
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) comprises 90% of pancreatic cancers, which is one of the most malignant cancers in the world. The 5-year survival rate of patients with PDAC is < 5% and is only < 20% in patients who undergo curative resection [1]. The reason for poor prognosis is mainly due to that most patients are diagnosed at an advanced stage, when the tumors are considered unresectable, and most of them have a chemoradiation resistance profile [2]. There are increasing attentions on the tumor microenvironment (TME), owing to the vital role it plays in PDAC progression [3]. Histologically, the percentage of malignant cells is much lower than the stroma in pancreatic cancer which forms a desmoplastic fibrotic network containing immune cells, endothelial cells, cancer-associated fibroblasts, pericytes, and pancreatic stellate cells (PSCs) [3–6].
PSCs were first discovered in 1982 in mouse pancreas, which stored lipid droplets containing vitamin A [7]. In 1991, PSCs were identified in healthy rats and humans [8]. In 1998, PSC isolation and culture techniques were invented [9], followed by rapid development of study in the mechanisms of PSC function in the progression of PDAC. However, the origin of PSCs is still controversial. Previously, it was believed that PSCs were derived from the neuroectoderm, but recent studies have reported that PSCs could have a mesodermal origin, as PSCs and hepatic stellate cells have similar biological features [10]. We now know that most proliferating PSCs are from resident PSCs in the pancreas; however, some studies have demonstrated a different source: bone marrow [11]. Furthermore, infiltrating monocytes can be converted into PSCs under specific conditions, e.g., following stimulation by monocyte chemoattractant protein-1 (MCP-1) [12]. Therefore, clarifying the actual source of PSCs could be helpful for PSC-targeting treatment, however, there is no evidence to support that PSCs from different sources have different functions or can be identified and targeted specifically in PDAC and it needs to study further in the future.
Generally, there are two main PSC statuses: quiescent and activated. Most quiescent PSCs store fat and retinoids in perinuclear droplets and express glial fibrillary acidic protein (GFAP). And the quiescent phenotype of PSCs could be defined via presence of lipid droplets in the cytoplasm [8, 13, 14]. Quiescent PSCs play a role in maintaining ECM growth and keeping the balance between secreted matrix metalloproteinase (MMP) and tissue inhibitors of MMP (TIMP). Some have argued that quiescent PSCs may also have immune and intermediary functions [14–17]. Quiescent PSCs could be activated in harmful conditions such as stress or inflammation, and the activation was demonstrated to be related to autophagy [18], an alternative metabolic pathway allowing tumor cells to obtain energy [19]. In addition, a recent study found that PSC activation levels could vary according to the microenvironment [20]. The typical features of activation are the expression of α-smooth muscle actin (α-SMA) and a significant quantity expression of ECM proteins (including collagen, laminin, fibronectin) [21]. In PDAC, the activation of PSC contributes to improved tumor cell initiation, development, evasion of immune surveillance, invasion, metastasis, and resistance to chemoradiation [22–24]. Moreover, activated PSCs can produce soluble factors such as transforming growth factor β (TGF-β); interleukin 1, 6, 8 (IL-1, IL-6, IL-8); stromal cell-derived factor-1 (SDF-1); hepatocyte growth factor (HGF), galectin-1 etc. to participate in PDAC cell formation and malignant behaviors. Chronic pancreatitis (CP), which is characterized by massive fibrosis, was demonstrated to be the potential precursors of PDAC [25, 26]. Previous study illustrated that fibrosis is the common feature of PDAC and CP, and PSCs are responsible for the promotion and maintenance of fibrosis [27]. Some PSC-derived cytokines such as TGF-β and fibroblast growth factor (FGF) could contribute to the synthesis of ECM, which leads to the development of fibrosis in CP [28]. Moreover, TGF-β, FGF, platelet-derived growth factor (PDGF) were demonstrated to contribute to the malignant transformation in CP [29], and that could be the reason for carcinogenesis of activated PSC in CP. A recent study argued that TGF-β, FGF and interleukin could activate autophagy in PDAC cells [18], and autophagy dependent alanine secreted by PSCs played a role in pancreatic cancer metabolism. This autophagy process was proven to be stimulated by PDAC cells in turn [30]. Furthermore, PDAC cells can interact with PSCs via the similar soluble factors, stimulating the PSC inflammatory profile, proliferation, and ECM and MMP synthesis, forming a vicious cycle [27, 31] in PDAC (Figure 1).
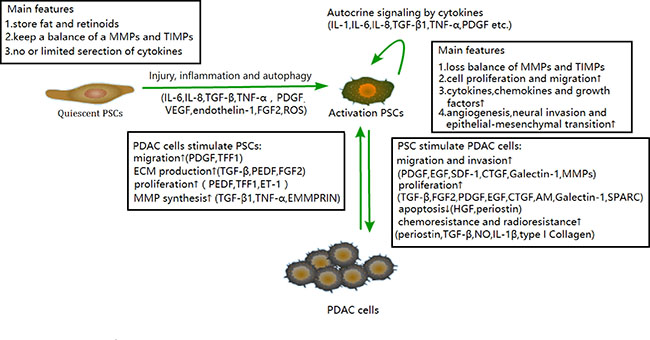
Figure 1: Various features of quiescent PSCs and activated PSCs. Activated PSCs can regulate PDAC cell migration, invasion, proliferation, apoptosis, chemoresistance, and radioresistance. Conversely, PDAC cells can stimulate PSC migration, ECM production, and MMP synthesis, which forms a positive loop based on cytokines secreted by PSCs or tumor cells in TMEs. PDGF, platelet-derived growth factor; EGF, epidermal growth factor; CTGF, connective tissue growth factor; AM, adrenomedullin; SPARC, secreted protein acidic and rich in cysteine; HGF, hepatocyte growth factor; NO, nitric oxide; PEDF, pigment epithelium-derived factor; TFF1, trefoil factor 1; ET-1, endothelin-1; EMMPRIN, extracellular MMP inducer.
The role of PSC-derived TGF-β in PDAC malignant progression
In PDAC, TGF-β is a key signaling mediator involved in stroma–tumor cross-talk, epithelial–mesenchymal transition (EMT), and tumor invasion, in addition, TGF-β was found to be produced by PSCs in the tumor stroma [31–33]. In the classic TGF-β/SMAD signaling pathway, TGF-β combines with its receptor in PSCs, and the activated receptor phosphorylates SMAD2/SMAD3, which combines with SMAD4 and this combination will be translocated to the PSC nucleus [34]. Then, PSCs produce ECM proteins, i.e., collagen, which can promote desmoplastic stroma in PDAC. However, its functions in tumor stroma are various and its characteristics depend on the microenvironment [35, 36]. In non-neoplastic epithelium, TGF-β can be a potential tumor growth suppressor; but in advanced cancer, TGF-β can be a tumor promoter, and this paradoxical switch during tumorigenesis has been linked to EMT process [37, 38]. TGF-β could inhibit stroma related cancer progression, and it could also induce proliferation and migration of pancreatic cancer cells [39, 40]. Here, we discuss the actual mechanistic basis of the novel functions of TGF-β, which could provide ideas for treatment targeting TGF-β or its related signaling pathways in PDAC.
The role of TGF-β in EMT in PDAC
EMT is a developmental process wherein the cell phenotype shifts from epithelial to motile, fibroblast-like morphology [41], which has been widely studied in various field such as tissue fibrosis and cancer progression [42, 43].
Tumor cells undergoing EMT have reduced intercellular adhesion, with decreased E-cadherin expression or β-catenin translocation and increased expression of mesenchymal markers such as vimentin, fibronectin, and N-cadherin [41], which is reversible and is typically believed to promote invasiveness, metastasis, resistance to chemotherapeutic agents, and sometimes, EMT could induce the emergence of cancer stem cell (CSC) phenotypes in cancers including PDAC [36, 41]. As the first identified cytokine that induces EMT in PDAC, much attention has been paid to PSC-derived TGF-β recently. There are several known TGF-β signaling pathways involving EMT in PDAC, dividing into SMAD-dependent and SMAD-independent signals such as the novel NADPH oxidase 4 (NOX4)-derived reactive oxygen species (ROS) signaling [44] and the cross-talk with Ras/Rad/ mitogen-activated sprotein kinase [45]. The repressors of these pathways are promising for treating PDAC.
TGF-β/SMAD pathway is a predominant promoter of EMT [46]. It is traditionally illustrated that TGF-β could be a growth suppressor in early-stage cancer and typically function as a tumor promoter in advanced cancers [36]. Interestingly, David et al. demonstrated that the PDAC cell phenotypes depend on whether SMAD4, a common transactivator protein, is present or absent [47]. The authors clarified that, in SMAD4-positive tumor cells, the SMAD2/3/4 complex can induce the expression of EMT-associated transcription factors such as SNAIL, which contributes to EMT. On the other hand, it can repress another transcription factor, Krüppel-like factor 5 (KLF5), the presence of which can promote SMAD2/3 pathway leading to tumor progression. In the absence of KLF5, the same SMAD2/3 pathway can cause PDAC cell apoptosis, which can repress tumor development. This could explain why loss of SMAD4 expression is generally associated with worse prognosis [48]. However, it remains to clarify whether EMT initiated by other ligands in SMAD4-positive cells would similarly lead to apoptosis. As mentioned above, it is easy to surmise that small-molecule inhibitors KLF5 could become another anti-metastatic agent in PDAC.
MicroRNAs (miRNAs), small, noncoding RNAs that target mRNAs, have become a current cancer research hotspot. There are some valuable findings about miRNAs in relation to EMT in PDAC, and they could regulate the progression of this cancer [49]. Their downstream targets determine the miRNA function. Some miRNAs were involved in regulating TGF-β-induced EMT, e.g., the miR-200 family (miR-200a, miR-200b, miR-200c), which is downregulated in cells that have undergone TGF-β-induced EMT, and EMT was prevented by their artificial expression [50]. Table 1 lists several recent findings on TGF-β-related miRNAs that could play a role in regulating EMT in PDAC.
