
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common solid tumor and leads to large amounts of deaths around the world [1]. The occurrence of HCC often comes from chronic infection with hepatitis virus B or C [2]. Up to now, no effective therapy for HCC complete cure has been developed. Patients with HCC show a poor prognosis and a high recurrence rate. Quite a lot of effort has been made to explore the mechanism of HCC progression and develop effective treatments. However, the molecular mechanisms in HCC are still largely unknown.
LDB2, also known as CLIM-1, was identified as an LIM domain-associated cofactor and functions as a transcriptional regulatory factor [3-5]. LDB2 belongs to the LIM-domain binding family including LDB1 and LDB2. Though the function of LDB1, the homologous protein of LDB2, has been widely explored, the biology role of LDB2 is largely unknown. LDB1 and LDB2 bear 78% identity and 89% similarity to each other [6]. LDB1 is ubiquitously expressed while LDB2 is expressed more regionally. LIM-HDs (LIM homeodomain transcription factors) associating with LDB proteins are essential to exert biological and transcriptional activity [7, 8]. Previous study has shown that LDB2 drivers transendothelial migration of leukocytes and atherosclerosis [9]. LDB2 targets CAD (carotid artery disease) by inhibiting the activity of TEML (the transendothelial migration of leukocyte). Johnsen and colleagues reported that LDB2 has a negative effect on ERα activity and impairs ERE-dependent transcription in breast cancer [6]. Nevertheless, the roles of LDB2 in HCC remain to be elucidated. Bromodomain containing 7 (BRD7) that is a member of the bromodomain-containing protein family was acknowledged as a component of one form of the SWI/SNF chromatin remodeling complex [10, 11]. Recent studies indicated that BRD7 functioned as a chromatin remodeler and inhibited gene expression [11, 12]. And more and more evidences showed that BRD7 were downregulated in some tumor cells including epithelial ovarian carcinoma, breast cancer and colorectal carcinoma [13-15]. BRD7 was demonstrated to act as a tumor suppressor [16-18]. For example, Chen et al. demonstrated that BRD7 was a potential tumor suppressor in hepatocellular carcinoma [17]. However, the molecular mechanism of BRD7 function in HCC was still unknown. HEY1 was a classic target gene of NOTCH signaling that was shown to promote many kinds of tumor progression [19-21]. The Expression of HEY1 can also be regulated in a NOTCH-independent way [22]. Previous study identified that HEY1 was a putative oncogene in HCC [23]. However, the function of HEY1 in HCC needs further investigation.
In this study, we showed that LDB2 inhibited proliferation and migration of hepatocarcinoma cells. Mechanistically, we found that LDB2 can promote BRD7 binding to HEY1 promoter and then inhibits HEY1 expression. HEY1 whose expression is upregulated in HCC functions as an oncogene. In brief, our research reveals a new regulatory mechanism for hepatocarcinoma cell proliferation and migration.
RESULTS
LDB2 is weakly expressed in HCC tumors
To understand the mechanism that regulates the occurrence of hepatocellular carcinoma, we analyzed previous reported microarray datasets [24-27]. We found that the expression of LDB2 was downregulated in HCC samples compared with normal tissues according to the microarray data in Wang’s cohort (GSE14520) and Zhang’s cohort (GSE25097) (Figure 1A). To confirm this observation, we examined 20 pairs of HCC samples and peritumor tissues and found that LDB2 was downregulated in most HCC sample cells (Figure 1B), which was further demonstrated by western blot (WB) and immunohistochemistry (IHC) (Figure 1C and 1D). What’s more, LDB2 was expressed in HCC patients of stage III least according to Wang’s cohort (GSE14520) (Figure 1E). Importantly, HCC patients with more LDB2 expression levels had higher survival rate, and vice versa according to Wang’s cohort (GSE14520) (Figure 1F). In sum, LDB2 is downregulated in HCC samples and negatively correlated with clinical severity and poor prognosis.
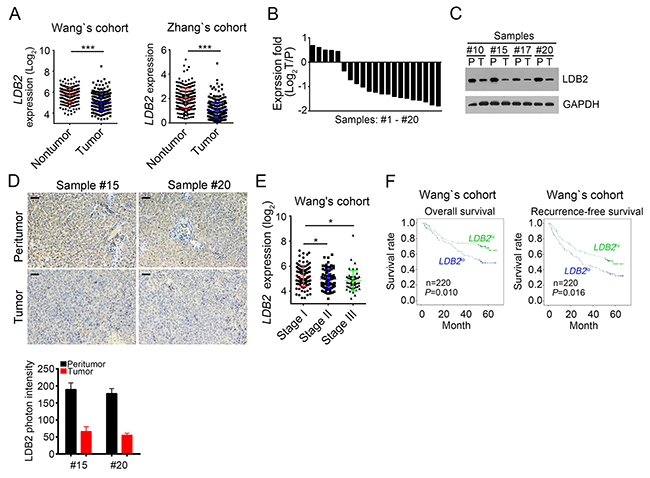
Figure 1: LDB2 is weakly expressed in HCC tumors. (A) Expressionanalysis ofLDB2 in normal (n=215) and tumor tissues (n=224) according to Wang’s cohort and Zhang’s cohort. (B) Total RNAs were extracted from human HCC samples. Then expression levels ofLDB2 were analyzed in 20 pairs of peritumor and HCC samples by RT-qPCR. Fold changes were normalized to endogenous Actb. T, tumor; P, peritumor. (C) Expression levels ofLDB2 in peritumor and HCC samples were examined by Western blot. GAPDH was chosen for sample loading control. (D) Protein levels of LDB2 were analyzed by IHC in peritumor and tumor samples. Scale bars, 100μm. (E) Expression level of LDB2 was negatively correlated with clinical stages according to Wang’s cohort. (F) Expression level of LDB2 was negatively correlated with poor prognosis by Kaplan–Meier survival analysis according to Wang’s cohort. The mean expression levels of LDB2 served as cut-off values. *p<0.05, **p<0.01 and ***p<0.001 by two-tailed Student’s t test. All data presented are shown as means ± SD collected from three independent experiments.
