
Introduction
Cyclooxygenase-2 (COX-2) was first found to be upregulated during the early stage of Alzheimer’s disease (AD) two decades ago [1]. Because AD is pathologically characterized by β-amyloid protein (Aβ) deposition in amyloid plaques (APs) and by tau phosphorylation in neurofibrillary tangles (NFTs) [2], numerous investigations have since been carried out to evaluate the roles of COX-2 and its metabolic products, prostaglandins (PGs), in Aβ deposition. However, the effects of COX-2 and PGs on tau phosphorylation were largely overlooked. To the best of our knowledge, COX-2 suppression by ibuprofen treatment decreases the phosphorylation of tau in APP/PS1/Tau Tg mice [3]. In addition, COX-2 overexpression induces the formation of NFTs in patients with Fukuyama-type congenital muscular dystrophy [4]. As the metabolic product of COX-2, 15d-PGJ2 treatment increases the cleavage of tau protein and results in the formation of ∆tau, which accelerates the formation of NFTs in human neuroblastoma SK-N-SH cells [5]. These observations clearly indicated that COX-2 and PGs might be able to regulate the phosphorylation of tau and the formation of NFTs during the development of AD.
Although we could not find more direct evidence showing the relationship between COX-2/PGs and tau phosphorylation, we cannot exclude the possibility that other molecules, such as cytokines, may induce tau phosphorylation via enhancing the expression of COX-2 and PGs. For example, in mice, TNF-α injection induces cognitive decline via increasing the expression of COX-2 [6]. In addition, IL-1β treatment has also been reported to impair the learning ability of mice via upregulating the production of PGE2 [7, 8]. Therefore, it is highly plausible that cytokines, including TNF-α, IL-1β and IL-6, can increase the phosphorylation of tau via COX-2 and PGs in different AD experimental models [9-14].
Related to the possible roles of cytokines in tau phosphorylation via COX-2 and PGs, it has recently been reported that Zn2+ dyshomeostasis can regulate cytokine expression. For instance, Chu et al. [15, 16] reported that TNF-α gene expression is increased following Zn2+ treatment in type 2 diabetes mellitus, Zn2+ treatment has ability to induce the expression of TNF-α in wistar rats [17]. In addition, the levels of Zn2+ and TNF-α have been reported as potential blood biomarkers for disease severity in the Taiwanese population with AD [18]. These observations also indicate that Zn2+ might be able to regulate tau phosphorylation. Notably, Zn2+ has been reported to be significantly increased within the cerebral cortex of AD patient [19, 20]. Moreover, tau hyperphosphorylation has been observed to be correlated with brain Zn2+ metabolism disorders [21]. Indeed, treatment with low concentrations of Zn2+ can induce the aggregation of tau protein in vitro. Mechanistically, previous studies revealed that Zn2+ induced tau hyperphosphorylation by activating the ERK1/2, JNK, GSK-3β, and p38 pathways [19]. Further, Zn2+ stimulated tau phosphorylation by inhibiting the protein phosphatase PP2A in cultured rat hippocampal slice [21]. In addition, the Zn2+ chelator clioquinol (CQ) significantly and reversibly reduced the phosphorylation of tau in rats. Actually, Zn2+ is not free to pass through the cell membrane and require specific transporters and membrane channels to participate in cell metabolism. The regulation of Zn2+ homeostasis is closely related to zinc transporter (Zinc transporter, ZnT) in brain. Specifically, ZnT3 is mainly distributed in the central nervous system, and located in synaptic vesicles of axon terminals in zinc glutamate neurons. Its function is to transport Zn2+ into in synaptic vesicles. It is also suggested that the abnormal expression and distribution of ZnT3 may be the main cause of the disorder of Zn2+ metabolism in AD brain [22]. Consistent with the critical roles of Zn2+ in tau phosphorylation, the abnormal expression and distribution of zinc transporter 3 (ZnT3) have also been reported to cause AD via disrupting Zn2+ metabolism in synaptic vesicles [23-26]. Furthermore, knockout of ZnT3 in APP mice significantly decreased the production of Aβ and formation of APs [27, 28].
Although all these studies indicated the involvement of COX-2, PGs, TNF-α and Zn2+ in regulating tau phosphorylation, the relationships and mechanisms linking these bioactive molecules remain unknown. Therefore, the purpose of the current study is to decipher the mechanisms of tau phosphorylation in TauP301S Tg mice. These TauP301S Tg mice express the P301S mutant form of human microtubule-associated protein tau (MAPT). At three months of age, transgenic mice exhibit clasping and limb retraction when lifted by the tail, which progresses to limb weakness. By ten months of age, the mice exhibit a hunched back and paralysis, followed by inability to feed. The onset of neurofibrillay tangles (NFTs) formation in the neocortex, amygdala, hippocampus, brain stem and spinal cord is five months of age. Transgenic mice display neuroinflammation with microglial activation and astrogliosis. The ultrastructure of the NFTs-like lesions detected is similar to that found in brain lesions of human Alzheimer’s disease patients. Specifically, our data demonstrated that the production of PGI2/PGF2α and the expression of COX-2 were induced by Zn2+ via the TNF-α pathway in a ZnT3-dependent manner. In addition, high levels of PGI2 and F2α stimulated the phosphorylation of tau at both Ser 202 and Ser 400/Thr 403/Ser 404 via the PI3-K/AKT, ERK1/2 and JNK/c-Jun pathways. These observations provide novel insights into the mechanisms of tau phosphorylation and may be instrumental for identifying therapeutic targets for combating AD.
Results
The expression of COX-2, TNF-α, ZnT3 and phosphorylation of tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404 were elevated in 6-month-old TauP301S Tg mice
In view of the possible roles of COX-2, TNF-α and ZnT3 in tau phosphorylation, we initially determined the expression of COX-2, TNF-α and ZnT3 in 6-month-old TauP301S Tg mice. As shown in Figure 1A-1C, the mRNA and protein expression of COX-2, TNF-α and ZnT3 were elevated in 6-months-old TauP301S Transgenic (Tg) mice compared to the paired wild type (WT) controls. In addition, immunoreactivity of p-Tau (Ser 202 and Ser 400/Thr 403/Ser 404) was progressively upregulated during the course of AD development (Figure 1D). Of note, the phosphorylated tau protein concentrated in the NFTs of late stage of AD patients, and the number of NFTs was increased gradually as the disease progresses (Figure 1D). As the limited accessibility of human samples from AD patients, we further found that the phosphorylation of tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404 was increased in the CA3 region of 6-month-old TauP301S Tg mice compared to that of the paired WT controls (Figure 1E). To further reveal the relationship of these molecules, we double stained the brains of AD patients with ZnT3, TNF-α, COX-2 and phosphorylated tau. The results demonstrated that phosphorylated tau colocalized with ZnT3, TNF-α and COX-2, respectively (Figure 1F). Collectively, these observations indicate the internal relationship among these molecules, which result in AD development and progression.