Table 1: MicroRNAs involved in TGF-β-induced EMT in PDAC
MicroRNA |
Target |
Functional role |
|---|---|---|
miR-367 [139] |
SMAD7 |
High level miR-367 downregulated SMAD7 expression and involved with poor progronsis in PDAC; miR-367 promoted EMT by increasing the expression of TGF-β |
miR-10b [140] |
Tat-interacting protein 30 (TIP30) |
MiR-10b overexpression accelerated PCC proliferation and tumor growth; miR-10b enhanced the stimulatory effects of EGF and TGF-β on cell migration and EMT, decreasing the expression of RAP2A, EPHB2, KLF4 and NF1. |
miR-323-3p [141] |
SMAD2 and SMAD3 |
Silencing of miR-323-3p increased the migration and invasion of PDAC cells, which could be promoted by loss of inhibition of TGF-β-induced EMT; low levels of miR-323-3p predicted poor prognosis in patients with PDAC. |
miR-663a and miR-4787-5p [142] |
not clear |
Lentiviral overexpression of miR-663a and miR-4787-5p reduced TGF-β1 synthesis and secretion in PDAC cells, which presented a EMT-resist phenotype just like being stimulated by 3-deazaneplanocin-A (DZNep). |
miR-655 [143] |
ZEB1 and TGFBR2 |
Researchers used a reporter system based on a stable clone derived from a pancreatic cancer cell line (Panc1) and they found that overexpression of miR-655 could upregulate E-cadherin and downregulate typical EMT-inducers accompanying the suppression of migration and invasion of mesenchymal-like cancer cells. |
As mentioned above, PSC could induce the EMT process of cancer cells [51], however, Tian L et al. recently demonstrated that EMT is a vital process during PSC activation as well, followed by significant alterations in migration, morphology capacity, and the expression of EMT-related gene in vitro [52]. Bone morphogenetic protein 7 (BMP7), a TGF-β-induced EMT antagonist [53], was a positive regulator of mesenchymal–epithelial transition (MET, the reverse of EMT) and was significantly decreased in mice of chronic renal injury [54], which was demonstrated in prostate and breast cancer cells too [55, 56]. In breast cancer, therapeutic administration of BMP7 could diminish breast cancer metastasis to bone [56]. Accordingly, BMP7 could be a promising antagonist to induce MET to restore quiescence in activated PSCs, which would be a potential therapeutic strategy for pancreatic cancer.
TGF-β and chemoradiation resistance in PDAC
Chemoresistance is one reason for the poor prognosis of PDAC, and this is a major problem during the treatment of this lethal cancer as well. Unfortunately, even well-known first-line agents, i.e., the existing standard gemcitabine therapy, have little effect and can only modestly prolong survival [57, 58].
Compared with other canonical cytokines contributing to chemoradiation resistance, such as IL-1β and nuclear factor-κB (NF-κB), PSC-derived TGF-β is a novel cytokine involved in the promotion of chemoradiation resistance in PDAC [27]. As discussed above, TGF-β participates in EMT process, which is linked with CSC development [59]. CSCs are considered a determining factor in chemoresistance and radioresistance, and they are enhanced by PSCs [60]. In fact, TGF-β could promote the CSC development, and this was proven in PANC-1 and PSN-1 pancreatic cancer cell lines. The researchers found that tumor cells were sensitized to radiation via the inhibition of EMT and CSC process following the use of multi-dose TGF-β-neutralizing antibody. Therefore, we may conclude that PSC-derived TGF-β expression in TMEs could play a vital role in PDAC chemoradiation resistance, and this effect might be based on TGF-β-induced EMT and CSC process. We have discussed that ECM proteins produced by PSCs are a considerable part of the desmoplastic stroma in the PDAC microenvironment, and they play an important role in initiating stromal–cancer cell cross-talk and limit chemotherapeutic drugs’ delivery and effectiveness, inducing chemoresistance. As TGF-β is one of the most important factors stimulating PSC secretion of ECM [61], more attention has been focused on it recently. There is cross-talk between TGF-β and Sonic Hh (SHH) signaling, another pathway involved in PDAC chemoresistance [62]; to be exact, the SHH pathway can be the downstream signaling pathway of TGF-β during pancreatic fibrosis, which can be the precursor to PDAC [27, 63]. Kenneth P. Olive et al. found that inhibiting SHH pathway could enhance the effectiveness of gemcitabine for a short term in mice [2]. However, recent clinical trials demonstrated that SHH inhibitors(such as vismodegib; Genentech, South San Francisco, Calif) combined with gemcitabine yielded no significant improvement on progression or overall survival, even increased mortality [64, 65]. Nevertheless, novel mechanisms such as autophagy [66, 67], and novel pathways involving TGF-β have been found recently, providing new ideas for overcoming chemoresistance in PDAC (Table 2).
Table 2: TGF-β-mediated agents or potential targets for chemoresistance in PDAC
Agent or potential target |
Descsription |
Mechanism of action |
Influence on PDAC |
|---|---|---|---|
Ormeloxifene [58] |
a nonsteroidal triphenylethylene compound |
blocked the Hh signaling pathway by inhibiting the important effectors of this pathway, such as SHH, SMO, Gli-1, and SDF-1 (CXCL12) , inhibited desmoplasia and interrupted the tumor–stromal interactions. |
reduced tumor progression, invasion, metastasis, and chemoresistance and enhanced the antitumor effect of gemcitabine. |
Lumican [144] |
an extracellular matrix proteoglycan secreted by PSCs |
can significantly decrease AMP-activated kinase (AMPK) activity, inhibiting chemotherapy-induced autophagy in both in vitro and in vivo. TGF-β can negatively control lumican transcription in PSC through novel SMAD4-SBE binding. |
improved the cancer cells’ sensitivity to gemcitabine, which was reflected in increased mitochondrial damage, reactive oxygen species (ROS) production and cytochrome C release. |
Simvastatin [145] |
a member of Statins family |
attenuated the tumor-associated macrophages(TAM)-mediated gemcitabine resistance of PDAC by blocking the TGF-β1/Gfi-1 axis. |
functioned on preventing chemoresistance in PDAC and was reported to reduce 67% risk of pancreatic cancer. |
CYR61 [57] |
a matricellular protein, cysteine-rich angiogenic inducer 61 |
TGF-β induced the expression of CYR61 in PSCs through canonical TGF-β-ALK5-SMAD2/3 signaling. |
increased cellular uptake of gemcitabine and sensitize PDAC cells to gemcitabine-induced apoptosis. |
TAK1 [146] |
TGF-β-activated kinase-1, a mitogen-activated protein kinase kinase kinase |
increased the NF-kB- and AP-1-mediated transcription of cIAP-2. |
suppressed proapoptotic signaling pathways, resulting in chemoresistance in PDAC. |
miR-17-92 [147] |
a member of miRNAs |
inhibited NODAL/ACTIVIN/TGF-β1 pathway, which could increase cancer stem cells(CSC)’ chemoresistance. |
abrogated CSC phenotypes and their tumourigenicity in PDAC. |
PSC-derived TGF-β and Immunotherapy: combination medication and personalized treatment
Most of PDAC contents are ECM and non-neoplastic cells such as PSCs, which play a critical role in PDAC development; as a result, many tumor cell-targeting treatments fail to eradicate PDAC [68, 69]. However, simple removal of stromal cells may lead to immunosuppression and shortened survival of patients, because stromal cells have inhibitory effects on pancreatic cancer progression simultaneously [70, 71]. Therefore, understanding the underlying molecular mechanism could help researchers to seek suitable therapies involving immunotherapy for patients with PDAC.
The human immune system has protective factors against illness which can identify ‘enemies’ and create suitable ‘weapons’ for defense. This ingenious system includes CD8+ cytotoxic T cells, macrophages, dendritic cells, and natural killer cells [72]. However, multiple mechanisms remarkably suppress these tumor-specific immune responses in malignancies such as PDAC, even in the early stages, and this process including TME infiltration by immunosuppressive cells such as myeloid-derived suppressive cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T cells (Tregs). PSC-derived TGF-β could likewise play a role in immunosuppression via inhibiting the above immune cells and increasing Tregs’ numbers [73]. TGF-β could induce the expression of the transcription factor forkhead box 3 (FOXP3), which can be a determinant factor in Tregs’ development, and the Tregs can secrete immunosuppressive cytokines that can suppress CD8+ T cell killing of tumor cells [74].
Using low-dose cyclophosphamide could downregulate TGF-β-induced Tregs’ number and functionality. However, this therapy could have inevitable adverse effects because it could do harm to the tumor-suppressive immune cells as well [75]. Given the multiple factors and various conditions in tumor progression, combination medication has its edge in some contexts. Interestingly, Malvicini et al. found that combining subtherapeutic doses of an adenovirus expressing IL-12 gene and low cyclophosphamide doses could inhibit IL-10 and TGF-β, modifying Treg performance and prolonging the survival of pancreatic carcinoma in an animal model [76]. Similarly, Soares et al. found that TGF-β inhibition combined with a allogeneic pancreas tumor vaccine secreting granulocyte/macrophage colony–stimulating factor (GM-CSF) dramatically increased effector CD8+ T lymphocyte infiltration and inhibited Tregs more significantly, followed by a survival advantage in a mouse trial [73]. As the vital role TGF-β plays in immunosuppression, combining TGF-β inhibitors such as the small-molecule galunisertib [77] or other repressors targeting downstream of TGF-β signaling with existing immunotherapy strategies could be more effective for PDAC treatment.
Based on the individualized difference of gene mutations, TGF-β-inhibiting therapy for each patient with PDAC could have different effects, therefore, it is meaningful to implement personalized treatment in PDAC patients. A recent study has shown that the loss of transcriptionally active p73 (TAp73), a p53 family member, could be a determinant factor for selecting patients who can benefit from TGF-β-inhibiting therapy [78]. This study displayed that the absence of p73 was involved in TGF-β signaling activation through a SMAD-independent pathway, followed by oncogenic effects such as EMT induced by TGF-β; in the presence of TAp73, the opposite effects were observed. However, eligible patients for TGF-β-inhibiting therapy are restrained at present [78] and the lack of more precise selection biomarkers for patient populations is the main barrier to its practice [79].