LDB2 deletion promotes liver cancer cell proliferation and migration
To explore the function of LDB2 in liver cancer, we deleted it in HCC samples (Figure 2A). Through MTT assays and colony formation assays, we found that LDB2-deficient cells displayed definite increased ability of proliferation (Figure 2B and 2C). Moreover, more LDB2-deficient tumor cells entered into cell cycle (Figure 2D). Besides, LDB2 knockout led to decreased cell apoptosis (Figure 2E). To evaluate the influence of LDB2 deletion on cell migration, we performed migration assays and found that there were more migrated cells after LDB2 deletion (Figure 2F). This result was further validated by the observation of increased expression of MPP2 and MPP9 in LDB2-deleted tumor cells (Figure 2G). Consider the heterogeneity of the tumor cells, we used HCC cell line HepG2 for experiments. We found that LDB2 knockdown promoted cell proliferation in HepG2 cells (Supplementary Figure 1A). To define the function of LDB2 in vivo, we injected 1×106 LDB2-deficient sample cells or control into BALB/c nude mice. Then tumor weights were measured 5 weeks post injection. LDB2 deletion remarkably promoted tumor formation in vivo (Figure 2H). Collectively, LDB2-dificiency promotes tumor proliferation in vitro and in vivo.
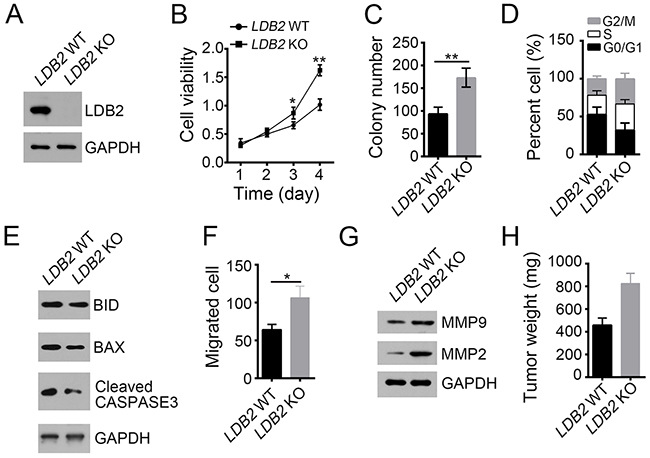
Figure 2: LDB2 deletion promotes liver cancer cell proliferation and migration. (A) LDB2 was deleted in HCC samples using two individual sgRNAs through CRISPR/Cas9 technology. The deletion efficiency was validated by Western blot. (B and C) Deletion of LDB2 enhanced proliferation of HCC sample cells as examined by MTT assays and colony formation assays. (D) Percentages of cell-cycle distribution of WT or LDB2-deleted HCC sample cells were checked by FACS. (E) Percentages of cell apoptosis in LDB2-depleted HCC samples were measured by WB. (F and G) LDB2-deleted cells showed increased migration ability as measured by migration assays and WB. (H) The weights of tumors were measured 5 weeks post injection. 2×106 WT or LDB2-deleted sample cells were injected into nude mice. *p<0.05 and **p<0.01 by two-tailed Student’s t test. All data presented are shown as means ± SD collected from three independent experiments.
LDB2 overexpression inhibits tumor cell proliferation and migration
We next overexpressed LDB2 in HCC primary tumor cells (Figure 3A). LDB2 overexpression dramatically impaired the ability of cell proliferation (Figure 3B and 3C). LDB2-overexpressing tumor cells expressed increased P21 (Figure 3D). Besides, overexpression of LDB2 significantly inhibited cell migration and promoted cell apoptosis (Figure 3E and 3F). Consequently, LDB2 overexpression remarkably reduced xenograft tumor growth (Figure 3G). Summarily, these data indicate that LDB2 plays an important role on regulating proliferation and survival of tumor cells.