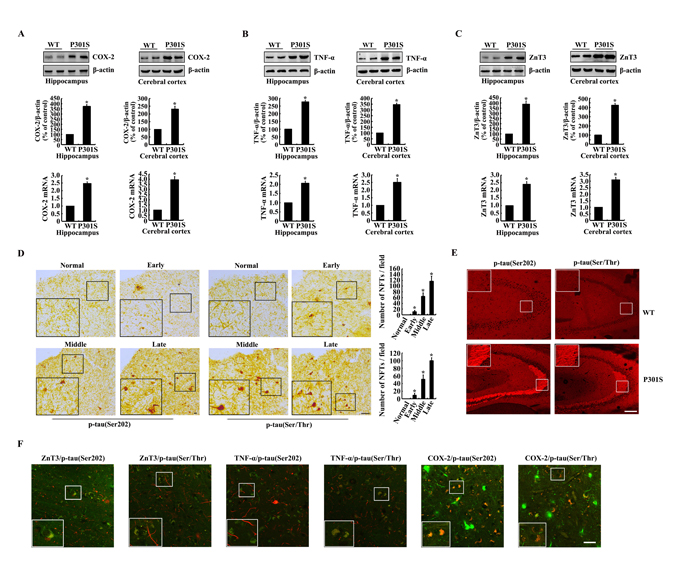
Figure 1: The expression of COX-2, TNF-α, ZnT3 and phosphorylation of tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404 were upregulated in 6-month-old TauP301S Tg mice. (A-C, E) The brains of WT or TauP301S Tg mice at the age of 6-month-old were collected following anesthesia and perfusion (n=8). (A-C) The mRNA and protein expression of COX-2, TNF-α and ZnT3 in the hippocampus and cerebral cortex of WT and Tg mice was determined by western blots. (E) The immunoreactivity of p-tau was determined by immunohistochemistry using an anti-tau (Ser202) or anti-tau (Ser400/Thr403/Ser404) antibody. (D, F) The tissue blocks of human brains at different stages of AD were collected by the New York Brain Bank at Columbia University. Free-floating slices (40 µm) were prepared by flat slicer (n=3). (D) The phosphorylation of tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404 were immunostained by immunohistochemistry using specific antibodies. (F) The slices of AD patients were double stained with p-tau and ZnT3, TNF-α and COX-2, respectively. The data represent the means ± S.E. *, p<0.05 with respect to WT controls.
Critical roles of Zn2+ and TNF-α in upregulating the expression of COX-2 in mice.
As the possible roles of Zn2+ and TNF-α in tau phosphorylation via COX-2, we determined the effects of Zn2+ and TNF-α on the expression of COX-2. To this purpose, we injected (i.c.v, intracerebroventricular injection) TNF-α (1 ng/5 µl) to the ventricles of 6-month-old WT mice. The results demonstrated that TNF-α injection (i.c.v) stimulated the mRNA and protein expression of COX-2 (Figure 2A, 2B). Similarly, ZnSO4 (10 µg/5 µl) injection (i.c.v) to the ventricles of 6-month-old WT mice induces the mRNA and protein expression of COX-2 in the hippocampus and cerebral cortex of 6-month-old WT mice (Figure 2C, 2D). More interestingly, ZnSO4 injection (i.c.v) clearly enhances the expression of ZnT3 or TNF-α in the hippocampus and cerebral cortex of 6-month-old WT mice (Figure 3A, 3B). When we blocked the biological function of ZnT3 by antibody, the expression of COX-2 was reduced in n2a cells (Figure 3C, 3D). These observations clearly indicated that ZnT3 mediated the effects of Zn2+ on inducing the expression of COX-2.
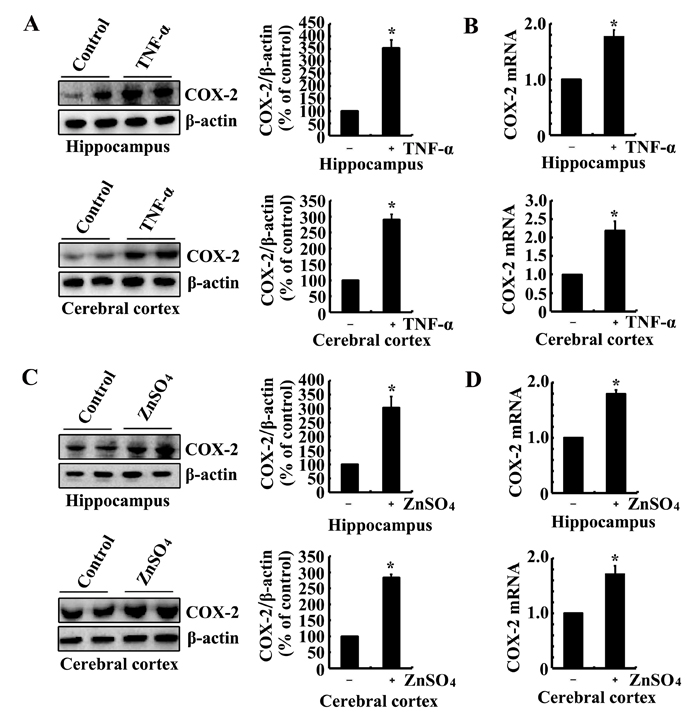
Figure 2: Intracerebroventricular injection of ZnSO4 and TNF-α to the ventricles of WT mice increases the expression of COX-2. (A-D) The WT mice at the age of 6 months were injected (i.c.v) with ZnSO4 (10 µg/5 µl) or TNF-α (1 ng/5 µl) (n=12). The brains were then collected after 24 h. mRNA and protein levels of COX-2 were determined by qRT-PCR and western blot, respectively. Total amounts of GAPDH and β-actin served as an internal control. The data represent the means ± S.E.. *, p<0.05 with respect to PBS (-)-injected (i.c.v) controls.
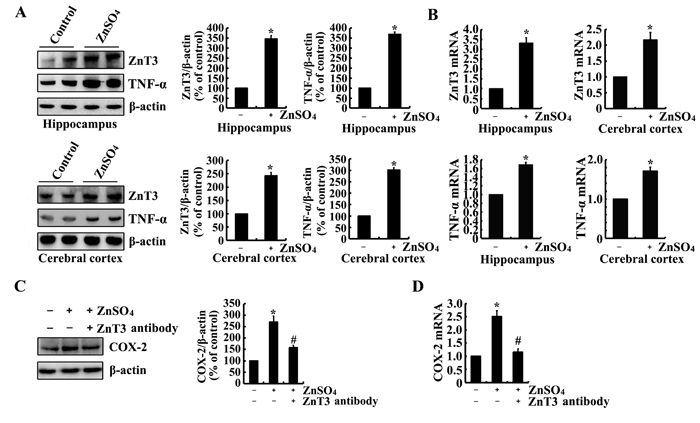
Figure 3: ZnSO4 stimulates the expression of COX-2 via inducing the expression of ZnT3 and TNF-α. (A, B) The WT mice at the age of 6 months were injected (i.c.v.) with ZnSO4 (10 µg/5 µl) (n=12). The brains were then collected after 24 h. mRNA and protein levels of ZnT3 and TNF-α were determined by qRT-PCR and western blot, respectively. Total amounts of GAPDH and β-actin served as an internal control. (C-D) In select experiments, n2a cells were treated with ZnSO4 (10 µM) in the absence or presence of ZnT3 antibody (1 µg/ml). (C, D) mRNA and protein levels of COX-2 were determined by qRT-PCR and western blots, respectively. Total amounts of GAPDH and β-actin served as an internal control. The data represent the means ± S.E.. *, p<0.05 with respect to PBS (-) or vehicle-treated controls. #, p<0.05 with respect to ZnSO4-treated alone.
