
INTRODUCTION
ICa is the key mediator for the excitation-contraction (E-C) coupling in cardiomyocytes. Ca2+ entry through L-type calcium channel (LTCC) triggers a release of Ca2+ from sarcoplasmic reticulum (SR), initiating myofilament contraction. The amplitude and the kinetics of ICa determine the amount of Ca2+ entry and consequently the contractile force and relaxation. In addition, Ca2+ entry and SR Ca2+ release activates a number of Ca2+-sensitive signaling cascades, most important among them is Ca2+/calmodulin-dependent protein kinase II (CaMKII). CaMKII plays a key role in regulation of E-C coupling by phosphorylation of a number of proteins involved in Ca2+ handling, such as LTCC, ryanodine receptor (RyR2), sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) and phospholamban (PLB) [1]. It is also a known powerful stimulator of the excitation-transcription (E-T) coupling mediated by class II histone deacetylases and myocyte enhancer factor 2 [2] and a arrhythmogenic molecule by regulation of several important membrane ion channels [3], of which the ICa, transient outward current (Ito) and late sodium current (INaL) are most extensively studied [4, 5]. In myocytes, CaMKII activation is initiated by Ca2+/CaM binding and can be mediated by β-AR agonist stimulation and oxidative stress. In the setting of heart failure (HF), reduction of Ito and increase of INaL prolong action potential duration (APD) which increases Ca2+ entry through LTCC and the subsequent SR Ca2+ release, leading to Ca2+ overload and CaMKII activation, and this process is facilitated by the enhanced β-AR adrenergic stimulation and oxidative stress, which causes excessive CaMKII activation. On the other hand, increased CaMKII activity hyperphosphorylates LTCC, leading to an increase in ICa magnitude and slowing of the inactivation time course. This induces a consequent increase in Ca2+ entry and further CaMKII activation. In other words, HF-related ICa remodeling is a CaMKII-dependent and self-exacerbated process. Indeed, we have recently reported that HF-related ICa remodeling is a result of increased CaMKII-dependent Ca2+ channel phosphorylation, whereas other regulatory pathways, e.g. PKA, may only play a minor role (if any) [6].
In fact, in the normal heart, a beat-to-beat CaMKII activation is delicately regulated, which effectively prevents CaMKII from over-activation. For instant, we have recently reported an important mechanism which can prevent calcium-induced CaMKII hyperphosphorylation [7]. We demonstrated that a significant amount of inactive CaMKII in myocytes is preserved in the CaMKII-Kv4.3 molecular complex. The Kv4.3-coupled CaMKII cannot be activated by elevated [Ca2+]i. This finding revealed that the CaMKII-Kv4.3 units are important intrinsic CaMKII inhibitor for the delicate regulation of CaMKII activity in ventricular myocytes. However, a growing body of evidence has elucidated that a reduction of Ito is a well-known feature of ventricular myocytes from HF animal models and patients, and this remodeling occurs at the early developing stage [8, 9]. Importantly, the majority of experiments in animal models and HF patients have correlated the decrease in Ito with the reduction of Kv4.3 expression [8, 10, 11], implicating Kv4.3 down-regulation in excessive CaMKII activation and the consequent remodeling of cellular Ca2+ handling in HF. In the current study, we tested whether Kv4.3 expression (restoration) can reverse ICa remodeling in HF ventricular myocytes, which is known mainly caused by excessive CaMKII activation.
RESULTS
Kv4.3 transfection in HF ventricular myocytes
HF was successfully produced 2 weeks after sTAB in mice confirmed by echocardiogram, with an increase in LV end-diastolic volume (from 38.2 ± 0.7 to 90.5 ± 3.0 μl) and decrease in ejection fraction (from 83.1 ± 0.5% to 38.8 ± 1.2%) (p <0.05, n=31). Ad-Kv4.3 was then injected to the LV myocardium in the HF mice. 7 days after in vivo adenovirus injection, HF mice transfected with Ad-Kv4.3 (n = 10) was used for myocyte isolation. Consistent with our previous results [7], multisite Ad-v4.3 injection in adult mouse LV produced an overall transfection rate of 70%, and the transfected myocytes showed normal shape and clear striations (Figure 1A & 1B). As a limit of multisite Ad-v4.3 injection, Kv4.3 transfection was heterogeneously distributed in the LV wall, with a higher transfection rate in the endocardium than epicardium [7]. Transient outward current was recorded in HF ventricular myocytes with and without Ad-Kv4.3 expression, respectively. The GFP positive myocytes under the fluorescence microscope indicate successful Kv4.3 expression. As expected, myocytes transfected with Ad-K4.3 manifested a nearly 2-fold increase in the transient outward current density, indicating successful expression of Kv4.3 channel (Figure 1B & 1C). The transient outward current density at +60 mV was increased from 11.09 ± 0.72 A/pF to 19.26 ± 1.19 pA/pF, a level similar to the that recorded in the endocardial myocytes isolated from the normal mice (19.2±1.1 pA/pF) [12]. In line with this, action potentials recorded in the Kv4.3 transfected myocytes manifested significant shortening of APD (Figure 1D).
