
Introduction
Neurofibromatosis type 1, also known as NF1 or von Recklinghausen’s disease, is a tumour predisposition syndrome characterized by the development of multiple neurofibromas, café-au-lait spots and Lisch nodules. Initially described by Professor Von Recklinghausen, a German pathologist back in 1882, NF1 is one of the most common genetic disorders worldwide [1, 2]. The NF1 gene is a classic tumour suppressor gene on chromosome 17. Its product neurofibromin is an important negative regulator of Ras cellular proliferation pathways [3-7]. Individuals with NF1 are at increased risk of developing various tumours, including malignant peripheral nerve sheath tumour (MPNST), phaeochromocytoma, leukaemia, glioma, rhabdomyosarcoma and breast cancer [8, 9]. Neurofibromatosis type 1 or NF1 is distinct from neurofibromatosis type 2 (NF2), which is less common. NF2 syndrome is related to mutations in NF2 on chromosome 22, with a different spectrum of tumours, notably schwannomas, meningiomas and ependymomas [10].
More recently, somatic NF1 aberrations have been increasingly reported in various sporadic tumours, including brain, lung, breast, ovarian tumours and melanomas. Significant challenges remain in the detection of both germline and somatic aberrations. A better understanding of the implications of these aberrations is critical for the improvement of treatment outcomes of tumours with NF1 aberrations.
NF1 syndrome
NF1 is a relatively common genetic condition, with an incidence of approximately 1 in 2,000 to 1 in 5,000 individuals worldwide [2]. Although it is an autosomal dominant genetic disorder, approximately half of the cases have no family history, with the condition arising from sporadic mutations of the NF1 gene. The germline NF1 mutation rate is ten-fold higher than that observed in other inherited disease genes, with estimates from 1/7,800 to 1/23,000 gametes [2, 11].
The condition has 100% penetrance but its degree of expression varies considerably, even within the same family with the identical mutation [12]. NF1 is diagnosed clinically for most patients, with genetic testing reserved for equivocal cases or in the context of research studies. The National Institutes of Health (NIH) diagnostic criteria stipulate that at least 2 of the criteria in Table 1 must be fulfilled to make the clinical diagnosis of NF1 [13].
Loss-of-function mutations in the NF1 gene can also lead to the development of a wide range of abnormalities in the cardiovascular, musculoskeletal and nervous systems, in addition to the predisposition to benign and malignant tumours. Hypertension, vasculopathy, valvular dysfunction, skeletal anomalies, dysmorphic features, osteoprorosis, cognitive impairment and epilepsy may occur as part of the NF1 syndrome [14].
Table 1: National Institutes of Health (NIH) diagnostic criteria for neurofibromatosis type 1 (NF1)
- Six or more café-au-lait macules >5mm in greatest diameter in prepubertal individuals, and >15mm in postpubertal individuals
- Two or more neurofibromas of any type or one plexiform neurofibroma
- Freckling in the axillary or inguinal regions
- Optic glioma
- Two or more iris hamartoma (Lisch nodules)
- Distinctive bony lesion such as sphenoid dysplasia, or thinning of the long bone cortex with or without pseudoarthrosis
- A first-degree relative (parent, sibling or offspring) with NF1 based on the above criteria
The NF1 phenotype is highly variable, ranging from a very mild manifestation of the disease in certain individuals, to a very severe form in some others [12]. In general, there is no definite correlation between a particular alteration and phenotype. Exceptions include deletion of the entire NF1 gene which is associated with a severe form of the disease [15], a recurrently ascertained 3-bp in-frame deletion of exon 17 (c.2970-2972 delAAT) that is associated with the typical pigmentary NF1 features but without cutaneous or surface plexiform neurofibromas [16], and duplication of the NF1 locus which usually leads to intellectual impairment and epilepsy without the other NF1 features [17, 18]. Intra- and interfamilial variation in severity of the phenotype suggests that expression of the same genotype may be influenced by epigenetic or environmental factors [12, 19]. Females with NF1 often experience an exacerbation of the condition following pregnancy, possibly related to changes in the hormonal milieu [20].
This overview will focus on mainly the oncological aspects of NF1 aberrations, given the recent discovery of somatic NF1 aberrations in various cancers in individuals without germline NF1.
Biology of NF1 and neurofibromin
Identified and cloned in 1990, the NF1 gene is located at chromosome 17q11.2 [4, 21], and is one of the largest genes in the human genome, with 60 exons spanning over 350kb of genomic DNA [4, 22]. Another distinctive feature of the gene is the presence of 3 genes in intron 27b on the antisense strand: OMGP (oligodendrocyte-myelin glycoprotein), a membrane glycoprotein, and EVI2A and EVI2B (ecotropic viral integration sites), which are involved in the development of mouse leukemia [23, 24].
NF1 encodes the protein neurofibromin, which has an estimated molecular mass of 327kDa and consists of 2818 amino acids. Neurofibromin is ubiquitously expressed, but most highly in the central nervous system, especially in neurons, astrocytes, oligodendrocytes and Schwann cells [25]. As might be expected for such a large gene, alternate exons, splice variants and alternate start sites have been reported. The major reported functional isoforms are derived from the insertion of extra exons that preserve the open reading frame and show tissue restricted expression.
The two major isoforms are neurofibromin types I and II. Neurofibromin type I is expressed predominantly in the brain, and has significant Ras regulatory activity. Neurofibromin type II, also known as GRD2 (domain II-related GAP) is the product of the insertion of exon 23a. In contrast to neurofibromin type 1, it has limited GAP regulatory function [26, 27]. It is expressed mainly in Schwann cells, and is essential for learning and memory in mouse models. In studies on sporadic colon, ovarian and breast cancers as well as gastric cancer cell lines, expression of the type I isoform relative to type II isoform is increased in tumour samples compared to normal tissue [28-31].
Information on other neurofibromin isoforms is limited. Neurofibromin types III and IV, which contain exon 48a and both exons 23a and 48a respectively, are expressed in mainly cardiac and skeletal muscles. They appear to be essential for normal muscle and cardiac development [32, 33]. Apart from neurofibromin types I-IV, two other isoforms have been described. An isoform which contains exon 9a is expressed mainly in neurons of the forebrain, and may be involved in memory and learning mechanisms [34, 35]. Another isoform has alternative exon 10a-2 inserted, introducing a transmembrane domain. The function of this variant, which is observed in a majority of human tissues, is unclear, but may perform a housekeeping function in intracellular membranes [36].
Roles of NF1 and neurofibromin in tumour suppression
NF1 is considered a classical tumour suppressor gene, with both copies of the NF1 gene reported to be inactivated in benign and malignant tumours in NF1 patients [37-39]. The first hit is inherited or acquired as a germline mutation, and the second hit occurs from a somatic event. Loss of heterozygosity (LOH) due to large somatic rearrangements, deletions and somatic recombination may affect the wild-type NF1 allele. This can also potentially affect other genes on chromosome 17, which include the tumour suppressor protein p53 at 17p13.2, human epidermal growth factor receptor 2 (HER2) at 17q21.1, topoisomerase II alpha (TOP2A)(17q21.1), signal transducer and activator of transcription 3 (STAT3)(17q21.2) and breast cancer gene 1 (BRCA1 )(17q21.2) [40].
Various Nf1+/- mouse models show predisposition to tumour formation, including phaeochromocytomas, leukaemias and malignant peripheral nerve sheath tumours (MPNST), similar to the spectrum of NF1-associated malignancies observed in human counterparts [41-43].
The tumour suppressor function of neurofibromin is largely attributed to a small central region which comprises 360 amino acids encoded by exons 20-27a. This critical region has marked structural and sequence similarity to ras-guanosine-triphosphate(GTP)ase activation proteins (GAPs) and is known as the GAP-related domain (GRD). GAPs inactivate Ras by accelerating the conversion of active Ras-GTP to its inactive guanosine diphosphate (GDP)-bound form. The downregulation of oncogene Ras by neurofibromin prevents the downstream activation of mitogen-activated protein kinase (MAPK) and the PI3K/Akt/mTOR (mammalian target of rapamycin) cell proliferation and differentiation pathways, as demonstrated in Figure 1 below [3, 5-7, 44].
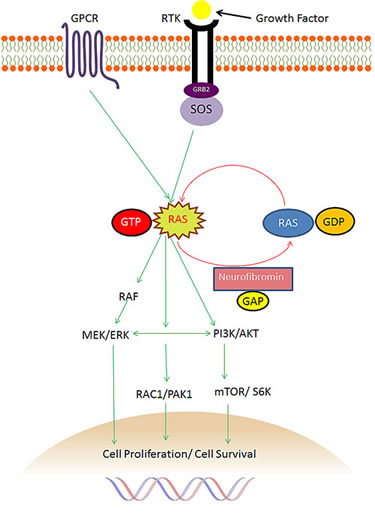
Figure 1: The role of NF1 and neurofibromin in the Ras pathway. G-protein coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs), when activated by ligand, promote guanine nucleotide exchange to form activated Ras-GTP complex. Neurofibromin inactivates Ras by accelerating the conversion of active Ras-GTP to inactive GDP-bound Ras with its Ras-GTPase activity. Consequently, neurofibromin suppresses activation of the downstream effectors of Ras, including PI3K, Akt, mTOR, S6 kinase and RAF, MEK, ERK as well as RAC1 and PAK1. RTKs=receptor tyrosine kinases. Grb2=growth factor receptor bound 2. SOS=mammalian homolog of the Drosophila son of sevenless. RAS=rat sarcoma viral oncogene homologue. GDP=guanosine diphosphate. GTP=guanosine triphosphate. RAF=murine sarcoma viral oncogene homologue. MEK=MAPK-ERK kinase. PI3K=phosphatidylinositol-3–kinase. AKT=V-akt murine thymoma viral oncogene homologue 1. mTOR=mammalian target of rapamycin. Rac1=Ras-related C3 botulinum toxin substrate 1. PAK1=P21-Activated Kinase.
The Ras-GAP function of neurofibromin may be enhanced by protein kinase C (PKC) phosphorylation of the cystein-serine rich domain (CSRD) of the neurofibromin domain encoded by exons 11-17. The clustering of missense mutations in these regions among NF1 patients indicate the importance of PKC phosphorylation in sustaining normal neurofibromin function [14,45- 48].
Neurofibromin has also been demonstrated to bind to caveolin-1 (Cav-1), a membrane protein which regulates signalling molecules such as p21ras, protein kinase C and growth factor receptors. Formation of the neurofibromin-Cav-1 complex may lead to inactivation of p21ras-GTP and modulation of the p21ras/MAPK, PI3K/Akt pathways, controlling cell proliferation and differentiation [14, 49].
Apart from downregulation of Ras via the homology to GAPs, there are several other postulated mechanisms for the tumour suppressor function of neurofibromin (Table 2).
Table 2: Mechanisms of Tumour Suppression by Neurofibromin
Mechanisms of Tumour Suppression Reported
• Downregulation of Ras
• Positive regulation of adenyl cyclase (AC)
• Pro-apoptotic effect (ras-dependent and ras-independent)
• Regulation of cell adhesion and motility
• Suppression of epithelial mesenchymal transition (EMT)
• Suppression of heat shock factor (HSF)
Neurofibromin is a positive regulator of the enzyme adenylyl cyclase (AC), which generates intracellular cyclic AMP (cAMP). cAMP-dependent signaling appears to be important in learning and memory, but also provides a possible mechanism for tumour suppressor function as it regulates Ras activity [50, 51]. Increased cAMP leads to activation of Rap1, an anti-mitogenic RAS pathway antagonist, which can result in inhibition of RAF activation in astrocytes [52, 53]. cAMP-mediated regulation of MAPK may have differential effects in different tissues; the mechanisms of cAMP-mediated tumorigenesis in tissues outside the nervous system have not yet been elucidated.
