: 179-193.
120. Williams EE, Trout LJ, Gallo RM, Pitfield SE, Bryant I, Penington DJ, Riese DJ 2nd. A constitutively active ErbB4 mutant inhibits drug-resistant colony formation by the DU-145 and PC-3 human prostate tumor cell lines. Cancer Lett. 2003;192:67-74.
121. Mill CP, Gettinger KL, Riese DJ., II Ligand stimulation of ErbB4 and A constitutively-active ErbB4 mutant result in different biological responses in human pancreatic tumor cell lines. Exp Cell Res. 2011; 317: 392-404.
122. Vidal GA, Clark DE, Marrero L, Jones FE. A constitutively active ERBB4/HER4 allele with enhanced transcriptional coactivation and cell-killing activities. Oncogene. 2007; 26: 462-466.
123. Burgess AW. EGFR family: structure physiology signalling and therapeutic targets. Growth Factors. 2008; 26: 263-74
124. Gilbertson RJ, Clifford SC, MacMeekin W, Meekin W, Wright C, Perry RH, Kelly P, Pearson AD, Lunec J. Expression of the ErbB-neuregulin signaling network during human cerebellar development: implications for the biology of medulloblastoma. Cancer Res. 1998; 58: 3932-3941.
125. Abd El-Rehim DM, Pinder SE, Paish CE, Bell JA, Rampaul RS, Blamey RW. Expression and co-expression of the members of the epidermal growth factor receptor (EGFR) family in invasive breast carcinoma. Br J Cancer. 2004; 91: 1532-1542.
126. Tang CK, Concepcion XZ, Milan M, Gong X, Montgomery E, Lippman ME. Ribozyme-mediated down-regulation of ErbB-4 in estrogen receptor–positive breast cancer cells inhibits proliferation both in vitro and in vivo. Cancer Res. 1999; 59: 5315-5322.
127. Wallace MD, Pfefferle AD, Shen L, McNairn AJ, Cerami EG, Fallon BL, Rinaldi VD, Southard TL, Perou CM, Schimenti JC. Comparative oncogenomics implicates the neurofibromin 1 gene (NF1) as a breast cancer driver. Genetics. 2012; 192: 385-396.
128. Marrero D, Peralta R, Valdivia A, De la Mora A, Romero P, Parra M, Mendoza N, Mendoza M, Rodriguez D, Camacho E, Duarte A, Castelazo G, Vanegas E, Garcia I, Vargas C, Arenas D, et al. The neurofibromin 1 type I isoform predominance characterises female population affected by sporadic breast cancer: preliminary data. J Clin Pathol. 2012; 65: 419-423.
129. McLaughlin SK1, Olsen SN, Dake B, De Raedt T, Lim E, Bronson RT, Beroukhim R, Polyak K, Brown M, Kuperwasser C, Cichowski K. The RasGAP gene, RASAL2, is a tumor and metastasis suppressor. Cancer Cell. 2013; 24: 365-378.
130. Li Y, Wei Q, Cao F, Cao X. Expression and promoter methylation of the RASSF1A gene in sporadic breast cancers in Chinese women. Oncol Rep. 2008; 19: 1149-1153.
131. Hollestelle A, Elstrodt F, Nagel JH, Kallemeijn WW, Schutte M. Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Mol Cancer Res. 2007; 5:195-201.
132. Hollestelle A, Nagel JH, Smid M, Lam S, Elstrodt F, Wasielewski M, Ng SS, French PJ, Peeters JK, Rozendaal MJ, Riaz M, Koopman DG, Ten Hagen TL, de Leeuw BH, Zwarthoff EC, Teunisse A, et al. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res Treat. 2010; 121: 53-64.
133. Eckert LB, Repasky GA, Ulkü AS, McFall A, Zhou H, Sartor CI, Der CJ. Involvement of Ras activation in human breast cancer cell signaling, invasion, and anoikis. Cancer Res. 2004; 64: 4585-4592.
134. Loboda A, Nebozhyn M, Klinghoffer R, Frazier J, Chastain M, Arthur W, Roberts B, Zhang T, Chenard M, Haines B, Andersen J, Nagashima K, Paweletz C, Lynch B, Feldman I, Dai H, et al. A gene expression signature of RAS pathway dependence predicts response to PI3K and RAS pathway inhibitors and expands the population of RAS pathway activated tumors. BMC Med Genomics. 2010; 3:26. doi: 10.1186/1755-8794-3-26.
135. Habashy HO, Powe DG, Glaab E, Ball G, Spiteri I, Krasnogor N, Garibaldi JM, Rakha EA, Green AR, Caldas C, Ellis IO. RERG (Ras-like, oestrogen-regulated, growth-inhibitor) expression in breast cancer: a marker of ER-positive luminal-like subtype. Breast Cancer Res Treat. 2011; 128: 315-326.
136. Zhang J, Liu X, Datta A, Govindarajan K, Tam WL, Han J, George J, Wong C, Ramnarayanan K, Phua TY, Leong WY, Chan YS, Palanisamy N, Liu ET, Karuturi KM, Lim B, et al. RCP is a human breast cancer-promoting gene with Ras-activating function. J Clin Invest. 2009; 119: 2171-2183.
137. Datta S, Hoenerhoff MJ, Bommi P, Sainger R, Guo WJ, Dimri M, Band H, Band V, Green JE, Dimri GP. Bmi-1 cooperates with H-Ras to transform human mammary epithelial cells via dysregulation of multiple growth-regulatory pathways. Cancer Res. 2007; 67: 10286-10295.
138. Hoenerhoff MJ, Chu I, Barkan D, Liu ZY, Datta S, Dimri GP, Green JE. BMI1 cooperates with H-RAS to induce an aggressive breast cancer phenotype with brain metastases. Oncogene. 2009; 28: 3022-3032.
139. Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C, Liu W, Stivers D, Baggerly K, Carey M, Lluch A, Monteagudo C, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009; 69: 4116-4124.
140. Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, Horlings H, Berns K, Hung MC, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008; 68: 6084-6091.
141. Cizkova M, Susini A, Vacher S, Cizeron-Clairac G, Andrieu C, Driouch K, Fourme E, Lidereau R, Bièche I. PIK3CA mutation impact on survival in breast cancer patients and in ERα, PR and ERBB2-based subgroups. Breast Cancer Res. 2012; 14: R28.
142. Cizkova M, Dujaric ME, Lehmann-Che J, Scott V, Tembo O, Asselain B, Pierga JY, Marty M, de Cremoux P, Spyratos F, Bieche I. Outcome impact of PIK3CA mutations in HER2-positive breast cancer patients treated with trastuzumab. Br J Cancer. 2013; 108: 1807-1809.
143. Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, Collins C, Pinkel D, Powell B, Mills GB, Gray JW. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet. 1999; 21: 99-102.
144. Aleskandarany MA, Rakha EA, Ahmed MA, Powe DG, Paish EC, Macmillan RD, Ellis IO, Green AR. PIK3CA expression in invasive breast cancer: a biomarker of poor prognosis. Breast Cancer Res Treat. 2010; 122: 45-53.
145. Chakrabarty A, Rexer BN, Wang SE, Cook RS, Engelman JA, Arteaga CL. H1047R phosphatidylinositol 3-kinase mutant enhances HER2-mediated transformation by heregulin production and activation of HER3. Oncogene. 2010; 29: 5193-5203.
146. Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011; 13:224. doi: 10.1186/bcr3039.
147. Young CD, Pfefferle AD, Owens P, Kuba MG, Rexer BN, Balko JM, Sánchez V, Cheng H, Perou CM, Zhao JJ, Cook RS, Arteaga CL. Conditional loss of ErbB3 delays mammary gland hyperplasia induced by mutant PIK3CA without affecting mammary tumor latency, gene expression, or signaling. Cancer Res. 2013; 73: 4075-4085.
148. Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, Bernards R. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007; 12: 395-402.
149. Gonzalez-Angulo AM, Ferrer-Lozano J, Stemke-Hale K, Sahin A, Liu S, Barrera JA, Burgues O, Lluch AM, Chen H, Hortobagyi GN, Mills GB, Meric-Bernstam F. PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol Cancer Ther. 2011; 10: 1093-1101.
150. Hill VK, Ricketts C, Bieche I, Vacher S, Gentle D, Lewis C, Maher ER, Latif F. Genome-wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res. 2011; 71: 2988-2999.
151. Veeck J, Noetzel E, Bektas N, Jost E, Hartmann A, Knuchel R, Dahl E. Promoter hypermethylation of the SFRP2 gene is a high-frequent alteration and tumor-specific epigenetic marker in human breast cancer. Mol Cancer 2008; 7: 83.
152. McCubrey JA, Steelman LS, Abrams SL, Misaghian N, Chappell WH, Basecke J, Nicoletti F, Libra M, Ligresti G, Stivala F, Maksimovic-Ivanic D, Mijatovic S, Montalto G, Cervello M, Laidler P, Bonati A, et al. Targeting the cancer initiating cell: the ultimate target for cancer therapy. Curr Pharm Des. 2012; 18: 1784-1795.
153. McCubrey JA, Abrams SL, Umezawa K, Cocco L, Martelli AM, Franklin RA, Chappell WH, Steelman LS. Novel approaches to target cancer initiating cells-Eliminating the root of the cancer. Adv Enzyme Regul. 2012; 52: 249-264.
154. McCubrey JA, Steelman LS, Chappell WH, Sun L, Davis NM, Abrams SL, Franklin RA, Cocco L, Evangelisti C, Chiarini F, Martelli AM, Libra M, Candido S, Ligresti G, Malaponte G, Mazzarino MC, et al. Advances in Targeting Signal Transduction Pathways. Oncotarget 2012; 3: 1505-1521.
155. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M, Candido S, Malaponte G, Mazzarino MC, Fagone P, Nicoletti F, Bäsecke J, Mijatovic S, Maksimovic-Ivanic D et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Cascade Inhibitors: How Mutations Can Result in Therapy Resistance and How to Overcome Resistance. Oncotarget. 2012; 3: 1068-1111.
156. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, Nicoletti F, Fagone P, Malaponte G, Mazzarino MC, Candido S, Libra M, Bäsecke J, Mijatovic S, Maksimovic-Ivanic D, Milella M et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012; 3: 954-987.
157. Martelli AM, Chiarini F, Evangelisti C, Ognibene A, Bressanin D, Billi AM, Manzoli L, Cappellini A, McCubrey JA. Targeting the liver kinase B1/AMP-dependent kinase pathway as a therapeutic strategy for hematological malignancies. Exp Opin Thera Targets 2012; 16: 729-742.
158. Martelli AM, Chiarini F, Evangelisti C, Cappellini A, Buontempo F, Bressanin D, Fini M, McCubrey JA. Dual inhibitors of phosphatidylinositol 3-kinase and mammalian target of rapamycin: A novel therapeutic strategy for acute leukemia treatment? Oncotarget 2012; 3: 371-394.
159. Bressanin D, Evangelisti C, Ricci F, Tabellini G, Chiarini F, Tazzari PL, Melchionda F, Buontempo F, Pagliaro P, Pession A, McCubrey JA, Martelli AM. Harnessing the PI3K/Akt/mTOR pathway in T-cell acute lymphoblastic leukemia: Eliminating activity by targeting at different levels. Oncotarget. 2012; 3:811-823.
160. McCubrey JA, Davis NM, Abrams SL, Montalto G, Cervello M, Basecke J, Libra M, Nicoletti F, Cocco L, Martelli AM, Steelman LS. Diverse roles of GSK-3: Tumor promoter-tumor suppressor, target in cancer therapy. Adv Biol Regul. 2014; 54: 176-196.
161. Follo MY, Faenza I, Piazzi M, Blalock WL, Manzoli L, McCubrey JA, Cocco L. Nuclear PI-PLCβ1: An appraisal on targets and pathology. Adv Biol Regul. 2014; 54:2-11.
162. Neri LM, Cani A, Martelli AM, Simioni C, Junghanss C, Tabellini G, Ricci F, Tazzari PL, Pagliaro P, McCubrey JA, Capitani S. Targeting the PI3K/Akt/mTOR signaling pathway in B-precursor acute lymphoblastic leukemia and its therapeutic potential. Leukemia. 2014; 28: 739-748.
163. Martelli AM, Lonetti A, Amadori S, McCubrey JA, Chiarini F. Enhancing the effectiveness of nucleoside analogs with mTORC1 blockers to treat acute myeloid leukemia patients. Cell Cycle. 2013; 12: 1815-1816.
164. D’Assoro AB, Liu T, Quatraro C, Amato A, Opyrchal M, Leontovich A, Ikeda Y, Ohmine S, Lingle W, Suman V, Ecsedy J, Iankov I, Di Leonardo D, Ayers-Inglers J, Degnim D, Billadeau D, et al. The mitotic kinase Aurora-A promotes distant metastases by inducing epithelial-to-mesenchymal transition in ERalpha+ breast cancer cells. Oncogene 2014; 33:599-610.
165. Chiarini F, Lonetti A, Teti G, Orsini E, Bressanin D, Cappellini A, Ricci F, Tazzari PL, Ognibene A, Falconi M, Pagliaro P, Iacobucci I, Martinelli G, Amadori S, McCubrey JA, Martelli AM. A combination of temsirolimus, an allosteric mTOR inhibitor, with clofarabine as a new therapeutic option for patients with acute myeloid leukemia. Oncotarget. 2012; 3: 1615-1628.
166. Chappell WH, Lehmann BD, Terrian DM, Abrams SL, Steelman LS, McCubrey JA. p53 expression controls prostate cancer sensitivity to chemotherapy and the MDM2 inhibitor Nutlin-3. Cell Cycle. 2012; 11: 4579-4588.
167. Chappell WH, Abrams SL, Franklin RA, Lahair MM, Montalto G, Cervello M, Martelli AM, Nicoletti F, Candido S, Libra M, Polesel J, Talamini R, Milella M, Tafuri A, Steelman LS, McCubrey JA. Ectopic NGAL expression can alter sensitivity of breast cancer cells to EGFR, Bcl-2, CaM-K inhibitors and the plant natural product berberine. Cell Cycle. 2012; 11: 4447-4461.
168. Chappell WH, Abrams SL, Montalto G, Cervello M, Martelli AM, Candido S, Libra M, Polesel J, Talamini R, Arlinghaus R, Steelman LS, McCubrey JA. Effects of ectopic expression of NGAL on doxorubicin sensitivity. Oncotarget. 2012; 3: 1236-1245.
169. Chappell WH, Abrams SL, Stadelman KM, Lahair MM, Franklin RA, Cocco L, Evangelisti C, Chiarini F, Martelli AM, Steelman LS, McCubrey JA. Increased NGAL (Lnc2) expression after chemotherapeutic drug treatment. Adv Biol Regul. 2013; 53:146-155.
170. Follo MY, Marmiroli S, Faenza I, Fiume R, Ramazzotti G, Martelli AM, Gobbi P, McCubrey JA, Finelli C, Manzoli FA, Cocco L. Nuclear phospholipase C β1 signaling, epigenetics and treatments in MDS. Adv Biol Regul. 2013; 52: 2-6.