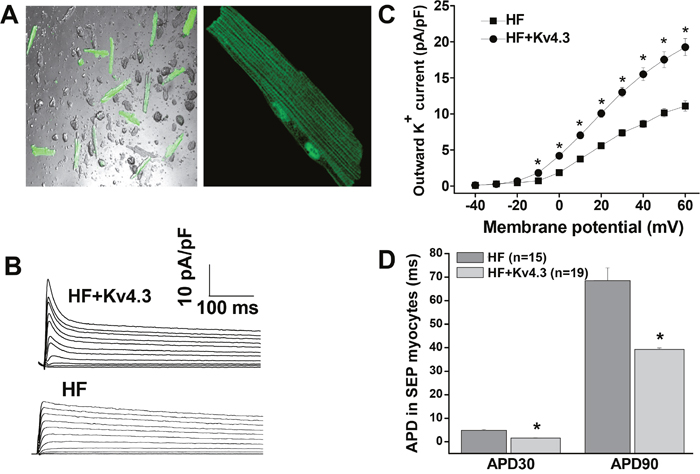
Figure 1: In vivo adenovirus-mediated transfection of Kv4.3 in HF ventricular myocytes. (Panel A) Isolated LV myocytes from HF mice 7 days after Ad-Kv4.3 transfection (n=5). Green cells indicate successful Kv4.3 expression. (Panel B) Representative current traces of transient outward current elicited by 5000 ms test pulses in 10 mV increments from a holding potential of -80 mV to +60 mV at pulse intervals of 10 s. (Panel C) Voltage relationship of transient outward current recorded in HF myocytes with (n=10) and without Ad-Kv4.3 (n=10) transfection, respectively. Current amplitudes were normalized to cell capacitance and plotted as mean values. (Panel D) Action potential durations recorded in the isolated LV subepicardial myocytes with and without Ad-Kv4.3 transfection. Vertical bars represent S.E.M. * denotes p < 0.05, compared to the controls.
Inhibition of CaMKII activity by Kv4.3 expression in HF ventricular myocytes
We have recently demonstrated that in vivo Kv4.3 transfection in LV ventricular myocytes in the normal mice significantly inhibited CaMKII activity by direct binding to the CaM binding sites and forming a CaMKII-Kv4.3 molecular complex [7]. Here, we tested the effect of Kv4.3 transfection on CaMKII inhibition in HF ventricular myocytes. We found that in the Ad-Kv4.3 transfected HF LV, CaMKII autophosphorylation was significantly reduced with a much larger reduction in the compartmentalized CaMKII activity in the SR region which was manifested by a much greater inhibition of the PLB phosphorylation at Thr17, a CaMKII-dependent phosphorylation site (Figure 2). These results suggest that Kv4.3 expression in failing ventricle caused a predominant inhibition of the membrane compartmentalized CaMKII activity, with only minor inhibition of the overall intracellular CaMKII activity. This is consistent with the membrane distribution of CaMKII-Kv4.3 units, i.e. CaMKII-Kv4.3 units protect against activation of CaMKII in the sarcolemmal membrane microdomains by trapping inactive CaMKII to CaMKII-Kv4.3 units and preventing it from being activated by elevated Ca2+ in the microdomain (e.g. LTCC-SR microdomain [13]).
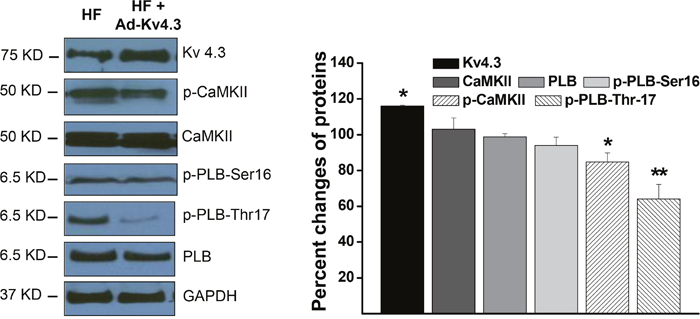
Figure 2: Western blot results showing protein expressions in the HF LV myocytes with and without Ad-Kv4.3 transfection, respectively. The left panel shows representative examples of Western blots. The statistic values shown in the right panel were from 3 mice and 3 independent experiments for each protein. * denotes p < 0.05, ** denotes p < 0.01, compared to the controls.