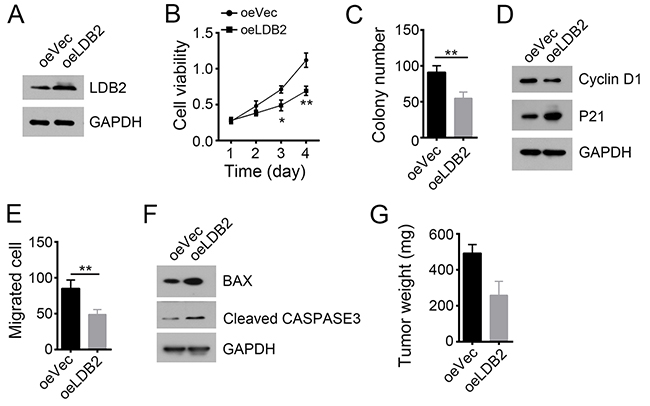
Figure 3: LDB2 overexpression inhibits tumor cell proliferation and migration. (A) Overexpression of LDB2 was confirmed by WB in HCC samples. GAPDH was chosen for loading control. (B and C) Cell proliferation ability was analyzed by MTT assays and colony formation assays. (D) LDB2 overexpression arrested cell cycle. (E) Overexpression of LDB2 inhibited cell migration. (F) LDB2 overexpression promotes cell apoptosis as shown by WB. (G) LDB2 overexpression inhibited tumor formation in vivo. 2×106 WT or LDB2-overexpressing sample cells were injected into nude mice. 5 weeks post injection, tumor weight was measured. *p<0.05, **p<0.01 and ***p<0.001 by two-tailed Student’s t test. All data presented are shown as means ± SD collected from three independent experiments.
LDB2 inhibits HEY1 expression
Many transcription factors (TFs) have been reported to be important for HCC occurrence such as SOX4, MYC, HEY1 and ZIC2 [28-30]. To explore the molecular mechanism of LDB2 function, we checked the effect of LDB2 deletion on representative TFs by RT-qPCR. Among them, HEY1 and SOX4 were the most up-regulated transcription factors (Figure 4A). However, HEY1 knockdown inhibited proliferation in LDB2 KO cells while SOX4 knockdown had no effect on proliferation (Supplementary Figure 1B). We then validated that LDB2 knockout significantly enhanced HEY1 expression in HCC sample cells (Figure 4B). Besides, we analyzed the correlation between the expression levels of LDB2 and HEY1. LDB2 expression was negatively related with that of HEY1 in HCC samples (Figure 4C). Then we knocked out HEY1 in LDB2-deficient cells, and found that LDB2 deletion promoted cell proliferation but HEY1 deletion reversed it (Figure 4D). Moreover, HEY1 deletion also impaired migration ability of tumor cells (Figure 4E). To further validate that HEY1 was a target gene of LDB2, we conducted luciferase assays and found that LDB2 overexpression inhibited the luciferase activity (Figure 4F). Altogether, LDB2 inhibits tumor cell proliferation and migration by targeting HEY1.
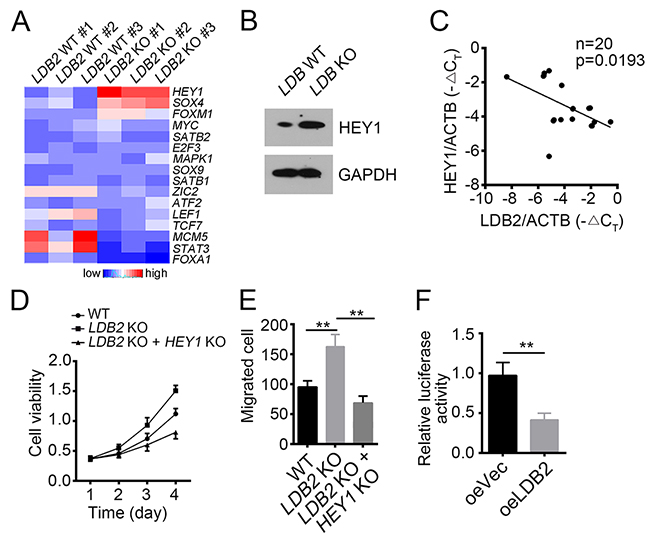
Figure 4: LDB2 inhibits HEY1 expression. (A) Expression patterns of indicative transcription factors in WT and LDB-deficient HCC samples. Fold changes were normalized to endogenous Actb. (B) Protein levels of LDB2 and HEY1 were checked by WB. GAPDH was chosen for loading control. (C) Expression level of LDB2 was negatively correlated with HEY1 expression in HCC samples (n=20). (D and E) HEY1 deletion inhibited cell proliferation and migration. (F) LDB2 inhibited HEY1 transcription as measured by luciferase assays. **p<0.01 by two-tailed Student’s t test. All data presented are shown as means ± SD collected from three independent experiments.
LDB2 recruits BRD7 to HEY1 promoter
To further define how LDB2 regulates HEY1 expression, we performed co-IP assays and mass spectrometry (MS). HCC sample cells were lysed and anti-LDB2 or IgG control was added. Then Protein A/G beads were used for enrichment of proteins/antibody complex, followed by SDS-PAGE and silver staining. Then the differential bands in the lane of anti-LDB2 were cut for MS. We identified BRD7 as a potential interactive protein of LDB2 (Figure 5A). Then we lysed sample cells and conducted co-IP assays to validate their interaction in vivo (Figure 5B). We also checked the effect of LDB2 on BRD7 expression. We found that overexpressing LDB2 had little effect on BRD7 mRNA levels in HCC (Supplementary Figure 1C). BRD7 has been reported to act as a chromatin remodeler and repress gene expression [11, 12]. To explore whether BRD7 is involved in the regulation of HEY1 expression, we conducted ChIP assays. We found that BRD7 enriched on HEY1 promoter (Figure 5C). Moreover, LDB2 deletion impaired the enrichment of BRD7 on HEY1 promoter (Figure 5D). Then we deleted BRD7 in sample cells and found that BRD7-deficiency promoted HEY1 expression (Figure 5E). On the contrary, overexpression of LDB2 or BRD7 inhibited HEY1 expression (Figure 5F). Finally, we analyzed the expression correlation between BRD7 and HEY1 according to Park’s cohort (GSE36376), and found that their expression was negative correlated (Figure 5G). Overall, LDB2 associates with BRD7 to inhibit HEY1 expression.