171. Dulińska-Litewka J, McCubrey JA, Laidler P. Increased AKT signaling resulting from the loss of androgen responsiveness in prostate cancer. Curr Med Chem. 2013; 20: 144-157.
172. Buontempo F, Chiarini F, Bressanin D, Tabellini G, Melchionda F, Pession A, Fini M, Neri LM, McCubrey JA, Martelli AM. Activity of the selective IκB kinase inhibitor BMS-345541 against T-cell acute lymphoblastic leukemia: involvement of FOXO3a. Cell Cycle. 2012; 11: 2467-2475.
173. Simioni C, Neri LM, Tabellini G, Ricci F, Bressanin D, Chiarini F, Evangelisti C, Cani A, Tazzari PL, Melchionda F, Pagliaro P, Pession A, McCubrey JA, Capitani S, Martelli AM. Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia. 2012; 26: 2336-2342.
174. Leontovich AA, Zhang S, Quatraro C, Iankov I, Veroux PF, Gambino MW, Degnim A, McCubrey J, Ingle J, Galanis E, D’Assoro AB. Raf-1 oncogenic signaling is linked to activation of mesenchymal to epithelial transition pathway in metastatic breast cancer cells. Int J Oncol. 2012; 40: 1858-1864.
175. Britton KM, Eyre R, Harvey IJ, Stemke-Hale K, Browell D, Lennard TW, Meeson AP. Breast cancer, side population cells and ABCG2 expression. Cancer Lett. 2012; 323: 97-105.
176. Korkaya H, Wicha MS. HER2 and breast cancer stem cells: more than meets the eye. Cancer Res. 2013; 73: 3489-3493.
177. Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008; 27:6120-6130..
178. Ithimakin S Day KC Malik F Zen Q Dawsey SJ Bersano-Begey TF, Quraishi AA, Ignatoski KW, Daignault S, Davis A, Hall CL, Palanisamy N, Heath AN, Tawakkol N, Luther TK, Clouthier SG, et al. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: Implications for efficacy of adjuvant trastuzumab. Cancer Res. 2013; 73: 1635–1646.
179. Paik S, Kim C, Wolmark N. HER2 status and benefit from adjuvant trastuzumab in breast cancer. N Engl J Med. 2008; 358: 1409-1411.
180. Korkaya H, Wicha MS. HER-2, notch, and breast cancer stem cells: targeting an axis of evil. Clin Cancer Res. 2009; 15: 1845-1847.
181. Korkaya H, Wicha MS. Selective targeting of cancer stem cells: a new concept in cancer therapeutics. BioDrugs.2007; 21: 299-310.
182. Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C, Liu W, Stivers D, Baggerly K, Carey M, Lluch A, Monteagudo C, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009; 69: 4116-4124.
183. Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, Margolick JB, Liotta LA, Petricoin E 3rd, Zhang Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U S A. 2007; 104: 16158-16163.
184. Santarpia L, Qi Y, Stemke-Hale K, Wang B, Young EJ, Booser DJ, Holmes FA, O’Shaughnessy J, Hellerstedt B, Pippen J, Vidaurre T, Gomez H, Valero V, Hortobagyi GN, Symmans WF, Bottai G, et al. Mutation profiling identifies numerous rare drug targets and distinct mutation patterns in different clinical subtypes of breast cancers. Breast Cancer Res Treat. 2012; 134: 333-343.
185. Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P, Kim SY, Dunn IF, Schinzel AC, Sandy P, Hoersch S, Sheng Q, Gupta PB, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009; 16: 21-32.
186. Wang Y, Zhou D, Phung S, Masri S, Smith D, Chen S. SGK3 is an estrogen-inducible kinase promoting estrogen-mediated survival of breast cancer cells. Mol Endocrinol. 2011; 25: 72-82.
187. Xu J, Wan M, He Q, Bassett RL Jr, Fu X, Chen AC, Shi F, Creighton CJ, Schiff R, Huo L, Liu D. SGK3 is associated with estrogen receptor expression in breast cancer. Breast Cancer Res Treat. 2012; 134: 531-541.
188. Bruhn MA, Pearson RB, Hannan RD, Sheppard KE. AKT-independent PI3-K signaling in cancer - emerging role for SGK3.Cancer Manag Res. 2013; 5: 281-292.
189. Yang SX, Costantino JP, Kim C, Mamounas EP, Nguyen D, Jeong JH, Wolmark N, Kidwell K, Paik S, Swain SM. Akt phosphorylation at Ser473 predicts benefit of paclitaxel chemotherapy in node-positive breast cancer. J Clin Oncol. 2010; 28: 2974-2981.
190. Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, Clouthier SG, Wicha MS. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009; 7(6):e1000121. doi: 10.1371/journal.pbio.1000121.
191. Rexer BN, Shyr Y, Arteaga CL. Phosphatase and tensin homolog deficiency and resistance to trastuzumab and chemotherapy. J Clin Oncol. 2013; 31: 2073-2075.
192. Korkaya H, Wicha MS. Breast cancer stem cells: we’ve got them surrounded. Clin Cancer Res. 2013; 19: 511-513.
193. Shostak K, Chariot A. NF-κB, stem cells and breast cancer: the links get stronger. Breast Cancer Res. 2011; 13: 214.
194. Generali D, Fox SB, Brizzi MP, Allevi G, Bonardi S, Aguggini S, Milani M, Bersiga A, Campo L, Dionisio R, Vergoni F, Giardini R, Dogliotti L, Bottini A, Harris AL, Berruti A. Down-regulation of phosphatidylinositol 3’-kinase/AKT/molecular target of rapamycin metabolic pathway by primary letrozole-based therapy in human breast cancer. Clin Cancer Res. 2008; 14: 2673-2680.
195. Cavazzoni A, Bonelli MA, Fumarola C, La Monica S, Airoud K, Bertoni R, Alfieri RR, Galetti M, Tramonti S, Galvani E, Harris AL, Martin LA, Andreis D, Bottini A, Generali D, Petronini PG. Overcoming acquired resistance to letrozole by targeting the PI3K/AKT/mTOR pathway in breast cancer cell clones. Cancer Lett. 2012; 323: 77-87.
196. Loi S, Haibe-Kains B, Majjaj S, Lallemand F, Durbecq V, Larsimont D, Gonzalez-Angulo AM, Pusztai L, Symmans WF, Bardelli A, Ellis P, Tutt AN, Gillett CE, Hennessy BT, Mills GB, Phillips WA, et al. PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. Proc Natl Acad Sci U S A. 2010; 107: 10208-10213.
197. Creighton CJ, Fu X, Hennessy BT, Casa AJ, Zhang Y, Gonzalez-Angulo AM, Lluch A, Gray JW, Brown PH, Hilsenbeck SG, Osborne CK, Mills GB, Lee AV, Schiff R. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res. 2010; 12: R40. doi: 10.1186/bcr2594.
198. Miller TW, Hennessy BT, González-Angulo AM, Fox EM, Mills GB, Chen H, Higham C, García-Echeverría C, Shyr Y, Arteaga CL. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010; 120: 2406-2413.
199. Steelman LS, Navolanic PM, Sokolosky ML, Taylor JR, Lehmann BD, Chappell WH, Abrams SL, Wong EW, Stadelman KM, Terrian DM, Leslie NR, Martelli AM, Stivala F, Libra M, Franklin RA, McCubrey JA. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity to mTOR inhibitors. Oncogene. 2008; 27: 4086-4095.
200. Miller TW, Pérez-Torres M, Narasanna A, Guix M, Stål O, Pérez-Tenorio G, Gonzalez-Angulo AM, Hennessy BT, Mills GB, Kennedy JP, Lindsley CW, Arteaga CL. Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009; 69: 4192-4201.