Roles of SDF-1 in PDAC invasion and metastasis
Chemokines are a small chemotactic cytokine family. SDF-1 (or, CXCL12) is a chemokine expressed in certain cancers, involved in tumor cell migration and metastasis [80, 81]. In particular, the SDF-1/CXCR4 axis, which was demonstrated by many recent studies, plays a crucial role in tumor–stromal interactions [82]. In PDAC, PSC-derived SDF-1 could promote PDAC cell invasion and gemcitabine chemoresistance by virtue of this axis [83, 84]. TAM recruitment to the tissue via CXCR4 expression in response to SDF-1 is an essential process of cancer initiation, formation, progression, and migration [85]. Particularly, in tumor metastasis, the formation of the pre-metastatic niche was recently established, and was considered the metastatic cancer prodromal stage [86]. It was recently proven that SDF-1 is involved with this process. In a murine model of Lewis lung carcinoma, SDF-1 was elevated prior to cancer cell infiltration to the lymph nodes, and inhibition of this SDF-1 signaling axis (cyclooxygenase 2/prostaglandin E receptor 3-dependent induction of SDF-1) is promising for suppressing pre-metastatic niche formation [87]. Furthermore, an inhibitor of the SDF-1 receptor CXCR4 and anti-CXCR4 antibody treatment was proven to reduce SDF-1-mediated neutrophil recruitment to the liver, and its interaction with TIMP1 could contribute to the formation of the pre-metastatic niche [88]. However, the explicit mechanisms of pre-metastatic niche formation in PDAC have yet to be determined.
There are several signaling pathways of SDF-1-induced invasion and metastasis in PDAC, and their mechanisms are diverse [89]. A recent study found that a SDF-1-targeting miRNA, miR-454, plays a role in TAM recruitment, which has value for understanding the mechanisms underlying tumor growth [90]. Some studies have shown that high SDF-1 expression levels are related to poor outcome of PDAC [91]. Interestingly, for many years, it was assumed that CXCR4 was the only SDF-1 receptor; however, recent studies have demonstrated that CXCR7 is a novel receptor of SDF-1 involved in several aspects of tumor invasion and metastasis in PDAC [81, 91, 92]; Liu et al. reported that SDF-1 and CXCR7’s expression in the ductal cells were related with poor prognosis, as the median survival time of SDF-1+CXCR7+ patients was 6 months while that of SDF-1−CXCR7− patients was 10 months [81]. In addition, there is cross-talk between the SDF-1 subtype SDF-1α and IL-6 signaling, which promoted tumor cell proliferation and chemoresistance in PDAC [84]. Moreover, a recent study asserted that PSC-secreted IL-6 and SDF-1α were responsible for nuclear factor erythroid 2 (NRF2) activation in PDAC cells. The expression of metabolic genes was upregulated by NRF2, which then promoted the detoxification of ROS and the synthesis of purine nucleotides, leading to PDAC cell proliferation [84, 93]. Another meaningful cross-talk is between the SDF-1/CXCR4 axis and the non-canonical Hh pathway [94]; the latter was usually considered to function in developing embryos [95]. It has been illustrated that the SDF-1/CXCR4 axis could promote the expression of smoothened, a vital protein in the Hh pathway whose overexpression can induce tumor cell EMT and invasion. To conclude, the cross-talk between SDF-1 and other signaling pathways provides a novel platform for deeper understanding of the mechanisms involved in SDF-1-induced invasion and metastasis in PDAC (Figure 2).
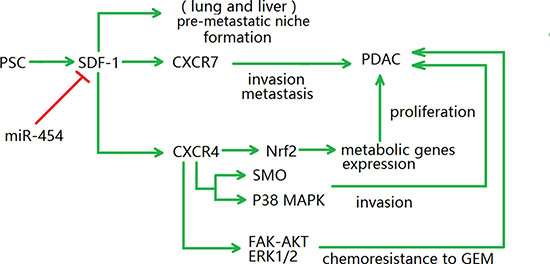
Figure 2: Diagram illustrating the SDF-1 signaling pathways. There are several SDF-1 signaling pathways involving with the malignant development of PDAC, which play the role in invasion, metastasis, metabolic genes expression and chemoresistance to GEM. In addition, SDF-1 participates in the formation of pre-metastatic niche in lung and liver cancers, but this process is not yet clear in PDAC. NRF2, nuclear factor erythroid 2; SMO, Smoothened; FAK, focal adhesion kinase; ERK, extracellular signal-regulated protein kinases; GEM, gemcitabine.
PSC-secreted IL-6 and potential targeted therapies
In PDAC microenvironment, IL-6 is one of the most abundant proinflammatory cytokines secreted by PSCs and tumor cells. In addition to the inflammatory response, IL-6 is associated with numerous tumor cell biological behaviors, including growth, survival, metastasis, angiogenesis, EMT, and chemoresistance [84, 97], which plays a role in the promotion of pancreatic cancer development. Recent research has shown that several IL-6 pathways regulated the relationship between PSCs and PDAC cells and specific inhibitors or antagonists could block the pathways for targeted therapy. Previous studies have stated that elevated MDSC levels were associated with reduced overall survival in patients with pancreatic cancer [97], and IL-6 produced by PSCs in TME could activate MDSC via JAK/STAT3 (Janus kinase/signal transducer and activator of transcription 3) signaling [98]. The activated MDSCs play a role in immunosuppression in the TME, which can protect PDAC cells from the immune cells’ attack and this could explain why high MDSC levels are usually related to poor prognosis. Recently, Hamada et al. found that the STAT3 pathway also regulated PSC-induced EMT in pancreatic cancer cells, [96, 99]. Furthermore, the NRF2 pathway was demonstrated to be induced by STAT3 to promote EMT in PDAC [96]. In short, IL-6/JAK/Stat3/NRF2 signaling pathway is a valuable target to seek suitable therapy options for PDAC patients. In addition, it has been proven that IL-6 is involved in the activation of macrophage phenotype switch, which takes place in the TME, followed by stimulated expression of EMT markers such as N-cadherin and vimentin in pancreatic cancer cells [100]. Finally, recent study argued that PSC-derived IL-6 was regulated by autophagy, and persistent PSC activation might be caused by an autocrine IL-6 loop [18]. In summary, further researches involving IL-6 in PDAC should focus on: 1). IL-6-related inflammatory response, 2). IL-6/JAK/Stat3/NRF2 signaling pathway, 3). IL-6-induced EMT process and the activation of macrophage phenotype switch, 4). IL-6-related autophagy.
All of the above forms a solid foundation for treating PDAC by targeting IL-6. Currently, the value of clinically available small molecules such as trametinib, regorafenib, sorafenib, and sunitinib as antineoplastic drugs is reflected in the suppression of the expression of IL-6 or IL-6 mRNA [101, 102]. However, these drug experiments were based on mouse models and require further investigation in the future. Table 3 lists some novel potential therapies targeting IL-6 pathways that are promising for treating PDAC.
Table 3: The potential therapies targeting IL-6 pathways for treatment of PDAC
Agent or candidate target |
Descriptions |
Impacts |
mechanism |
|---|---|---|---|
Saha [100] |
Histone Deacetylase HDAC I/II inhibitor |
prevented neoplastic lesion formation, fbrosis, and M2 macrophage in the KC mice exposed to cigarette smoke. |
inhibited HDAC3 to decrease the level of IL-6 produced by the cancer cells. |
JQ1 and I-BET 762 [148] |
Bromodomain inhibitors, small molecule inhibitors that target BET (bromodomain and extra terminal) proteins |
decreased c-Myc and p-Erk 1/2 protein levels and inhibited proliferation in pancreatic cancer cells. |
suppressed the production of nitric oxide and inflammatory cytokines including IL-6. |
GV1001 [149] |
a telomerase-based cancer vaccine |
the combination treatment of GV1001 and gemcitabine could signifcantly reduce the fbrosis in tumor tissue and induce apoptosis. |
the combination can suppress the IL-6, however, the actual mechanism remains to be investigated. |
HIC1 [150] |
Hypermethylated in cancer 1 ,a tumour suppressor gene |
negative HIC1 expression predicted poor dignosis; inhibited the invasion and metastasis of pancreatic cancer cells both in vitro and in vivo; repressed the expression of STAT3 target genes, including c-Myc, VEGF, CyclinD1, MMP2 and MMP9. |
inhibited STAT3 activity and it was likely to function via inhibition of IL-6/JAK/STAT3 signaling pathway. |
ANXA2 [151] |
Annexin A2,a negatively charged phospholipidbinding protein |
mediated resistance to gemcitabine. |
increased the activity of NF-κB, whose downstream target genes including that encoding IL-6. |
RA [152] |
Retinoic acid, a small molecular derivative of vitamin A |
inhibited pancreatic cancer cell migration and EMT. |
reduced IL-6 secreted by CAFs(cancer associated fibroblasts). |
Galectin-1 functions as a tumor promoter in PDAC
A 29-kDa β-galactoside-binding protein, galectin-1 is a member of the galectin lectin family. It functions both inside and outside the cell; however, its carbohydrate-binding role is extracellular [103, 104]. And the extracellular effects of galectin-1 in PDAC progression will be discussed in this section. The previous literature has shown that galectin-1 was secreted by activated PSCs in PDAC stroma [105]. In the TME, persistent PSC activation promotes tumor cell malignant behavior, and galectin-1 plays a crucial role in PSC activation [27] [106].
Recent studies have demonstrated that chemokine production, PSC proliferation, collagen and fibronectin synthesis could be induced by galectin-1 [107, 108], and they were responsible for the tumor desmoplastic reaction around cancer cells [109]. Moreover, these effects could lead to immunosuppression in PDAC [106]. In a healthy human body, the immune system maintains a balance between tumor immunosuppression and anti-tumor activity [110]. However, in PDAC, galectin-1 could help tumor cells to escape from immune surveillance [106]. The actual function profile of the immunosuppressive is that galectin-1 can induce effector T cell apoptosis and anergy via the “caspase first” or “mitochondria first” pathways and alter Th1/Th2 balance by stimulating Th2 cytokine (IL-6 and IL-10) secretion but decreasing Th1 cytokine (tumor necrosis factor-β and interferon-γ) secretion [106]. Moreover, tumor-related process such as invasion, angiogenesis, proliferation, MMP2, MMP9 expression, and EMT process could be induced by galectin-1 in PDAC [51, 104, 111, 112]. Acinar–ductal metaplasia (ADM) is a significant process in PDAC development, which is triggered by PSC-secreted galectin-1 via the epidermal growth factor receptor and pancreatic and duodenal homeobox 1 pathways [113]. In addition, the Hh/Gli axis could be another pathway involved in ADM, but its mechanism has not been elucidated. As described hereinbefore, SDF-1 is a critical tumor–stromal interaction mediator and could promote PDAC progression. It was proven to be upregulated by endogenous galectin-1, promoting tumor metastasis [82]. Furthermore, PSC-secreted galectin-1 is upregulated by TGF-β1, which is produced by PDAC cell, and TGF-β1 can stimulate PSC activation simultaneously, inducing more galectin-1 secretion. The malignant behavior of PDAC is related to this vicious cycle of mutually reinforcing mechanism [112].