Kv4.3 expression in HF ventricular myocytes reversed ICa density and inactivation time course
We have recently reported a transmural gradient of ICa density in the normal and HF LV, with a smaller ICa density in the subendocardial (SEN) than in the subepicardial (SEP) myocytes [6]. We also demonstrated a proportional increase in ICa density and a slowed inactivation in HF LV which is caused by the excessive CaMKII-dependent phosphorylation of LTCC. Consistent with our recent findings that Kv4.3 expression can selectively inhibit membrane associated CaMKII activation by binding to the Calmodulin-binding site [7], here, we found that Kv4.3 expression reversed ICa remodeling in HF LV by reducing ICa density and acceleration of ICa inactivation, while the transmural gradient was preserved. For instance, peak ICa (recorded at test potential of +10 mV) was reduced from 7.48 ± 0.31 pA/pF (n=23) to 6.34 ± 0.35 pA/pF (n=15) for SEP and from 6.51 ± 0.25 pA/pF (n=23) to 4.9 ± 0.24 pA/pF (n=15) for SEN cells, respectively (p < 0.05)(Figure 3A). Interestingly, Kv4.3 expression reduced ICa density in HF ventricular myocytes to a level close to that we previously reported in the sham mouse ventricular myocytes [6].
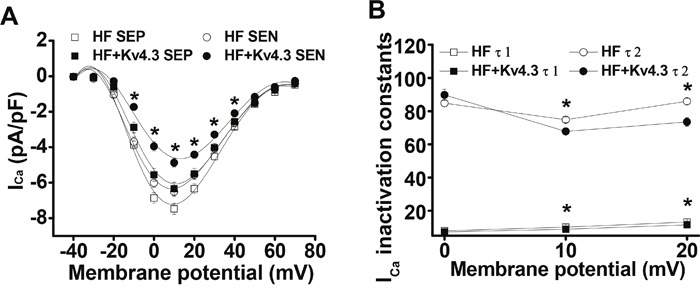
Figure 3: (Panel A) ICa-voltage relationship recorded in the SEP and SEN myocytes from the HF LV with and without Ad-Kv4.3 transfection, respectively. Current densities were plotted as mean values. Vertical bars represent S.E.M. Peak ICa (recorded at test potential of +10 mV) was reduced from 7.48 ± 0.31 pA/pF (n=23) to 6.34 ± 0.35 pA/pF (n=15) for SEP and from 6.51 ± 0.25 pA/pF (n=23) to 4.9 ± 0.24 pA/pF (n=15) for SEN cells, respectively. (Panel B) Statistic analysis of ICa inactivation time constants recorded from the HF LV myocytes with (n=36) and without Ad-Kv4.3 (n=46) transfection, respectively. The decay phase was fit by biexponential function. Mean vales were obtained by fitting currents recorded from each individual cell. τ1 and τ2 refer to the fast and slow inactivation time constants, respectively. Vertical bars represent S.E.M. * denotes p < 0.05, compared to the controls. SEP and SEN represent myocytes isolated from subepicardial and subendocardial myocardium, respectively.
In addition to the proportionately reduction in ICa density by Kv4.3 expression, we also evaluated the kinetics of ICa inactivation (current decay). We observed that ICa inactivation was significantly accelerated in HF myocytes transfected with Kv4.3. To quantify these changes, ICa inactivation was modeled as an exponential decay and fit by 2 exponential functions [11]. The mean values of fast (τ1) and slow (τ2) time constants in Ad-Kv4.3 transfected (n=18) and untransfected HF myocytes (n=19) were shown in Figure 3B. ICa inactivation was significantly accelerated at potentials of +10mV and above in Kv4.3 transfected HF myocytes. For example, at +10 mV, τ1 and τ2 for the Kv4.3 transfected myocytes (n=36) were 8.9 ± 0.7 ms and 67.8 ± 1.9 ms respectively, compared with 10.2 ± 0.4 ms and 74.9 ± 1.7 ms for untransfected myocytes (n=46, p < 0.05).