Neurofibromin has also been reported to exert tumour suppressor function via a proapoptotic effect by Ras-dependent and Ras-independent pathways. Nf1-/-, Nf1+/-, and Nf1+/+ mouse embryonic fibroblasts (MEFs) exhibited gene-dosage-related resistance to apoptosis. Neurofibromin-deficient MEFs and human NF1 malignant peripheral nerve sheath tumour (MPNST) cells were more resistant to apoptosis than neurofibromin-expressing MEFs and schwannoma cells. Administration of farnesylthiosalicyic acid (FTS), a Ras inhibitor, increased apoptosis of the neurofibromin-deficient SV40 MEFs and MPNST cells, indicating dependence on the Ras pathway. However, the resistance of neurofibromin-deficient SV40 MEFs and MPNST cells to staurosporine (protein kinase C inhibitor which induces apoptosis), UV irradiation, and vincristine was independent of Ras and cAMP, as demonstrated by the inability of Ras inhibitors or agents that elevate cAMP levels to overcome the resistance. Expression levels of key apoptotic components such as Bcl-2 family proteins, caspases and the X-linked inhibitor of apoptosis (XIAP) were similar in neurofibromin-expressing and neurofibromin-deficient MEFs. The exact mechanism of the Ras-independent proapoptotic effects of neurofibromin remains unclear [54].
The role of neurofibromin in cell motility is important not only for the functioning in neurons, but may also contribute to its tumour suppressor function. Neurofibromin regulates the dynamics and reorganisation of actin filaments via the Rho-ROCK-LIMK2-cofillin pathway, and may be involved in adhesion and signalling at neuronal synapses through its interaction (via its GRD and C-terminal domains) with the transmembrane heparin sulphate proteoglycan syndecan. Lack of neurofibromin triggers the Rho-ROCK-LIMK2-cofilin pathway to alter the organization of actin cytoskeleton, promoting cell motility, invasiveness, and cell-cell adhesion, resulting in the formation of large cell aggregates. This may lead to the formation of multiple neurofibromas in NF1 patients, which consist of aggregates of various cell types, including Schwann cells, fibroblasts, endothelial cells and mast cells on a background of excessive extracellular matrix deposition [55, 56].
Another mechanism of tumour suppression by neurofibromin relates to its association with the N-terminal of focal adhesion kinase (FAK), a protein localised at contact sites of cells with extracellular matrix known as focal adhesions. This interaction helps to regulate cellular events including adhesion, proliferation, motility, cellular migration and survival. Nf1+/+ mouse embryonic fibroblast (MEF) cells exhibited less growth under serum deprivation conditions with reduced adherence on collagen and fibronectin-treated plates, compared to Nf1–/– cells [57].
There is also data to suggest that loss of neurofibromin leads to epithelial-mesenchymal transition (EMT). EMT is implicated in tumorigenesis and cancer metastasis. Immunohistochemical analysis and real-time quantitative reverse transcription polymerase chain reaction showed increased expression of EMT-related transcription factors including Snail, Slug, Twist, ZEB1 and ZEB2 in NF1-associated neurofibroma specimens and NF1-derived Schwann cells. Knockdown of NF1 with siRNA induced the expression of these transcription factors in normal human Schwann cells as well as epithelial-like breast cancer cell lines [58].
More recently, loss of NF1 has been reported to promote carcinogenesis by activating heat shock factor 1 (HSF1), the master transcriptional regulator of the heat shock response. Knockout of NF1 in MEFs triggered activation of HSF1, increasing HSF1 levels. This resulted in Nf1–/– cells becoming tolerant to proteotoxic stress with proteasome inhibitors and HSP90 inhibitor. This activation of HSF1 relied on dysregulated MAPK signaling. HSF1, in turn, supported MAPK signaling. In NPcis+/- mouse models where Trp53 and Nf1 genes are disrupted on the same chromosome to develop soft tissue sarcomas resembling human MPNSTs, Hsf1 knockout impeded NF1-associated carcinogenesis by attenuating oncogenic RAS/MAPK signaling. In cell lines from human malignant peripheral nerve sheath tumors (MPNSTs) driven by NF1 loss and in surgically excised human MPNSTs, HSF1 was also overexpressed and activated or phosphorylated [59].
Tumours associated with NF1
Individuals with NF1 are predisposed to developing both benign and malignant tumours throughout life. The risk of malignancy is increased 2.5 to fourfold in NF1 compared to the general population [8, 60]. Average life expectancy is reduced by 10-15 years, with cancer being the most common cause of death [2].
The tumour types individuals with NF1 are at increased risk of developing include both nervous system and non-nervous system tumours. The characteristics of the more common NF1-associated tumours are listed in Table 3. Accurate estimation of the relative frequencies of the various tumour types is challenging, as different studies based on hospital data may overestimate the frequency of specific tumours compared to population-based studies. This partly accounts for the wide range of prevalence or incidence figures reported in the literature for various tumours.
Malignant peripheral nerve sheath tumours (MPNSTs), previously referred to as neurofibrosarcomas, are a major cause of morbidity and mortality in NF1. MPNSTs typically arise from malignant transformation of plexiform neurofibromas, and occasionally spinal nerve root or subcutaneous neurofibromas. In NF1 the lifetime risk of developing MPNST is 8-13%, with estimated annual incidence at 0.16%, compared to 0.001% in the general population [9, 61, 62].
There is a wide range of other NF1-associated tumours including optic pathway gliomas (OPGs), rhabdomyosarcomas, neuroblastomas and juvenile myelomonocytic leukaemias (JMML) in the paediatric setting, as well as gastrointestinal stromal tumour (GIST), phaeochromocytomas and carcinoid tumours in adults. OPGs, like MPNSTs, may occur in both children and adults [9, 61, 62]. More recently, an increased risk of breast cancer among women with NF1 has also been reported [63, 64]. Breast cancer in NF1 patients appears to have an aggressive phenotype in the two reported case series [65, 66].
NF1 patients are also at an increased risk of developing radiation-induced malignancies. In a study of NF1 patients treated with radiotherapy for optic glioma, the relative risk of second CNS tumour was 3.04 (95% CI, 1.29 to 7.15) [67]. Hence radiotherapy should be avoided in children with NF1, unless it is absolutely essential. In mouse model studies, Nf1+/- mice subjected to irradiation developed in-field tumours associated with NF1 such as phaeochromocytomas, as well as typical second malignant neoplasms such as sarcomas and breast cancers [68-70]. This may be related to upregulated, perturbed cell cycle and DNA repair pathways with NF1-haploinsufficiency, as observed in human lymphoblastoid cell lines from NF1-affected and normal individuals, as well as in lymphocytes from wildtype and Nf1+/- mice. Activation of DNA damage response (DDR) genes can paradoxically trigger oncogene-induced DNA damage and genomic instability, resulting in carcinogenesis [71, 72]. Interestingly, somatic monoallelic loss of NF1 and TP53 in the adjacent allele was observed in radiation-induced malignancies arising in both wildtype and Nf1+/- mice in one study [68]. NF1 loss appears to be a critical event in mutagen-induced malignancies beyond the classical NF1-associated tumour types.
Table 3: Tumours associated with NF1 syndrome
Tumour Type Associated with NF1 |
Age category |
Frequency |
Mechanism(s) |
Differences compared to sporadic tumours |
References |
Malignant peripheral nerve sheath tumour (MPNST) |
Adult, Paediatric |
Lifetime risk 8-13% |
LOH of NF1, mutation in TP53, copy number alterations, including deletion of CDKN2A, loss of PTEN |
Earlier onset; central rather than peripheral location |
|
Optic pathway glioma (OPG) |
Adult (usually young), Paediatric |
Incidence 1.5%-7.5% (Patil) prevalence 5-25% |
LOH of NF1, mutation in TP53, deletion of CDKN2A |
Earlier onset; anterior rather than posterior optic pathway |
|
Rhabdomyosarcoma |
Paediatric |
Prevalence 1.4-6% |
unknown |
Earlier onset; urinary tract rather than head and neck |
|
Neuroblastoma |
Paediatric |
Unknown |
LOH of NF1, amplification of MYCN, deletion of 1p36 |
||
Juvenile myelomonocytic leukaemia (JMML) |
Paediatric |
Lifetime risk 200-fold increased |
LOH of NF1, or compound heterozygous microlesions. |
||
Gastrointestinal Stromal Tumours (GISTs) |
Adult |
Lifetime risk 6% |
LOH of NF1, some copy number alterations |
Small intestine and multiple rather than gastric origin; lack of response to imatinib with lack of KIT and PDGFRA mutations |
|
Phaeochromocytoma |
Adult |
Prevalence 1% |
LOH of NF1 |
Earlier onset; occasionally bilateral or extradrenal |
|
Carcinoid |
Adult |
Prevalence 1% |
LOH of NF1 |
Earlier onset; periampullary rather than small intestine |
|
Breast Cancer |
Adult |
Standardised incidence ratio of 3.5 to 5.2 |
Unknown |
Earlier onset; possibly more aggressive |
Somatic NF1 Aberrations in Sporadic Tumours and Effects of NF1 Deficiency
With the recent cancer genome sequencing projects, the heterogeneity of cancer genomes has been unraveled. Somatic NF1 aberrations are increasingly reported in various sporadic tumours, including brain, lung, breast, ovarian tumours as well melanomas and leukemias (Figure 2). This is particularly relevant with the advent of novel molecular therapies which can potentially be targeted at aberrations in the NF1 pathway. Improved understanding of the mechanisms of carcinogenesis is critical for the optimisation of these targeted therapies.
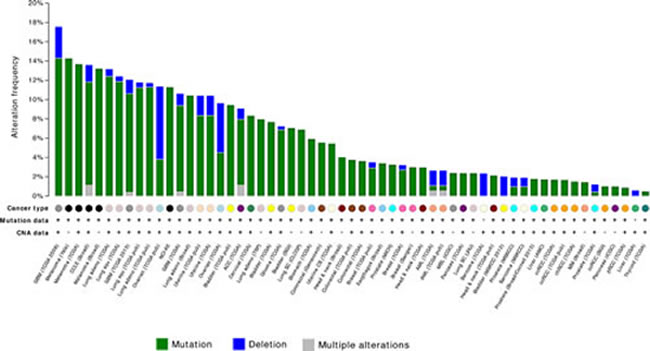
Figure 2: Frequency of NF1 mutation and homozygous deletion in human neoplasms (Source: The cBio Cancer genomics Portal; http://www.cbioportal.org)
Brain Tumours
In glioblastoma multiforme (GBM), NF1 is one of the most frequently mutated or deleted genes. The prevalence of NF1 somatic mutations in sporadic GBMs was initially estimated to be approximately 15%, with a subsequent study by The Cancer Genome Atlas (TCGA) network reporting aberrations in at least 23% (47 out of 206) of human GBM samples when both NF1 inactivating mutations and deletions (including heterozygous deletions) were analysed [93, 94]. However, when only mutations and homozygous deletions are considered, the frequency of alterations ranges from 12.1 to 17.6% [94, 95].