Effects of PGI2 and F2α on stimulating the phosphorylation of tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404
As the metabolic products of COX-2, immunostaining experiments were carried out to determine the effects of PGI2 and F2α on the phosphorylation of tau. As a consequence, PGI2 and F2α injection (i.c.v) for 24 h to the ventricles of mice clearly increases the immunoreactivity of p-tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404 (Figure 4A). To validate the critical roles of TNF-α and ZnSO4 in upregulating the expression of COX-2, we further determined the effects of TNF-α and ZnSO4 on the phosphorylation of tau. As expected, the results showed that injection (i.c.v) of TNF-α (1 ng/5 µl) and ZnSO4 (10 µg/5 µl) to the ventricles of mice obviously induced the phosphorylation of tau at the sites of Ser 202 and Ser 400/Thr 403/Ser 404 (Figure 4A). These immunostaining results were further validated by qRT-PCR and western blots. In detail, PGI2 and F2α injection (i.c.v) were able to stimulate the phosphorylation of tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404 in the himmpocampus and cerebral cortex of mice (Figure 4B-4E). In addition, the phosphorylation of tau at the sites of Ser 202 and Ser 400/Thr 403/Ser 404 was also induced by the stimulation of ZnSO4 and TNF-α in the hippocampus and cerebral cortex of mice (Figure 4B-4G). Based on these observations, our data have verified the fact that PGI2 and F2α potentially mediated the effects of COX-2 on inducing the phosphorylation of tau at the sites of Ser 202 and Ser 400/Thr 403/Ser 404 in mice, which in turn potentially contribute the effects of Zn2+ and TNF-α on the phosphorylation of tau.
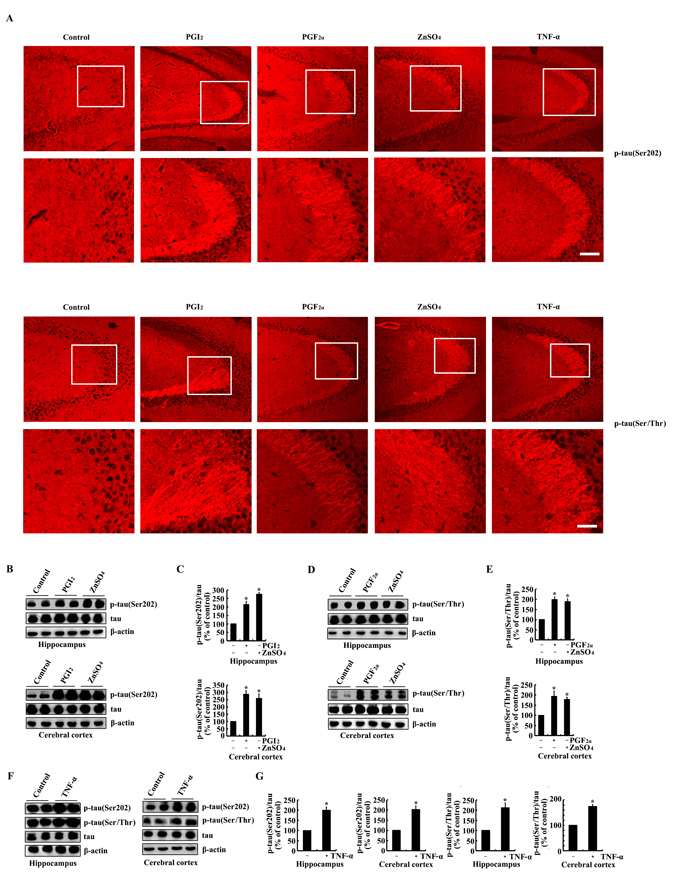
Figure 4: PGI2 and F2α mediated the effects of ZnSO4and TNF-α on stimulating the phosphorylation of tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404. The WT mice at the age of 6 months were injected (i.c.v) with PGI2 (1 µg/5 µl), PGF2α (1 µg/5 µl), ZnSO4 (10 µg/5 µl) or TNF-α (1 ng/5 µl) (n=12). (A) The phosphorylation of tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404 were determined by immunostaining using the correspondent antibody. (B-G) Western blots were further used to determine the phosphorylation of tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404. The intensity of bands was analyzed by Bio-Rad imaging software. The data represent the means ± S.E.. *, p<0.05 with respect to PBS (-)-injected (i.c.v) controls.
Key roles of COX-2 in mediating the effects of Zn2+ and TNF-α on stimulating the phosphorylation of tau at the sites of both Ser 202 andSer 400/Thr 403/Ser 404
As the potential roles of Zn2+ and TNF-α in inducing the phosphorylation of tau via upregulating the expression of COX-2, it is necessary to verify if COX-2 mediates the effects of Zn2+ and TNF-α on stimulating the phosphorylation of tau. As a first step, experiments were carried out to inject (i.c.v) into the ventricles of 6-month-old TauP301S Tg mice. The results demonstrated that NS398, a COX-2 inhibitors (1 µg/5 µl) treatment clearly decreased the phosphorylation of tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404 in the hippocampus and cerebral cortex of TauP301S Tg mice (Figure 5A-5D). In addition, NS398 (100 µM) treatment blocked the effects of TNF-α (10 ng/ml) and ZnSO4 (10 µM) on stimulating the phosphorylation of tau at the sites of Ser 202 and Ser 400/Thr 403/Ser 404 in n2a cells (Figure 5E-5H). Therefore, it is clear that Zn2+ and TNF-α regulate the phosphorylation of tau via COX-2 activation.
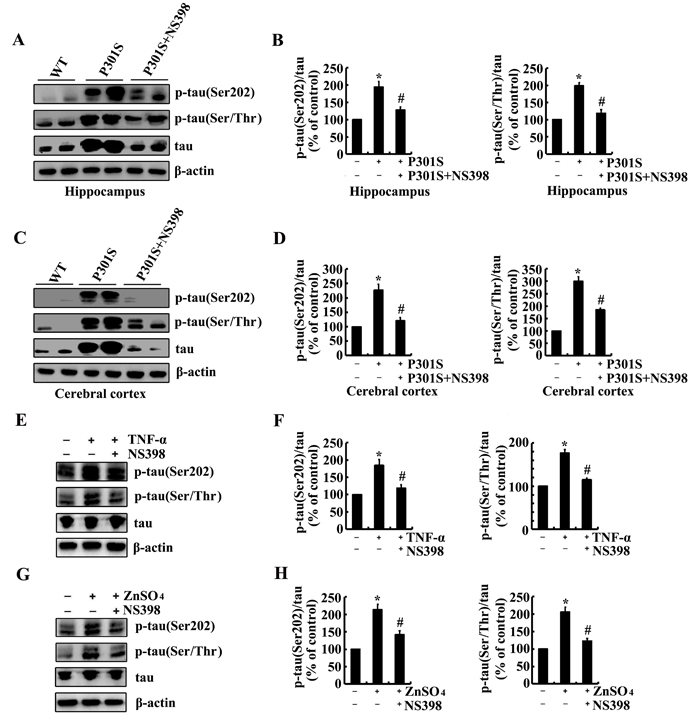
Figure 5: NS398 treatment decreases the phosphorylation of tau at the sites of Ser 202 and Ser 400/Thr 403/Ser 404 in 6-month-old TauP301S Tg mice or TNF-α- or ZnSO4-activated n2a cells. (A-D) The TauP301S Tg mice at the age of 6 months were injected (i.c.v) with NS398 (1 µg/5 µl) and the brains were then collected after 24 h. (E-H) In select experiments, the n2a cells were treated with TNF-α (10 ng/ml) or ZnSO4 (10 µΜ) in the absence or presence of NS398 (100 µΜ) for 24 h. The phosphorylation of tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404 were determined by western blots. The intensity of bands was analyzed by Bio-Rad imaging software. The data represent the means ± S.E. *, p<0.05 with respect to WT or vehicle-treated controls. #, p<0.05 with respect to TauP301S Tg mice or TNF-α-, ZnSO4-treated alone.