There is a positive association between galectin-1 expression and PDAC tumor size, lymph node metastasis, perineural invasion, differentiation, Union for International Cancer Control stage, and survival [106, 112, 114]. As various promoters influence galectin-1 expression during tumor formation, galectin-1 might be a promising drug target and biomarker for PDAC [115, 116]. These valuable preclinical evidence showed that inhibiting galectin-1 could be efficient for treating PDAC. For example, β-lactose, a competitive inhibitor of galectin-1, could inhibit the immunosuppressive effect induced by galectin-1 in vivo [106]. In addition, monoclonal antibodies are a promising therapeutic approach, e.g., 0118 (PTX008, OTX008), as they have high specificity for galectin-1 and are small molecules [116]. However, most of the galectin-1-targeting therapies are in preclinical or early clinical development and their prolonged clinical application remain far on the horizon (Figure 3).
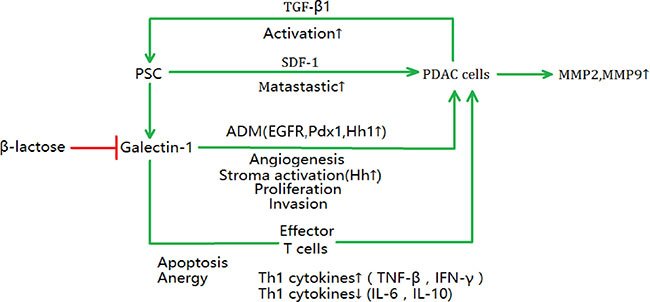
Figure 3: The effects of PSC-induced galectin-1 on PDAC cell progression. There are various extracellular effects of galectin-1 in PDAC progression, such as metastasis, ADM (acinar–ductal metaplasia), angiogenesis, stroma activation, proliferation, invasion of PDAC cells. In addition, the galectin-1 contributes to the apoptosis and anergy of effector T cell in immune system and alters Th1/Th2 balance. SDF-1, stromal cell-derived factor-1; TGF-β, transforming growth factor β; EGFR, epidermal growth factor receptor; Pdx1, pancreatic and duodenal homeobox 1; Hh1, hedgehog1; IFN-γ, interferon-γ; IL-6, interleukin 6; IL-10, interleukin 10.
HGF: a novel target for PDAC treatment
HGF is a 90-kDa glycoprotein, mainly originating from stromal cells such as blood endothelial cells, macrophages, fat-storing cells, neutrophils, and fibroblasts [117]. In PDAC, HGF is mainly produced by PSCs and plays a critical role in the cross-talk between the PDAC cells and PSCs [118–120]. The various functions of HGF are realized mainly via binding to its specific tyrosine-kinase receptor c-MET [121, 122], and the HGF-c-MET pathway recently has become a research hotspot involving PDAC progression. Upon binding to HGF, c-MET could activate its multiple downstream pathways, such as PI3K/Akt, MAPK, and STAT3, promoting invasion, proliferation, migration, mitogenesis, DNA-synthesis and chemotherapy resistance of PDAC cells [123–125].
In PDAC, the angiogenesis is a crucial process involving tumor growth, progression, and metastasis, which could be evaluated by the proliferation of vascular endothelial cell and the formation of tube [125, 126]. Vascular endothelial growth factor (VEGF) is a well-known pro-angiogenic growth factor, and the inhibitor of VEGF and its receptor have been approved by the FDA (Food and Drug Administration, USA) in PDAC treatment [127]. Nevertheless, recent researches showed VEGF inhibitors could promote the metastasis and invasion of tumor cell, followed by decreased survival [128–130]. Interestingly, the anti-HGF therapy showed its edge when conflicting with PDAC. Patel MB et al. demonstrated that targeting HGF/c-MET and urokinase-type plasminogen activator (uPA) pathways could be beneficial for inhibiting endothelial cell proliferation and closed tube formation [125]. AMG102 and amiloride, the specific inhibitors of HGF and uPA, were proven to have potent antiangiogenic influences when combined, and they were independent of patient heterogeneity, which has more practical and clinical value. However, not all the combination therapy could achieve expected outcome, and we should figure out the exact mechanism underlying [131–133]. As illustrated before, gemcitabine is a standard chemotherapeutic agent for PDAC. When combined with HGF inhibition, the antimetastatic, antiangiogenic and antiproliferative effects were reduced and there have even been cases of tumor progression [119]. This could be explained by the theory that gemcitabine can select out treatment-resistant and aggressive cancer cells, and could promote EMT process and metastasis of cancer cells [119, 134].
When using HGF inhibition therapy, we should take functional heterogeneity into consideration as well. The p53 gene is an anti-oncogene whose mutation is related to more than half of human cancer [135]. Yang et al. found that P53 deficiency could increase the invasion and migration of Panc-1 cells, which could upregulate the expression of c-MET [120]. Furthermore, a member of the inhibitor of apoptosis protein, Survivin, was proven to be upregulated by HGF-c-MET pathway, which could promote the tumor progression. However, in the same research, another cell line SW1990 showed low sensitivity to HGF, and that could be explained by the difference of the expression level of c-MET in two cell lines. This finding suggested that c-MET could be a novel biomarker for choosing suitable patients who can benefit the most from the treatment targeting HGF. Similarly, the various PSC populations were proven to be heterogeneous, and their capacity to stimulate the migration and DNA-synthesis of PDAC cells and expression level of HGF were different according to the research based on eight different PDAC patients [124], which was in agreement with the previous study about heterogeneity of PSCs [136]. Furthermore, IL-1α and TGF-β were demonstrated to play a role in regulating the expression of HGF secreted by PSC and the multifunction induced by HGF. Recent studies demonstrated that approximately 50% TGF-β signaling pathway exists mutations [137, 138], and the heterogeneity of TGF-β could contribute to functional heterogeneity of PSCs. As the complexity of various signaling pathways between PSCs and PDAC cell is further complicated by the diverse expression level among different patients, inhibiting HGF could have different outcomes in different subsets of patients.
Outlook: The inhibition of PSC-derived soluble factors is promising for PDAC treatment
The interaction between PSCs and PDAC cells play an important role in the promotion of tumor development. Nowadays, there are increasing attentions on researches of the cytokines or proteins in PDAC microenvironment, i.e., TGF-β, IL-6, SDF-1, HGF and galectin-1, which have their own regulatory downstream signaling pathways of PSCs and PDAC cells. Furthermore, there are usually cross-talks between these cytokines or proteins, such as between IL-6 and SDF-1α, which has a synergistic effect during PDAC formation. Intervening in these pathways and blocking their critical processes could be valuable for treating PDAC. However, it needs more work to clarify the definite mechanisms of these pathways, and to discover more effective targeting agents.
Abbreviations
α-SMA, α-smooth muscle actin; ADM, acinar–ductal metaplasia; BMP7, bone morphogenetic protein 7; CAF, carcinoma-associated fibroblasts, COX-2/EP3, cyclooxygenase 2/prostaglandin E receptor 3; CSC, cancer stem cell; ECM, extracellular matrix; EGFR, epidermal growth factor receptor; EMT, epithelial–mesenchymal transition; FGF2, fibroblast growth factor 2; FOXP3, forkhead box 3; GFAP, glial fibrillary acidic protein; GM-CSF, granulocyte/macrophage colony-stimulating factor; Hh, Hedgehog; IFN-γ, interferon-γ; IL-1, interleukin 1; IL-6, interleukin 6; IL-8, interleukin 8; JAK/STAT3, Janus kinase/signal transducer and activator of transcription 3; KLF5, Krüppel-like factor 5; MAPK, mitogen-activated protein kinase; MCP-1, monocyte chemoattractant protein-1; MDSC, myeloid-derived suppressive cell; MET, mesenchymal–epithelial transition; miRNA, microRNA; MMP, matrix metalloproteinase; NF-κB, nuclear factor-κB; NOX4, NADPH oxidase 4; NRF2, nuclear factor erythroid 2; OV, oncolytic virus; PDAC, pancreatic ductal adenocarcinoma; PDX1, pancreatic and duodenal homeobox 1; PSC, pancreatic stellate cell; PDGF, platelet-derived growth factor; ROS, reactive oxygen species; SDF-1, stromal cell-derived factor-1; SHH, Sonic Hh; SMO, Smoothened; TAM, tumor-associated macrophage; TAp73, transcriptionally active p73; TGF-β, transforming growth factor β; TIMP, tissue inhibitors of MMP; TME, tumor microenvironment; TNF-β, tumor necrosis factor-β; Treg, regulatory T cells; UICC, Union for International Cancer Control; EFGR, Epidermal growth factor receptor; uPA, urokinase-type plasminogen activator
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
FUNDING
This work was supported by grants from the National Natural Science Funding of China (81572344), the Jiangsu Province Natural Science Foundation of China (BK20140495), the Postdoctoral Science Foundation of China (2013M530243), the Six Big Talent Peak Projects of Jiangsu Province (2014-WSW-078), training project of key talents of youth medicine in Jiangsu province, China (QNRC2016330), the Science and Technology Development Funding of Yangzhou City (2012123), the Postdoctoral Science Foundation of Jiangsu Province (2013), and the “Promote health development by science and technology” Program of Jiangsu Province ( KF201225), and the academic science and technology innovation fund for college students (x20160750, x20160753, x20160774, x20160783).
REFERENCES
1. Egawa S, Toma H, Ohigashi H, Okusaka T, Nakao A, Hatori T, Maguchi H, Yanagisawa A, Tanaka M. Japan Pancreatic Cancer Registry; 30th year anniversary: Japan Pancreas Society. Pancreas. 2012; 41:985–92. https://doi.org/10.1097/MPA.0b013e318258055c.
2. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009; 324:1457–61. https://doi.org/10.1126/science.1171362.
3. Giannuzzo A, Pedersen SF, Novak I. The P2X7 receptor regulates cell survival, migration and invasion of pancreatic ductal adenocarcinoma cells. Mol Cancer. 2015; 14:203. https://doi.org/10.1186/s12943-015-0472-4.
4. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012; 196:395–406. https://doi.org/10.1083/jcb.201102147.
5. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004; 432:332–7. https://doi.org/10.1038/nature03096.
6. Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM, Tuveson DA. Stromal biology and therapy in pancreatic cancer. Gut. 2011; 60:861–8. https://doi.org/10.1136/gut.2010.226092.
7. Watari N, Hotta Y, Mabuchi Y. Morphological studies on a vitamin A-storing cell and its complex with macrophage observed in mouse pancreatic tissues following excess vitamin A administration. Okajimas Folia Anat Jpn. 1982; 58:837–58.
8. Ikejiri N. The vitamin A-storing cells in the human and rat pancreas. Kurume Med J. 1990; 37:67–81.
9. Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998; 43:128–33.
10. Asahina K, Zhou B, Pu WT, Tsukamoto H. Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology. 2011; 53:983–95. https://doi.org/10.1002/hep.24119.
11. Sparmann G, Kruse ML, Hofmeister-Mielke N, Koczan D, Jaster R, Liebe S, Wolff D, Emmrich J. Bone marrow-derived pancreatic stellate cells in rats. Cell Res. 2010; 20:288–98. https://doi.org/10.1038/cr.2010.10.
12. Ino K, Masuya M, Tawara I, Miyata E, Oda K, Nakamori Y, Suzuki K, Ohishi K, Katayama N. Monocytes Infiltrate the Pancreas via the MCP-1/CCR2 Pathway and Differentiate into Stellate Cells. Plos One. 2014; 9. https://doi.org/ARTN e8488910.1371/journal.pone.0084889.
13. Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, Siech M, Beger H, Grunert A, Adler G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998; 115:421–32.
14. Bynigeri RR, Jakkampudi A, Jangala R, Subramanyam C, Sasikala M, Rao GV, Reddy DN, Talukdar R. Pancreatic stellate cell: Pandora's box for pancreatic disease biology. World J Gastroenterol. 2017; 23:382–405. https://doi.org/10.3748/wjg.v23.i3.382.
15. Shimizu K, Kobayashi M, Tahara J, Shiratori K. Cytokines and peroxisome proliferator-activated receptor gamma ligand regulate phagocytosis by pancreatic stellate cells. Gastroenterology. 2005; 128:2105–18.
16. Berna MJ, Seiz O, Nast JF, Benten D, Blaker M, Koch J, Lohse AW, Pace A. CCK1 and CCK2 receptors are expressed on pancreatic stellate cells and induce collagen production. J Biol Chem. 2010; 285:38905–14. https://doi.org/10.1074/jbc.M110.125534.
17. Phillips PA, Yang L, Shulkes A, Vonlaufen A, Poljak A, Bustamante S, Warren A, Xu ZH, Guilhaus M, Pirola R, Apte MV, Wilson JS. Pancreatic stellate cells produce acetylcholine and may play a role in pancreatic exocrine secretion. Proc Natl Acad Sci USA. 2010; 107:17397–402. https://doi.org/10.1073/pnas.1000359107.
18. Endo S, Nakata K, Ohuchida K, Takesue S, Nakayama H, Abe T, Koikawa K, Okumura T, Sada M, Horioka K, Zheng B, Mizuuchi Y, Iwamoto C, et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated With Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology. 2017; 152:1492–506.e24.
19. Fujii S, Mitsunaga S, Yamazaki M, Hasebe T, Ishii G, Kojima M, Kinoshita T, Ueno T, Esumi H, Ochiai A. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci. 2008; 99:1813–9. https://doi.org/10.1111/j.1349-7006.2008.00893.x.
20. Strobel O, Dadabaeva N, Felix K, Hackert T, Giese NA, Jesenofsky R, Werner J. Isolation and culture of primary human pancreatic stellate cells that reflect the context of their tissue of origin. Langenbecks Arch Surg. 2016; 401:89–97. https://doi.org/10.1007/s00423-015-1343-6.
21. Wehr AY, Furth EE, Sangar V, Blair IA, Yu KH. Analysis of the human pancreatic stellate cell secreted proteome. Pancreas. 2011; 40:557–66. https://doi.org/10.1097/MPA.0b013e318214efaf.
22. Masamune A, Kikuta K, Watanabe T, Satoh K, Hirota M, Shimosegawa T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am J Physiol Gastrointest Liver Physiol. 2008; 295:G709-17. https://doi.org/10.1152/ajpgi.90356.2008.
23. Jiang X, Abiatari I, Kong B, Erkan M, De Oliveira T, Giese NA, Michalski CW, Friess H, Kleeff J. Pancreatic islet and stellate cells are the main sources of endocrine gland-derived vascular endothelial growth factor/prokineticin-1 in pancreatic cancer. Pancreatology. 2009; 9:165–72. https://doi.org/10.1159/000178888.
24. Hamada S, Masamune A, Takikawa T, Suzuki N, Kikuta K, Hirota M, Hamada H, Kobune M, Satoh K, Shimosegawa T. Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem Biophys Res Commun. 2012; 421:349–54. https://doi.org/10.1016/j.bbrc.2012.04.014.
25. Pandol S, Gukovskaya A, Edderkaoui M, Dawson D, Eibl G, Lugea A. Epidemiology, risk factors, and the promotion of pancreatic cancer: role of the stellate cell. J Gastroenterol Hepatol. 2012; 27 Suppl 2:127–34. https://doi.org/10.1111/j.1440-1746.2011.07013.x.
26. Chu GC, Kimmelman AC, Hezel AF, DePinho RA. Stromal biology of pancreatic cancer. J Cell Biochem. 2007; 101:887–907. https://doi.org/10.1002/jcb.21209.
27. Tang D, Wang D, Yuan Z, Xue X, Zhang Y, An Y, Chen J, Tu M, Lu Z, Wei J, Jiang K, Miao Y. Persistent activation of pancreatic stellate cells creates a microenvironment favorable for the malignant behavior of pancreatic ductal adenocarcinoma. Int J Cancer. 2013; 132:993–1003. https://doi.org/10.1002/ijc.27715.
28. Manohar M, Verma AK, Venkateshaiah SU, Sanders NL, Mishra A. Pathogenic mechanisms of pancreatitis. World J Gastrointest Pharmacol Ther. 2017; 8:10–25. https://doi.org/10.4292/wjgpt.v8.i1.10.
29. Algul H, Treiber M, Lesina M, Schmid RM. Mechanisms of disease: chronic inflammation and cancer in the pancreas--a potential role for pancreatic stellate cells? Nat Clin Pract Gastroenterol Hepatol. 2007; 4:454–62. https://doi.org/10.1038/ncpgasthep0881.
30. Sousa CM, Biancur DE, Wang XX, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying HQ, Asara JM, Evans RM, Cantley LC, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion (vol 536, pg 479, 2016). Nature. 2016; 540:150–. https://doi.org/10.1038/nature19851.
31. Tjomsland V, Pomianowska E, Aasrum M, Sandnes D, Verbeke CS, Gladhaug IP. Profile of MMP and TIMP Expression in Human Pancreatic Stellate Cells: Regulation by IL-1alpha and TGFbeta and Implications for Migration of Pancreatic Cancer Cells. Neoplasia. 2016; 18:447–56. https://doi.org/10.1016/j.neo.2016.06.003.
32. Lohr M, Schmidt C, Ringel J, Kluth M, Muller P, Nizze H, Jesnowski R. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001; 61:550–5.
33. Satoh K, Shimosegawa T, Hirota M, Koizumi M, Toyota T. Expression of transforming growth factor beta1 (TGFbeta1) and its receptors in pancreatic duct cell carcinoma and in chronic pancreatitis. Pancreas. 1998; 16:468–74.
34. Glazer ES, Welsh E, Pimiento JM, Teer JK, Malafa MP. TGFbeta1 overexpression is associated with improved survival and low tumor cell proliferation in patients with early-stage pancreatic ductal adenocarcinoma. Oncotarget. 2017; 8:999–1006. https://doi.org/10.18632/oncotarget.13533.
35. Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer. 2013; 13:788–99. https://doi.org/10.1038/nrc3603.
36. Gupta S, Maitra A. EMT: Matter of Life or Death? Cell. 2016; 164:840–2. https://doi.org/10.1016/j.cell.2016.02.024.
37. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74. https://doi.org/10.1016/j.cell.2011.02.013.
38. Morrison CD, Parvani JG, Schiemann WP. The relevance of the TGF-β Paradox to EMT-MET programs. Cancer Lett. 2013; 341:30.
39. Oyanagi J, Kojima N, Sato H, Higashi S, Kikuchi K, Sakai K, Matsumoto K, Miyazaki K. Inhibition of transforming growth factor-β signaling potentiates tumor cell invasion into collagen matrix induced by fibroblast-derived hepatocyte growth factor. Exp Cell Res. 2014; 326:267–79.
40. Tjomsland V, Sandnes D, Pomianowska E, Cizmovic ST, Aasrum M, Brusevold IJ, Christoffersen T, Gladhaug IP. The TGFβ-SMAD3 pathway inhibits IL-1α induced interactions between human pancreatic stellate cells and pancreatic carcinoma cells and restricts cancer cell migration. J Exp Clin Cancer Res.2016; 35:122.
41. Satoh K, Hamada S, Shimosegawa T. Involvement of epithelial to mesenchymal transition in the development of pancreatic ductal adenocarcinoma. J Gastroenterol. 2015; 50:140–6. https://doi.org/10.1007/s00535-014-0997-0.
42. Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003; 112:1776–84. https://doi.org/10.1172/JCI20530.
43. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006; 7:131–42. https://doi.org/10.1038/nrm1835.
44. Hiraga R, Kato M, Miyagawa S, Kamata T. Nox4-derived ROS signaling contributes to TGF-beta-induced epithelial-mesenchymal transition in pancreatic cancer cells. Anticancer Res. 2013; 33:4431–8.
45. Gui T, Sun Y, Shimokado A, Muragaki Y. The Roles of Mitogen-Activated Protein Kinase Pathways in TGF-beta-Induced Epithelial-Mesenchymal Transition. J Signal Transduct. 2012; 2012:289243. https://doi.org/10.1155/2012/289243.
46. Xu M, Cai J, Wei H, Zhou M, Xu P, Huang H, Peng W, Du F, Gong A, Zhang Y. Scoparone Protects Against Pancreatic Fibrosis via TGF-β/Smad Signaling in Rats. Cell Physiol Biochem. 2016; 40:277–86.