Data from mouse models support the importance of NF1 as a glioblastoma suppressor gene. Inactivation of TP53 and PTEN may cooperate with NF1 loss to induce the malignant transformation [96]. Haploinsufficiency for the NF1 tumour suppressor may have functional consequences, such as increased astrocyte proliferation and augmentation of angiogenesis in Nf1+/- heterozygous mouse models [97, 98]. Integrated genomic analysis of the TCGA data identified GBMs with NF1 and PTEN alterations to have a distinct mesenchymal-like expression profile. This mesenchymal subtype was characterised by the expression of mesenchymal markers such as CHI3L1 (also known as YKL40) and MET, as well as astrocytic markers (CD44, MERTK), reflecting the epithelial-to-mesenchymal transition. There was also high expression of genes in the tumour necrosis factor (TNF) and NK-ĸB pathway, related to the greater necrosis and associated inflammatory response in this subtype [99, 100].
Melanoma
Loss of NF1 in malignant melanoma cell lines was reported soon after discovery of the NF1 gene in the early 1990s [101, 102], but it was only recently that comprehensive genomic characterization of melanomas was performed. Melanomas may be classified into 3 major classes: 1) sun-shielded melanomas with wild type BRAF and NRAS which have low mutation load but high number of copy gains, 2) sun-exposed melanomas with BRAF or NRAS mutations and 3) sun-exposed melanomas with wild-type BRAF and NRAS, few copy number alterations but high mutation load. The last subtype of melanoma was typically associated with more advanced age, and 30% of melanomas from this class (10/33 samples) carried deleterious NF1 mutations. TP53, ARID2 and PTPRK were frequently mutated in these melanomas, suggesting that inactivation of tumour suppressors contribute to the pathogenesis of these BRAF and NRAS independent tumours [103, 104]. The overall frequency of NF1 mutations is estimated at 14% of cutaneous melanomas, with total of 475 specimens analysed so far in 3 separate studies (Figure 2) [103, 105].
Somatic NF1 mutations have also been reported in melanoma specimens harboring BRAF mutations [105, 106]. In a mouse model study, NF1 mutations cooperate with BRAF mutations in the pathogenesis of melanomas by preventing oncogene-induced senescence [106]. Loss of neurofibromin expression and NF1 loss-of-function mutations have been reported in melanomas from patients with de novo as well as acquired resistance to BRAF inhibitors [106, 107]. A pooled RNA interference screen targeting >16,500 genes in a BRAF inhibitor-sensitive melanoma cell line identified NF1 as the highest ranking gene whose knockdown abrogated the growth inhibitory effects of PLX4720, a BRAF inhibitor [107]. NF1-mutant melanomas are unlikely to respond to standard BRAF-targeted therapies but may benefit from drugs targeting the MEK and PI3K pathway instead. In mice injected with BRAF/NF1-mutated melanoma cells, there was resistance to vemurafenib, a BRAF inhibitor. In contrast, there was greater sensitivity to MEK inhibitor PD0325901, PI3K inhibitor GDC-0941 and rapamycin, an mTOR inhibitor. Importantly, rapamycin synergized with PD0325901, resulting in tumour regression in the allografts [106].
Lung Cancer
Whole exome or genome sequencing of primary lung adenocarcinomas identified NF1 as one of the most frequently mutated genes, with an estimated frequency of 11-12% [108, 109]. The clinical significance of NF1 mutations in the lung adenocarcinoma sequencing studies is not reported, but reduced NF1 mRNA expression was recently found to confer both intrinsic and acquired resistance to EGFR inhibitors in another recently reported study. However, somatic NF1 mutations were not found in the specimens from these patients with resistant tumours (more details in section on challenges of molecular diagnosis of NF1 and NF1 somatic aberrations) [110].
Approximately 12% of squamous cell lung cancers have alterations in NF1, according to a recently published TCGA study on squamous cell carcinomas. mRNA expression profiling identified 4 distinct subtypes of squamous cell lung cancers – classical, primitive, basal and secretory expression subtypes. The basal expression subtype of squamous cell lung carcinoma characteristically showed alterations in NF1 [111].
There is less data on small cell lung cancer, but the frequency of mutations in NF1 was reported as 2.4% and 6.9% in two separate studies of a smaller scale [112, 113].
Ovarian Carcinoma
The importance of NF1 in ovarian cancer was first reported by Sangha et al [114]. Initial genome-wide screen of DNA copy number alterations (CNAs) identified apparent NF1 homozygous deletions in 2 out of 36 primary ovarian serous carcinomas. This led to the discovery that 6 out of 18 ovarian carcinoma-derived cell lines had markedly reduced or lacked expression of NF1 protein, with 5 of the 6 cell lines harbouring NF1 mutations. Alterations in NF1, including splicing mutations and homozygous deletions, were identified in 22% (9/41) of the primary ovarian serous carcinomas studied. There was evidence of Ras pathway activation in these tumours and cell lines with NF1 defects, in the absence of KRAS or BRAF mutations. NF1 appears to cooperate with TP53 mutations which are present in virtually all ovarian serous carcinomas, in carcinogenesis [114].
In the large scale integrated genomic analyses of 489 high grade serous ovarian carcinomas by the TCGA cooperative group, NF1 has been recognized as one of the most frequently altered genes, with aberrrations in 12% of the cases (8% homozygous deletions, 4% mutations) [115]. These alterations affect signaling in the PI3K/Ras pathway, and may have therapeutic implications as discussed later in this review.
Breast Tumours
Although a computational biology study on gene expression datasets had previously reported associations between the activity levels of regulatory pathways linked with NF1 to clinical outcome in breast cancer [116], the importance of NF1 in the pathogenesis of breast cancer was not investigated further until recently. Absence of neurofibromin protein and lack of expression of NF1 mRNA type 1 isoform have been reported in the highly aggressive human breast cancer MDA-MB-231 cell line which is resistant to endocrine and cytotoxic agents. This was associated with accumulation of phosphorylated MAPK and activated Ras [117]. More recently, this Claudin-low subtype cell line was found to harbour NF1 mutation [118]. The Cancer Genome Project led by the Sanger Institute and The Cancer Genome Atlas (TCGA) projects reported NF1 mutations in approximately 3% of the breast cancers sequenced. Proportionally more NF1 mutations were found in luminal or ER+HER2- subtypes, although they were also present in selected HER2-overexpressing and triple negative or basal tumours [119, 120]. This may have therapeutic implications, given that knockdown of NF1 in MCF7 cells conferred resistance to tamoxifen in a genome-wide functional study [121].
NF1 has also been implicated as a breast cancer driver in a recent mouse model study. Chaos3 mice, which are engineered with a point mutation in the minichromosome maintenance 4 (Mcm4) gene, are highly unstable genomically, leading to the development of mammary tumours which resemble human breast cancers [122]. NF1 was found to be deleted in nearly all the mammary tumours from these mouse models. This led to re-examination of the TCGA data. 27.7% of human breast cancers in the TCGA project were subsequently found to harbour NF1 aberrations, majority of which were heterozygous deletions. Over 40% of HER2-overexpressing and basal subtypes showed these aberrations. This highlights the importance of investigating for the loss of NF1, in addition to the mutations [122].
Loss of heterozygosity of NF1 has been detected in radiation-induced breast cancers from patients without NF1 syndrome. The monoallelic loss of NF1 is likely to increase the potential for cooperating with other pathways such as TP53 pathways to promote cellular proliferation and carcinogenesis [68]. Loss of NF1 gene has also been reported in malignant phyllodes tumour of the breast [123] .
Haematological Malignancies
NF1 was previously implicated as one of the important drivers in certain sporadic haematological malignancies. Myeloid malignanices frequently harbor mutations in the Ras pathway. It is likely that NRAS/KRAS/NF1 aberrations cooperate with mutations in transcription factors and genes that regulate the epigenome in complex events leading to the development of AML [124]. In earlier studies, NF1 mutations were reported in up to 7% of acute myeloid leukemia (AML) cases, while 12% of 95 cases studied had copy number alterations in NF1 with mainly heterozygous deletions. Complete absence of NF1 expression was reported in 7% of adult AML, and this was associated with increased Ras-bound GTP [125]. In another study on a subset of AML with CBFB–MYH11 rearrangements, 16% of cases showed deletion of NF1 [126]. However, two recent large scale studies suggested that NF1 aberrations are not as frequent in de novo AML, although it may occur as a secondary event in disease progression [127, 128]. After taking into account the size of the gene in the test for significantly mutated genes, NF1 is not one of the significantly mutated genes in AML, with the gene altered in 2.7% of 187 cases [128].
Limited data suggests the frequency of NF1 alterations in myelodysplastic syndrome (MDS) varies from 0% to 9% [129, 130]. Recurrent cryptic alterations or deletions of the NF1 locus have been detected in 3 out of 35 patients in one of the studies [129]. The frequency of NF1 mutations in sporadic acute lymphoid leukemia (ALL) was recently reported as 3-8% [131, 132]. The prevalence of NF1 aberrations in other haematological malignancies such as multiple myeloma is currently unclear.
Colorectal Carcinoma
Data on the nature and the frequency of NF1 aberrations in colorectal carcinoma vary widely. After the initial report by Li et al that 1 out of 22 sporadic colon adenocarcinomas (4.5%) harboured the amino acid substitution altering Lys-1423 in the NF1 GRD [133], loss of heterozygosity (LOH) involving the NF1 gene in 14-57% of colorectal carcinomas was reported in two small studies [134, 135]. In addition to NF1 missense mutations, Ahlquist et al also found duplication of the whole NF1 gene or parts of it in 4 out of 24 specimens (17%) [136]. Nine out of ten NF1 mutations detected in this study occurred in introns likely involved in exon splicing. Notably, 8 of these 10 carcinomas showed microsatellite instability [136]. In contrast, NF1 was found to be altered in approximately 3.8-5.6% of colorectal carcinomas in two recent next generation sequencing studies [137, 138].
Other Sporadic Tumours
As displayed in Figure 2; there are several other tumours in which NF1 aberrations have been reported.
Aberrations of NF1 have also been reported in sporadic soft tissue sarcomas. Up to 10.5% of myxofibrosarcomas and 8% of pleomorphic liposarcomas harbor NF1 mutations [139, 140]. In a study on embryonal rhabdomyosarcoma, loss of NF1 occurred in 35%(9/26) of tumours (heterozygous or homozygous deletion of NF1 or heterozygous chromosomal loss), and were mutually exclusive with Ras mutations, suggesting NF1 loss as an alternative and potentially common driver of Ras activation in this major subtype of soft tissue sarcoma in young children [140].
A few studies reported somatic NF1 aberrations or inactivation in 26-41% of sporadic phaeochromocytomas from individuals without NF1. In keeping with the observation that NF1 individuals are at increased risk of developing phaeochromocytomas, these findings suggest that loss of NF1 function is a crucial event in the pathogenesis of both sporadic and NF1-associated phaeochromocytomas [91, 141, 142] .
Challenges of molecular diagnosis of NF1 and detection of NF1 somatic aberrations
The diagnosis of NF1 syndrome is usually established clinically in individuals with constitutional features of the syndrome. Germline NF1 testing is reserved mainly for equivocal cases, for prenatal diagnosis and in the research setting. Detection of NF1 mutations or deletions can be highly challenging due to several factors. NF1 is one of the largest genes, with 60 exons spanning over 350kb of DNA. The gene also has one of the highest mutation rates, with up to half of the mutations being novel mutations. In addition to the myriad of possible lesions with more than 1,200 different germline mutations reported so far (source: The Human Gene Mutation Database; http://www.hgmd.org) and the lack of mutation hotspots, the presence of several pseudogenes can complicate the molecular diagnosis further [11, 143-147]. A multi-step protocol involving analysis of genomic DNA and mRNA with RT-PCR, direct sequencing, multiplex ligation-dependent probe amplification (MLPA), and previously using also microsatellite marker analysis and FISH, was required to identify up to 95% of pathogenic mutations in individuals fulfilling the clinical NIH diagnostic criteria [148-150]. Analysis of RNA is essential as splicing mutations may be present in more than 20% of individuals with NF1 syndrome [144, 149, 150], and may be located deep in introns which may be missed when only exons are studied.