Voltage-dependence of ICa activation and inactivation
To measure the voltage dependence of ICa inactivation, we used a two-pulse protocol with a 300 ms conditioning pulse at potentials ranging from -70 mV to +30 mV (from holding potential of –50 mV), followed by a 300 ms test pulse to +10 mV. To determine the voltage dependence of ICa activation, we used a holding potential of –50 mV and steps of 300 ms duration test pulses from –40 to +60 in 10 mV steps. These protocols have been described in detail in our previous studies [7]. For both activation and inactivation of ICa, no significant difference in voltage-dependence has been observed between SEP and SEN myocytes and between the Ad-Kv4.3 transfected and untransfected HF LV myocytes (Figure 4A).
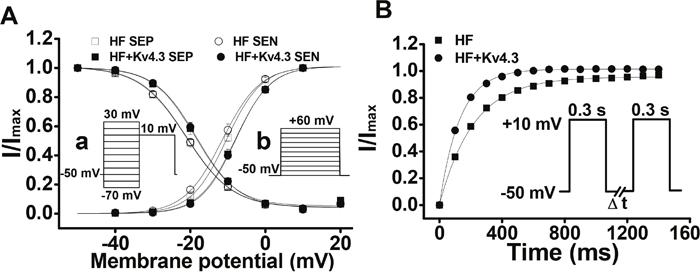
Figure 4: (Panel A) Voltage-dependent activation and inactivation of ICa recorded in the HF LV myocytes with (n=18 for SEP, n=13 for SEN) and without Ad-Kv4.3 (n=23 for both SEP and SEN) transfection, respectively. Currents elicited by each test pulse were normalized to the maximum current. The mean values are plotted versus conditioning pulse potentials (for inactivation, inset a) or test pulse potentials (for activation, inset b). Vertical bars represent S.E.M. (Panel B) Mean values of ICa recovery from inactivation. Paired 300 ms pulses at +10 mV were applied from a holding potential of -50 mV with the time interval (ΔT) varied from 50 ms to 1500 ms in 50 ms steps (insert). The protocol was repeated every 10 s. The mean recovery time constants were 128.2 ± 1.5 ms (n=28) for HF myocytes transfected with Ad-Kv4.3 and 216.3 ± 3.9 ms (n=31) for untransfected HF myocytes, respectively.
Accelerated ICa recovery from inactivation in HF myocytes transfected with Kv4.3
To record the time course of ICa recovery from inactivation, we held cells at a holding potential of –50 mV and applied a 300 ms pulse to +10 mV, followed by a variable time period marked as DT at –50 mV and a test pulse of 300 ms to +10 mV (insert of Figure 4B). The time constants of recovery from inactivation were evaluated by fitting data for each cell with a mono-exponential equation. In HF myocytes transfected with Kv4.3, the recovery time course was significantly accelerated (Figure 4B). The mean recovery time constants were 128.2 ± 1.5 ms (n=28) for HF myocytes transfected with Ad-Kv4.3 and 216.3 ± 3.9 ms (n=31) for untransfected HF myocytes, respectively (p<0.05). These data demonstrated that Kv4.3 expression reversed the HF-related delay of ICa recovery from inactivation to a level we reported in ventricular myocytes isolated from the normal mice [7].
Kv4.3 expression recovered the Ca2+-induced ICa facilitation in HF LV myocytes
In addition to the Increased peak ICa, slowed inactivation and recovery from inactivation, another hallmark change of ICa in the HF myocytes is the blunted frequency-dependent Ca2+-induced ICa facilitation [6]. Here, we measured ICa facilitation in HF LV myocytes that have been transfected with Ad-Kv4.3. Consistent with our previous results recorded in HF ventricular myocytes [6], Ca2+-induced ICa facilitation was blunted and a frequency-dependent ICa suppression was recorded in all of the untransfected HF LV myocytes (n=32). However, Ca2+-induced ICa facilitation was observed in each recorded HF LV myocytes transfected with Ad-Kv4.3 (n=39) (Figure 5). These data, then, are consistent with the reduced CaMKII activity and a consequent reversion of ICa facilitation.