47. David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, Massague J. TGF-beta Tumor Suppression through a Lethal EMT. Cell. 2016; 164:1015–30. https://doi.org/10.1016/j.cell.2016.01.009.
48. Fan J, Yang MX, Ouyang Q, Fu D, Xu Z, Liu X, Mino-Kenudson M, Geng J, Tang F. Phosphatase PPM1A is a novel prognostic marker in pancreatic ductal adenocarcinoma. Hum Pathol. 2016; 55:151–8. https://doi.org/10.1016/j.humpath.2016.05.002.
49. Yu J, Li A, Hong SM, Hruban RH, Goggins M. MicroRNA alterations of pancreatic intraepithelial neoplasias. Clin Cancer Res. 2012; 18:981–92. https://doi.org/10.1158/1078-0432.CCR-11-2347.
50. Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The mir-200 family and mir-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008; 10:593–601. https://doi.org/10.1038/ncb1722.
51. Kikuta K, Masamune A, Watanabe T, Ariga H, Itoh H, Hamada S, Satoh K, Egawa S, Unno M, Shimosegawa T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem Biophys Res Commun. 2010; 403:380–4. https://doi.org/10.1016/j.bbrc.2010.11.040.
52. Tian L, Lu ZP, Cai BB, Zhao LT, Qian D, Xu QC, Wu PF, Zhu Y, Zhang JJ, Du Q, Miao Y, Jiang KR. Activation of pancreatic stellate cells involves an EMT-like process. Int J Oncol. 2016; 48:783–92. https://doi.org/10.3892/ijo.2015.3282.
53. Zeisberg M, Shah AA, Kalluri R. Bone morphogenic protein-7 induces mesenchymal to epithelial transition in adult renal fibroblasts and facilitates regeneration of injured kidney. J Biol Chem. 2005; 280:8094.
54. Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003; 9:964.
55. Buijs JT, Rentsch CA, van der Horst G, van Overveld PG, Wetterwald A, Schwaninger R, Henriquez NV, Ten Dijke P, Borovecki F, Markwalder R, Thalmann GN, Papapoulos SE, Pelger RC, et al. BMP7, a putative regulator of epithelial homeostasis in the human prostate, is a potent inhibitor of prostate cancer bone metastasis in vivo. Am J Pathol. 2007; 171:1047–57.
56. Buijs JT, Henriquez NV, van Overveld PG, van der Horst G, Que I, Schwaninger R, Rentsch C, Ten Dijke P, Cleton-Jansen AM, Driouch K, Lidereau R, Bachelier R, Vukicevic S, et al. Bone morphogenetic protein 7 in the development and treatment of bone metastases from breast cancer. Cancer Res. 2007; 67:8742–51.
57. Hesler RA, Huang JJ, Starr MD, Treboschi VM, Bernanke AG, Nixon AB, McCall SJ, White RR, Blobe GC. TGF-beta-induced stromal CYR61 promotes resistance to gemcitabine in pancreatic ductal adenocarcinoma through downregulation of the nucleoside transporters hENT1 and hCNT3. Carcinogenesis. 2016; 37:1041–51. https://doi.org/10.1093/carcin/bgw093.
58. Khan S, Ebeling MC, Chauhan N, Thompson PA, Gara RK, Ganju A, Yallapu MM, Behrman SW, Zhao HT, Zafar N, Singh MM, Jaggi M, Chauhan SC. Ormeloxifene Suppresses Desmoplasia and Enhances Sensitivity of Gemcitabine in Pancreatic Cancer. Cancer Res. 2015; 75:2292–304. https://doi.org/10.1158/0008-5472.Can-14-2397.
59. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704–15. https://doi.org/10.1016/j.cell.2008.03.027.
60. Al-Assar O, Demiciorglu F, Lunardi S, Gaspar-Carvalho MM, McKenna WG, Muschel RM, Brunner TB. Contextual regulation of pancreatic cancer stem cell phenotype and radioresistance by pancreatic stellate cells. Radiother Oncol. 2014; 111:243–51. https://doi.org/10.1016/j.radonc.2014.03.014.
61. Nakerakanti S, Trojanowska M. The Role of TGF-beta Receptors in Fibrosis. Open Rheumatol J. 2012; 6:156–62. https://doi.org/10.2174/1874312901206010156.
62. Kelleher FC. Hedgehog signaling and therapeutics in pancreatic cancer. Carcinogenesis. 2011; 32:445–51. https://doi.org/10.1093/carcin/bgq280.
63. Tsang SW, Zhang H, Lin C, Xiao H, Wong M, Shang H, Yang ZJ, Lu A, Yung KK, Bian Z. Rhein, a natural anthraquinone derivative, attenuates the activation of pancreatic stellate cells and ameliorates pancreatic fibrosis in mice with experimental chronic pancreatitis. PLoS One. 2013; 8:e82201. https://doi.org/10.1371/journal.pone.0082201.
64. Ko AH, LoConte N, Tempero MA, Walker EJ, Kate Kelley R, Lewis S, Chang WC, Kantoff E, Vannier MW, Catenacci DV, Venook AP, Kindler HL. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas. 2016; 45:370–75.
65. Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, Marsh R, Wallace J, Kozloff M, Rajdev L, Cohen D, Wade J, Sleckman B, et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J Clin Oncol. 2015; 33:4284–92.
66. Li X, Roife D, Kang Y, Dai B, Pratt M, Fleming JB. Extracellular lumican augments cytotoxicity of chemotherapy in pancreatic ductal adenocarcinoma cells via autophagy inhibition. Oncogene. 2016; 35:4881–90. https://doi.org/10.1038/onc.2016.20.
67. Xu Y, An Y, Wang X, Zha W, Li X. Inhibition of the Hedgehog pathway induces autophagy in pancreatic ductal adenocarcinoma cells. Oncol Rep. 2014; 31:707.
68. Lucas T, Benihoud K, Vigant F, Schmidt CQA, Bachem MG, Simmet T, Kochanek S. Hexon Modification to Improve the Activity of Oncolytic Adenovirus Vectors against Neoplastic and Stromal Cells in Pancreatic Cancer. Plos One. 2015; 10. https://doi.org/ARTN e011725410.1371/journal.pone.0117254.
69. Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK, LeBleu VS, Roy N, Novitskiy SV, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016; 22:497–505. https://doi.org/10.1038/nm.4082.
70. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Fernandez-Barrena MG, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014; 25:735–47.
71. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, De Jesus-Acosta A, Sharma P, Heidari P, et al, and BC Ö. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014; 25:719–34.
72. Sánchez-Elsner T, Botella LM, Velasco B, Corbí A, Attisano L, Bernabéu C. Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J Biol Chem. 2001; 276:38527–35. https://doi.org/10.1074/jbc.M104536200.
73. Soares KC, Rucki AA, Kim V, Foley K, Solt S, Wolfgang CL, Jaffee EM, Zheng L. TGF-beta blockade depletes T regulatory cells from metastatic pancreatic tumors in a vaccine dependent manner. Oncotarget. 2015; 6:43005–15. https://doi.org/10.18632/oncotarget.5656.
74. Chalmin F, Mignot G, Bruchard M, Chevriaux A, Vegran F, Hichami A, Ladoire S, Derangere V, Vincent J, Masson D, Robson SC, Eberl G, Pallandre JR, et al. Stat3 and Gfi-1 Transcription Factors Control Th17 Cell Immunosuppressive Activity via the Regulation of Ectonucleotidase Expression. Immunity. 2012; 36:362–73. https://doi.org/10.1016/j.immuni.2011.12.019.
75. Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood. 2005; 105:2862–8. https://doi.org/10.1182/blood-2004-06-2410.
76. Malvicini M, Ingolotti M, Piccioni F, Garcia M, Bayo J, Atorrasagasti C, Alaniz L, Aquino JB, Espinoza JA, Gidekel M, Scharovsky OG, Matar P, Mazzolini G. Reversal of gastrointestinal carcinoma-induced immunosuppression and induction of antitumoural immunity by a combination of cyclophosphamide and gene transfer of IL-12. Mol Oncol. 2011; 5:242–55. https://doi.org/10.1016/j.molonc.2011.03.007.
77. Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, Cleverly AL, Desaiah D, Guba SC, Benhadji KA, Slapak CA, Lahn MM. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther. 2015; 9:4479–99. https://doi.org/10.2147/DDDT.S86621.
78. Thakur AK, Nigri J, Lac S, Leca J, Bressy C, Berthezene P, Bartholin L, Chan P, Calvo E, Iovanna JL, Vasseur S, Guillaumond F, Tomasini R. TAp73 loss favors Smad-independent TGF-beta signaling that drives EMT in pancreatic ductal adenocarcinoma. Cell Death Differ. 2016; 23:1358–70. https://doi.org/10.1038/cdd.2016.18.
79. Strimpakos AS, Syrigos KN, Saif MW. Novel agents and new combination treatments on phase I studies on solid tumors and pancreatic cancer. JOP. 2012; 13:345–8. https://doi.org/10.6092/1590-8577/947.
80. Miao Z, Luker KE, Summers BC, Berahovich R, Bhojani MS, Rehemtulla A, Kleer CG, Essner JJ, Nasevicius A, Luker GD, Howard MC, Schall TJ. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci USA. 2007; 104:15735–40. https://doi.org/10.1073/pnas.0610444104.
81. Liu Z, Teng XY, Meng XP, Wang BS. Expression of stromal cell-derived factor 1 and CXCR7 ligand receptor system in pancreatic adenocarcinoma. World J Surg Oncol. 2014; 12:348. https://doi.org/10.1186/1477-7819-12-348.
82. Qian D, Lu Z, Xu Q, Wu P, Tian L, Zhao L, Cai B, Yin J, Wu Y, Staveley-O'Carroll KF, Jiang K, Miao Y, Li G. Galectin-1-driven upregulation of SDF-1 in pancreatic stellate cells promotes pancreatic cancer metastasis. Cancer Lett. 2017. https://doi.org/10.1016/j.canlet.2017.03.024.
83. Gao Z, Wang X, Wu K, Zhao Y, Hu G. Pancreatic stellate cells increase the invasion of human pancreatic cancer cells through the stromal cell-derived factor-1/CXCR4 axis. Pancreatology. 2010; 10:186–93. https://doi.org/10.1159/000236012.