Signaling pathways involved in regulating the phosphorylation of tau
We next aimed to determine the possible signaling pathways in regulating the phosphorylation of tau. As a consequence, experiments were carried out to inject NS398 (1 µg/5 µl) to the ventricles of 6-month-old TauP301S Tg mice. The results demonstrated that the phosphorylation of ERK1/2, AKT and c-Jun were highly induced in 6-month-old TauP301S Tg mice (Figure 6A-6D). In addition, NS398 treatment significantly decreased the phosphorylation of ERK1/2, AKT and c-Jun in 6-month-old TauP301S Tg mice (Figure 6A-6D). In line with these in vivo results, we further treated n2a cells with TNF-α (10 ng/ml) and ZnSO4 (10 µM) in the absence or presence of NS398 (100 µM) for 24 h. The results demonstrated that NS398 clearly decreased the phosphorylation of ERK1/2, AKT and c-Jun in TNF-α- or ZnSO4-treated n2a cells (Figure 6E-6H). To further verify the involvement of these possible signaling pathways, we treated the n2a cells with PGI2 or PGF2α in the absence or presence of inhibitors of signaling pathways, respectively. The results demonstrated that treatment with inhibitors of PI3-K/AKT, ERK1/2 and JNK/c-Jun obviously suppressed the PGI2- and F2α-activated tau phosphorylation (Figure 7A-7D).
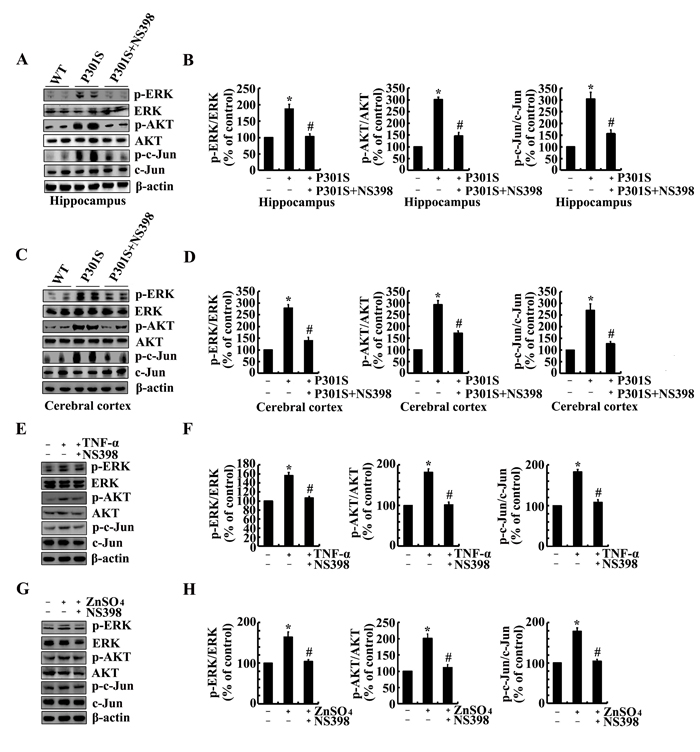
Figure 6: NS398 treatment decreases the activity of PI3-K/AKT, ERK1/2 and JNK/c-Jun pathways in 6-month-old TauP301S Tg mice and TNF-α- or ZnSO4-stimulated n2a cells. (A-D) The TauP301S Tg mice at the age of 6 months were injected (i.c.v) with NS398 (1 µg/5 µl) and the brains were then collected after 24 h. (E-H) In select experiments, the n2a cells were treated with TNF-α (10 ng/ml) or ZnSO4 (10 µΜ) in the absence or presence of NS398 (100 µΜ) for 24 h. The phosphorylation of AKT, ERK1/2 and c-Jun were determined by western blots. The intensity of bands was analyzed by Bio-Rad imaging software. The data represent the means ± S.E. *, p<0.05 with respect to WT or vehicle-treated controls. #, p<0.05 with respect to TauP301S Tg mice or TNF-α-, ZnSO4-treated alone.
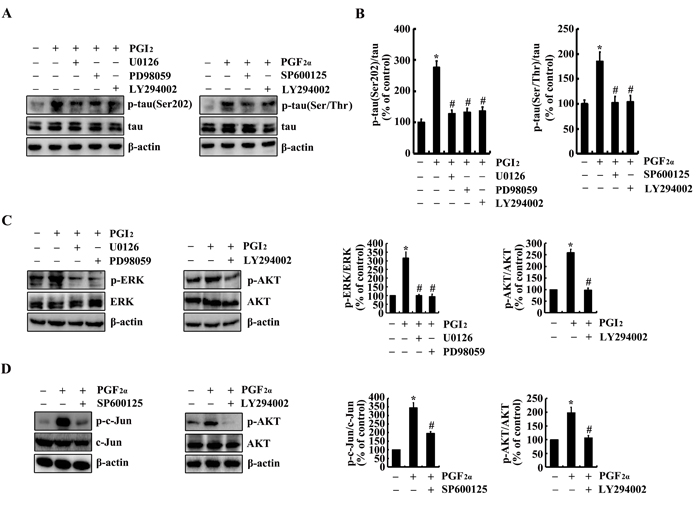
Figure 7: Involvement of PI3-K/AKT, ERK1/2 and JNK/c-Jun pathways in regulating tau phosphorylation at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404 in PGI2- or PGF2α-stimulated n2a cells. n2a cells were treated with PGI2 (10 µΜ) or PGF2α (1 µΜ) in the absence or presence of PI3-K inhibitor, LY294002 (20 µΜ), ERK1/2 inhibitor, PD98059 (20 µΜ) and U0126(20 µΜ), JNK inhibitor, SP600125 (10 µΜ), for 24 h. (A, B) The phosphorylation of tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404 were determined by western blots. (C, D) The phosphorylation of AKT, ERK1/2 and c-Jun were determined by western blots. The intensity of bands was analyzed by Bio-Rad imaging software. The data represent the means ± S.E. *, p<0.05 with respect to vehicle-treated controls. #, p<0.05 with respect to PGI2 or F2α-treated alone.
NS398 treatment decreases the cognitive decline of TauP301S Tg mice
As the pivotal roles of COX-2 in regulating the phosphorylation of tau, the questions are easily raised if COX-2 overexpression will finally affect the learning ability of TauP301S Tg mice. In accordance with this hypothesis, nest construction assays were designed to determine the learning ability of mice [29]. As expected, the learning ability of TauP301S Tg mice appeared to be impaired at the age of 6-month-old (Figure 8A, 8B). In addition, NS398 treatment improves the ability of nest construction for 6-month-old TauP301S Tg mice (Figure 8A, 8B). To further verify these behavioral deficiencies, limb clasping experiments were carried out as to determine the hallmark of AD. The results demonstrated that the TauP301S Tg mice showed limb-clasping reflexes at 6 months old, which was partially reversed by NS398 treatment (Figure 8C, 8D). These observations further emphasize the pivotal roles of COX-2 in regulating the development or progression of AD.
In summary, our data revealed that ZnSO4 played critical effects on stimulating the expression of TNF-α in a ZnT3-dependent mechanism. Ηighly expressed TNF-α induced the expression of COX-2. COX-2 induced the phosphorylation of tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404 via PGI2- and PGF2α-dependent PI3-K/AKT, ERK1/2 and JNK/c-Jun activation pathways. Finally, the phosphorylation of tau protein will impair the learning ability of TauP301S Tg mice (Figure 9).
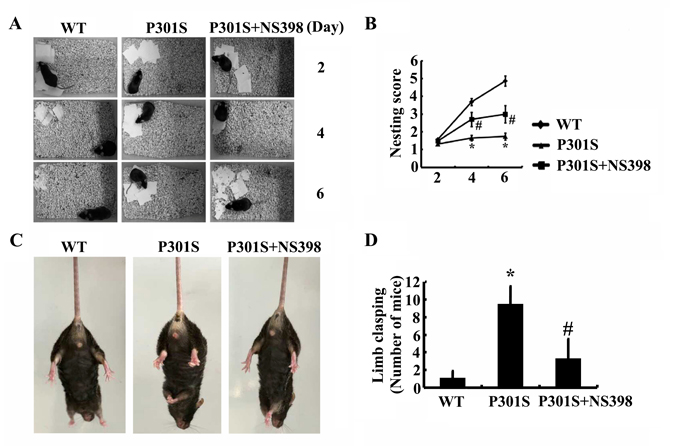
Figure 8: COX-2 plays pivotal roles in impairing the learning ability of TauP301S Tg mice. (A, B) TauP301S Tg mice at the age of 1 months old were treated with NS398 (1 mg/kg/d) for 5 months (n=12). The learning ability of mice was then determined by nest construction. Baseline data were obtained on d 0, after the addition of paper towels in clean cages. Habituation behavior was recorded everyday and the nesting score was obtained as described in “Materials and Methods”. (C, D) Abnormal limb clasping of the mice were determined by lifting the tail. Each group was tested six times.