201. Gonzalez-Angulo AM, Stemke-Hale K, Palla SL, Carey M, Agarwal R, Meric-Berstam F, Traina TA, Hudis C, Hortobagyi GN, Gerald WL, Mills GB, Hennessy BT. Androgen receptor levels and association with PIK3CA mutations and prognosis in breast cancer. Clin Cancer Res. 2009; 15: 2472-2478.
202. Kaaks R, Lukanova A, Kurzer MS. Obesity, endogenous hormones, and endometrial cancer risk: a synthetic review. Cancer Epidemiol Biomarkers Prev. 2002; 11: 1531-1543.
203. Chan DW, Mak CS, Leung TH, Chan KK, Ngan HY. Down-regulation of Sox7 is associated with aberrant activation of Wnt/b-catenin signaling in endometrial cancer. Oncotarget. 2012; 3: 1546-1556.
204. Casaburi I, Avena P, Lanzino M, Sisci D, Giordano F, Maris P, Catalano S, Morelli C, Ando S. Chenodeoxycholic acid through a TGR5-dependent CREB signaling activation enhances cyclin D1 expression and promotes human endometrial cancer cell proliferation. Cell Cycle. 2012; 11: 2699-2710.
205. Li W, Tai Y, Zhou J, Gu W, Bai Z, Zhou T, Zhong Z, McCue PA, Sang N, Ji JY, Kong B, Jiang J, Wang C. Repression of endometrial tumor growth by targeting SREBP1 and lipogenesis. Cell Cycle. 2012; 11: 2348-2358.
206. Lal A, Panos R, Marjanovic M, Walker M, Fuentes E, Kapp DS, Henner WD, Buturovic LJ, Halks-Miller M. A gene expression profile test for the differential diagnosis of ovarian versus endometrial cancers. Oncotarget. 2012; 3: 212-223.
207. Slomovitz BM, Coleman RL.The PI3K/AKT/mTOR pathway as a therapeutic target in endometrial cancer. Clin Cancer Res. 2012; 18: 5856-5864.
208. Myers AP. New strategies in endometrial cancer: Targeting the PI3K/mTOR Pathway--The devil is in the details. Clin Cancer Res. 2013; 19: 5264-5274.
209. Weigelt B, Warne PH, Lambros MB, Reis-Filho JS, Downward J. PI3K pathway dependencies in endometrioid endometrial cancer cell lines. PI3K pathway dependencies in endometrioid endometrial cancer cell lines. Clin Cancer Res. 2013; 19: 3533-3544.
210. Carvalho S, Milanezi F, Costa JL, Amendoeira I, Schmitt F. PIKing the right isoform: the emergent role of the p110beta subunit in breast cancer. Virchows Arch. 2010; 456: 235-243.
211. Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, Ju Z, Cantley LC, Scherer SE, Liang H, Lu KH, Broaddus RR, et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011; 1: 170-185.
212. Meric-Bernstam F, Chen H, Akcakanat A, Do KA, Lluch A, Hennessy BT, Hortobagyi GN, Mills GB, Gonzalez-Angulo AM. Aberrations in translational regulation are associated with poor prognosis in hormone receptor-positive breast cancer. Breast Cancer Res. 2012; 14: R138.
213. Steelman LS. Navolanic P. Chappell WH. Abrams SL. Wong EW. Martelli AM. Cocco L. Stivala F. Libra M. Nicoletti F. Drobot LB. Franklin RA. McCubrey JA. Involvement of Akt and mTOR in chemotherapeutic- and hormonal-based drug resistance and response to radiation in breast cancer cells. Cell Cycle. 2011; 10: 3003-3015.
214. Ma C, Wang J, Gao Y, Gao TW, Chen G, Bower KA, Odetallah M, Ding M, Ke Z, Luo J. The role of glycogen synthase kinase 3beta in the transformation of epidermal cells. Cancer Res. 2007; 67: 7756-7764.
215. Dong J, Peng J, Zhang H, Mondesire WH, Jian W, Mills GB, Hung MC, Meric-Bernstam F. Role of glycogen synthase kinase 3beta in rapamycin-mediated cell cycle regulation and chemosensitivity. Cancer Res 2005; 65: 1961-1972.
216. Dal Col J, Dolcetti R: GSK-3beta inhibition: at the crossroad between Akt and mTOR constitutive activation to enhance cyclin D1 protein stability in mantle cell lymphoma. Cell Cycle 2008, 7: 2813-2816.
217. Wang Y, Lam JB, Lam KS, Liu J, Lam MC, Hoo RL, Wu D, Cooper GJ, Xu A: Adiponectin modulates the glycogen synthase kinase-3beta/betacatenin signaling pathway and attenuates mammary tumorigenesis of MDA-MB-231 cells in nude mice. Cancer Res. 2006, 66: 11462-11470.
218. Sokolosky M, Chappell WH, Stadelman K, Abrams SL, Davis NM, Steelman LS, McCubrey JA. Inhibition of GSK-3β activity can result in drug and hormonal resistance and alter sensitivity to targeted therapy in MCF-7 breast cancer cells. Cell Cycle. 2014; 2014; 13: 820-833.
219. Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol. 2005; 168: 29-33.
220. Prasad CP, Rath G, Mathur S, Bhatnagar D, Parshad R, Ralhan R. Expression analysis of E-cadherin, slug and GSK3beta in invasive ductal carcinoma of breast. BMC Cancer. 2009; 9, 325. doi:10.1186/1471-2407-9-325.
221. Belletti B, Baldassarre G. New light on p27(kip1) in breast cancer. Cell Cycle. 2012; 11: 3701-3702.
222. Holz MK. The role of S6K1 in ER-positive breast cancer. Cell Cycle. 2012; 11: 3159-3165.
223. Farina AK, Bong YS, Feltes CM, Byers SW. Post-transcriptional regulation of cadherin-11 expression by GSK-3 and beta-catenin in prostate and breast cancer cells. PloS One. 2009; 4(3), e4797. doi:10.1371/journal.pone.0004797.
224. Cesi V, Casciati A, Sesti F, Tanno B, Calabretta B, Raschella G. TGF-induced c-Myb affects the expression of EMT-associated genes and promotes invasion of ER+ breast cancer cells. Cell Cycle. 2011; 10: 4149-4161.
225. Ramsay R. Myb: getting involved in EMT. Cell Cycle. 2012; 11: 433-434,.
226. Sala A. c-MYB and TGF: EMT’s dynamic duo in breast cancer. Cell Cycle. 11(1):17, 2012
227. Basu D, Montone KT, Wang LP, Gimotty PA, Hammond R, Diehl JA, Rustgi AK, Lee JT, Rasanen K, Weinstein GS, Herlyn M. Detecting and targeting mesenchymal-like subpopulations within squamous cell carcinomas. Cell Cycle. 2011; 10: 2008-2016.
228. Plotnikov A, Li Y, Tran TH., Tang W, Palazzo JP, Rui H, Fuchs SY.Oncogene-mediated inhibition of glycogen synthase kinase 3 beta impairs degradation of prolactin receptor. Cancer Res, 2008; 68: 1354-1361.
229. Hartz AM, Madole EK, Miller DS, Bauer B. Estrogen receptor beta signaling through phosphatase and tensin homolog/phosphoinositide 3-kinase/Akt/glycogen synthase kinase 3 down-regulates blood-brain barrier breast cancer resistance protein. J Pharmacology Exp Therapeutics. 2010; 334: 467-476.
230. Takahashi RU, Takeshita F, Honma K, Ono M, Kato K, Ochiya T. Ribophorin II regulates breast tumor initiation and metastasis through the functional suppression of GSK3beta. Sci Rep. 2013; 3:2474. doi: 10.1038/srep02474.
231. Ougolkov AV. Billadeau DD. Targeting GSK-3: A promising approach for cancer therapy? Future Oncology. 2006; 2: 91-100.