84. Zhang H, Wu H, Guan J, Wang L, Ren X, Shi X, Liang Z, Liu T. Paracrine SDF-1alpha signaling mediates the effects of PSCs on GEM chemoresistance through an IL-6 autocrine loop in pancreatic cancer cells. Oncotarget. 2015; 6:3085–97. https://doi.org/10.18632/oncotarget.3099.
85. Meng F, Li C, Li W, Gao Z, Guo K, Song S. Interaction between pancreatic cancer cells and tumor-associated macrophages promotes the invasion of pancreatic cancer cells and the differentiation and migration of macrophages. IUBMB Life. 2014; 66:835–46.
86. Geiger TR, Peeper DS. Metastasis mechanisms. Biochim Biophys Acta. 2009; 1796:293–308.
87. Ogawa F, Amano H, Eshima K, Ito Y, Matsui Y, Hosono K, Kitasato H, Iyoda A, Iwabuchi K, Kumagai Y, Satoh Y, Narumiya S, Majima M. Prostanoid induces premetastatic niche in regional lymph nodes. J Clin Invest. 2014; 124:4882–94.
88. Seubert B, Grünwald B, Kobuch J, Cui H, Schelter F, Schaten S, Siveke JT, Lim NH, Nagase H, Simonavicius N. TIMP-1 creates a pre-metastatic niche in the liver through SDF-1/CXCR4-dependent neutrophil recruitment in mice. Hepatology. 2015; 61:238–48.
89. Pan F, Ma S, Cao W, Liu H, Chen F, Chen X, Shi R. SDF-1α upregulation of MMP-2 is mediated by p38 MAPK signaling in pancreatic cancer cell lines. Mol Biol Rep. 2013; 40:4139–46.
90. Fan Y, Xu LL, Shi CY, Wei W, Wang DS, Cai DF. MicroRNA-454 regulates stromal cell derived factor-1 in the control of the growth of pancreatic ductal adenocarcinoma. Sci Rep. 2016; 6:22793. https://doi.org/10.1038/srep22793.
91. Liang JJ, Zhu S, Bruggeman R, Zaino RJ, Evans DB, Fleming JB, Gomez HF, Zander DS, Wang H. High levels of expression of human stromal cell-derived factor-1 are associated with worse prognosis in patients with stage II pancreatic ductal adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 2010; 19:2598–604. https://doi.org/10.1158/1055-9965.EPI-10-0405.
92. Zheng K, Li HY, Su XL, Wang XY, Tian T, Li F, Ren GS. Chemokine receptor CXCR7 regulates the invasion, angiogenesis and tumor growth of human hepatocellular carcinoma cells. J Exp Clin Cancer Res. 2010; 29:31. https://doi.org/10.1186/1756-9966-29-31.
93. Wu YS, Looi CY, Subramaniam KS, Masamune A, Chung I. Soluble factors from stellate cells induce pancreatic cancer cell proliferation via Nrf2-activated metabolic reprogramming and ROS detoxification. Oncotarget. 2016; 7: 36719–32. https://doi.org/10.18632/oncotarget.9165.
94. Li X, Ma Q, Xu Q, Liu H, Lei J, Duan W, Bhat K, Wang F, Wu E, Wang Z. SDF-1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial-mesenchymal transition in vitro through non-canonical activation of Hedgehog pathway. Cancer Lett. 2012; 322:169–76. https://doi.org/10.1016/j.canlet.2012.02.035.
95. Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001; 15:3059–87. https://doi.org/10.1101/gad.938601.
96. Wu YS, Chung I, Wong WF, Masamune A, Sim MS, Looi CY. EMT Paracrine IL-6 signaling mediates the effects of pancreatic stellate cells on epithelial-mesenchymal transition via Stat3/Nrf2 pathway in pancreatic cancer cells. Biochim Biophys Acta. 2017; 1861:296–306. https://doi.org/10.1016/j.bbagen.2016.10.006.
97. Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011; 60:1419–30. https://doi.org/10.1007/s00262-011-1028-0.
98. Mace TA, Bloomston M, Lesinski GB. Pancreatic cancer-associated stellate cells: A viable target for reducing immunosuppression in the tumor microenvironment. OncoImmunology. 2013; 2:e24891. https://doi.org/10.4161/onci.24891.
99. Hamada S, Masamune A, Yoshida N, Takikawa T, Shimosegawa T. IL-6/STAT3 Plays a Regulatory Role in the Interaction Between Pancreatic Stellate Cells and Cancer Cells. Dig Dis Sci. 2016; 61:1561–71. https://doi.org/10.1007/s10620-015-4001-5.
100. Edderkaoui M, Xu S, Chheda C, Morvaridi S, Hu RW, Grippo PJ, Mascariñas E, Principe DR, Knudsen B, Xue J, Habtezion A, Uyeminami D, Pinkerton KE, Pandol SJ. HDAC3 mediates smoking-induced pancreatic cancer. Oncotarget. 2016; 7:7747–60. https://doi.org/10.18632/oncotarget.6820.
101. Witteck L, Jaster R. Trametinib and dactolisib but not regorafenib exert antiproliferative effects on rat pancreatic stellate cells. Hepatobiliary Pancreat Dis Int. 2015; 14:642–50. https://doi.org/10.1016/s1499-3872(15)60032-7.
102. Elsner A, Lange F, Fitzner B, Heuschkel M, Krause BJ, Jaster R. Distinct antifibrogenic effects of erlotinib, sunitinib and sorafenib on rat pancreatic stellate cells. World J Gastroenterol. 2014; 20:7914–25. https://doi.org/10.3748/wjg.v20.i24.7914.
103. Cousin JM, Cloninger MJ. The Role of Galectin-1 in Cancer Progression, and Synthetic Multivalent Systems for the Study of Galectin-1. Int J Mol Sci. 2016; 17. https://doi.org/10.3390/ijms17091566.
104. Camby I, Le Mercier M, Lefranc F, Kiss R. Galectin-1: a small protein with major functions. Glycobiology. 2006; 16:137r-57r. https://doi.org/10.1093/glycob/cwl025.
105. Xue X, Lu Z, Tang D, Yao J, An Y, Wu J, Li Q, Gao W, Xu Z, Qian Z, Dai C, Wei J, Miao Y, et al. Galectin-1 secreted by activated stellate cells in pancreatic ductal adenocarcinoma stroma promotes proliferation and invasion of pancreatic cancer cells: an in vitro study on the microenvironment of pancreatic ductal adenocarcinoma. Pancreas. 2011; 40:832–9. https://doi.org/10.1097/MPA.0b013e318217945e.
106. Tang D, Gao J, Wang S, Yuan Z, Ye N, Chong Y, Xu C, Jiang X, Li B, Yin W, Miao Y, Wang D, Jiang K. Apoptosis and anergy of T cell induced by pancreatic stellate cells-derived galectin-1 in pancreatic cancer. Tumour Biol. 2015; 36:5617–26. https://doi.org/10.1007/s13277-015-3233-5.
107. Masamune A, Satoh M, Hirabayashi J, Kasai K, Satoh K, Shimosegawa T. Galectin-1 induces chemokine production and proliferation in pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2006; 290:G729–36. https://doi.org/10.1152/ajpgi.00511.2005.
108. Wu MH, Hong HC, Hong TM, Chiang WF, Jin YT, Chen YL. Targeting galectin-1 in carcinoma-associated fibroblasts inhibits oral squamous cell carcinoma metastasis by downregulating MCP-1/CCL2 expression. Clin Cancer Res. 2011; 17:1306–16. https://doi.org/10.1158/1078-0432.CCR-10-1824.
109. Berberat PO, Friess H, Wang L, Zhu ZW, Bley T, Frigeri L, Zimmermann A, Buchler MW. Comparative analysis of galectins in primary tumors and tumor metastasis in human pancreatic cancer. J Histochem Cytochem. 2001; 49:539–49.
110. Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006; 90:1–50. https://doi.org/10.1016/S0065-2776(06)90001-7.
111. Dings RP, Van Laar ES, Loren M, Webber J, Zhang Y, Waters SJ, Macdonald JR, Mayo KH. Inhibiting tumor growth by targeting tumor vasculature with galectin-1 antagonist anginex conjugated to the cytotoxic acylfulvene, 6-hydroxylpropylacylfulvene. Bioconjug Chem. 2010; 21:20–27. https://doi.org/10.1021/bc900287y.
112. Tang D, Zhang J, Yuan Z, Gao J, Wang S, Ye N, Li P, Gao S, Miao Y, Wang D, Jiang K. Pancreatic satellite cells derived galectin-1 increase the progression and less survival of pancreatic ductal adenocarcinoma. PLoS One. 2014; 9:e90476. https://doi.org/10.1371/journal.pone.0090476.
113. Martinez-Bosch N, Fernandez-Barrena MG, Moreno M, Ortiz-Zapater E, Munne-Collado J, Iglesias M, Andre S, Gabius HJ, Hwang RF, Poirier F, Navas C, Guerra C, Fernandez-Zapico ME, et al. Galectin-1 drives pancreatic carcinogenesis through stroma remodeling and Hedgehog signaling activation. Cancer Res. 2014; 74:3512–24. https://doi.org/10.1158/0008-5472.CAN-13-3013.
114. Chen R, Pan S, Ottenhof NA, de Wilde RF, Wolfgang CL, Lane Z, Post J, Bronner MP, Willmann JK, Maitra A, Brentnall TA. Stromal galectin-1 expression is associated with long-term survival in resectable pancreatic ductal adenocarcinoma. Cancer Biol Ther. 2012; 13:899–907. https://doi.org/10.4161/cbt.20842.
115. Rosenberger I, Strauss A, Dobiasch S, Weis C, Szanyi S, Gil-Iceta L, Alonso E, Gonzalez Esparza M, Gomez-Vallejo V, Szczupak B, Plaza-Garcia S, Mirzaei S, Israel LL, et al. Targeted diagnostic magnetic nanoparticles for medical imaging of pancreatic cancer. J Control Release. 2015; 214:76–84. https://doi.org/10.1016/j.jconrel.2015.07.017.
116. Astorgues-Xerri L, Riveiro ME, Tijeras-Raballand A, Serova M, Neuzillet C, Albert S, Raymond E, Faivre S. Unraveling galectin-1 as a novel therapeutic target for cancer. Cancer Treat Rev. 2014; 40:307–19. https://doi.org/10.1016/j.ctrv.2013.07.007.