Given the potential difficulties of detecting the pathogenic mutation in individuals with clinical features of NF1, the identification of somatic NF1 aberrations in sporadic tumours can also pose a significant challenge. It is possible that the frequency of somatic NF1 alterations in various tumours is higher than what is currently recognized.
Although next generation sequencing (NGS) may be less laborious than direct sequencing, there are also limitations with NGS techniques. Decreased specificity of the capture probes may lead to the capture and enrichment of off-target sequences, including those from pseudogenes and closely related genes [143]. Exome sequencing alone may not detect splicing mutations or gene rearrangements. Whole genome sequencing combined with transcriptome analysis may be superior, but there are limitations to its applicability in the clinical setting currently due to the general requirement for fresh frozen tissue, complexity of data analysis and cost.
Downregulation of NF1 and neurofibromin via other mechanisms
Epigenetic factors, such as gene silencing by microRNAs and DNA methylation, may also influence the expression of NF1 and neurofibromin, as described below.
microRNAs are endogenous, small noncoding RNAs which can influence their target gene expression post-transcription. Downregulation of NF1 by microRNA-193b, which is overexpressed in sporadic head and neck squamous cell carcinomas (HNSCC), led to activation of ERK and resulted in tumour progression. Survival outcomes in HNSCC patients whose tumours expressed high levels of miR-193b were inferior compared to patients with low miR-193b expression. Knockdown of miR-193b in HNSCC cells increased NF1 transcript and protein expression levels, decreased ERK phosphorylation with reduction in cell viability, migration, invasion and tumour formation [151].
There is limited data on methylation changes, but methylation of NF1 was recently found to be the cause of a somatic second-hit inactivation in pilocytic astrocytoma from a patient with NF1 [152].
Excessive proteasomal degradation of neurofibromin can also result in deficiency of this critical tumour suppressor protein [153]. The ubiquitin ligase complex which controls both the regulated destruction and pathogenic destabilisation of neurofibromin was recently identified in glioblastomas as a Cullin 3(Cul3)/kelch repeat and BTB domain-containing 7 complex. Inhibition of Cul3 with Cul3-specific shRNAs suppressed Ras/ERK signaling; agents aimed at blocking neurofibromin destruction may be a potential therapeutic strategy for further development [154].
Given that the expression of NF1 may be influenced by epigenetic factors, microRNAs [155] and proteasomal degradation [153, 154], a proteomics-based approach may help to detect deficiency of neurofibromin. The utility of immunohistochemical staining of neurofibromin has not been fully explored. Complete absence of neurofibromin staining on immunohistochemistry was found in 15-18% of melanomas [106]. However, quantitation of protein expression correlating with treatment outcomes has not been well studied. This is also complicated by the fact that current antibodies available may not be able to distinguish between the normal and mutant neurofibromin protein. Functional studies of “mutant neurofibromin” will be challenging with the huge protein size and myriad abnormalities possible.
The challenges of elucidating the mechanisms of NF1 deficiency are demonstrated in the recent study on reduced NF1 expression as a driver of resistance to EGFR inhibitor in lung cancer. NF1 mRNA expression was reduced in EGFR TKI-resistant lung cancer specimens, but somatic mutations and methylation changes involving NF1 were not detected. To account for the downregulation of NF1 mRNA, immunohistochemistry using multiple antibodies was performed, but none of them demonstrated adequate specificity to detect neurofibromin in human lung tissue [110].
Therapeutic Strategies for NF1 and NF1-associated/deficient malignancies
Management Options for NF1 syndrome and neurofibromas
The management of individuals with NF1 consists mainly of surgical resection of neurofibromas when they cause discomfort or impingement of neighbouring structures such as nerves or spinal cord. There is an unmet need for novel molecular therapies to treat the systemic manifestations in NF1.
Early clinical trials using thalidomide, 13-cisretinoic acid (CRA) or interferon α-2a to target angiogenesis and differentiation in NF1 patients with plexiform neurofibromas induced at best a minor response in a minority of patients [156, 157]. Early phase trials using pirfenidone, an antifibrotic agent drug which targets the stromal contributions, showed similar limited activity in plexiform neurofibromas in adults and children [158, 159].
Since Ras is overactivated with dysfunction of NF1, subsequent NF1 trials focused on inhibition of Ras. Farnesylation and geranylgeranylation of Ras proteins is essential for translocation to the cell membrane with subsequent activation of the Ras pathway. The activity of tipifarnib, a farnesyl transferase inhibitor, was reported in a phase 1 trial on children with solid tumours or NF1 and plexiform neurofibromas. Stable disease was the best response; no significant regressions were observed [160]. More recently, in a phase 2 placebo-controlled study on children and young adults with NF1 and progressive plexiform neurofibromas, tipifarnib did not prolong the time to progression compared to placebo [161]. Similarly, results from a phase 2 study using sirolimus (rapamycin), an mTOR inhibitor, in NF1 patients with plexiform neurofibroma, did not report any regression of the lesions [162]. Clinical trials using everolimus, a newer generation mTOR inhibitor and other therapies are in progress (http://www.clinicaltrials.gov). The MEK inhibitor PD0325901 was effective in shrinking plexiform neurofibromas in more than 80% of genetically engineered mice, but data on clinical activity in human subjects is awaited [163].
Pegylated interferon-α-2b, which has antiproliferative, antiangiogenic and immunomodulatory properties, induced minor response in 29% of young patients with plexiform neurofibromas in a phase I trial [164]. Tumour stabilization or prevention of new lesions may be a more realistic endpoint as dramatic regression of established “benign” tumours is less likely. Although neurofibromas may show LOH in a subset of Schwann cells, the mode of pathogenesis is different from that of malignant tumours [165]. However, imatinib mesylate, an oral kinase inhibitor targeting c-kit and PDGFRβ, was recently reported to decrease plexiform neurofibromas by 20% or more in 6 out of 36 NF1 patients in a phase 2 trial. This effect may partially be related to targeting cellular phosphor-signalling cascades [166-168]. In contrast, sorafenib which targets c-kit and PDGFRβ as well as RAF, VEGFR2, was poorly tolerated and did not show any tumour response in a phase 1 trial on children with NF1 and plexiform neurofibromas [169]. The clinical efficacy of these compounds in treating neurofibromas remains to be tested in larger clinical trials.
Potential therapeutic strategies for NF1-deficient malignancies
Data on the efficacy of molecular therapies in NF1-deficient malignancies is currently limited to results from preclinical studies (Figure 3). Much of this research has been conducted on models of MPNST derived from NF1 patients. This is set to change with emerging clinical trials where the molecular therapy is matched to the genomic profile of each individual’s tumour. A one-size fits all approach may not always deliver an optimal outcome. For instance, although imatinib is standard-of-care for most patients with sporadic GIST, KIT/PDGFRA mutations are uncommon in GISTs arising in NF1 individuals, so response to imatinib is poor in these patients [88].
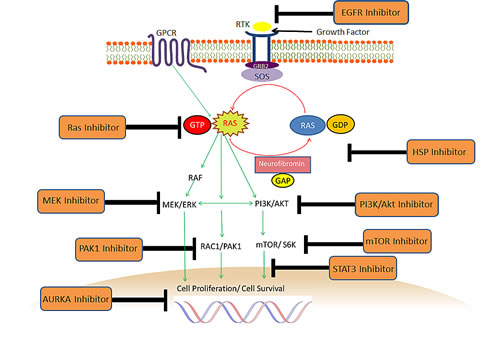
Figure 3: Potential therapeutic strategies for NF1-deficient malignancies. The molecular therapies above have been tested in the preclinical setting, largely for MPNSTs. There is also data on some of the inhibitors for neurofibromin-deficient breast cancer, glioblastoma, AML, soft tissue sarcoma, lung cancer and melanoma. Combination therapy targeting more than one checkpoint may be required for optimal inhibition.
There is preclinical data to support the activity of MEK inhibitors, Ras inhibitor farnesylthiosalicylic acid, sirolimus, everolimus and PI3K/Akt/mTOR inhibitors in MPNST cell lines or xenografts derived from NF1 patients [163, 170-174] (Figure 3). The addition of erlotinib, an epithelial growth factor receptor (EGFR) inhibitor to everolimus, inhibited growth and induced apoptosis further in 4 NF1-derived and 1 sporadic MPNST cell lines as well as the STS26T sporadic MPNST xenograft [172]. EGFR expression is present in most MPNST cell lines, and the EGFR signaling pathways were found to be associated with tumorigenesis in the Nf1:p53 mouse tumor model [172, 175].
Signal transducer and activator of transcription-3 (STAT3) is a potential target for treating NF1-associated or NF1-deficient cancers, as STAT3 is activated downstream in the PI3K/mTOR pathway. The natural product cucurbitacin-I, a potent STAT3 inhibitor, was found to inhibit the growth of NF1-deficient MPNST cells in vitro and in vivo in xenografts [176] .
Since heat shock factor is activated with loss of NF1, it is not surprising that the addition of HSP90 inhibitor IPI-504, to rapamycin, led to synergistic activity with damage of endoplasmic reticulum and mitochondria in NF1-deficient MPNST mouse models [177].
More recently, integrative transcriptome analyses have identified Aurorakinase A(AURKA) as a potential therapeutic target. AURKA was overexpressed and amplified in NF1-related MPNST, but not neurofibromas. MLN8237, an AURKA selective inhibitor, was effective in stabilizing tumour volume and prolonged survival of mice with MPNST xenografts [178].
Inhibitors of PAK1, a downstream effector in the Ras pathway, have also been reported to suppress the growth of NF1-deficient MPNST cells as well as neurofibromin-deficient human breast cancer (MDA-MB-231) xenografts in mice. There is evidence that many tumours, including breast cancers, are addicted to abnormal activation of PAK1, a Ser/Thr kinase which in turn stimulates cyclin D1, for their growth [179, 180].
In sporadic tumours harbouring NF1 aberrations, MEK inhibitors have been found to be effective in treating neurofibromin-deficient sporadic glioblastoma cell lines, NF1-deficient AMLs and NF1-deleted soft tissue sarcomas in mouse models [181-183]. Following the discovery that NF1 deficiency confers intrinsic and acquired resistance to EGFR inhibitor in lung cancer, treatment of neurofibromin-deficient lung cancers in vitro and in xenografts with MEK inhibitory drugs (AZD-6244, CI-1040 and PD0325901) restored sensitivity to erlotinib when given in combination [110].
Combination therapies targeting more than one checkpoint in the cell proliferation pathway, such as blocking both the PI3K/mTOR and MEK pathways in the allografts of NF1/BRAF-mutated melanomas and dual EGFR, MEK inhibition concurrently in TKI-resistant NF1-deficient lung adenocarcinomas, may be superior to monotherapy [106, 110]. Inhibiting a single checkpoint may lead to activation of compensatory negative feedback pathways.
Future strategies may include inhibition of excessive destruction of neurofibromin and other epigenetic therapies. In Nf2-mutant Schwann cells, inhibition of SIRT2, a class III histone deacetylase, triggered necrosis [184]. The role of HDAC inhibitors, which may decrease Akt phosphorylation, has not been fully explored for Nf1-mutant cells. Inhibition of LIM kinase in the Rho-ROCK-LIMK-cofilin pathway regulated by neurofibromin is another potential strategy. In Nf1–/– MEFs, novel LIMK inhibitors blocked the phosphorylation of cofilin, resulting in actin severance and inhibition of cell migration and growth [185]. The utility of these drugs in NF1-deficient tumours may be worth investigating, especially in combination with Ras or AURKA inhibitors, which may have synergistic effects [185, 186]. Improved understanding of the biology of NF1 and neurofibromin in normal cells and cancer is critical for the development of novel treatment strategies.