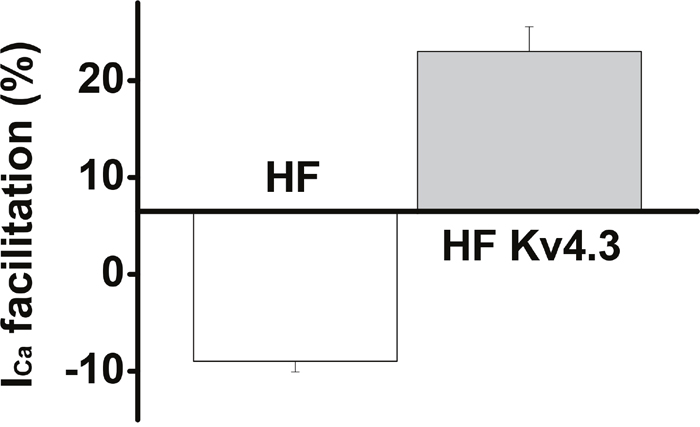
Figure 5: Mean values of the frequency-dependent Ca2+-induced ICa facilitation in HF LV myocytes with (n=39) and without Ad-Kv4.3 (n=32) transfection, respectively, showing a recovery of ICa facilitation in HF LV myocytes with Kv4.3 expression.
DISCUSSION
There is growing body of evidence suggesting that CaMKII is an important sensor for the altered Ca2+ handling in HF myocytes. We have reported that in pressure-overload HF mouse LV myocytes, CaMKII activity increased by ≈2.6 fold [6]. Accordingly, ICa was significantly potentiated with increased current density and slowed inactivation (i.e. changing LTCC gating to mode 2) [6]. These changes facilitate Ca2+ influx during diastole. On the other hand, excessive CaMKII activation induces diastolic SR Ca2+ leak, which further increases diastolic Ca2+, especially in the LTCC-SR microdomain, causing a persistent CaMKII activation. The excessive activation of CaMKII in turn hyperphosphorylates LTCC, causing further remodeling of ICa. Meanwhile, CaMKII-dependent RyR hyperphosphorylation causes SR Ca2+ leak and increase in the diastolic Ca2+ level, which facilitates the Ca2+ efflux function of sodium-calcium exchanger (INCX), which, in combination with altered LTCC gating, leads to prolongation of APD and triggers abnormal impulses [14]. Therefore, ICa remodeling is a result of increased CaMKII activation and also a mediator for the disrupted calcium homeostasis in HF. Correction of ICa remodeling is an important measure contributing to normalization of calcium handling in the failing heart.
Our recent work demonstrated that Kv4.3 is an intrinsic CaMKII inhibitor which inhibits predominantly the membrane-associated CaMKII with less inhibition of the overall CaMKII [7]. In the setting of HF, Kv4.3 down-regulation directly contributes to the excessive activation of CaMKII in the membrane LTCC microdomian, causing HF-related ICa remodeling. This has been further demonstrated by a most recent study which showed that an increase in the open probability of LTCC is linked to enhanced CaMKII-mediated phosphorylation of LTCC in the membrane microdomains, contributing to the development of early afterdepolarizations [15]. Taken together, the down-regulation of Kv4.3 is an important mechanism underlying membrane-associated CaMKII activation and CaMKII-mediated ICa remodeling in HF. The main innovation of this study is to determine whether reestablishing Kv4.3 expression can effectively reverse ICa remodeling in HF.
In this study, we report a significant reduction in ICa density in the HF LV myocytes transfected with Ad-Kv4.3, leaving the transmural gradient intact. Interestingly, the mean level of ICa density recorded in the HF LV myocytes with Kv4.3 expression was quite close to the value recorded in the sham mouse LV myocytes [6]. In addition to the current density, Kv4.3 expression induced a regression of the biophysical properties of ICa in HF myocytes, i.e. the time courses of ICa inactivation and recovery from inactivation were accelerated. Furthermore, the frequency-dependent Ca2+-induced ICa facilitation which is known mediated by beat-to-beat activation of CaMKII [16], a common feature of ventricular myocytes but has been blunted in HF [6], was recovered by Kv4.3 expression. Altogether, these results suggest that the expression of Kv4.3 can successfully reverse ICa remodeling in ventricular myocytes of HF.
Although CaMKII is proposed to be a novel therapeutic target for HF and HF-related ventricular arrhythmias [17-21], due to the ubiquitous distribution of CaMKII and lack of cardiac specific CaMKII inhibitor, the pharmacological CaMKII inhibition is actually not clinically applicable. To selectively inhibit the compartmentalized CaMKII activity, we propose to re-establish the intrinsic CaMKII-Kv4.3 units by in vivo expression of Kv4.3 in HF ventricular myocytes. Due to the membrane distribution [7], CaMKII-Kv4.3 units prevent Ca2+-induced CaMKII activation (i.e. the Ca2+ entered through LTCC and the Ca2+ released from RyR at each heart beat) and thus largely inhibit CaMKII activity in the LTCC-SR microdomain with less inhibition to the overall CaMKII. Our results revealed this pattern of CaMKII inhibition in Kv4.3 transfected HF myocytes, demonstrated by a greater reduction in p-Thr17-PLB (local CaMKII activity) than p-CaMKII (overall bulk CaMKII activity). It is predictable that restoration of the down-regulated Kv4.3 in HF LV is of great importance in normalizing Ca2+ homeostasis and membrane-associated ion channel remodeling via specific inhibition of the compartmentalized CaMKII activity, thus improving E-C coupling and inhibiting ventricular arrhythmias in HF. This opens a direction of future study.