232. Ougolkov AV, Bone ND, Fernandez-Zapico ME, Kay NE, Billadeau DD. Inhibition of glycogen synthase kinase-3 activity leads to epigenetic silencing of nuclear factor kappaB target genes and induction of apoptosis in chronic lymphocytic leukemia B cells. Blood 2007; 110: 735-742.
233. Ougolkov AV, Fernandez-Zapico ME, Bilim VN, Smyrk TC, Chari ST, Billadeau DD. Aberrant nuclear accumulation of glycogen synthase kinase-3beta in human pancreatic cancer: Association with kinase activity and tumor dedifferentiation. Clinical Cancer Res. 2006; 12: 5074-5081.
234. Radojicic J. Zaravinos A. Vrekoussis T. Kafousi M. Spandidos DA. Stathopoulos EN. MicroRNA expression analysis in triple-negative (ER, PR and Her2/neu) breast cancer. Cell Cycle. 10: 507-517.
235. Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA. Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle. 2011; 10: 3871-3885.
236. Zhou J, Freeman TA, Ahmad F, Shang X, Mangano E, Gao E, Farber J, Wang Y, Ma XL, Woodgett J, Vagnozzi RJ, Lal H, Force T. GSK-3 is a central regulator of age-related pathologies in mice. J Clin Inv. 2013; 123: 1821-1832.
237. Meares GP, Jope RS. (2007). Resolution of the nuclear localization mechanism of glycogen synthase kinase-3: Functional effects in apoptosis. J Biol Chem. 2007; 282: 16989-17001.
238. Cizkova M, Cizeron-Clairac G, Vacher S, Susini A, Andrieu C, Lidereau R, Bièche I. Gene expression profiling reveals new aspects of PIK3CA mutation in ERalpha-positive breast cancer: major implication of the Wnt signaling pathway. PLoS One. 2010; 5:e15647.
239. Weinstein-Oppenheimer CR, Henríquez-Roldán CF, Davis J, Navolanic PM, Saleh OA, Steelman LS, Franklin RA, Robinson PJ, McMahon M, McCubrey JA. Role of the Raf signal transduction cascade in the in vitro resistance to the anticancer drug doxorubicin. Clin Cancer Res. 2001; 7: 2892-2907.
240. Davis JM, Weinstein-Oppenheimer CR, Steelman LS, Navolanic PN, Hu W, Konopleva M, Blagosklonny MV, McCubrey JA. Raf-1 and Bcl-2 induce distinct and common pathways which contribute to breast cancer drug resistance. Clin Cancer Res. 2003; 9: 1161-1170.
241. Balko JM, Cook RS, Vaught DB, Kuba MG, Miller TW, Bhola NE, Sanders ME, Granja-Ingram NM, Smith JJ, Meszoely IM, Salter J, Dowsett M, Stemke-Hale K, González-Angulo AM, Mills GB, Pinto JA, et al. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat Med. 2012; 18: 1052-1059.
242. Generali D, Buffa FM, Berruti A, Brizzi MP, Campo L, Bonardi S, Bersiga A, Allevi G, Milani M, Aguggini S, Papotti M, Dogliotti L, Bottini A, Harris AL, Fox SB. Phosphorylated ERalpha, HIF-1alpha, and MAPK signaling as predictors of primary endocrine treatment response and resistance in patients with breast cancer. J Clin Oncol. 2009; 27: 227-234.
243. Opyrchal M, Salisbury JL, Zhang S, McCubrey J, Hawse J, Goetz MP, Lomberk GA, Haddad T, Degnim A, Lange C, Ingle JN, Galanis E, D’Assoro AB. Aurora-A Mitotic Kinase Induces Endocrine Resistance through Down-Regulation of ERα Expression in Initially ERα+ Breast Cancer Cells. PLoS One. 2014; 9:e96995
244. Leontovich AA, Salisbury JL, Veroux M, Tallarita T, Billadeau D, McCubrey J, Ingle J, Galanis E, D’Assoro AB. Inhibition of Cdk2 activity decreases Aurora-A kinase centrosomal localization and prevents centrosome amplification in breast cancer cells. Oncol Rep. 2013; 29:1785-1788.
245. Steelman LS, Navolanic P, Franklin RA, Bonati A, Libra M, Stivala F, Martelli AM, McCubrey JA. Combining chemo-, hormonal and targeted therapies to treat breast cancer. Mol Med Reports. 2008; 1: 139-160.
246. Ligresti G, Libra M, Militello L, Clementi S, Donia M, Imbesi R, Malaponte G, Capellani A, McCubrey JA, Stivala F. Breast cancer: molecular basis and therapeutic strategies. Mol Med Reports 2008; 1: 451-458.
247. Murphy JO, Sacchini VS. New innovative techniques in radiotherapy for breast cancer. Minerva Chirurgica. 2013; 68: 139-154.
248. Twelves C, Vahdat LT, Cortes J. Novel treatment options in the management of metastatic breast cancer. Clin Adv Hem & Oncology. 2011; 9: 1-16.
249. Li Q, Jiang Y, Wei W, Yang H, Liu J. Clinical efficacy of including capecitabine in neoadjuvant chemotherapy for breast cancer: a systematic review and meta-analysis of randomized controlled trials. PLoS ONE. 2013; 8:e53403.
250. Corominas-Faja B, Quirantes-Pine R, Oliveras-Ferraros C, Vazquez-Martin A, Cufi S, Martin-Castillo B, Micol V, Joven J, Segura-Carretero A, Menendez JA. Metabolomic fingerprint reveals that metformin impairs one-carbon metabolism in a manner similar to the antifolate class of chemotherapy drugs. Aging. 2012; 4: 480-498.
251. Gabriel EM, Jatoi I. Breast cancer chemoprevention. Exp Rev Anticancer Ther. 2012; 12: 223-228.
252. Navolanic PM, McCubrey JA. Pharmacological breast cancer therapy. Int J Oncol 2005; 27: 1341-1344.
253. Mackey JR, Martin M, Pienkowski T, Rolski J, Guastalla JP, Sami A, Glaspy J, Juhos E, Wardley A, Fornander T, Hainsworth J, Coleman R, Modiano MR, Vinholes J, Pinter T, Rodríguez-Lescure A, et al. Adjuvant docetaxel, doxorubicin, and cyclophosphamide in node-positive breast cancer: 10-year follow-up of the phase 3 randomised BCIRG 001 trial. Lancet Oncol. 2013; 14:72-80.
254. Martín M, Seguí MA, Antón A, Ruiz A, Ramos M, Adrover E, Aranda I, Rodríguez-Lescure A, Grosse R, Calvo L, Barnadas A, Isla D, Martinez del Prado P, Ruiz Borrego M, Zaluski J, Arcusa A, et al. Adjuvant docetaxel for high-risk, node-negative breast cancer. N Engl J Med. 2010; 363: 2200-2210.
255. Bria E, Carlini P, Cuppone F, Vaccaro V, Milella M, Cognetti F. Early recurrence risk: aromatase inhibitors versus tamoxifen. Exp Rev Anticancer Ther. 2010; 10: 1239-1253.
256. Guo WZ, Shiina I, Wang Y, Umeda E, Watanabe C, Uetake S, Ohashi Y, Yamori T, Dan S. Ridaifen-SB8, a novel tamoxifen derivative, induces apoptosis via reactive oxygen species-dependent signaling pathway. Biochem Pharmacology. 2013; 86: 1272-1284.
257. Droog M, Beelen K, Linn S, Zwart W. Tamoxifen resistance: From bench to bedside. Eur J Pharmacology. 2013; 717: 47-57.