Conclusions
NF1 and neurofibromin play critical roles in tumour suppression. The frequency of somatic NF1 aberrations in sporadic tumours is increasingly recognized. These alterations are associated with distinct subtypes in certain cancers, and may be associated with poorer treatment outcomes. Significant challenges remain in unravelling the complexity of the large NF1 gene and its product neurofibromin. Improved molecular diagnosis techniques are essential for detecting these aberrations. There is also an unmet need to develop novel systemic therapies for treating NF1-deficient tumours.
Acknowledgements
This work was supported in part by SingHealth Foundation Research Grant, Singapore (SHF/FG426S/2009).
References
1. Brosius S. A history of von Recklinghausen’s NF1. J Hist Neurosci. 2010; 19(4):333-348.
2. Rasmussen SA and Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000; 151(1):33-40.
3. Xu GF, O’Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, Dunn D, Stevens J, Gesteland R and White R. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990; 62(3):599-608.
4. Cawthon RM, Weiss R, Xu GF, Viskochil D, Culver M, Stevens J, Robertson M, Dunn D, Gesteland R and O’Connell P. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990; 62(1):193-201.
5. Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, Conroy L, Clark R, O’Connell P and Cawthon RM. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell. 1990; 63(4):843-849.
6. Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M and Collins F. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990; 63(4):851-859.
7. Bollag G and McCormick F. Differential regulation of rasGAP and neurofibromatosis gene product activities. Nature. 1991; 351(6327):576-579.
8. Sorensen SA, Mulvihill JJ and Nielsen A. Long-term follow-up of von Recklinghausen neurofibromatosis. Survival and malignant neoplasms. N Engl J Med. 1986; 314(16):1010-1015.
9. Brems H, Beert E, de Ravel T and Legius E. Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. The Lancet Oncology. 2009; 10(5):508-515.
10. Schroeder RD, Angelo LS and Kurzrock R. NF2/merlin in hereditary neurofibromatosis 2 versus cancer: biologic mechanisms and clinical associations. Oncotarget. 2014; 5(1):67-77.
11. Griffiths S, Thompson P, Frayling I and Upadhyaya M. Molecular diagnosis of neurofibromatosis type 1: 2 years experience. Fam Cancer. 2007; 6(1):21-34.
12. Easton DF, Ponder MA, Huson SM and Ponder BA. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): evidence for modifying genes. Am J Hum Genet. 1993; 53(2):305-313.
13. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988; 45(5):575-578.
14. Larizza L, Gervasini C, Natacci F and Riva P. Developmental abnormalities and cancer predisposition in neurofibromatosis type 1. Curr Mol Med. 2009; 9(5):634-653.
15. Upadhyaya M, Ruggieri M, Maynard J, Osborn M, Hartog C, Mudd S, Penttinen M, Cordeiro I, Ponder M, Ponder BA, Krawczak M and Cooper DN. Gross deletions of the neurofibromatosis type 1 (NF1) gene are predominantly of maternal origin and commonly associated with a learning disability, dysmorphic features and developmental delay. Hum Genet. 1998; 102(5):591-597.
16. Upadhyaya M, Huson SM, Davies M, Thomas N, Chuzhanova N, Giovannini S, Evans DG, Howard E, Kerr B, Griffiths S, Consoli C, Side L, Adams D, Pierpont M, Hachen R, Barnicoat A, et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet. 2007; 80(1):140-151.
17. Grisart B, Rack K, Vidrequin S, Hilbert P, Deltenre P, Verellen-Dumoulin C and Destrée A. NF1 microduplication first clinical report: association with mild mental retardation, early onset of baldness and dental enamel hypoplasia? Eur J Hum Genet. 2008; 16(3):305-311.
18. Moles KJ, Gowans GC, Gedela S, Beversdorf D, Yu A, Seaver LH, Schultz RA, Rosenfeld JA, Torchia BS and Shaffer LG. NF1 microduplications: identification of seven nonrelated individuals provides further characterization of the phenotype. Genet Med. 2012; 14(5):508-514.
19. Pasmant E, Vidaud M, Vidaud D and Wolkenstein P. Neurofibromatosis type 1: from genotype to phenotype. J Med Genet. 2012; 49(8):483-489.
20. Roth TM, Petty EM and Barald KF. The role of steroid hormones in the NF1 phenotype: focus on pregnancy. Am J Med Genet A. 2008; 146A(12):1624-1633.
21. Wallace MR, Marchuk DA, Andersen LB, Letcher R, Odeh HM, Saulino AM, Fountain JW, Brereton A, Nicholson J and Mitchell AL. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990; 249(4965):181-186.
22. Li Y, O’Connell P, Breidenbach HH, Cawthon R, Stevens J, Xu G, Neil S, Robertson M, White R and Viskochil D. Genomic organization of the neurofibromatosis 1 gene (NF1). Genomics. 1995; 25(1):9-18.
23. Viskochil D, Cawthon R, O’Connell P, Xu GF, Stevens J, Culver M, Carey J and White R. The gene encoding the oligodendrocyte-myelin glycoprotein is embedded within the neurofibromatosis type 1 gene. Mol Cell Biol. 1991; 11(2):906-912.
24. Cawthon RM, Andersen LB, Buchberg AM, Xu GF, O’Connell P, Viskochil D, Weiss RB, Wallace MR, Marchuk DA and Culver M. cDNA sequence and genomic structure of EV12B, a gene lying within an intron of the neurofibromatosis type 1 gene. Genomics. 1991; 9(3):446-460.
25. Nordlund M, Gu X, Shipley MT and Ratner N. Neurofibromin is enriched in the endoplasmic reticulum of CNS neurons. J Neurosci. 1993; 13(4):1588-1600.
26. Suzuki H, Takahashi K, Kubota Y and Shibahara S. Molecular cloning of a cDNA coding for neurofibromatosis type 1 protein isoform lacking the domain related to ras GTPase-activating protein. Biochem Biophys Res Commun. 1992; 187(2):984-990.
27. Andersen LB, Ballester R, Marchuk DA, Chang E, Gutmann DH, Saulino AM, Camonis J, Wigler M and Collins FS. A conserved alternative splice in the von Recklinghausen neurofibromatosis (NF1) gene produces two neurofibromin isoforms, both of which have GTPase-activating protein activity. Mol Cell Biol. 1993; 13(1):487-495.
28. Cacev T, Radosević S, Spaventi R, Pavelić K and Kapitanović S. NF1 gene loss of heterozygosity and expression analysis in sporadic colon cancer. Gut. 2005; 54(8):1129-1135.
29. Iyengar TD, Ng S, Lau CC, Welch WR, Bell DA, Berkowitz RS and Mok SC. Differential expression of NF1 type I and type II isoforms in sporadic borderline and invasive epithelial ovarian tumors. Oncogene. 1999; 18(1):257-262.
30. Marrero D, Peralta R, Valdivia A, De la Mora A, Romero P, Parra M, Mendoza N, Mendoza M, Rodriguez D, Camacho E, Duarte A, Castelazo G, Vanegas E, Garcia I, Vargas C, Arenas D, et al. The neurofibromin 1 type I isoform predominance characterises female population affected by sporadic breast cancer: preliminary data. J Clin Pathol. 2012; 65(5):419-423.
31. Uchida T, Matozaki T, Suzuki T, Matsuda K, Wada K, Nakano O, Konda Y, Nishisaki H, Nagao M and Sakamoto C. Expression of two types of neurofibromatosis type 1 gene transcripts in gastric cancers and comparison of GAP activities. Biochem Biophys Res Commun. 1992; 187(1):332-339.
32. Gutmann DH, Geist RT, Rose K and Wright DE. Expression of two new protein isoforms of the neurofibromatosis type 1 gene product, neurofibromin, in muscle tissues. Dev Dyn. 1995; 202(3):302-311.
33. Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, Buchberg AM, Jenkins NA, Parada LF and Copeland NG. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994; 8(9):1019-1029.
34. Danglot G, Régnier V, Fauvet D, Vassal G, Kujas M and Bernheim A. Neurofibromatosis 1 (NF1) mRNAs expressed in the central nervous system are differentially spliced in the 5’ part of the gene. Hum Mol Genet. 1995; 4(5):915-920.
35. Gutmann DH, Zhang Y and Hirbe A. Developmental regulation of a neuron-specific neurofibromatosis 1 isoform. Ann Neurol. 1999; 46(5):777-782.
36. Kaufmann D, Müller R, Kenner O, Leistner W, Hein C, Vogel W and Bartelt B. The N-terminal splice product NF1-10a-2 of the NF1 gene codes for a transmembrane segment. Biochem Biophys Res Commun. 2002; 294(2):496-503.
37. Rasmussen SA, Overman J, Thomson SA, Colman SD, Abernathy CR, Trimpert RE, Moose R, Virdi G, Roux K, Bauer M, Rojiani AM, Maria BL, Muir D and Wallace MR. Chromosome 17 loss-of-heterozygosity studies in benign and malignant tumors in neurofibromatosis type 1. Genes Chromosomes Cancer. 2000; 28(4):425-431.
38. John AM, Ruggieri M, Ferner R and Upadhyaya M. A search for evidence of somatic mutations in the NF1 gene. J Med Genet. 2000; 37(1):44-49.
39. Eisenbarth I, Beyer K, Krone W and Assum G. Toward a survey of somatic mutation of the NF1 gene in benign neurofibromas of patients with neurofibromatosis type 1. Am J Hum Genet. 2000; 66(2):393-401.
40. Reinholz MM, Bruzek AK, Visscher DW, Lingle WL, Schroeder MJ, Perez EA and Jenkins RB. Breast cancer and aneusomy 17: implications for carcinogenesis and therapeutic response. The Lancet Oncology. 2009; 10(3):267-277.
41. Gutmann DH and Giovannini M. Mouse Models of Neurofibromatosis 1 and 2. Neoplasia. 2002; 4(4):279-290.
42. Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A and Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet. 1994; 7(3):353-361.
43. Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME, Bronson RT and Jacks T. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999; 286(5447):2172-2176.
44. Johannessen CM, Reczek EE, James MF, Brems H, Legius E and Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci USA. 2005; 102(24):8573-8578.
45. Mangoura D, Sun Y, Li C, Singh D, Gutmann DH, Flores A, Ahmed M and Vallianatos G. Phosphorylation of neurofibromin by PKC is a possible molecular switch in EGF receptor signaling in neural cells. Oncogene. 2006; 25(5):735-745.
46. Xu H and Gutmann DH. Mutations in the GAP-related domain impair the ability of neurofibromin to associate with microtubules. Brain research. 1997; 759(1):149-152.
47. Mattocks C, Baralle D, Tarpey P, ffrench-Constant C, Bobrow M and Whittaker J. Automated comparative sequence analysis identifies mutations in 89% of NF1 patients and confirms a mutation cluster in exons 11–17 distinct from the GAP related domain. J Med Genet. 2004; 41(4).
48. Fahsold R, Hoffmeyer S, Mischung C, Gille C, Ehlers C, Kücükceylan N, Abdel-Nour M, Gewies A, Peters H, Kaufmann D, Buske A, Tinschert S and Nürnberg P. Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am J Hum Genet. 2000; 66(3):790-818.