Limitations
As an endogenous CaMKII inhibitor, Kv4.3 mainly affects CaMKII activity in the sarcolemmal compartment due to their membrane localization in myocytes. Meanwhile, Kv4.3 expression also leads to APD shortening and a consequent reduction in [Ca2+]i, which decreases the overall intracellular CaMKII activity. This is exactly what we observed in this study. However, since the LTCC is a membrane protein, the membrane-localized CaMKII activity is accordingly much more important for the regulation of ICa than the bulk CaMKII activity. In this study, we have demonstrated the key role of Kv4.3 expression in the regression of ICa remodeling in HF ventricular myocytes. We cannot, however, dissect the contribution of the overall CaMKII activation which is associated with the APD prolongation. This portion of contribution (if any) would be minor due to the fact of small change in the bulk CaMKII activity which is also away from the LTCC location. As an advantage, we recorded ICa in the myocytes with and without successful Kv4.3 expression in the same heart, which avoided the influence of heart condition and the variations of the dissociation-related cell quality.
In addition to ICa, other membrane currents, such as INa, Ik1 and INCX may also be modified by Kv4.3 expression since they are direct substrates of CaMKII. This may lead to a complex change in the cellular Ca2+ homeostasis and electrical property. These are certainly the interest of future study.
MATERIALS AND METHODS
All the animal experiments were performed conforming the NIH guidelines (Guide for the care and use of laboratory animals) with the protocols approved by the institutional Animal Care and Use Committee.
Severe thoracic aortic banding
Increased pressure in the proximal aorta was induced by severe thoracic aortic banding (sTAB) [6, 22]. Briefly, male C57BL6 mice (6-8 weeks old) were anesthetized with ketamine (100 mg/kg IP) plus xylazine (5 mg/kg IP). The mice were then ventilated at 120 breaths per minute at 0.1 mL tidal volume. A 3-mm left-sided thoracotomy was created at the second intercostal space. The transverse aortic arch was ligated (7-0 Prolene) between the innominate and left common carotid arteries with an overlying 28-gauge needle. The needle was then removed, leaving a discrete region of stenosis. The chest was closed using tissue adhesive.
Isolation of individual ventricular myocytes
Mice were euthanized with pentobarbital (150 mg/kg IP) and hearts were then removed through open chest surgery. LV myocytes were isolated enzymatically by a protocol we described previously [6]. In brief, after retrograde perfusion with Krebs-Ringer solution (2 mL/min for 5 min), the heart was perfused by a fresh solution containing 1 mg/mL Worthington type II collagenase for another 15 - 20 minutes. The heart was then removed and the LV wall was cut and put into a culture dish filled with “KB” solution. A small 90-degree curved forceps was used to carefully dissect a very thin layer of endomyocardium. The mid-region myocardium was dissected and discarded, leaving the remainder as epimyocardium. Endomyocardium and epimyocardium were minced into small pieces in “KB” solution, triturated and filtered. Only calcium-tolerant, quiescent and rod-shaped cells showing clear cross striations were studied.
In vivo Kv4.3 gene transfection
Multi-site adenovirus injection was performed through a 1 cm incision in the left side sixth intercostal space in HF mice. Delivery of adenoviral vectors into the LV myocardium can be performed safely under visual control. In order to increase the transfection rate and reduce the toxicity, adenoviruses Ad-Kv4.3 was purified before use [7]. A total volume of 12 μL adenovirus at 1.6 x 1012 plaque forming unit was distributed into the LV apex, anterior, lateral, and posterior walls with two injections for each region and 1.5 μl for each injection. 7 days after injection, mice were used for myocytes isolation.
Electrophysiological recordings
Currents were recorded in the LV myocytes isolated from HF mice 7 days after in vivo LV myocardium injection with Ad-Kv4.3. GFP positive myocytes were selected for ICa and outward K+ current recording under fluorescence microscope (Ad-K4.3 contains GFP reporter gene), while the currents in the GFP negative myocytes were recorded as control. Because ICa has transmural gradient in HF LV [6], we have separated the subepicardial and subendocardial myocytes for data acquisition and analysis.