Discussion
COX-2 and its metabolic products, prostaglandins (PGs), have been reported to be upregulated during the early stage of AD [30, 31]. Although the interest in investigating COX-2 was disrupted by the failure of clinical trials [32], the results of these clinical trials have been called into question based on the lack of a mechanistic study for guidance. Therefore, we investigated the mechanisms by which COX-2 metabolic products, PGI2 and F2α might regulate the phosphorylation of tau. Specifically, TNF-α and ZnSO4 stimulated the expression of COX-2 and the production of PGs, such as PGI2 and F2α. Notably, ZnT3 mediated the induction of COX-2 expression by ZnSO4. Then, high COX-2 expression increases the phosphorylation of tau by producing PGI2 and F2α through the PI3-K/AKT, ERK1/2 and JNK/c-Jun pathways. Consistent with the pivotal roles of COX-2 and PGs in regulating tau phosphorylation, treatment with the COX-2-specific inhibitor NS398 decreases the cognitive decline of TauP301S Tg mice (Figure 9).
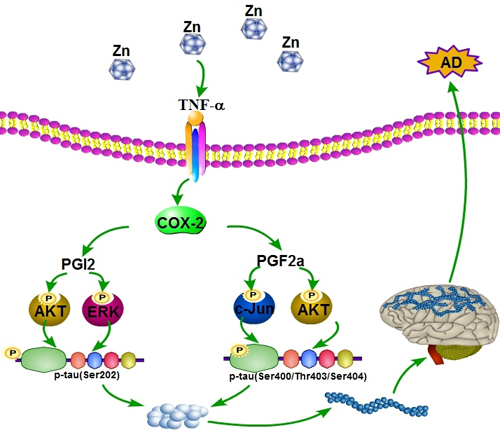
Figure 9: Proposed events of the signaling cascade regulate the pathogenesis of AD. In detail, ZnSO4 have ability to upregulate the expression of COX-2 and the production of PGI2 and F2α via inducing the expression of TNF-α. Highly accumulated PGI2 and F2α induce the phosphorylation of tau at the sites of both Ser 202 and Ser 400/Thr 403/Ser 404 via PI3-K/AKT, ERK1/2 and JNK/c-Jun pathways. These actions finally result in the cognitive decline of TauP301S Tg mice.
COX-2 is tightly regulated under physiological conditions. As an inducible enzyme, COX-2 mRNA and protein synthesis can be induced in numerous cell types, including neurons, in a time- and dose-dependent manner by metal ions, cytokines, oncogenes and growth factors [33-38]. It is thus presumed that COX-2 is primarily responsible for the elevated production of PGs at the sites of disease. Consistent with this hypothesis, Montine et al. [31] reported that COX-2 and PGE2 were upregulated during the early stage of AD. These observations indicate that the early stage of AD must include stimulators that result in the upregulation of COX-2. Among various stimulators for COX-2 expression, we found that Zn2+ and TNF-α can enhance the expression of COX-2. In line with our observation, Wu et al. [39]reported that Zn2+ exposure resulted in pronounced increases in COX-2 mRNA and protein expression in human airway epithelial cells. Further, Choi et al. [40] reported that Zn2+ chelation inhibits the expression of COX-2 in ischemia-activated retinal neurons. Mechanistically, Choi et al. [40] reported that the p38- and epithelial growth factor (EGF) receptor-activated PI3-K/AKT pathway is necessary for Zn2+-induced COX-2 expression. In contrast to these observations, Taccioli et al. [41] reported that Zn2+ replenishment decreases the overexpression of COX-2 in Zn2+ deficient rats. However, this study should be interpreted with caution because their experiments do not focus on neurons.
Apart from Zn2+, TNF-α also increases the expression of COX-2. In line with our observation, Tyagi et al. [42] reported that TNF-α can induce the expression of COX-2 in tumorigenic mouse lung epithelial LM2 cells. In addition, Kuwano et al. [43] reported that TNF-α enhances the production of COX-2 protein in HUVECs. These observations were further validated in human NCI-H292 epithelial cells, suggesting that TNF-α induced COX-2 expression via the phospholipase C-γ2, protein kinase C-α, tyrosine kinase, NF-κB-inducing kinase, and IκB1/2 pathways [44]. Further, Medeiros et al. [6] showed that TNF-α mediated the effects of Aβ1-40 in upregulating the expression of COX-2, which results in cognitive decline in mice. Reciprocally, Xing et al. [45] reported the involvement of COX-2 in hypoxia-induced TNF-α expression in osteoblasts. However, they report that TNF-α is not involved in regulating COX-2 expression in osteoblasts [45]. Nevertheless, this discrepancy might be due to cell-specific responses to TNF-α treatment. Along these lines and with our data, we can conclude that TNF-α is responsible for upregulating the expression of COX-2 during the course of AD development.
COX-2 exhibits multiple biological functions and is generally thought to regulate the pathogenesis of AD via its metabolic products, including PGE2, PGD2 [and its dehydration end product 15-deoxy-∆12,14-PGJ2 (15d-PGJ2)], PGI2, PGF2α and TXA2 [46]. For example, 15d-PGJ2 treatment increases the cleavage of tau protein and results in the formation of ∆tau, which accelerates the formation of NFTs in human neuroblastoma SK-N-SH cells [5]. Although COX-2 inhibition or overexpression has been indicated to be involved in tau phosphorylation or the formation of NFTs [3, 4], to the best of our knowledge, there is no further evidence showing a relationship between PGs and tau phosphorylation. Therefore, we first investigated whether PGI2 and F2α had the ability to induce the phosphorylation of tau. Consistent with this hypothesis, we found that PGI2 and F2α could increase the phosphorylation of tau at the sites of Ser202 and Ser 400/Thr 403/Ser 404.
In view of these novel findings, we further found that the PI3-K/AKT, ERK1/2 and JNK/c-Jun pathways mediated the effects of PGI2 and F2α in stimulating the phosphorylation of tau at both Ser 202 and Ser 400/Thr 403/Ser 404. Consistent with our findings, Baki et al. [47] reported that the PI3-K/AKT pathway mediated the effects of PS1 in stimulating the phosphorylation of tau via a glycogen synthase kinase-3 (GSK-3)-dependent mechanism. In addition, tau phosphorylation was also regulated by PI3-K/AKT-mediated inactivation of GSK-3β in EphB2-stimulated tau Tg mice [48]. Apart from the PI3-K/AKT pathway, hyperphosphorylation of tau is also mediated by ERK in neuroblastoma SK-N-SH cells [49]. Similarly, Carlyle et al. [50] reported that increased cAMP/PKA signaling is responsible for upregulating the phosphorylation of tau, which increases the risk of degeneration in aging association cortex. In addition, JNK plays a key role in tau hyperphosphorylation in AD models [51]. Consistent with these reports, our data revealed the involvement of the PI3-K/AKT, ERK1/2 and JNK/c-Jun pathways in augmenting the phosphorylation of tau.