49. Boyanapalli M, Lahoud OB, Messiaen L, Kim B, Anderle de Sylor MS, Duckett SJ, Somara S and Mikol DD. Neurofibromin binds to caveolin-1 and regulates ras, FAK, and Akt. Biochem Biophys Res Commun. 2006; 340(4):1200-1208.
50. Tong J, Hannan F, Zhu Y, Bernards A and Zhong Y. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat Neurosci. 2002; 5(2):95-96.
51. Dasgupta B, Dugan LL and Gutmann DH. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J Neurosci. 2003; 23(26):8949-8954.
52. Dugan LL, Kim JS, Zhang Y, Bart RD, Sun Y, Holtzman DM and Gutmann DH. Differential effects of cAMP in neurons and astrocytes. Role of B-raf. J Biol Chem. 1999; 274(36):25842-25848.
53. Brown JA, Gianino SM and Gutmann DH. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J Neurosci. 2010; 30(16):5579-5589.
54. Shapira S, Barkan B, Friedman E, Fridman E, Kloog Y and Stein R. The tumor suppressor neurofibromin confers sensitivity to apoptosis by Ras-dependent and Ras-independent pathways. Cell Death Differ. 2007; 14(5):895-906.
55. Ozawa T, Araki N, Yunoue S, Tokuo H, Feng L, Patrakitkomjorn S, Hara T, Ichikawa Y, Matsumoto K, Fujii K and Saya H. The neurofibromatosis type 1 gene product neurofibromin enhances cell motility by regulating actin filament dynamics via the Rho-ROCK-LIMK2-cofilin pathway. J Biol Chem. 2005; 280(47):39524-39533.
56. Hsueh YP, Roberts AM, Volta M, Sheng M and Roberts RG. Bipartite interaction between neurofibromatosis type I protein (neurofibromin) and syndecan transmembrane heparan sulfate proteoglycans. J Neurosci. 2001; 21(11):3764-3770.
57. Kweh F, Zheng M, Kurenova E, Wallace M, Golubovskaya V and Cance WG. Neurofibromin physically interacts with the N-terminal domain of focal adhesion kinase. Mol Carcinog. 2009; 48(11):1005-1017.
58. Arima Y, Hayashi H, Kamata K, Goto TM, Sasaki M, Kuramochi A and Saya H. Decreased expression of neurofibromin contributes to epithelial-mesenchymal transition in neurofibromatosis type 1. Exp Dermatol. 2010; 19(8):e136-141.
59. Dai C, Santagata S, Tang Z, Shi J, Cao J, Kwon H, Bronson RT, Whitesell L and Lindquist S. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J Clin Invest. 2012; 122(10):3742-3754.
60. Zöller ME, Rembeck B, Odén A, Samuelsson M and Angervall L. Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer. 1997; 79(11):2125-2131.
61. Patil S and Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012; 17(1):101-116.
62. Laycock-van Spyk S, Thomas N, Cooper DN and Upadhyaya M. Neurofibromatosis type 1-associated tumours: their somatic mutational spectrum and pathogenesis. Hum Genomics. 2011; 5(6):623-690.
63. Sharif S, Moran A, Huson SM, Iddenden R, Shenton A, Howard E and Evans DGR. Women with neurofibromatosis 1 are at a moderately increased risk of developing breast cancer and should be considered for early screening. J Med Genet. 2007; 44(8):481-484.
64. Wang X, Levin AM, Smolinski SE, Vigneau FD, Levin NK and Tainsky MA. Breast cancer and other neoplasms in women with neurofibromatosis type 1: A retrospective review of cases in the Detroit metropolitan area. American Journal of Medical Genetics Part A. 2012; 158A(12):3061-3064.
65. Teh BT, Birrell G, Farrell A, Leonard JH, Walters MK, Palmer JM, Ramsay JR, Schlect DJ, Furnival C, Lavin MF, Bennett I and Hayward NK. Breast cancer in six women with neurofibromatosis type 1. Breast. 1997; 6(3):155-160.
66. Yap YS, Wong J, Chan M, Yong WS, Ong KW, Ngeow J, Tan B, Madhukumar P, Tan MH, Ang P, Teh BT, Tan PH, Lee A. Clinical and Pathological Characteristics of Breast Cancer in Women with Neurofibromatosis Type 1. Cancer Research. 2012; 72(24S;1024): P3-08-09.
67. Sharif S, Ferner R, Birch JM, Gillespie JE, Gattamaneni HR, Baser ME and Evans DGR. Second Primary Tumors in Neurofibromatosis 1 Patients Treated for Optic Glioma: Substantial Risks After Radiotherapy. JCO. 2006; 24(16):2570-2575.
68. Choi G, Huang B, Pinarbasi E, Braunstein SE, Horvai AE, Kogan S, Bhatia S, Faddegon B and Nakamura JL. Genetically mediated Nf1 loss in mice promotes diverse radiation-induced tumors modeling second malignant neoplasms. Cancer Res. 2012; 72(24):6425-6434.
69. Chao RC, Pyzel U, Fridlyand J, Kuo Y-M, Teel L, Haaga J, Borowsky A, Horvai A, Kogan SC, Bonifas J, Huey B, Jacks TE, Albertson DG and Shannon KM. Therapy-induced malignant neoplasms in Nf1 mutant mice. Cancer Cell. 2005; 8(4):337-348.
70. Nakamura JL, Phong C, Pinarbasi E, Kogan SC, Vandenberg S, Horvai AE, Faddegon BA, Fiedler D, Shokat K, Houseman BT, Chao R, Pieper RO and Shannon K. Dose-dependent effects of focal fractionated irradiation on secondary malignant neoplasms in Nf1 mutant mice. Cancer Res. 2011; 71(1):106-115.
71. Pemov A, Park C, Reilly KM and Stewart DR. Evidence of perturbations of cell cycle and DNA repair pathways as a consequence of human and murine NF1-haploinsufficiency. BMC Genomics. 2010; 11.
72. Halazonetis TD, Gorgoulis VG and Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008; 319(5868):1352-1355.
73. Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM and Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986; 57(10):2006-2021.
74. Riccardi VM and Powell PP. Neurofibrosarcoma as a complication of von Recklinghausen neurofibromatosis. Neurofibromatosis. 1989; 2(3):152-165.
75. Ferner RE and Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002; 62(5):1573-1577.
76. Nielsen GP, Stemmer-Rachamimov AO, Ino Y, Moller MB, Rosenberg AE and Louis DN. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am J Pathol. 1999; 155(6):1879-1884.
77. Legius E, Marchuk DA, Collins FS and Glover TW. Somatic deletion of the neurofibromatosis type 1 gene in a neurofibrosarcoma supports a tumour suppressor gene hypothesis. Nat Genet. 1993; 3(2):122-126.
78. Glover TW, Stein CK, Legius E, Andersen LB, Brereton A and Johnson S. Molecular and cytogenetic analysis of tumors in von Recklinghausen neurofibromatosis. Genes Chromosomes Cancer. 1991; 3(1):62-70.
79. Legius E, Dierick H, Wu R, Hall BK, Marynen P, Cassiman JJ and Glover TW. TP53 mutations are frequent in malignant NF1 tumors. Genes Chromosomes Cancer. 1994; 10(4):250-255.
80. Gregorian C, Nakashima J, Dry SM, Nghiemphu PL, Smith KB, Ao Y, Dang J, Lawson G, Mellinghoff IK, Mischel PS, Phelps M, Parada LF, Liu X, Sofroniew MV, Eilber FC and Wu H. PTEN dosage is essential for neurofibroma development and malignant transformation. Proc Natl Acad Sci USA. 2009; 106(46):19479-19484.
81. Sung L, Anderson JR, Arndt C, Raney RB, Meyer WH and Pappo AS. Neurofibromatosis in children with Rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma study IV. J Pediatr. 2004; 144(5):666-668.
82. Martinsson T, Sjöberg RM, Hedborg F and Kogner P. Homozygous deletion of the neurofibromatosis-1 gene in the tumor of a patient with neuroblastoma. Cancer Genet Cytogenet. 1997; 95(2):183-189.
83. Origone P, Defferrari R, Mazzocco K, Lo Cunsolo C, De Bernardi B and Tonini GP. Homozygous inactivation of NF1 gene in a patient with familial NF1 and disseminated neuroblastoma. Am J Med Genet A. 2003; 118A(4):309-313.
84. Stiller CA, Chessells JM and Fitchett M. Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. British journal of cancer. 1994; 70(5):969-972.
85. Flotho C, Steinemann D, Mullighan CG, Neale G, Mayer K, Kratz CP, Schlegelberger B, Downing JR and Niemeyer CM. Genome-wide single-nucleotide polymorphism analysis in juvenile myelomonocytic leukemia identifies uniparental disomy surrounding the NF1 locus in cases associated with neurofibromatosis but not in cases with mutant RAS or PTPN11. Oncogene. 2007; 26(39):5816-5821.
86. Steinemann D, Arning L, Praulich I, Stuhrmann M, Hasle H, Stary J, Schlegelberger B, Niemeyer CM and Flotho C. Mitotic recombination and compound-heterozygous mutations are predominant NF1-inactivating mechanisms in children with juvenile myelomonocytic leukemia and neurofibromatosis type 1. Haematologica. 2010; 95(2):320-323.
87. Maertens O, Prenen H, Debiec-Rychter M, Wozniak A, Sciot R, Pauwels P, De Wever I, Vermeesch JR, de Raedt T, De Paepe A, Speleman F, van Oosterom A, Messiaen L and Legius E. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet. 2006; 15(6):1015-1023.
88. Mussi C, Schildhaus H-U, Gronchi A, Wardelmann E and Hohenberger P. Therapeutic consequences from molecular biology for gastrointestinal stromal tumor patients affected by neurofibromatosis type 1. Clin Cancer Res. 2008; 14(14):4550-4555.
89. Xu W, Mulligan LM, Ponder MA, Liu L, Smith BA, Mathew CG and Ponder BA. Loss of NF1 alleles in phaeochromocytomas from patients with type I neurofibromatosis. Genes Chromosomes Cancer. 1992; 4(4):337-342.
90. Gutmann DH, Cole JL, Stone WJ, Ponder BA and Collins FS. Loss of neurofibromin in adrenal gland tumors from patients with neurofibromatosis type I. Genes Chromosomes Cancer. 1994; 10(1):55-58.
91. Bausch B, Borozdin W, Mautner VF, Hoffmann MM, Boehm D, Robledo M, Cascon A, Harenberg T, Schiavi F, Pawlu C, Peczkowska M, Letizia C, Calvieri S, Arnaldi G, Klingenberg-Noftz RD, Reisch N, et al. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with pheochromocytoma and neurofibromatosis type 1. J Clin Endocrinol Metab. 2007; 92(7):2784-2792.
92. Stewart W, Traynor JP, Cooke A, Griffiths S, Onen NF, Balsitis M, Shah AA, Upadhyaya M and Tobias ES. Gastric carcinoid: germline and somatic mutation of the neurofibromatosis type 1 gene. Fam Cancer. 2007; 6(1):147-152.
93. Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008; 321(5897):1807-1812.
94. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455(7216):1061-1068.
95. Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu C-J, Genovese G, Shmulevich I, Barnholtz-Sloan J, et al. The somatic genomic landscape of glioblastoma. Cell. 2013; 155(2):462-477.
96. Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, Messing A and Parada LF. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer cell. 2005; 8(2):119-130.
97. Gutmann DH, Loehr A, Zhang Y, Kim J, Henkemeyer M and Cashen A. Haploinsufficiency for the neurofibromatosis 1 (NF1) tumor suppressor results in increased astrocyte proliferation. Oncogene. 1999; 18(31):4450-4459.