117. Jiang WG, Martin TA, Parr C, Davies G, Matsumoto K, Nakamura T. Hepatocyte growth factor, its receptor, and their potential value in cancer therapies. Crit Rev Oncol Hematol. 2005; 53:35–69.
118. Gholamin S, Fiuji H, Maftouh M, Mirhafez R, Shandiz FH, Avan A. Targeting c-MET/HGF signaling pathway in upper gastrointestinal cancers: rationale and progress. Curr Drug Targets. 2014; 15:1302–11.
119. Pothula SP, Xu Z, Goldstein D, Biankin AV, Pirola RC, Wilson JS, Apte MV. Hepatocyte growth factor inhibition: a novel therapeutic approach in pancreatic cancer. Br J Cancer. 2016; 114:269–80. https://doi.org/10.1038/bjc.2015.478.
120. Yang XP, Liu SL, Xu JF, Cao SG, Li Y, Zhou YB. Pancreatic stellate cells increase pancreatic cancer cells invasion through the hepatocyte growth factor /c-Met/survivin regulated by P53/P21. Exp Cell Res. 2017; 357:79–87. https://doi.org/10.1016/j.yexcr.2017.04.027.
121. Goyal L, Muzumdar MD, Zhu AX. Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res. 2013; 19:2310–8. https://doi.org/10.1158/1078-0432.CCR-12-2791.
122. Fajardo-Puerta AB, Mato Prado M, Frampton AE, Jiao LR. Gene of the month: HGF. J Clin Pathol. 2016; 69:575–79. https://doi.org/10.1136/jclinpath-2015-203575.
123. Delitto D, Vertes-George E, Hughes SJ, Behrns KE, Trevino JG. c-Met signaling in the development of tumorigenesis and chemoresistance: potential applications in pancreatic cancer. World J Gastroenterol. 2014; 20:8458–70. https://doi.org/10.3748/wjg.v20.i26.8458.
124. Tjomsland V, Aasrum M, Christoffersen T, Gladhaug IP. Functional heterogeneity in tumor-derived human pancreatic stellate cells: differential expression of HGF and implications for mitogenic signaling and migration in pancreatic cancer cells. Oncotarget. 2017; 8:71672–84. https://doi.org/10.18632/oncotarget.17800.
125. Patel MB, Pothula SP, Xu Z, Lee AK, Goldstein D, Pirola RC, Apte MV, Wilson JS. The role of the hepatocyte growth factor/c-MET pathway in pancreatic stellate cell-endothelial cell interactions: antiangiogenic implications in pancreatic cancer. Carcinogenesis. 2014; 35:1891–900. https://doi.org/10.1093/carcin/bgu122.
126. Lowery FJ, Yu D. Growth factor signaling in metastasis: current understanding and future opportunities. Cancer Metastasis Rev. 2012; 31:479–91. https://doi.org/10.1007/s10555-012-9380-x.
127. Mankal P, O'Reilly E. Sunitinib malate for the treatment of pancreas malignancies--where does it fit? Expert Opin Pharmacother. 2013; 14:783–92. https://doi.org/10.1517/14656566.2013.776540.
128. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009; 15:220–31. https://doi.org/10.1016/j.ccr.2009.01.027.
129. Tijeras-Raballand A, Neuzillet C, Couvelard A, Serova M, de Gramont A, Hammel P, Raymond E, Faivre S. Resistance to targeted therapies in pancreatic neuroendocrine tumors (PNETs): molecular basis, preclinical data, and counteracting strategies. Target Oncol. 2012; 7:173–81. https://doi.org/10.1007/s11523-012-0229-6.
130. Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009; 15:232–9. https://doi.org/10.1016/j.ccr.2009.01.021.
131. Kindler HL, Niedzwiecki D, Hollis D, Sutherland S, Schrag D, Hurwitz H, Innocenti F, Mulcahy MF, O'Reilly E, Wozniak TF, Picus J, Bhargava P, Mayer RJ, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J Clin Oncol. 2010; 28:3617–22. https://doi.org/10.1200/JCO.2010.28.1386.
132. Van Cutsem E, Vervenne WL, Bennouna J, Humblet Y, Gill S, Van Laethem JL, Verslype C, Scheithauer W, Shang A, Cosaert J, Moore MJ. Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J Clin Oncol. 2009; 27:2231–7. https://doi.org/10.1200/JCO.2008.20.0238.
133. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, et al, and National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007; 25:1960–6. https://doi.org/10.1200/JCO.2006.07.9525.
134. Quint K, Tonigold M, Di Fazio P, Montalbano R, Lingelbach S, Ruckert F, Alinger B, Ocker M, Neureiter D. Pancreatic cancer cells surviving gemcitabine treatment express markers of stem cell differentiation and epithelial-mesenchymal transition. Int J Oncol. 2012; 41:2093–102. https://doi.org/10.3892/ijo.2012.1648.
135. Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013; 15:2–8. https://doi.org/10.1038/ncb2641.
136. Ikenaga N, Ohuchida K, Mizumoto K, Cui L, Kayashima T, Morimatsu K, Moriyama T, Nakata K, Fujita H, Tanaka M. CD10+ pancreatic stellate cells enhance the progression of pancreatic cancer. Gastroenterology. 2010; 139:1041–51, 51 e1-8. https://doi.org/10.1053/j.gastro.2010.05.084.
137. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, Nourse C, Murtaugh LC, Harliwong I, et al, and Australian Pancreatic Cancer Genome Initiative. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016; 531:47–52.
138. Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, Choti MA, Yeo CJ, McCue P, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015; 6:6744.
139. Zhu Z, Xu Y, Zhao J, Liu Q, Feng W, Fan J, Wang P. miR-367 promotes epithelial-to-mesenchymal transition and invasion of pancreatic ductal adenocarcinoma cells by targeting the Smad7-TGF-beta signalling pathway. Br J Cancer. 2015; 112:1367–75. https://doi.org/10.1038/bjc.2015.102.
140. Ouyang H, Gore J, Deitz S, Korc M. microRNA-10b enhances pancreatic cancer cell invasion by suppressing TIP30 expression and promoting EGF and TGF-beta actions. Oncogene. 2014; 33:4664–74. https://doi.org/10.1038/onc.2013.405.
141. Wang C, Liu P, Wu H, Cui P, Li Y, Liu Y, Liu Z, Gou S. MicroRNA-323-3p inhibits cell invasion and metastasis in pancreatic ductal adenocarcinoma via direct suppression of SMAD2 and SMAD3. Oncotarget. 2016; 7:14912–24. https://doi.org/10.18632/oncotarget.7482.
142. Mody HR, Hung SW, AlSaggar M, Griffin J, Govindarajan R. Inhibition of S-Adenosylmethionine-Dependent Methyltransferase Attenuates TGF beta 1-Induced EMT and Metastasis in Pancreatic Cancer: Putative Roles of miR-663a and miR-4787-5p. Mol Cancer Res. 2016; 14:1124–35. https://doi.org/10.1158/1541-7786.Mcr-16-0083.
143. Harazono Y, Muramatsu T, Endo H, Uzawa N, Kawano T, Harada K, Inazawa J, Kozaki K. miR-655 Is an EMT-Suppressive MicroRNA Targeting ZEB1 and TGFBR2. Plos One. 2013; 8. https://doi.org/ARTN e6275710.1371/journal.pone.0062757.
144. Kang Y, Roife D, Lee Y, Lv H, Suzuki R, Ling J, Rios Perez MV, Li X, Dai B, Pratt M, Truty MJ, Chatterjee D, Wang H, et al. Transforming Growth Factor-beta Limits Secretion of Lumican by Activated Stellate Cells within Primary Pancreatic Adenocarcinoma Tumors. Clin Cancer Res. 2016; 22:4934–46. https://doi.org/10.1158/1078-0432.CCR-15-2780.
145. Xian G, Zhao J, Qin C, Zhang Z, Lin Y, Su Z. Simvastatin attenuates macrophage-mediated gemcitabine resistance of pancreatic ductal adenocarcinoma by regulating the TGF-beta1/Gfi-1 axis. Cancer Lett. 2017; 385:65–74. https://doi.org/10.1016/j.canlet.2016.11.006.
146. Melisi D, Xia Q, Paradiso G, Ling J, Moccia T, Carbone C, Budillon A, Abbruzzese JL, Chiao PJ. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J Natl Cancer Inst. 2011; 103:1190–204. https://doi.org/10.1093/jnci/djr243.
147. Cioffi M, Trabulo SM, Sanchez-Ripoll Y, Miranda-Lorenzo I, Lonardo E, Dorado J, Reis Vieira C, Ramirez JC, Hidalgo M, Aicher A, Hahn S, Sainz B Jr, Heeschen C. The miR-17-92 cluster counteracts quiescence and chemoresistance in a distinct subpopulation of pancreatic cancer stem cells. Gut. 2015; 64:1936–48. https://doi.org/10.1136/gutjnl-2014-308470.
148. Leal AS, Williams CR, Royce DB, Pioli PA, Sporn MB, Liby KT. Bromodomain inhibitors, JQ1 and I-BET 762, as potential therapies for pancreatic cancer. Cancer Lett. 2017; 394:76–87. https://doi.org/10.1016/j.canlet.2017.02.021.
149. Park JK, Kim Y, Kim H, Jeon J, Kim TW, Park JH, Hwnag YI, Lee WJ, Kang JS. The anti-fibrotic effect of GV1001 combined with gemcitabine on treatment of pancreatic ductal adenocarcinoma. Oncotarget. 2016; 7:75081–93. https://doi.org/10.18632/oncotarget.12057.
150. Zhu H, Qin H, Li DM, Liu J, Zhao Q. Effect of PPM1H on malignant phenotype of human pancreatic cancer cells. Oncol Rep. 2016; 36:2926–34. https://doi.org/10.3892/or.2016.5065.
151. Jung H, Kim JS, Kim WK, Oh KJ, Kim JM, Lee HJ, Han BS, Kim DS, Seo YS, Lee SC, Park SG, Bae KH. Intracellular annexin A2 regulates NF-kappaB signaling by binding to the p50 subunit: implications for gemcitabine resistance in pancreatic cancer. Cell Death Dis. 2015; 6:e1606. https://doi.org/10.1038/cddis.2014.558.
152. Guan J, Zhang H, Wen Z, Gu Y, Cheng Y, Sun Y, Zhang T, Jia C, Lu Z, Chen J. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer Lett. 2014; 345:132–9. https://doi.org/10.1016/j.canlet.2013.12.006.