In conclusion, this study provides new evidence for the synergistic roles of PGI2 and F2α in regulating the phosphorylation of tau both in vitro and in vivo. Specifically, Zn2+ stimulated the expression of TNF-α, highly expressed TNF-α induced the expression of COX-2 and the production of PGI2 and F2α. High accumulation of PGI2 and F2α induces the phosphorylation of tau at both Ser 202 and Ser 400/Thr 403/Ser 404 via the PI3-K/AKT, ERK1/2 and JNK/c-Jun pathways. These observations provide new insights into the mechanisms of tau phosphorylation in a PGI2- and F2α-dependent mechanism in the brain during AD development.
Materials and Methods
Reagents
ZnSO4, PGI2, PGF2α and the inhibitors including NS398, U0126, PD98059, LY294002 and SP600125 were obtained from Sigma-Aldrich Corp (St. Louis, MO, USA). Antibodies against β-actin, ERK1/2, p-ERK1/2, c-Jun, p-c-Jun, AKT, p-AKT, COX-2, TNF-α, p-tau (Ser 202), p-tau (Ser400/Thr403/Ser404) and tau were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). ZnT3 antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). TNF-α was purchased from Raybiotech, Inc. (Norcross, GA, USA). All reagents for the qRT-PCR and SDS-PAGE experiments were purchased from Bio-Rad Laboratories. All other reagents were from Invitrogen (Carlsbad, CA, USA) unless otherwise specified.
Cell culture
Mouse neuroblastoma 2a (n2a) cells were grown (37 °C and 5% CO2) on 6-cm tissue culture dishes (106 cells per dish) in appropriate medium. In a separate set of experiments, the cells were grown in serum-free medium for an additional 12 h before incubation with inhibitors in the absence or presence of PGI2 or PGF2α, as previously described [52].
Transgenic mice and treatments
The wild type (WT) or TauP301S Tg mice [B6;C3-Tg(Prnp-MAPT*P301S)PS19Vle/J (Stock Number: 008169)] were obtained from The Jackson laboratory (Bar Harbor, ME, USA). Genotyping was performed at 3-4 weeks after birth. The mice were housed in a controlled environment under a standard room temperature, relative humidity and 12-h light/dark cycle with free access to food and water. Mice were randomly separated into several groups and each group contains 12 mice. Mice at 6 months of age were injected (i.c.v) with PGI2 (1 µg/5 µl), PGF2α (1 µg/5 µl), ZnSO4 (10 µg/5 µl), TNF-α (1 ng/5 µl), or NS398 (1 µg/5 µl) for 24 h before determining the phosphorylation of tau. In select experiments, TauP301S Tg mice at the age of 1-months-old were treated with NS398 (1 mg/kg/d) for 5 months before determining the learning ability by nest construction test. The general health and body weights of animals were monitored every day. The brains of animals from the different groups were collected under anesthesia and perfusion as previously described [53-56].
Real-Time PCR
qRT-PCR assays were performed with the MiniOpticon Real-Time PCR detection system (Bio-Rad) using total RNA and the GoTaq one-step Real-Time PCR kit with SYBR green (Promega) and the appropriate primers as previously described [57-59]. The GenBank accession number and forward and reverse primers for mouse COX-2 and GAPDH are provided in our previous publications [57]: mouse ZnT3 (NM_011773) F-GTCTCCCTCTGGATAGTCACTGG, R-GCATACTCTGCACCTGTAGATCC; TNF-α (NM_013693) F-cagaaaagcaagcagccaac, R-gggaacttctcatccctttg. The gene expression values were normalized to those of GAPDH.
Western blots
Tissues or cells were lysed in radio-immune precipitation assay buffer (25 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% SDS) containing protease inhibitor cocktail (Pierce Chemical Company). The protein content of the cell lysates was determined using a bicinchoninic acid (BCA) protein assay reagent (Pierce Chemical Company). The total cell lysates (4 μg) were subjected to SDS-PAGE, transferred to a membrane, and probed with a panel of specific antibodies. Each membrane was only probed with one antibody. β-actin was used as a loading control. All western hybridizations were performed at least in triplicate using a different cell preparation each time.
Intracerebroventricular injection
NS398, PGI2, PGF2α, ZnSO4, TNF-α or vehicle (PBS) were injected (i.c.v) to WT or TauP301S Tg mice as previously described [53-56]. Briefly, stereotaxic injections were placed at the following coordinates from the bregma: mediolateral: -1.0 mm; anteroposterior: -0.22 mm; dorsoventral: -2.8 mm. Following injection, each mouse recovered spontaneously on a heated pad. The reliability of injection sites were validated by injecting trypan blue dye (Invitrogen) in separate cohorts of mice and observing staining in the cerebral ventricles. Twenty-four hours after injection, mice were harvested after anaesthesia and perfusion [53-56].
Immunofluorescence
Brain tissues were collected from WT or TauP301S Tg mice. Serial sections (10-µM thick) were cut by cryostats (Leica, CM1850, Germany). After fixation with 4% paraformaldehyde, slides were stained with p-Tau (Ser 202), p-Tau (Ser 400/Thr 403/Ser 404), TNF-α, COX-2 or ZnT3 antibody with Alexa Fluor 555 secondary antibodies (Cell Signaling Technology, Inc., Danvers, MA, USA) or Alexa Fluor 488 secondary antibodies (Jackson ImmunoResearch, Inc., West Grove, PA, USA) before observing under confocal microscopy (Leica, TCS-SP8, Leica).
Nest construction
Nest construction experiments were performed as previously described [56]. One week after corn cob nest-building test, the corn cob was replaced with clean ones. Two hours prior to the onset of the dark phase of the lighting cycle, 8 pieces of paper (5×5 cm2) were introduced in the home cage to create conditions for nesting. The nests were scored the following morning along a 4-point system: (1) no biting/rearing with random dispersion of the paper, (2) no biting/tearing of paper with gathering in a corner/side of the cage, (3) moderate biting/bearing on paper with gathering in a corner/side of the cage, and (4) extensive biting/tearing on paper with gathering in a corner/side of the cage.
Animal committee
All animals were handled according to the care and use of medical laboratory animals (Ministry of Health, Peoples Republic of China, 1998) and all experimental protocols were approved by the Laboratory Ethics Committees of College of Life and Health Sciences of Northeastern University.
Human brain samples
Human brain samples were obtained from New York Brain Bank, serial numbers TT4263 (early stage of AD, the patient is 73-years-old man who was diagnosed as a mild AD patient), T4308 (middle stage of AD, the patient is 86-years-old man who was diagnosed as moderate AD patient), T4339 and T4304 (late stage of AD, the patients are 88-years-old woman and 84 years-old woman who were diagnosed as severe and end stage of AD patients).
Statistical analysis
All data are represented as the mean ± S.E. of at least three independent experiments. The statistical significance of the differences between the means was determined either using Student’s t-test [52-56].
Author contributions
P.W., and Y. W. conceived and performed all of the experiments, participated in the design of the study and wrote the manuscript. P.P.G., X.Y., Y.S.G. and Y.J.Z. carried out select experiments. P.W. (along with Z.Y.W.) interpreted the data and wrote the manuscript.
ACKNOWLEDGMENTS AND FUNDING
This work was supported in part or in whole by the National Natural Science Foundation of China (CN) (81500934, 31571064, 31300777, 31371091 and 81600941), the Fundamental Research Funds of China (N152004004, N120520001, N120320001, N130120002, N142004002, N141008001/7 and L1520001), the National Natural Science Foundation of Liaoning, China (CN) (2015020662) and the Liaoning Provincial Talent Support Program (LJQ2013029).
Conflicts of Interest
The authors declare no competing financial interests.
References
1. Luo D, Long Y, Chen GJ. Cyclooxygenase-2 gene polymorphisms and risk of Alzheimer’s disease: A meta-analysis. J Neurol Sci. 2015; 359:100–05.