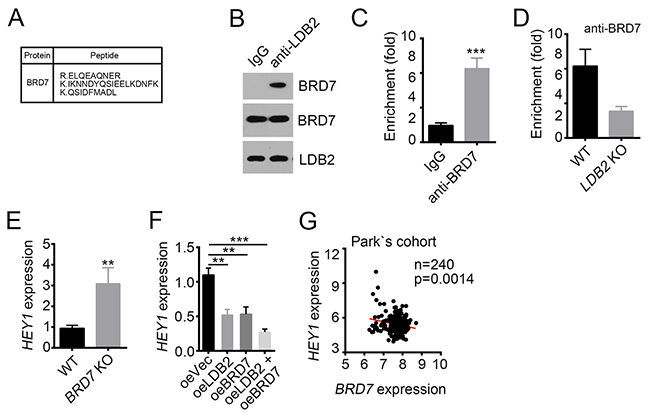
Figure 5: LDB2 recruits BRD7 to HEY1 promoter. (A) BRD7 was identified as a potential interactive protein of LDB2 by MS. (B) LDB2 directly associated with BRD7 as shown by co-IP assays. 1×107 sample cells were lyzed and supernatant was used for incubation with antibody. (C) BRD7 enriched on HEY1 promoter. (D) LDB2 deletion impaired the enrichment of BRD7 on HEY1 promoter. (E) BRD7 deletion promoted HEY1 expression. Total RNA was extracted from WT or BRD7-deleted sample cells and RT-qPCR was conducted. Fold changes were normalized to endogenous Actb. (F) Overexpression of LDB2 or BRD7 inhibited HEY1 expression. Fold changes were normalized to endogenous Actb. (G) BRD7 expression was negatively correlated with HEY1 expression according to Park’s cohort. *p<0.05, **p<0.01 and ***p<0.001 by two-tailed Student’s t test. All data presented are shown as means ± SD collected from three independent experiments.
HEY1 is correlated with clinical severity and prognosis of HCC patients
To explore the physiological role of HEY1 in HCC, we analyzed the expression levels of HEY1 and found that HEY1 was highly expressed in tumor tissues (Figure 6A and 6B). HEY1 was also highly expressed in HCC tumor cells according to Wang’s cohort (GSE14520) (Figure 6C). To further define the clinical significance of HEY1 expression in HCC, we analyzed the data set of Wang’s cohort (GSE14520). We found that samples of stage III display higher expression of HEY1 (Figure 6D). To further explore the relation of HEY1 expression with HCC prognosis, we divided HCC samples into two groups according to HEY1 expression levels (mean expression level was chosen for cut-off value). Then Kaplan–Meier survival analysis was conducted and results showed that HCC patients with HEY1 higher expression had a poor prognosis and lower overall or recurrence-free survival rate, and vice versa (Figure 6E). Collectively, LDB2 and BRD7 inhibit HEY1 expression in HCC sample cells. The expression levels of HEY1 were correlated with clinical severity and prognosis.
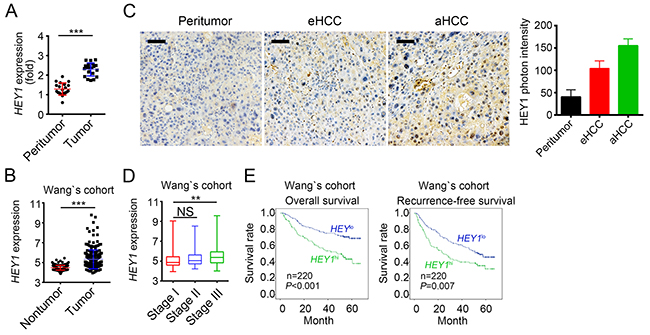
Figure 6: HEY1 is correlated with clinical severity and prognosis of HCC patients. (A) HEY1 was highly expressed in HCC samples. Total RNAs were extracted from tumor and peritumor tissues. mRNA levels of HEY1 were analyzed by RT-qPCR. Fold changes were nomalized to endogenous Actb. (B) HEY1 was highly expressed in tumor cells according to Wang’s cohort. (C) The expression levels of HEY1 were higher in eHCC (early HCC) cells and highest in aHCC (advanced HCC) cells as measured by IHC. Scale bar, 100μm. (D) Higher expression of HEY1 in serious HCC samples according to Wang’s cohort. (E) HEY1 expression level was positively correlated with HCC poor prognosis by Kaplan–Meier survival analyses according to Wang’s cohort. The mean expression levels of HEY1 served as cut-off values. **p<0.01 and ***p<0.001 by two-tailed Student’s t test. All data presented are shown as means ± SD collected from three independent experiments.