ICa and outward K+ current were recorded by whole-cell voltage clamp technique, with pipette resistances of 2-3 M when filled with internal solution. ICa was recorded at room temperature. Cells were depolarized every 10 s from a holding potential of -50 mV to test potentials between -40 to +60 mV (10mV steps) for 300 ms. To minimize the impact of ICa run-down, we added 5 mmol/L Mg-ATP to the pipette solution and conducted data acquisition after 5 - 10 minutes of equilibration between pipette solution and intracellular contents [6]. Cells showing continuous current run-down were excluded from the analysis. For recording of outward K+ current, CdCl2 (0.2 mM) was used to block Ca2+ currents and the calcium-dependent transient outward current (Ito2) [6]. Myocytes were depolarized for 5000 ms from a holding potential of -80 mV to potentials from -40 mV to +60 mV in 10 mV voltage steps at a pulse interval of 15s. INa was eliminated by applying a 30 ms pre-pulse from holding potential to -40 mV.
Solutions
The Krebs-Ringer solution for cell isolation contained (mmol/L): NaCl 35, KCl 4.75, KH2P04 1.19, Na2HP04 16, Sucrose 134, NaCO3 25, Glucose 10, HEPES 10, pH 7.4 with NaOH. The “KB” solution for storage of cells contained (mmol/L): taurine 10, glutamic acid 70, KCl 25, KH2PO4 10, glucose 22, EGTA 0.5, pH adjusted to 7.2 with KOH. Perfusion solution for ICa recording contained (mmol/L): TEA 135, MgCl2 0.53, CaCl2 1.8, CsCl 20, HEPES 5. pH 7.4 with CsOH. Pipette solution for ICa recording contained (mmol/L): CsOH 110, aspartic acid 90, CsCl 20, tetraethylammonium Cl (TEA-Cl) 10, HEPES 10, EGTA 10, Mg-ATP 5, Na2 creatine phosphate 5, GTP (Tris) 0.4, leupeptin 0.1, pH 7.2 with CsOH. Tyrode’s solution for outward K+ current recording (mM): NaCl 135, MgCl2 1.1, CaCl2 1.8, KCl 5.4, HEPES 10, glucose 10, pH 7.4 with NaOH. Pipette solution for outward K+ current recordings (mM): KCl 130, MgCl2 1, HEPES 10, EGTA 5, Mg-ATP 5, Na2-creatine phosphate 5, pH 7.2 with KOH.
Western blot analysis
Myocytes were homogenized in 1% TX-100 buffer [50 mM Tris-HCl, pH 7.4, 4% glycerol, 1 mM DTT, 1% Triton X-100, 1 mM EDTA, Mini-Complete® protease inhibitor cocktail (Roche), and phosphatase inhibitor cocktails I and II (Sigma-Aldrich)] and immunoblotted for Kv4.3, CaMKII, phospho-CaMKII (p-CaMKII), p-PLB-Ser16, Phospho-Thr17-PLB, PLB, and GAPDH, respectively. Primary antibodies used in this study include rabbit polyclonal and mouse monoclonal anti-CaMKII antibodies (M-176 and G-1) (Santa Cruz Biotechnology, Santa Cruz, CA), polyclonal anti-phospho-CaMKII (Thr287) antibody (Cell Signaling solutions, Lake Placid, NY), goat polyclonal anti-PLB antibody (Santa Cruz Biotechnology), rabbit polyclonal anti-P-Ser16PLB antibodies (Badrilla, Leeds, UK), rabbit polyclonal anti-P-Thr17PLB antibodies (Badrilla, Leeds, UK), rabbit polyclonal antibody against Kv4.3 (Alomone Labs, Jerusalem, Isarael) and mouse monoclonal anti-GAPDH antibody (Millipore Corporation).
Statistical analysis
ICa amplitude was evaluated as the peak inward current. Biexponential functions were used to model current decay kinetics. Peak outward K+ current amplitude was measured as the difference between the peak current and the sustained current remaining at the end of a 5000 ms pulse. In order to correct for variability in cell size, current were expressed as current density by dividing the absolute current amplitude by cell capacitance (pA/pF). Results are expressed as mean ± SEM. Statistical analysis was performed using Sigmastat for Windows (Version 3.5).). The paired and unpaired t-test was used for the comparisons and p<0.05 was regarded as significant.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by grants to Yanggan Wang from the National Natural Science Foundation of China (NSFC, Grant Nos. 81270304 and 81420108004) and Zhongnan Hospital of Wuhan University (No. 7).