258. Hsu PY, Hsu HK, Lan X, Juan L, Yan PS, Labanowska J, Heerema N, Hsiao TH, Chiu YC, Chen Y, Liu Y, Li L, Li R, Thompson IM, Nephew KP, Sharp ZD, Amplification of distant estrogen response elements deregulates target genes associated with tamoxifen resistance in breast cancer. Cancer Cell. 2013; 24: 197-212.
259. Marotta LL, Polyak K. Unraveling the complexity of basal-like breast cancer. Oncotarget. 2011; 2: 588-599.
260. Borgna S, Armellin M, di Gennaro A, Maestro R, Santarosa M. Mesenchymal traits are selected along with stem features in breast cancer cells grown as mammospheres. Cell Cycle. 11: 4242-4251.
261. Marty M, Fumoleau P, Adenis A, Rousseau Y, Merrouche Y, Robinet G, Senac I, Puozzo C. Oral vinorelbine pharmacokinetics and absolute bioavailability study in patients with solid tumors. Ann Oncol 2001; 12: 1643–1649.
262. Pritchard KI. Endocrine therapy: is the first generation of targeted drugs the last? J Intern Med. 2013; 274: 144-152.
263. Nahta R, Esteva1 FJ. HER-2-targeted therapy –Lessons learned and future directions. Clin Cancer Res. 2003; 9: 5078–5048.
264. Fleck L. The costs of caring: Who pays? Who profits? Who panders?”. Hastings Cent Rep 2006; 36: 13–17.
265. McCormack PL. Pertuzumab: a review of its use for first-line combination treatment of HER2-positive metastatic breast cancer. Drugs. 2013; 73: 1491-1502.
266. Kümler I, Tuxen MK, Nielsen DL. A systematic review of dual targeting in HER2-positive breast cancer. Cancer Treat Rev. 2013. pii: S0305-7372(13)00191-6.
267. Metzger-Filho O, Winer EP, Krop I. Pertuzumab: Optimizing HER2 blockade. Clin Cancer Res. 2013; 19: 5552-5556.
268. Baselga J, Cortés J, Kim SB, and the CLEOPATRA Study Group. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012; 366: 109-119.
269. Blumenthal GM, Scher NS, Cortazar P, Chattopadhyay S, Tang S, Song P, Liu Q, Ringgold K, Pilaro AM, Tilley A, King KE, Graham L, Rellahan BL, Weinberg WC, Chi B, Thomas C, et al. First FDA approval of dual anti-HER2 regimen: Pertuzumab in combination with trastuzumab and docetaxel for HER2-positive metastatic breast cancer. Clin Cancer Res. 2013; 19: 4911-4916
270. Keating GM. Pertuzumab: in the first-line treatment of HER2-positive metastatic breast cancer. Drugs. 2012; 72: 353-360.
271. Teicher BA, Doroshow JH. The promise of antibody-drug conjugates. N. Engl. J. Med. 2012; 367: 1847–1848.
272. Verma S, Miles D, Gianni L, Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh DY, Diéras V, Guardino E, Fang L, Lu MW, Olsen S, Blackwell K; EMILIA Study Group. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012; 367: 1783–1791.
273. den Hollander P, Savage MI, Brown PH. Targeted therapy for breast cancer prevention. Front Oncol. 2013; 3:250.
274. Rexer BN, Ham AJ, Rinehart C, Hill S, Granja-Ingram Nde M, González-Angulo AM, Mills GB, Dave B, Chang JC, Liebler DC, Arteaga CL. Phosphoproteomic mass spectrometry profiling links Src family kinases to escape from HER2 tyrosine kinase inhibition. Oncogene. 2011; 30: 4163-4174.
275. Vicier C, Dieci MV, Andre F. New strategies to overcome resistance to mammalian target of rapamycin inhibitors in breast cancer. Curr Opin Oncol. 2013; 25: 587-593.
276. Joo WD, Visintin I, Mor G. Targeted cancer therapy - Are the days of systemic chemotherapy numbered? Maturitas. 2013. pii: S0378-5122(13)00292-2.
277. Sendur MA, Zengin N, Aksoy S, Altundag K. Everolimus: a new hope for patients with breast cancer. Curr Med Res Opin. 2014; 30:75-87.
278. Chakrabarty A, Bhola NE, Sutton C, Ghosh R, Kuba MG, Dave B, Chang JC, Arteaga CL. Trastuzumab-resistant cells rely on a HER2-PI3K-FoxO-Survivin axis and are sensitive to PI3K inhibitors. Cancer Res 2013; 73: 1190–1200.
279. Esteva FJ, Franco SX, Hagan MK, Brewster AM, Somer RA, Williams W, Florance AM, Turner S, Stein S, Perez A. An open-label safety study of lapatinib plus trastuzumab plus paclitaxel in first-line HER2-positive metastatic breast cancer. Oncologist. 2013; 18: 661-666.
280. Vinayak S, Carlson RW. mTOR inhibitors in the treatment of breast cancer. Oncology. 2013; 27: 38-44,
281. Bayraktar S, Glück S. Molecularly targeted therapies for metastatic triple-negative breast cancer. Breast Cancer Res Treat. 2013; 138: 21-35.
282. Lehn S. Ferno M. Jirstrom K. Ryden L. Landberg G. A non-functional retinoblastoma tumor suppressor (RB) pathway in premenopausal breast cancer is associated with resistance to tamoxifen. Cell Cycle. 2011; 10: 956-962.
283. Witkiewicz AK, Knudsen ES. RB pathway and therapeutic sensitivity: new insights in breast cancer and tamoxifen therapy. Cell Cycle 2011; 10: 1525.
284. Borgna S, Armellin M, di Gennaro A, Maestro R, Santarosa M. Mesenchymal traits are selected along with stem features in breast cancer cells grown as mammospheres. Cell Cycle 2012; 11: 4242-4251.
285. Anand A. Anand A. Paclitaxel in doxorubicin-resistant metastatic breast cancer patients. J Nat Cancer Inst. 1995; 87: 1642.
286. Osborne CK, Wiebe VJ, McGuire WL, Ciocca DR, DeGregorio MW. Tamoxifen and the isomers of 4-hydroxytamoxifen in tamoxifen-resistant tumors from breast cancer patients. J Clin Oncol. 1992; 10: 304-310.
287. McCubrey JA, Sokolosky ML, Lehmann BD, Taylor JR, Navolanic PM, Chappell WH, Abrams SL, Stadelman KM, Wong EWT, Misaghian N, Bäsecke J, Libra M, Stivala F, Ligresti G, Tafuri A, Milella M, et al. Alteration of Akt activity increases chemotherapeutic drug and hormonal resistance in breast cancer yet confers an Achilles heel by sensitization to targeted therapy Adv Enzyme Regul. 2008; 48: 113-135.
288. Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Corominas-Faja B, Urruticoechea A, Martin-Castillo B, Menendez JA. Autophagy-related gene 12 (ATG12) is a novel determinant of primary resistance to HER2-targeted therapies: utility of transcriptome analysis of the autophagy interactome to guide breast cancer treatment. Oncotarget. 2012; 3: 1600-1614.
289. Zhang F, Rothermund K, Gangadharan SB, Pommier Y, Prochownik EV, Lazo JS. Phenotypic screening reveals topoisomerase I as a breast cancer stem cell therapeutic target. Oncotarget. 2012; 3: 998-1010.
290. Chung S, Suzuki H, Miyamoto T, Takamatsu N, Tatsuguchi A, Ueda K, Kijima K, Nakamura Y, Matsuo Y. Development of an orally-administrative MELK-targeting inhibitor that suppresses the growth of various types of human cancer. Oncotarget. 2012; 3: 16-40.