DISCUSSION
HCC, the fifth most common cancer, has very low 5-year survival rate [31, 32]. Up to now, there was no good method for HCC complete treatment. So many efforts have been made to explore the molecular mechanism for hepatocarcinogenesis and the methods for HCC treatment. Nevertheless, very limited progress is achieved and the underlying mechanism of hepatocarcinogenesis is largely unknown. Therefore, it’s impending to define the mechanism underlying HCC progression. In our research, we found that LDB2 is weakly expressed in HCC sample cells. LDB2 deletion significantly promotes the proliferation and migration abilities of tumor cells. Moreover, LDB2 deficiency led to enhanced tumor propagation in mice. In mechanism, LDB2 associates with BRD7 and bound to HEY1 promoter. Then HEY1 transcription initiation was inhibited. Finally, high expression levels of HEY1 are linked to clinical severity and poor prognosis.
LDB2 belongs to LIM domain binding family. As a homologous protein of LDB2, the function of LDB1 has been largely studied because of the ubiquitously expressed in tissues [6]. However, the role of LDB2 remains to be elucidated. Especially, the relationship of LDB2 with HCC is largely unknown. Through analysis of online data set of microarray about HCC samples, we found that LDB2 was remarkably downregulated in tumor cells, which suggests a tumor suppressor of LDB2. To demonstrate it, we deleted LDB2 in tumor cells. We found that LDB2-deficient cells displayed enhanced proliferation and migration ability. Besides, LDB2 knockout promoted tumor growth in vivo. Hence our results will shed light on the clinical correlation between LDB2 and HCC. Previous research revealed that the co-factor of LIM domains promotes breast tumorigenesis by enhancing the expression of ErbB2 and ErbB3 [33]. However, our results showed that LDB2 is a tumor repressor in HCC, which indicated that there may be various roles of LDB proteins in different tumors.
The LIM domain is found in a variety protein families such as the LIM-homeodomain (Lhx) and LIM-only (LMO) transcription factors [34]. Previous studies most focused on the interaction of LDB proteins with LIM domain containing proteins [35-37]. LDB proteins can also bind to other DNA-binding proteins, such as Otx family members [38]. However, how LDB2 links chromatin remodeler and gene expression is still unknown. Our study revealed that LDB2 associates with BRD7 on HEY1 promoter and then regulate HEY1 expression.
BRD7 that is a subunit of polybromo-associated BRG1-associated factor (PBAF)- specific SWI/SNF chromatin remodeling complexes contains an evolutionally conserved bromodomain [11]. This domain containing proteins can regulate gene transcription [39]. [4]. BRD7 has been demonstrated to be important for the transcriptional activation or repression of target genes such as P53 [40, 41]. Accumulating evidence showed that BRD7 is involved in multiple cancers and serves as a tumor suppressor, including HCC [17]. However, the fashion of BRD7 function in HCC remains to be explored. We showed that BRD7 binds to HEY1 promoter and represses its transcription. HEY1 was a classical target gene of NOTCH signaling. It can also be regulated in a NOTCH-independent manner [22]. NOTCH signaling is involved in various cancers and has been extensively explored. Nevertheless, the function of HEY1 seems to be ignored. We showed that the expression of HEY1 was repressed by LDB2 and BRD7. Higher expression of HEY1 means poorer prognosis. Our study showed that overexpressing LDB2 and BRD7 in the meantime seems to further reduce the expression of HEY1. So other proteins may also interact with BRD7 to regulate HEY1 expression and function in HCC, which needs to be investigated further.
Summarily, our research demonstrates that LDB2 can inhibit tumor cell proliferation and migration by downregulating HEY1 expression. Therefore, this finding gives rise to a new insight into the mechanism of hepatocarcinogenesis.
MATERIALS AND METHODS
Patient samples
20 pairs of Peritumor and HCC tissues involved in this study were obtained by laser microdissection from the Third Affiliated Hospital of Harbin Medical University. For cell culture, sample tissues were cut into small pieces and then digested with collagenase IV for 60min at 37 °C. Consents approving the use of the tissues for this research were obtained from all patients. All the experiments were approved by the Third Affiliated Hospital of Harbin Medical University. The study protocol was approved by the Third Affiliated Hospital of Harbin Medical University.
Cell lines and cell culture
293T cells from ATCC and HCC sample cells were cultured in DMEM medium containing 10% fetal bovine serum (FBS; Invitrogen), 100 μg/ml penicillin and 100U/ml streptomycin.