98. Wu M, Wallace MR and Muir D. Nf1 haploinsufficiency augments angiogenesis. Oncogene. 2006; 25(16):2297-2303.
99. Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010; 17(1):98-110.
100. Shen R, Mo Q, Schultz N, Seshan VE, Olshen AB, Huse J, Ladanyi M and Sander C. Integrative subtype discovery in glioblastoma using iCluster. PLoS ONE. 2012; 7(4).
101. Andersen LB, Fountain JW, Gutmann DH, Tarlé SA, Glover TW, Dracopoli NC, Housman DE and Collins FS. Mutations in the neurofibromatosis 1 gene in sporadic malignant melanoma cell lines. Nat Genet. 1993; 3(2):118-121.
102. Johnson MR, Look AT, DeClue JE, Valentine MB and Lowy DR. Inactivation of the NF1 gene in human melanoma and neuroblastoma cell lines without impaired regulation of GTP.Ras. Proc Natl Acad Sci USA. 1993; 90(12):5539-5543.
103. Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, Ariyan S, Narayan D, Dutton-Regester K, Capatana A, Holman EC, Bosenberg M, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012; 44(9):1006-1014.
104. Mar VJ, Wong SQ, Li J, Scolyer RA, McLean C, Papenfuss AT, Tothill RW, Kakavand H, Mann GJ, Thompson JF, Behren A, Cebon JS, Wolfe R, Kelly JW, Dobrovic A and McArthur GA. BRAF/NRAS wild-type melanomas have a high mutation load correlating with histologic and molecular signatures of UV damage. Clin Cancer Res. 2013; 19(17):4589-4598.
105. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J-P, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, et al. A landscape of driver mutations in melanoma. Cell. 2012; 150(2):251-263.
106. Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L, Bronson RT, McMahon M, Granter S, Flaherty K, Wargo JA, Marais R and Cichowski K. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013; 3(3):338-349.
107. Whittaker SR, Theurillat J-P, Van Allen E, Wagle N, Hsiao J, Cowley GS, Schadendorf D, Root DE and Garraway LA. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013; 3(3):350-362.
108. Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008; 455(7216):1069-1075.
109. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, Stojanov P, Cibulskis K, Choi K, et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell. 2012; 150(6):1107-1120.
110. de Bruin EC, Cowell CF, Warne PH, Jiang M, Saunders RE, Melnick MA, Gettinger SN, Walther Z, Wurtz A, Heynen GJ, Heideman DAM, Gomez-Roman JJ, García-Castaño A, Gong Y, Ladanyi M, Varmus H, et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014.
111. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012; 489(7417):519-525.
112. Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, Rivers CS, Foo CK, Bhatt D, Stinson J, Gnad F, Haverty PM, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012; 44(10):1111-1116.
113. Peifer M, Fernández-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, Menon R, Koker M, Dahmen I, Müller C, Di Cerbo V, Schildhaus H-U, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012; 44(10):1104-1110.
114. Sangha N, Wu R, Kuick R, Powers S, Mu D, Fiander D, Yuen K, Katabuchi H, Tashiro H, Fearon ER and Cho KR. Neurofibromin 1 (NF1) Defects Are Common in Human Ovarian Serous Carcinomas and Co-occur with TP53 Mutations. Neoplasia. 2008; 10(12):1362-1372.
115. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474(7353):609-615.
116. Teschendorff AE, Journee M, Absil PA, Sepulchre R and Caldas C. Elucidating the altered transcriptional programs in breast cancer using independent component analysis. PLoS computational biology. 2007; 3(8):e161.
117. Ogata H, Sato H, Takatsuka J and De Luca LM. Human breast cancer MDA-MB-231 cells fail to express the neurofibromin protein, lack its type I mRNA isoform and show accumulation of P-MAPK and activated Ras. Cancer Lett. 2001; 172(2):159-164.
118. Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P, Ducar M, Van Hummelen P, Macconaill LE, Hahn WC, Meyerson M, Gabriel SB and Garraway LA. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012; 2(1):82-93.
119. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012; 486(7403):400-404.
120. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490(7418):61-70.
121. Mendes-Pereira AM, Sims D, Dexter T, Fenwick K, Assiotis I, Kozarewa I, Mitsopoulos C, Hakas J, Zvelebil M, Lord CJ and Ashworth A. Genome-wide functional screen identifies a compendium of genes affecting sensitivity to tamoxifen. Proc Natl Acad Sci USA. 2012; 109(8):2730-2735.
122. Wallace MD, Pfefferle AD, Shen L, McNairn AJ, Cerami EG, Fallon BL, Rinaldi VD, Southard TL, Perou CM and Schimenti JC. Comparative oncogenomics implicates the neurofibromin 1 gene (NF1) as a breast cancer driver. Genetics. 2012; 192(2):385-396.
123. Lee J, Wang J, Torbenson M, Lu Y, Liu QZ and Li S. Loss of SDHB and NF1 genes in a malignant phyllodes tumor of the breast as detected by oligo-array comparative genomic hybridization. Cancer Genet Cytogenet. 2010; 196(2):179-183.
124. Ward AF, Braun BS and Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. 2012; 120(17):3397-3406.
125. Parkin B, Ouillette P, Wang Y, Liu Y, Wright W, Roulston D, Purkayastha A, Dressel A, Karp J, Bockenstedt P, Al-Zoubi A, Talpaz M, Kujawski L, Liu Y, Shedden K, Shakhan S, et al. NF1 inactivation in adult acute myelogenous leukemia. Clin Cancer Res. 2010; 16(16):4135-4147.
126. Haferlach C, Dicker F, Kohlmann A, Schindela S, Weiss T, Kern W, Schnittger S and Haferlach T. AML with CBFB-MYH11 rearrangement demonstrate RAS pathway alterations in 92% of all cases including a high frequency of NF1 deletions. Leukemia. 2010; 24(5):1065-1069.
127. Boudry-Labis E, Roche-Lestienne C, Nibourel O, Boissel N, Terre C, Perot C, Eclache V, Gachard N, Tigaud I, Plessis G, Cuccuini W, Geffroy S, Villenet C, Figeac M, Leprêtre F, Renneville A, et al. Neurofibromatosis-1 gene deletions and mutations in de novo adult acute myeloid leukemia. Am J Hematol. 2013; 88(4):306-311.
128. Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013; 368(22):2059-2074.
129. Kolquist KA, Schultz RA, Furrow A, Brown TC, Han J-Y, Campbell LJ, Wall M, Slovak ML, Shaffer LG and Ballif BC. Microarray-based comparative genomic hybridization of cancer targets reveals novel, recurrent genetic aberrations in the myelodysplastic syndromes. Cancer Genet. 2011; 204(11):603-628.
130. Misawa S, Horiike S, Kaneko H and Kashima K. Genetic aberrations in the development and subsequent progression of myelodysplastic syndrome. Leukemia. 1997; 11 Suppl 3:533-535.
131. Balgobind BV, Van Vlierberghe P, van den Ouweland AMW, Beverloo HB, Terlouw-Kromosoeto JNR, van Wering ER, Reinhardt D, Horstmann M, Kaspers GJL, Pieters R, Zwaan CM, Van den Heuvel-Eibrink MM and Meijerink JPP. Leukemia-associated NF1 inactivation in patients with pediatric T-ALL and AML lacking evidence for neurofibromatosis. Blood. 2008; 111(8):4322-4328.
132. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen S-C, Wei L, Collins-Underwood JR, Ma J, Roberts KG, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012; 481(7380):157-163.
133. Li Y, Bollag G, Clark R, Stevens J, Conroy L, Fults D, Ward K, Friedman E, Samowitz W and Robertson M. Somatic mutations in the neurofibromatosis 1 gene in human tumors. Cell. 1992; 69(2):275-281.
134. Cawkwell L, Lewis FA and Quirke P. Frequency of allele loss of DCC, p53, RBI, WT1, NF1, NM23 and APC/MCC in colorectal cancer assayed by fluorescent multiplex polymerase chain reaction. British Journal of Cancer. 1994; 70(5):813-818.
135. Leggett B, Young J, Buttenshaw R, Thomas L, Young B, Chenevix-Trench G, Searle J and Ward M. Colorectal carcinomas show frequent allelic loss on the long arm of chromosome 17 with evidence for a specific target region. British Journal of Cancer. 1995; 71(5):1070-1073.
136. Ahlquist T, Bottillo I, Danielsen SA, Meling GI, Rognum TO, Lind GE, Dallapiccola B and Lothe RA. RAS signaling in colorectal carcinomas through alteration of RAS, RAF, NF1, and/or RASSF1A. Neoplasia. 2008; 10(7):680-686, 682 p following 686.
137. Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS, Guillory J, Ha C, Dijkgraaf GJP, Stinson J, Gnad F, Huntley MA, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012; 488(7413):660-664.
138. Network TCGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487(7407):330-337.
139. Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, Decarolis PL, Shah K, Socci ND, Weir BA, Ho A, Chiang DY, Reva B, Mermel CH, Getz G, Antipin Y, Beroukhim R, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010; 42(8):715-721.
140. Paulson V, Chandler G, Rakheja D, Galindo RL, Wilson K, Amatruda JF and Cameron S. High-resolution array CGH identifies common mechanisms that drive embryonal rhabdomyosarcoma pathogenesis. Genes Chromosomes Cancer. 2011; 50(6):397-408.
141. Burnichon N, Buffet A, Parfait B, Letouzé E, Laurendeau I, Loriot C, Pasmant E, Abermil N, Valeyrie-Allanore L, Bertherat J, Amar L, Vidaud D, Favier J and Gimenez-Roqueplo A-P. Somatic NF1 inactivation is a frequent event in sporadic pheochromocytoma. Hum Mol Genet. 2012; 21(26):5397-5405.
142. Welander J, Larsson C, Bäckdahl M, Hareni N, Sivlér T, Brauckhoff M, Söderkvist P and Gimm O. Integrative genomics reveals frequent somatic NF1 mutations in sporadic pheochromocytomas. Hum Mol Genet. 2012; 21(26):5406-5416.
143. Martín Y, Dopazo A and Hernández-Chico C. Progress and challenges in developing a molecular diagnostic test for neurofibromatosis type 1. Expert Rev Mol Diagn. 2011; 11(7):671-673.
144. Ars E, Serra E, García J, Kruyer H, Gaona A, Lázaro C and Estivill X. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet. 2000; 9(2):237-247.
145. Purandare SM, Huntsman Breidenbach H, Li Y, Zhu XL, Sawada S, Neil SM, Brothman A, White R, Cawthon R and Viskochil D. Identification of neurofibromatosis 1 (NF1) homologous loci by direct sequencing, fluorescence in situ hybridization, and PCR amplification of somatic cell hybrids. Genomics. 1995; 30(3):476-485.
146. Luijten M, Wang Y, Smith BT, Westerveld A, Smink LJ, Dunham I, Roe BA and Hulsebos TJ. Mechanism of spreading of the highly related neurofibromatosis type 1 (NF1) pseudogenes on chromosomes 2, 14 and 22. European journal of human genetics : EJHG. 2000; 8(3):209-214.
147. Yu H, Zhao X, Su B, Li D, Xu Y, Luo S, Xiao C and Wang W. Expression of NF1 pseudogenes. Hum Mutat. 2005; 26(5):487-488.
148. Messiaen LM, Callens T, Mortier G, Beysen D, Vandenbroucke I, Van Roy N, Speleman F and Paepe AD. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat. 2000; 15(6):541-555.