REFERENCES
1. Mattiazzi A, Bassani RA, Escobar AL, Palomeque J, Valverde CA, Vila PM, Bers DM. Chasing cardiac physiology and pathology down the CaMKII cascade. Am J Physiol Heart Circ Physiol. 2015; 308:H1177-H1191.
2. Mollova MY, Katus HA, Backs J. Regulation of CaMKII signaling in cardiovascular disease. Front Pharmacol. 2015; 6:178.
3. Hund TJ, Mohler PJ. Role of CaMKII in cardiac arrhythmias. Trends Cardiovasc Med. 2015; 25:392-397.
4. He Q, Feng Y, Wang Y. Transient outward potassium channel: a heart failure mediator. Heart Fail Rev. 2015; 20:349-362.
5. Wang Y, Hill JA. Electrophysiological remodeling in heart failure. J Mol Cell Cardiol. 2010; 48:619-632.
6. Wang Y, Tandan S, Cheng J, Yang C, Nguyen L, Sugianto J, Johnstone JL, Sun Y, Hill JA. Ca2+/calmodulin-dependent protein kinase II-dependent remodeling of Ca2+ current in pressure overload heart failure. J Biol Chem. 2008; 283:25524-25532.
7. Keskanokwong T, Lim HJ, Zhang P, Cheng J, Xu L, Lai D, Wang Y. Dynamic Kv4.3-CaMKII unit in heart: an intrinsic negative regulator for CaMKII activation. Eur Heart J. 2011; 32:305-315.
8. Kaab S, Dixon J, Duc J, Ashen D, Nabauer M, Beuckelmann DJ, Steinbeck G, McKinnon D, Tomaselli GF. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation. 1998; 98:1383-1393.
9. Bassani RA. Transient outward potassium current and Ca2+ homeostasis in the heart: beyond the action potential. Braz J Med Biol Res. 2006; 39:393-403.
10. Zicha S, Xiao L, Stafford S, Cha TJ, Han W, Varro A, Nattel S. Transmural expression of transient outward potassium current subunits in normal and failing canine and human hearts. J Physiol. 2004; 561:735-748.
11. Borlak J, Thum T. Hallmarks of ion channel gene expression in end-stage heart failure. FASEB J. 2003; 17:1592-1608.
12. Wang Y, Cheng J, Chen G, Rob F, Naseem RH, Nguyen L, Johnstone JL, Hill JA. Remodeling of outward K+ currents in pressure-overload heart failure. J Cardiovasc Electrophysiol. 2007; 18:869-875.
13. Bers DM. Cardiac excitation-contraction coupling. Nature. 2002; 415:198-205.
14. Sipido KR, Bito V, Antoons G, Volders PG, Vos MA. Na/Ca exchange and cardiac ventricular arrhythmias. Ann N Y Acad Sci. 2007; 1099:339-348.
15. Sanchez-Alonso JL, Bhargava A, O'Hara T, Glukhov AV, Schobesberger S, Bhogal N, Sikkel MB, Mansfield C, Korchev YE, Lyon AR, Punjabi PP, Nikolaev VO, Trayanova NA, Gorelik J. Microdomain-specific modulation of L-type calcium channels leads to triggered ventricular arrhythmia in heart failure. Circ Res. 2016; 119:944-955.
16. Yuan W, Bers DM. Ca-dependent facilitation of cardiac Ca current is due to Ca-calmodulin-dependent protein kinase. Am J Physiol. 1994; 267:H982-H993.
17. Maier LS. Role of CaMKII for signaling and regulation in the heart. Front Biosci. 2009; 14:486-496.
18. Maier LS, Bers DM, Brown JH. Calmodulin and Ca2+/calmodulin kinases in the heart - physiology and pathophysiology. Cardiovasc Res. 2007; 73:629-630.
19. Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J Cardiovasc Pharmacol. 2009; 54:180-187.
20. Bers DM. Beyond beta blockers. Nat Med. 2005; 11:379-380.
21. Anderson ME. Calmodulin kinase signaling in heart: an intriguing candidate target for therapy of myocardial dysfunction and arrhythmias. Pharmacol Ther. 2005; 106:39-55.
22. Wang Y, Cheng J, Joyner RW, Wagner MB, Hill JA. Remodeling of early-phase repolarization: a mechanism of abnormal impulse conduction in heart failure. Circulation. 2006; 113:1849-1856.