CRISPR/Cas9 mediated knockout
We used CRISPR/Cas9 approach for LDB2, BRD7 and HEY1 deletion. sgRNA was designed through online CRISPR Design Tool (http://tools.genome-engineering.org). Then we cloned sgRNAs into LentiCRISPRv2 plasmid (52961) purchased from Addgene. For lentivirus production, 293T cells were transfected with LentiCRISPRv2, pMD2.G (Addgene, 12259) and psPAX2 (Addgene, 12260). A pair of guide RNAs was used simultaneously for LDB, BRD7 or HEY1 deletion. The HCC cells infected with lentivirus were treated with puromycin and then monoclonalization was conducted. sgRNA sequences are as follows: LDB2 #1: 5'-GGCACACGTCCTCAATGCTC-3'; LDB2 #2:5'-GTGCCACACGCTGTAATTTG-3'; BRD7 #1: 5'-AAGCGCGTCGATTAAAATCG-3'; BRD7 #2: 5'- GGATTACAAGTGTGGGCTAC-3'; HEY1 #1: 5'- GTTATCATCGCGGAGCTTTT-3'; HEY1 #2: 5'- GAGTCCCGAGCCCCTACATT-3'.
Antibodies
Anti-LDB2 (sc-101088), anti-BRD7 (sc-376180) and anti-HEY1 (sc-134362) were purchased from Santa Cruz Biotechnology.
Apoptosis analysis
Apoptosis analysis was conducted through Annexin V-FITC/PI apoptosis detection kit (eBiosciences) by FACS analysis.
Xenograft tumor formation
We purchased six weeks old female BALB/c nude mice from HFK Biosciences and maintained under pathogen-free conditions with approval by the Institutional Committee of the Third Affiliated Hospital of Harbin Medical University. For tumor propagation analysis, 2×106 LDB2-deficient cells were subcutaneously injected into mice. Tumor weight was analyzed on week 5 after injection. Animal experiments were performed in accordance with relevant guidelines and regulations of the Institutional Animal Care and Use Committees at the Third Affiliated Hospital of Harbin Medical University, and protocols were approved by the Institutional Animal Care and Use Committees at the Third Affiliated Hospital of Harbin Medical University.
MTT assay
In MTT assays, 1×103 cells were seeded into 96-well plates. Cell proliferation was measured by MTT assays at different time points. MTT (20 μl, 5 mg/ml) (Sigma, USA) was added into each well and incubated for 4 h at 37°C. Then 150 μl DMSO was added to solubilize the crystals. To determine cell viability, the absorbance (540 nm) was measured.
Colony formation and migration assay
For colony formation assays, 2×103 cells were seeded into a 6-well plate and incubated for 12 days at 37°C. And then the cells were fixed in 90% ethanol and stained with crystal violet solution. The formed colonies were counted.
For cell Migration assays, the transwell filter chambers (Costar, Corning, NY) were used according to the manufacturers’ instructions. Briefly, 2×105 cells in serum-free medium were added into the top chamber. the lower chamber containing medium supplemented with 10% FBS. After cultured for 12 h, cells on the lower surface were stained and analyzed in six random fields for each group.
Real-time quantitative PCR
Total RNAs were extracted with TRIzol according to the manufacturer’s protocol. Then cDNA was synthesized with the M-MLV reverse transcriptase (Promega). Then mRNA transcripts were analyzed with ABI 7300 qPCR system using specific primer pairs. Relative expressions were calculated and normalized to endogenous Actb. Primers used were available if requested.
Chromatin immunoprecipitation (ChIP) assay
Cells were cross-linked with 1% formaldehyde at 37 °C for 10 min, washed twice with PBS, lysed with SDS lysis buffer and sonicated into 300–500 bp DNA fragments. Lysates were incubated with 4 μg antibody overnight at 4 °C. Then Protein A Agarose/Salmon Sperm DNA (50% Slurry) beads were added for another 4 h. After washed, DNA was eluted from beads and purified. Enriched DNA was checked by q-PCR. Primers used for HEY1 promoter analysis were described before [42] and as follows: forward: 5'-TCAGTGTGTGCGGAACGCAAG-3'; reward: 5'- TTCTTCACCTCGATGGTCTCGTC-3'.
Luciferase reporter gene assays
The Dual-Luciferase Reporter Assay System (Promega) was used for luciferase reporter assay. Cells were cultured on 24-well plates at 3×104 cells per well. The indicated regions of HEY1 promoter (-2000~0 bp: upstream of TSS region) were cloned into pGL3 plasmid. 1 ng pRL-TK was co-transfected into cells. After 24 hours, cells were lysed and detected according to manufacturer’s instructions.
Cohort analysis
Online-available data sets were available from NCBI (https://www.ncbi.nlm.nih.gov/gds/). Three online available cohorts used were as follows: Wang’s cohort (GSE14520) and Zhang’s cohort (GSE25097).
IHC staining
The method has been reported before [43]. In brief, HCC sections were deparaffinized, rehydrated and blocked with 3% H2O2 for 15 min and antigen retrieval with Tris-EDTA buffer (10 mM, pH 8.0) at 121°C for 5min. After 30 min incubation in 5% goat serum, the sections were incubated in primary antibodies overnight, washed with PBS and then incubated in HRP-conjugated secondary antibodies. The final detection was conducted with the substrate detection of HRP. Finally the sections were stained using haematoxylin.
Co-immunoprecipitation and mass spectrometry
HCC sample cells were lysed with RIPA lysis buffer and anti-LDB2 or IgG control was added for incubation at 4 °C overnight. Then ProteinA/G beads were added and incubated at 4 °C for 2h. Then beads were collected and IP components were separated by SDS-PAGE, followed by silver staining. Differential bands enriched by anti-LDB2 were collected for mass spectrometry (LTQ Orbitrap XL).