2. Shoghi-Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, Read S, Satyamurthy N, Petric A, Huang SC, Barrio JR. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002; 10:24-35.
3. McKee AC, Carreras I, Hossain L, Ryu H, Klein WL, Oddo S, LaFerla FM, Jenkins BG, Kowall NW, Dedeoglu A. Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res. 2008; 1207:225–3.
4. Oka A, Itoh M, Takashima S. The early induction of cyclooxygenase 2 associated with neurofibrillary degeneration in brains of patients with Fukuyama-type congenital muscular dystrophy. Neuropediatrics. 1999; 30:34-37.
5. Arnaud LT, Myeku N, Figueiredo-Pereira ME. Proteasome-caspase-cathepsin sequence leading to tau pathology induced by prostaglandin J2 in neuronal cells. J Neurochem. 2009; 110:328-342.
6. Medeiros R, Figueiredo CP, Pandolfo P, Duarte FS, Prediger RD, Passos GF, Calixto JB. The role of TNF-alpha signaling pathway on COX-2 upregulation and cognitive decline induced by beta-amyloid peptide. Behav Brain Res. 2010; 209:165-173.
7. Hein AM, Stutzman DL, Bland ST, Barrientos RM, Watkins LR, Rudy JW, Maier SF. Prostaglandins are necessary and sufficient to induce contextual fear learning impairments after interleukin-1 beta injections into the dorsal hippocampus. Neuroscience. 2007; 150:754-763.
8. Matsumoto Y, Yamaguchi T, Watanabe S, Yamamoto T. Involvement of arachidonic acid cascade in working memory impairment induced by interleukin-1 beta. Neuropharmacology. 2004; 46:1195-1200.
9. Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, O’Banion MK. Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci. 2013; 33:5053-5064.
10. Li Y, Liu L, Barger SW, Griffin WS. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci. 2003; 23:1605-1611.
11. Quintanilla RA, Orellana DI, Gonzalez-Billault C, Maccioni RB. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp Cell Res. 2004; 295:245-257.
12. Sheng JG, Jones RA, Zhou XQ, McGinness JM, Van Eldik LJ, Mrak RE, Griffin WS. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer’s disease: potential significance for tau protein phosphorylation. Neurochem Int. 2001; 39:341-348.
13. Sheng JG, Zhu SG, Jones RA, Griffin WS, Mrak RE. Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo. Exp Neurol. 2000; 163:388-391.
14. Tanji K, Mori F, Imaizumi T, Yoshida H, Satoh K, Wakabayashi K. Interleukin-1 induces tau phosphorylation and morphological changes in cultured human astrocytes. Neuroreport. 2003; 14:413-417.
15. Chu A, Foster M, Hancock D, Bell-Anderson K, Petocz P, Samman S. TNF-alpha gene expression is increased following zinc supplementation in type 2 diabetes mellitus. Genes Nutr. 2015; 10:440.
16. Foster M, Petocz P, Samman S. Inflammation markers predict zinc transporter gene expression in women with type 2 diabetes mellitus. J Nutr Biochem. 2013; 24:1655-1661.
17. Chauhan AK, Mittra N, Patel DK, Singh C. Cyclooxygenase-2 Directs Microglial Activation-Mediated Inflammation and Oxidative Stress Leading to Intrinsic Apoptosis in Zn-Induced Parkinsonism. Mol Neurobiol. 2017.
18. Huang CW, Wang SJ, Wu SJ, Yang CC, Huang MW, Lin CH, Cheng IH. Potential blood biomarker for disease severity in the Taiwanese population with Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2013; 28:75-83.
19. An WL, Bjorkdahl C, Liu R, Cowburn RF, Winblad B, Pei JJ. Mechanism of zinc-induced phosphorylation of p70 S6 kinase and glycogen synthase kinase 3beta in SH-SY5Y neuroblastoma cells. J Neurochem. 2005; 92:1104-1115.
20. Mo ZY, Zhu YZ, Zhu HL, Fan JB, Chen J, Liang Y. Low micromolar zinc accelerates the fibrillization of human tau via bridging of Cys-291 and Cys-322. J Biol Chem. 2009; 284:34648–57.
21. Sun XY, Wei YP, Xiong Y, Wang XC, Xie AJ, Wang XL, Yang Y, Wang Q, Lu YM, Liu R, Wang JZ. Synaptic released zinc promotes tau hyperphosphorylation by inhibition of protein phosphatase 2A (PP2A). J Biol Chem. 2012; 287:11174-11182.
22. Adlard PA, Parncutt JM, Finkelstein DI, Bush AI. Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J Neurosci. 2010; 30:1631-1636.
23. Lyubartseva G, Smith JL, Markesbery WR, Lovell MA. Alterations of zinc transporter proteins ZnT-1, ZnT-4 and ZnT-6 in preclinical Alzheimer’s disease brain. Brain Pathol. 2010; 20:343-350.
24. Zhang LH, Wang X, Stoltenberg M, Danscher G, Huang L, Wang ZY. Abundant expression of zinc transporters in the amyloid plaques of Alzheimer’s disease brain. Brain Res Bull. 2008; 77:55-60.
25. Zhang LH, Wang X, Zheng ZH, Ren H, Stoltenberg M, Danscher G, Huang L, Rong M, Wang ZY. Altered expression and distribution of zinc transporters in APP/PS1 transgenic mouse brain. Neurobiol Aging. 2010; 31:74-87.
26. Zheng W, Wang T, Yu D, Feng WY, Nie YX, Stoltenberg M, Danscher G, Wang ZY. Elevation of zinc transporter ZnT3 protein in the cerebellar cortex of the AbetaPP/PS1 transgenic mouse. J Alzheimers Dis. 2010; 20:323-331.
27. Friedlich AL, Lee JY, van Groen T, Cherny RA, Volitakis I, Cole TB, Palmiter RD, Koh JY, Bush AI. Neuronal zinc exchange with the blood vessel wall promotes cerebral amyloid angiopathy in an animal model of Alzheimer’s disease. J Neurosci. 2004; 24:3453-3459.
28. Lee JY, Cole TB, Palmiter RD, Suh SW, Koh JY. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc Natl Acad Sci USA. 2002; 99:7705-7710.
29. Morales-Corraliza J, Schmidt SD, Mazzella MJ, Berger JD, Wilson DA, Wesson DW, Jucker M, Levy E, Nixon RA, Mathews PM. Immunization targeting a minor plaque constituent clears beta-amyloid and rescues behavioral deficits in an Alzheimer’s disease mouse model. Neurobiol Aging. 2013; 34:137-145.
30. Ho L, Luterman JD, Aisen PS, Pasinetti GM, Montine TJ, Morrow JD. Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology. 2000; 55:323.
31. Montine TJ, Sidell KR, Crews BC, Markesbery WR, Marnett LJ, Roberts LJ 2nd, Morrow JD. Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology. 1999; 53:1495-1498.
32. Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ, and Alzheimer’s Disease Cooperative Study. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003; 289:2819–26.
33. Healy ZR, Zhu F, Stull JD, Konstantopoulos K. Elucidation of the signaling network of COX-2 induction in sheared chondrocytes: COX-2 is induced via a Rac/MEKK1/MKK7/JNK2/c-Jun-C/EBPbeta-dependent pathway. Am J Physiol Cell Physiol. 2008; 294:C1146-1157.
34. Howe LR, Crawford HC, Subbaramaiah K, Hassell JA, Dannenberg AJ, Brown AM. PEA3 is up-regulated in response to Wnt1 and activates the expression of cyclooxygenase-2. J Biol Chem. 2001; 276:20108-20115.