149. Valero MC, Martín Y, Hernández-Imaz E, Marina Hernández A, Meleán G, Valero AM, Javier Rodríguez-Álvarez F, Tellería D and Hernández-Chico C. A highly sensitive genetic protocol to detect NF1 mutations. J Mol Diagn. 2011; 13(2):113-122.
150. Sabbagh A, Pasmant E, Imbard A, Luscan A, Soares M, Blanché H, Laurendeau I, Ferkal S, Vidaud M, Pinson S, Bellanné-Chantelot C, Vidaud D, Parfait B and Wolkenstein P. NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: the French experience. Hum Mutat. 2013; 34(11):1510-1518.
151. Lenarduzzi M, Hui ABY, Alajez NM, Shi W, Williams J, Yue S, O’Sullivan B and Liu F-F. MicroRNA-193b enhances tumor progression via down regulation of neurofibromin 1. PLoS ONE. 2013; 8(1).
152. Gutmann DH, McLellan MD, Hussain I, Wallis JW, Fulton LL, Fulton RS, Magrini V, Demeter R, Wylie T, Kandoth C, Leonard JR, Guha A, Miller CA, Ding L and Mardis ER. Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res. 2013; 23(3):431-439.
153. McGillicuddy LT, Fromm JA, Hollstein PE, Kubek S, Beroukhim R, De Raedt T, Johnson BW, Williams SMG, Nghiemphu P, Liau LM, Cloughesy TF, Mischel PS, Parret A, Seiler J, Moldenhauer G, Scheffzek K, et al. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer cell. 2009; 16(1):44-54.
154. Hollstein PE and Cichowski K. Identifying the Ubiquitin Ligase complex that regulates the NF1 tumor suppressor and Ras. Cancer Discov. 2013; 3(8):880-893.
155. Sedani A, Cooper DN and Upadhyaya M. An emerging role for microRNAs in NF1 tumorigenesis. Hum Genomics. 2012; 6.
156. Gupta A, Cohen BH, Ruggieri P, Packer RJ and Phillips PC. Phase I study of thalidomide for the treatment of plexiform neurofibroma in neurofibromatosis 1. Neurology. 2003; 60(1):130-132.
157. Packer RJ, Gutmann DH, Rubenstein A, Viskochil D, Zimmerman RA, Vezina G, Small J and Korf B. Plexiform neurofibromas in NF1: toward biologic-based therapy. Neurology. 2002; 58(10):1461-1470.
158. Babovic-Vuksanovic D, Widemann BC, Dombi E, Gillespie A, Wolters PL, Toledo-Tamula MA, O’Neill BP, Fox E, MacDonald T, Beck H and Packer RJ. Phase I trial of pirfenidone in children with neurofibromatosis 1 and plexiform neurofibromas. Pediatr Neurol. 2007; 36(5):293-300.
159. Babovic-Vuksanovic D, Ballman K, Michels V, McGrann P, Lindor N, King B, Camp J, Micic V, Babovic N, Carrero X, Spinner R and O’Neill B. Phase II trial of pirfenidone in adults with neurofibromatosis type 1. Neurology. 2006; 67(10):1860-1862.
160. Widemann BC, Salzer WL, Arceci RJ, Blaney SM, Fox E, End D, Gillespie A, Whitcomb P, Palumbo JS, Pitney A, Jayaprakash N, Zannikos P and Balis FM. Phase I trial and pharmacokinetic study of the farnesyltransferase inhibitor tipifarnib in children with refractory solid tumors or neurofibromatosis type I and plexiform neurofibromas. J Clin Oncol. 2006; 24(3):507-516.
161. Widemann BC, Dombi E, Gillespie A, Wolters PL, Belasco J, Goldman S, Korf BR, Solomon J, Martin S, Salzer W, Fox E, Patronas N, Kieran MW, Perentesis JP, Reddy A, Wright JJ, et al. Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro-oncology. 2014; 16(5):707-718.
162. Weiss B, Widemann BC, Wolters P, Dombi E, Vinks AA, Cantor A, Korf B, Perentesis J, Gutmann DH, Schorry E, Packer R and Fisher MJ. Sirolimus for non-progressive NF1-associated plexiform neurofibromas: an NF clinical trials consortium phase II study. Pediatr Blood Cancer. 2014; 61(6):982-986.
163. Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME, Eaves D, Widemann B, Kim M-O, Dombi E, Sabo J, Hardiman Dudley A, Niwa-Kawakita M, Page GP, Giovannini M, Aronow BJ, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013; 123(1):340-347.
164. Jakacki RI, Dombi E, Potter DM, Goldman S, Allen JC, Pollack IF and Widemann BC. Phase I trial of pegylated interferon-α-2b in young patients with plexiform neurofibromas. Neurology. 2011; 76(3):265-272.
165. Zhu Y, Ghosh P, Charnay P, Burns DK and Parada LF. Neurofibromas in NF1: Schwann Cell Origin and Role of Tumor Environment. Science. 2002; 296(5569):920-922.
166. Robertson KA, Nalepa G, Yang F-C, Bowers DC, Ho CY, Hutchins GD, Croop JM, Vik TA, Denne SC, Parada LF, Hingtgen CM, Walsh LE, Yu M, Pradhan KR, Edwards-Brown MK, Cohen MD, et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: a phase 2 trial. The Lancet Oncology. 2012; 13(12):1218-1224.
167. Yang F-C, Ingram DA, Chen S, Zhu Y, Yuan J, Li X, Yang X, Knowles S, Horn W, Li Y, Zhang S, Yang Y, Vakili ST, Yu M, Burns D, Robertson K, et al. Nf1-dependent tumors require a microenvironment containing Nf1+/-- and c-kit-dependent bone marrow. Cell. 2008; 135(3):437-448.
168. Lasater EA, Bessler WK, Mead LE, Horn WE, Clapp DW, Conway SJ, Ingram DA and Li F. Nf1+/- mice have increased neointima formation via hyperactivation of a Gleevec sensitive molecular pathway. Hum Mol Genet. 2008; 17(15):2336-2344.
169. Kim A, Dombi E, Tepas K, Fox E, Martin S, Wolters P, Balis FM, Jayaprakash N, Turkbey B, Muradyan N, Choyke PL, Reddy A, Korf B and Widemann BC. Phase I trial and pharmacokinetic study of sorafenib in children with neurofibromatosis type I and plexiform neurofibromas. Pediatr Blood Cancer. 2013; 60(3):396-401.
170. Barkan B, Starinsky S, Friedman E, Stein R and Kloog Y. The Ras inhibitor farnesylthiosalicylic acid as a potential therapy for neurofibromatosis type 1. Clin Cancer Res. 2006; 12(18):5533-5542.
171. Bhola P, Banerjee S, Mukherjee J, Balasubramanium A, Arun V, Karim Z, Burrell K, Croul S, Gutmann DH and Guha A. Preclinical in vivo evaluation of rapamycin in human malignant peripheral nerve sheath explant xenograft. Int J Cancer. 2010; 126(2):563-571.
172. Johansson G, Mahller YY, Collins MH, Kim M-O, Nobukuni T, Perentesis J, Cripe TP, Lane HA, Kozma SC, Thomas G and Ratner N. Effective in vivo targeting of the mammalian target of rapamycin pathway in malignant peripheral nerve sheath tumors. Mol Cancer Ther. 2008; 7(5):1237-1245.
173. Zou CY, Smith KD, Zhu Q-S, Liu J, McCutcheon IE, Slopis JM, Meric-Bernstam F, Peng Z, Bornmann WG, Mills GB, Lazar AJ, Pollock RE and Lev D. Dual targeting of AKT and mammalian target of rapamycin: a potential therapeutic approach for malignant peripheral nerve sheath tumor. Mol Cancer Ther. 2009; 8(5):1157-1168.
174. Endo M, Yamamoto H, Setsu N, Kohashi K, Takahashi Y, Ishii T, Iida K-i, Matsumoto Y, Hakozaki M, Aoki M, Iwasaki H, Dobashi Y, Nishiyama K, Iwamoto Y and Oda Y. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin Cancer Res. 2013; 19(2):450-461.
175. Li H, Velasco-Miguel S, Vass WC, Parada LF and DeClue JE. Epidermal growth factor receptor signaling pathways are associated with tumorigenesis in the Nf1:p53 mouse tumor model. Cancer Res. 2002; 62(15):4507-4513.
176. Banerjee S, Byrd JN, Gianino SM, Harpstrite SE, Rodriguez FJ, Tuskan RG, Reilly KM, Piwnica-Worms DR and Gutmann DH. The neurofibromatosis type 1 tumor suppressor controls cell growth by regulating signal transducer and activator of transcription-3 activity in vitro and in vivo. Cancer Res. 2010; 70(4):1356-1366.
177. De Raedt T, Walton Z, Yecies JL, Li D, Chen Y, Malone CF, Maertens O, Jeong SM, Bronson RT, Lebleu V, Kalluri R, Normant E, Haigis MC, Manning BD, Wong K-K, Macleod KF, et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer cell. 2011; 20(3):400-413.
178. Patel AV, Eaves D, Jessen WJ, Rizvi TA, Ecsedy JA, Qian MG, Aronow BJ, Perentesis JP, Serra E, Cripe TP, Miller SJ and Ratner N. Ras-driven transcriptome analysis identifies aurora kinase a as a potential malignant peripheral nerve sheath tumor therapeutic target. Clin Cancer Res. 2012; 18(18):5020-5030.
179. Hirokawa Y, Nheu T, Grimm K, Mautner V, Maeda S, Yoshida M, Komiyama K and Maruta H. Sichuan pepper extracts block the PAK1/cyclin D1 pathway and the growth of NF1-deficient cancer xenograft in mice. Cancer Biol Ther. 2006; 5(3):305-309.
180. Hirokawa Y, Levitzki A, Lessene G, Baell J, Xiao Y, Zhu H and Maruta H. Signal therapy of human pancreatic cancer and NF1-deficient breast cancer xenograft in mice by a combination of PP1 and GL-2003, anti-PAK1 drugs (Tyr-kinase inhibitors). Cancer Lett. 2007; 245(1-2):242-251.
181. See WL, Tan IL, Mukherjee J, Nicolaides T and Pieper RO. Sensitivity of glioblastomas to clinically available MEK inhibitors is defined by neurofibromin 1 deficiency. Cancer Res. 2012; 72(13):3350-3359.
182. Lauchle JO, Kim D, Le DT, Akagi K, Crone M, Krisman K, Warner K, Bonifas JM, Li Q, Coakley KM, Diaz-Flores E, Gorman M, Przybranowski S, Tran M, Kogan SC, Roose JP, et al. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature. 2009; 461(7262):411-414.
183. Dodd RD, Mito JK, Eward WC, Chitalia R, Sachdeva M, Ma Y, Barretina J, Dodd L and Kirsch DG. NF1 deletion generates multiple subtypes of soft-tissue sarcoma that respond to MEK inhibition. Mol Cancer Ther. 2013; 12(9):1906-1917.
184. Petrilli A, Bott M and Fernandez-Valle C. Inhibition of SIRT2 in merlin/NF2-mutant Schwann cells triggers necrosis. Oncotarget. 2013; 4(12):2354-2365.
185. Mashiach-Farkash E, Rak R, Elad-Sfadia G, Haklai R, Carmeli S, Kloog Y and Wolfson HJ. Computer-based identification of a novel LIMK1/2 inhibitor that synergizes with salirasib to destabilize the actin cytoskeleton. Oncotarget. 2012; 3(6):629-639.
186. Rak R and Kloog Y. Targeting LIM kinase in cancer and neurofibromatosis. Cell cycle (Georgetown, Tex). 2014; 13(9):1360-1361.