Statistical analysis
All statistical analyses were performed using the Statistical Package for the Social Sciences version 20.0 software (SPSS Inc., Chicago, IL, USA). The results are shown as the mean ± SD and were analyzed using Student’s t-test. All tests were two-sided, and P < 0.05 was considered statistically significant.
Author contributions
H.Y. designed and performed experiments, analyzed data and wrote the paper; R.J., L.Z., S.S. and J.G. performed experiments; H.Z. initiated the study, organized, designed, and wrote the paper.
CONFLICTS OF INTEREST
The authors declare no conflicts interests.
FUNDING
This work was supported by the Scientific Research of the Health and Family Planning Commission of Heilongjiang Province of China (No.2013045).
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61: 69-90.
2. Xu X, Fan Z, Kang L, Han J, Jiang C, Zheng X, Zhu Z, Jiao H, Lin J, Jiang K, Ding L, Zhang H, Cheng L, et al. Hepatitis B virus X protein represses miRNA-148a to enhance tumorigenesis. J Clin Invest. 2013; 123: 630-45.
3. Bach I, Carriere C, Ostendorff HP, Andersen B, Rosenfeld MG. A family of LIM domain-associated cofactors confer transcriptional synergism between LIM and Otx homeodomain proteins. Genes Dev. 1997; 11: 1370-80.
4. Agulnick AD, Taira M, Breen JJ, Tanaka T, Dawid IB, Westphal H. Interactions of the LIM-domain-binding factor Ldb1 with LIM homeodomain proteins. Nature. 1996; 384: 270-2.
5. Visvader JE, Mao X, Fujiwara Y, Hahm K, Orkin SH. The LIM-domain binding protein Ldb1 and its partner LMO2 act as negative regulators of erythroid differentiation. Proc Natl Acad Sci U S A. 1997; 94: 13707-12.
6. Johnsen SA, Gungor C, Prenzel T, Riethdorf S, Riethdorf L, Taniguchi-Ishigaki N, Rau T, Tursun B, Furlow JD, Sauter G, Scheffner M, Pantel K, Gannon F, et al. Regulation of estrogen-dependent transcription by the LIM cofactors CLIM and RLIM in breast cancer. Cancer Res. 2009; 69: 128-36.
7. Becker T, Ostendorff HP, Bossenz M, Schluter A, Becker CG, Peirano RI, Bach I. Multiple functions of LIM domain-binding CLIM/NLI/Ldb cofactors during zebrafish development. Mech Dev. 2002; 117: 75-85.
8. Thaler JP, Lee SK, Jurata LW, Gill GN, Pfaff SL. LIM factor Lhx3 contributes to the specification of motor neuron and interneuron identity through cell-type-specific protein-protein interactions. Cell. 2002; 110: 237-49.
9. Shang MM, Talukdar HA, Hofmann JJ, Niaudet C, Asl HF, Jain RK, Rossignoli A, Cedergren C, Silveira A, Gigante B, Leander K, de Faire U, Hamsten A, et al. Lim domain binding 2: a key driver of transendothelial migration of leukocytes and atherosclerosis. Arterioscler Thromb Vasc Biol. 2014; 34: 2068-77.
10. Harte MT, O’Brien GJ, Ryan NM, Gorski JJ, Savage KI, Crawford NT, Mullan PB, Harkin DP. BRD7, a subunit of SWI/SNF complexes, binds directly to BRCA1 and regulates BRCA1-dependent transcription. Cancer Res. 2010; 70: 2538-47.
11. Kaeser MD, Aslanian A, Dong MQ, Yates JR 3rd, Emerson BM. BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. J Biol Chem. 2008; 283: 32254-63.
12. Liu H, Zhou M, Luo X, Zhang L, Niu Z, Peng C, Ma J, Peng S, Zhou H, Xiang B, Li X, Li S, He J, et al. Transcriptional regulation of BRD7 expression by Sp1 and c-Myc. BMC Mol Biol. 2008; 9: 111.
13. Park YA, Lee JW, Kim HS, Lee YY, Kim TJ, Choi CH, Choi JJ, Jeon HK, Cho YJ, Ryu JY, Kim BG, Bae DS. Tumor suppressive effects of bromodomain-containing protein 7 (BRD7) in epithelial ovarian carcinoma. Clin Cancer Res. 2014; 20: 565-75.
14. Pern F, Bogdanova N, Schurmann P, Lin M, Ay A, Langer F, Hillemanns P, Christiansen H, Park-Simon TW, Dork T. Mutation analysis of BRCA1, BRCA2, PALB2 and BRD7 in a hospital-based series of German patients with triple-negative breast cancer. PLoS One. 2012; 7: e47993.
15. Wu WJ, Hu KS, Chen DL, Zeng ZL, Luo HY, Wang F, Wang DS, Wang ZQ, He F, Xu RH. Prognostic relevance of BRD7 expression in colorectal carcinoma. Eur J Clin Invest. 2013; 43: 131-40.