35. Kawaguchi H, Pilbeam CC, Gronowicz G, Abreu C, Fletcher BS, Herschman HR, Raisz LG, Hurley MM. Transcriptional induction of prostaglandin G/H synthase-2 by basic fibroblast growth factor. J Clin Invest. 1995; 96:923-930.
36. Miller C, Zhang M, He Y, Zhao J, Pelletier JP, Martel-Pelletier J, Di Battista JA. Transcriptional induction of cyclooxygenase-2 gene by okadaic acid inhibition of phosphatase activity in human chondrocytes: co-stimulation of AP-1 and CRE nuclear binding proteins. J Cell Biochem. 1998; 69:392-413.
37. Sakai T, Kambe F, Mitsuyama H, Ishiguro N, Kurokouchi K, Takigawa M, Iwata H, Seo H. Tumor necrosis factor alpha induces expression of genes for matrix degradation in human chondrocyte-like HCS-2/8 cells through activation of NF-kappaB: abrogation of the tumor necrosis factor alpha effect by proteasome inhibitors. J Bone Miner Res. 2001; 16:1272-80.
38. Xie W, Herschman HR. v-src induces prostaglandin synthase 2 gene expression by activation of the c-Jun N-terminal kinase and the c-Jun transcription factor. J Biol Chem. 1995; 270:27622-27628.
39. Wu W, Silbajoris RA, Cao D, Bromberg PA, Zhang Q, Peden DB, Samet JM. Regulation of cyclooxygenase-2 expression by cAMP response element and mRNA stability in a human airway epithelial cell line exposed to zinc. Toxicol Appl Pharmacol. 2008; 231:260-266.
40. Choi JS, Kim KA, Yoon YJ, Fujikado T, Joo CK. Inhibition of cyclooxygenase-2 expression by zinc-chelator in retinal ischemia. Vision Res. 2006; 46:2721–27.
41. Taccioli C, Wan SG, Liu CG, Alder H, Volinia S, Farber JL, Croce CM, Fong LY. Zinc replenishment reverses overexpression of the proinflammatory mediator S100A8 and esophageal preneoplasia in the rat. Gastroenterology. 2009; 136:953-966.
42. Tyagi A, Agarwal C, Dwyer-Nield LD, Singh RP, Malkinson AM, Agarwal R. Silibinin modulates TNF-alpha and IFN-gamma mediated signaling to regulate COX2 and iNOS expression in tumorigenic mouse lung epithelial LM2 cells. Mol Carcinog. 2012; 51:832-842.
43. Kuwano T, Nakao S, Yamamoto H, Tsuneyoshi M, Yamamoto T, Kuwano M, Ono M. Cyclooxygenase 2 is a key enzyme for inflammatory cytokine-induced angiogenesis. FASEB J. 2004; 18:300–10.
44. Chen CC, Sun YT, Chen JJ, Chiu KT. TNF-alpha-induced cyclooxygenase-2 expression in human lung epithelial cells: involvement of the phospholipase C-gamma 2, protein kinase C-alpha, tyrosine kinase, NF-kappa B-inducing kinase, and I-kappa B kinase 1/2 pathway. J Immunol. 2000; 165:2719-2728.
45. Xing Y, Wang R, Chen D, Mao J, Shi R, Wu Z, Kang J, Tian W, Zhang C. COX2 is involved in hypoxia-induced TNF-alpha expression in osteoblast. Sci Rep. 2015; 5:10020.
46. Akarasereenont P, Techatrisak K, Chotewuttakorn S, Thaworn A. The induction of cyclooxygenase-2 in IL-1beta-treated endothelial cells is inhibited by prostaglandin E2 through cAMP. Mediators Inflamm. 1999; 8:287-294.
47. Baki L, Shioi J, Wen P, Shao Z, Schwarzman A, Gama-Sosa M, Neve R, Robakis NK. PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: effects of FAD mutations. EMBO J. 2004; 23:2586-2596.
48. Jiang J, Wang ZH, Qu M, Gao D, Liu XP, Zhu LQ, Wang JZ. Stimulation of EphB2 attenuates tau phosphorylation through PI3K/Akt-mediated inactivation of glycogen synthase kinase-3beta. Sci Rep. 2015; 5:11765.
49. Guise S, Braguer D, Carles G, Delacourte A, Briand C. Hyperphosphorylation of tau is mediated by ERK activation during anticancer drug-induced apoptosis in neuroblastoma cells. J Neurosci Res. 2001; 63:257-267.
50. Carlyle BC, Nairn AC, Wang M, Yang Y, Jin LE, Simen AA, Ramos BP, Bordner KA, Craft GE, Davies P, Pletikos M, Šestan N, Arnsten AF, Paspalas CD. cAMP-PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc Natl Acad Sci USA. 2014; 111:5036–41.
51. Ploia C, Antoniou X, Sclip A, Grande V, Cardinetti D, Colombo A, Canu N, Benussi L, Ghidoni R, Forloni G, Borsello T. JNK plays a key role in tau hyperphosphorylation in Alzheimer’s disease models. J Alzheimers Dis. 2011; 26:315-329.
52. Wang P, Guan PP, Wang T, Yu X, Guo JJ, Wang ZY. Aggravation of Alzheimer’s disease due to the COX-2-mediated reciprocal regulation of IL-1beta and Abeta between glial and neuron cells. Aging Cell. 2014; 13:605-615.
53. Wang P, Guan PP, Guo JW, Cao LL, Xu GB, Yu X, Wang Y, Wang ZY. Prostaglandin I2 upregulates the expression of anterior pharynx-defective-1alpha and anterior pharynx-defective-1beta in amyloid precursor protein/presenilin 1 transgenic mice. Aging Cell. 2016; 15:861-871.
54. Wang P, Guan PP, Yu X, Zhang LC, Su YN, Wang ZY. Prostaglandin I(2) Attenuates Prostaglandin E(2)-Stimulated Expression of Interferon gamma in a beta-Amyloid Protein- and NF-kappaB-Dependent Mechanism. Sci Rep. 2016; 6:20879.
55. Wang P, Yu X, Guan PP, Guo JW, Wang Y, Zhang Y, Zhao H, Wang ZY. Magnesium ion influx reduces neuroinflammation in Abeta precursor protein/Presenilin 1 transgenic mice by suppressing the expression of interleukin-1beta. Cell Mol Immunol. 2017; 14:451-464.
56. Yu X, Guan PP, Guo JW, Wang Y, Cao LL, Xu GB, Konstantopoulos K, Wang ZY, Wang P. By suppressing the expression of anterior pharynx-defective-1alpha and -1beta and inhibiting the aggregation of beta-amyloid protein, magnesium ions inhibit the cognitive decline of amyloid precursor protein/presenilin 1 transgenic mice. FASEB J. 2015; 29:5044–58.
57. Guan PP, Guo JW, Yu X, Wang Y, Wang T, Konstantopoulos K, Wang ZY, Wang P. The role of cyclooxygenase-2, interleukin-1beta and fibroblast growth factor-2 in the activation of matrix metalloproteinase-1 in sheared-chondrocytes and articular cartilage. Sci Rep. 2015; 5:10412.
58. Guan PP, Yu X, Guo JJ, Wang Y, Wang T, Li JY, Konstantopoulos K, Wang ZY, Wang P. By activating matrix metalloproteinase-7, shear stress promotes chondrosarcoma cell motility, invasion and lung colonization. Oncotarget. 2015; 6:9140–59. https://doi.org/10.18632/oncotarget.3274.
59. Wang P, Guan PP, Wang T, Yu X, Guo JJ, Konstantopoulos K, Wang ZY. Interleukin-1beta and cyclic AMP mediate the invasion of sheared chondrosarcoma cells via a matrix metalloproteinase-1-dependent mechanism. Biochim Biophys Acta. 2014; 1843:923-933.