291. Perez R, Schally AV, Vidaurre I, Rincon R, Block NL, Rick FG. Antagonists of growth hormone-releasing hormone suppress in vivo tumor growth and gene expression in triple negative breast cancers. Oncotarget. 2012; 3: 988-997.
292. Denmeade SR, Isaacs JT. Engineering enzymatically activated “molecular grenades” for cancer. Oncotarget. 2012; 3: 666-667.
293. Chen L, Aktas BH, Wang Y, He X, Sahoo R, Zhang N, Denoyelle S, Kabha E, Yang H, Freedman RY, Supko JG, Chorev M, Wagner G, Halperin JA. Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget. 2012; 3: 869-881.
294. Chen JJ, Shen HC, Rivera Rosado LA, Zhang Y, Di X, Zhang B. Mislocalization of death receptors correlates with cellular resistance to their cognate ligands in human breast cancer cells. Oncotarget. 2012; 3: 833-842.
295. Katz E, Sims AH, Sproul D, Caldwell H, Dixon MJ, Meehan RR, Harrison DJ. Targeting of Rac GTPases blocks the spread of intact human breast cancer. Oncotarget. 2012; 3: 608-619.
296. Blagosklonny MV. Wt p53 impairs response to chemotherapy: make lemonade to spare normal cells. Oncotarget. 2012; 3: 601-607.
297. Aceto N, Bentires-Alj M. Targeting protein-tyrosine phosphatases in breast cancer. Oncotarget. 2012; 3: 514-515
298. Cufi S, Corominas-Faja B, Vazquez-Martin A, Oliveras-Ferraros C, Dorca J, Bosch-Barrera J, Martin-Castillo B, Menendez JA. Metformin-induced preferential killing of breast cancer initiating CD44+CD24-/low cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget. 2012; 3: 395-398.
299. Wang M, Gartel AL. The suppression of FOXM1 and its targets in breast cancer xenograft tumors by siRNA. Oncotarget. 2011; 2: 1218-1226.
300. Yi T, Elson P, Mitsuhashi M, Jacobs B, Hollovary E, Budd TG, Spiro T, Triozzi P, Borden EC. Phosphatase inhibitor, sodium stibogluconate, in combination with interferon (IFN) alpha 2b: phase I trials to identify pharmacodynamic and clinical effects. Oncotarget. 2011; 2: 1155-1164.
301. Zhou XZ, PinX1: a sought-after major tumor suppressor at human chromosome 8p23. Oncotarget.2011; 2: 810-819.
302. Pasquier E, Ciccolini J, Carre M, Giacometti S, Fanciullino R, Pouchy C, Montero MP, Serdjebi C, Kavallaris M, Andre N. Propranolol potentiates the anti-angiogenic effects and anti-tumor efficacy of chemotherapy agents: implication in breast cancer treatment. Oncotarget. 2011; 2: 797-809.
303. Napoli M, Girardini JE, Piazza S, Del Sal G. Wiring the oncogenic circuitry: Pin1 unleashes mutant p53. Oncotarget. 2011; 2: 654-656.
304. Eckert MA, Yang J. Targeting invadopodia to block breast cancer metastasis. Oncotarget. 2011; 2: 562-568.
305. Baldwin RM, Morettin A, Paris G, Goulet I, Cote J. Alternatively spliced protein arginine methyltransferase 1 isoform PRMT1v2 promotes the survival and invasiveness of breast cancer cells. Cell Cycle. 2012; 11: 4597-4612.
306. Dean JL, McClendon AK, Hickey TE, Butler LM, Tilley WD, Witkiewicz AK, Knudsen ES. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle. 2012; 11: 2756-2761.
307. Minn AJ, Bevilacqua E, Yun J, Rosner MR. Identification of novel metastasis suppressor signaling pathways for breast cancer. Cell Cycle. 2012; 11: 2452-2457.
308. Zou C, Xu Q, Mao F, Li D, Bian C, Liu LZ, Jiang Y, Chen X, Qi Y, Zhang X, Wang X, Sun Q, Kung HF, Lin MC, Dress A, Wardle F, et al.. MiR-145 inhibits tumor angiogenesis and growth by N-RAS and VEGF. Cell Cycle. 2012; 11: 2137-2145.
309. Tarasewicz E, Jeruss JS. Phospho-specific Smad3 signaling: impact on breast oncogenesis. Cell Cycle. 2012; 11: 2443-2451.
310. Fornetti J, Martinson H, Borges V, Schedin P. Emerging targets for the prevention of pregnancy-associated breast cancer. Cell Cycle. 2012; 11: 639-640.
311. Lee EK, Diehl JA. Breast cancer go sMAD: cyclin towards aggressive phenotypes. Cell Cycle. 2011; 10: 187.
312. Liu F. Inhibition of Smad3 activity by cyclin D-CDK4 and cyclin E-CDK2 in breast cancer cells. Cell Cycle. 2011; 10: 186.
313. Liu B, Fan Z, Edgerton SM, Yang X, Lind SE, Thor AD. Potent anti-proliferative effects of metformin on trastuzumab-resistant breast cancer cells via inhibition of erbB2/IGF-1 receptor interactions. Cell Cycle. 2011; 10: 2959-2966.
314. Oliveras-Ferraros C, Cufi S, Vazquez-Martin A, Torres-Garcia VZ, Del Barco S, Martin-Castillo B, Menendez JA. Micro(mi)RNA expression profile of breast cancer epithelial cells treated with the anti-diabetic drug metformin: induction of the tumor suppressor miRNA let-7a and suppression of the TGF-induced oncomiR miRNA-181a. Cell Cycle. 2011; 10: 1144-1151.
315. Menendez JA, Oliveras-Ferraros C, Cufi S, Corominas-Faja B, Joven J, Martin-Castillo B, Vazquez-Martin A. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle. 2011; 11: 2782-2792.
316. Oliveras-Ferraros C, Cufi S, Vazquez-Martin A, Menendez OJ, Bosch-Barrera J, Martin-Castillo B, Joven J, Menendez JA. Metformin rescues cell surface major histocompatibility complex class I (MHC-I) deficiency caused by oncogenic transformation. Cell Cycle. 2012; 11: 865-870.
317. Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, Pavlides S, Tsirigos A, Ertel A, Pestell RG, Broda P, Minetti C, Lisanti MP, Sotgia F. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle. 2011; 10: 4047-4064.
318. Deng XS, Wang S, Deng A, Liu B, Edgerton SM, Lind SE, Wahdan-Alaswad R, Thor AD. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle. 2012; 11: 367-376.
319. Raven JF, Williams V, Wang S, Tremblay ML, Muller WJ, Durbin JE, Koromilas AE. Stat1 is a suppressor of ErbB2/Neu-mediated cellular transformation and mouse mammary gland tumor formation. Cell Cycle. 2011; 10: 794-804.
320. Leonessa F, Clarke R. ATP binding cassette transporters and drug resistance in breast cancer. Endo-Rel Cancer. 2003; 10: 43-73.
321. Lainey E, Sebert M, Thepot S, Scoazec M, Bouteloup C, Leroy C, De Botton S, Galluzzi L, Fenaux P, Kroemer G. Erlotinib antagonizes ABC transporters in acute myeloid leukemia. Cell Cycle. 2012; 11: 4079-4092.
322. McClendon AK, Dean JL, Rivadeneira DB, Yu JE, Reed CA, Gao E, Farber JL, Force T, Koch WJ, Knudsen ES. CDK4/6 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell Cycle. 2012; 11: 2747-2755.
323. Salem AF, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Mitochondrial biogenesis in epithelial cancer cells promotes breast cancer tumor growth and confers autophagy resistance. Cell Cycle. 2012; 11: 4174-4180.
324. Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Ketone body utilization drives tumor growth and metastasis. Cell Cycle. 2012; 11: 3964-3971.
325. Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Lisati MP, Sotgia F. Ketone bodies and two-compartment tumor metabolism: stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle. 2012; 11: 3956-3963.
326. Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, Philp NJ, Pestell RG, Lisanti MP. Mitochondrial metabolism in cancer metastasis: visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle. 2012; 11: 1445-1454.
327. Salem AF, Whitaker-Menezes D, Lin Z, Martinez-Outschoorn UE, Tanowitz HB, Al-Zoubi MS, Howell A, Pestell RG, Sotgia F, Lisanti MP. Two-compartment tumor metabolism: autophagy in the tumor microenvironment and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle. 2012; 11:2545-2556.
328. Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, Sneddon S, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle. 2012; 11: 1108-1117.
329. McGee AM, Douglas DL, Liang Y, Hyder SM, Baines CP. The mitochondrial protein C1qbp promotes cell proliferation, migration and resistance to cell death. Cell Cycle. 2011; 10: 4119-4127.
330. Mitra S, Stemke-Hale K, Mills GB, Claerhout S. Interactions between tumor cells and microenvironment in breast cancer: a new opportunity for targeted therapy. Cancer Sci. 2012; 103: 400-407.
331. Oplustilova L, Wolanin K, Mistrik M, Korinkova G, Simkova D, Bouchal J, Lenobel R, Bartkova J, Lau A, O’Connor MJ, Lukas J, Bartek J. Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle.2012; 11: 3837-3850.
332. Bonofiglio D, Cione E, Vizza D, Perri M, Pingitore A, Qi H, Catalano S, Rovito D, Genchi G, Ando S. Bid as a potential target of apoptotic effects exerted by low doses of PPAR and RXR ligands in breast cancer cells. Cell Cycle. 2011; 10: 2344-2354.
333. Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res Treat. 2011; 126: 355-364.
334. Oliveras-Ferraros C, Corominas-Faja B, Cufi S, Vazquez-Martin A, Martin-Castillo B, Iglesias JM, Lopez-Bonet E, Martin AG, Menendez JA. Epithelial-to-mesenchymal transition (EMT) confers primary resistance to trastuzumab (Herceptin).Cell Cycle. 2012; 11: 4020-4032.
335. Malik F, Korkaya H, Clouthier SG, Wicha MS. Lin28 and HER2: two stem cell regulators conspire to drive aggressive breast cancer. Cell Cycle. 2012. 11: 2780-2781.
336. Martin-Castillo B, Oliveras-Ferraros C, Vazquez-Martin A, Cufí S, Moreno JM, Corominas-Faja B, Urruticoechea A, Martín ÁG, López-Bonet E, Menendez JA. Basal/HER2 breast carcinomas: integrating molecular taxonomy with cancer stem cell dynamics to predict primary resistance to trastuzumab (Herceptin). Cell Cycle. 2013. 15; 12: 225-245.
337. Baselga, J. et al. Improved clinical and cell cycle response with an mTOR inhibitor, daily oral RAD001 (everolimus) plus letrozole versus placebo plus letrozole in a randomized phase II neoadjuvant trial in ER+ breast cancer. J Clin Oncol. 2008; 26: suppl; abstr 530.
338. Jerusalem G.H et al. Multicenter phase I clinical trial of daily and weekly RAD001 in combination with vinorelbine and trastuzumab in patients with HER2-overexpressing metastatic breast cancer with prior resistance to trastuzumab. J Clin Oncol. 2008; 26: suppl; abstr 1057.
339. Tanaka C, O’Reilly T, Kovarik JM, Shand N, Hazell K, Judson I, Raymond E, Zumstein-Mecker S, Stephan C, Boulay A, Hattenberger M, Thomas G, Lane HA. Identifying optimal biologic doses of everolimus (RAD001) in patients with cancer based on the modeling of preclinical and clinical pharmacokinetic and pharmacodynamic data. J Clin Oncol. 2008; 26: 1596-1602.
340. Jerusalem G, Fasolo A, Dieras V, Cardoso F, Bergh J, Vittori L, Zhang Y, Massacesi C, Sahmoud T, Gianni L. Phase I trial of oral mTOR inhibitor everolimus in combination with trastuzumab and vinorelbine in pre-treated patients with HER2-overexpressing metastatic breast cancer. Breast Cancer Res Treat. 2011 Jan;125(2):447-55.
341. von Minckwitz G, Eidtmann H, Rezai M, Fasching PA, Tesch H, Eggemann H, Schrader I, Kittel K, Hanusch C, Kreienberg R, Solbach C, Gerber B, Jackisch C, Kunz G, Blohmer JU, Huober J, et al., Neoadjuvant chemotherapy and bevacizumab for HER2-negative breast cancer. N Engl J Med. 2012; 366: 299-309.
342. Untch M, Loibl S, Bischoff J, Eidtmann H, Kaufmann M, Blohmer JU, Hilfrich J, Strumberg D, Fasching PA, Kreienberg R, Tesch H, Hanusch C, Gerber B, Rezai M, Jackisch C, Huober J, et al., Lapatinib versus trastuzumab in combination with neoadjuvant anthracycline-taxane-based chemotherapy (GeparQuinto, GBG 44): a randomised phase 3 trial. Lancet Oncol. 2012; 13: 135-44.
343. Patel SP, Lazar AJ, Papadopoulos NE, Liu P, Infante JR, Glass MR, Vaughn CS, LoRusso PM, Cohen RB, Davies MA, Kim KB. Clinical responses to selumetinib (AZD6244; ARRY-142886)-based combination therapy stratified by gene mutations in patients with metastatic melanoma. Cancer. 2013; 119: 799-805.
344. Morrow PK, Wulf GM, Ensor J, Booser DJ, Moore JA, Flores PR, Xiong Y, Zhang S, Krop IE, Winer EP, Kindelberger DW, Coviello J, Sahin AA, Nuñez R, Hortobagyi GN, Yu D, Esteva FJ. Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J Clin Oncol. 2011; 29: 3126-3132.
345. Massarweh S, Romond E, Black EP, Van Meter E, Shelton B, Kadamyan-Melkumian V, Stevens M, Elledge R. A phase II study of combined fulvestrant and everolimus in patients with metastatic estrogen receptor (ER)-positive breast cancer after aromatase inhibitor (AI) failure. Breast Cancer Res Treat. 2014; 143: 325-332.
346. Wolff AC, Lazar AA, Bondarenko I, Garin AM, Brincat S, Chow L, Sun Y, Neskovic-Konstantinovic Z, Guimaraes RC, Fumoleau P, Chan A, Hachemi S, Strahs A, Cincotta M, Berkenblit A, Krygowski M, Kang LL, Moore L, Hayes DF. Randomized phase III placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J Clin Oncol. 2013 Jan 10;31(2):195-202. doi: 10.1200/JCO.2011.38.3331. Epub 2012 Dec 10.
347. Patel SP, Lazar AJ, Papadopoulos NE, Liu P, Infante JR, Glass MR, Vaughn CS, LoRusso PM, Cohen RB, Davies MA, Kim KB. Clinical responses to selumetinib (AZD6244; ARRY-142886)-based combination therapy stratified by gene mutations in patients with metastatic melanoma. Cancer. 2013; 119: 799-805.
348. Kim J, Lim W, Kim EK, Kim MK, Paik NS, Jeong SS, Yoon JH, Park CH, Ahn SH, Kim LS, Han S, Nam SJ, Kang HS, Kim SI, Yoo YB, Jeong J, et al. Phase II randomized trial of neoadjuvant metformin plus letrozole versus placebo plus letrozole for estrogen receptor positive postmenopausal breast cancer (METEOR). BMC Cancer. 2014; 14: 170.