16. Yu X, Li Z, Shen J. BRD7: a novel tumor suppressor gene in different cancers. Am J Transl Res. 2016; 8: 742-8.
17. Chen CL, Wang Y, Pan QZ, Tang Y, Wang QJ, Pan K, Huang LX, He J, Zhao JJ, Jiang SS, Zhang XF, Zhang HX, Zhou ZQ, et al. Bromodomain-containing protein 7 (BRD7) as a potential tumor suppressor in hepatocellular carcinoma. Oncotarget. 2016; 7: 16248-61. doi: 10.18632/oncotarget.7637.
18. Zhang Q, Wei L, Yang H, Yang W, Yang Q, Zhang Z, Wu K, Wu J. Bromodomain containing protein represses the Ras/Raf/MEK/ERK pathway to attenuate human hepatoma cell proliferation during HCV infection. Cancer Lett. 2016; 371: 107-16.
19. Choy L, Hagenbeek TJ, Solon M, French D, Finkle D, Shelton A, Venook R, Brauer MJ, Siebel CW. Constitutive NOTCH3 Signaling Promotes the Growth of Basal Breast Cancers. Cancer Res. 2017; 77: 1439-52.
20. Xiao Y, Yang X, Miao Y, He X, Wang M, Sha W. Inhibition of cell proliferation and tumor growth of colorectal cancer by inhibitors of Wnt and Notch signaling pathways. Oncol Lett. 2016; 12: 3695-700.
21. Jung CW, Kong JS, Seol H, Park S, Koh JS, Lee SS, Kim MJ, Choi IJ, Myung JK. Expression of activated Notch1 and Hey1 in papillary thyroid carcinoma. Histopathology. 2017; 70: 301-8.
22. Woltje K, Jabs M, Fischer A. Serum induces transcription of Hey1 and Hey2 genes by Alk1 but not Notch signaling in endothelial cells. PLoS One. 2015; 10: e0120547.
23. Jia D, Wei L, Guo W, Zha R, Bao M, Chen Z, Zhao Y, Ge C, Zhao F, Chen T, Yao M, Li J, Wang H, et al. Genome-wide copy number analyses identified novel cancer genes in hepatocellular carcinoma. Hepatology. 2011; 54: 1227-36.
24. Roessler S, Jia HL, Budhu A, Forgues M, Ye QH, Lee JS, Thorgeirsson SS, Sun Z, Tang ZY, Qin LX, Wang XW. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010; 70: 10202-12.
25. Roessler S, Long EL, Budhu A, Chen Y, Zhao X, Ji J, Walker R, Jia HL, Ye QH, Qin LX, Tang ZY, He P, Hunter KW, et al. Integrative genomic identification of genes on 8p associated with hepatocellular carcinoma progression and patient survival. Gastroenterology. 2012; 142: 957-66 e12.
26. Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, Mulawadi FH, Wong KF, Liu AM, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012; 44: 765-9.
27. Lamb JR, Zhang C, Xie T, Wang K, Zhang B, Hao K, Chudin E, Fraser HB, Millstein J, Ferguson M, Suver C, Ivanovska I, Scott M, et al. Predictive genes in adjacent normal tissue are preferentially altered by sCNV during tumorigenesis in liver cancer and may rate limiting. PLoS One. 2011; 6: e20090.
28. Chen ZZ, Huang L, Wu YH, Zhai WJ, Zhu PP, Gao YF. LncSox4 promotes the self-renewal of liver tumour-initiating cells through Stat3-mediated Sox4 expression. Nat Commun. 2016; 7: 12598.
29. Wang X, Sun L, Wang X, Kang H, Ma X, Wang M, Lin S, Liu M, Dai C, Dai Z. Acidified bile acids enhance tumor progression and telomerase activity of gastric cancer in mice dependent on c-Myc expression. Cancer Med. 2017.
30. Zhu P, Wang Y, He L, Huang G, Du Y, Zhang G, Yan X, Xia P, Ye B, Wang S, Hao L, Wu J, Fan Z. ZIC2-dependent OCT4 activation drives self-renewal of human liver cancer stem cells. J Clin Invest. 2015; 125: 3795-808.
31. Allemani C, Weir HK, Carreira H, Harewood R, Spika D, Wang XS, Bannon F, Ahn JV, Johnson CJ, Bonaventure A, Marcos-Gragera R, Stiller C, Azevedo e Silva G, et al. Global surveillance of cancer survival 1995-2009: analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet. 2015; 385: 977-1010.
32. Altekruse SF, McGlynn KA, Dickie LA, Kleiner DE. Hepatocellular carcinoma confirmation, treatment, and survival in surveillance, epidemiology, and end results registries, 1992-2008. Hepatology. 2012; 55: 476-82.
33. Salmans ML, Yu Z, Watanabe K, Cam E, Sun P, Smyth P, Dai X, Andersen B. The co-factor of LIM domains (CLIM/LDB/NLI) maintains basal mammary epithelial stem cells and promotes breast tumorigenesis. PLoS Genet. 2014; 10: e1004520.
34. Zheng Q, Zhao Y. The diverse biofunctions of LIM domain proteins: determined by subcellular localization and protein-protein interaction. Biol Cell. 2007; 99: 489-502.
35. Caputo L, Witzel HR, Kolovos P, Cheedipudi S, Looso M, Mylona A, van IWF, Laugwitz KL, Evans SM, Braun T, Soler E, Grosveld F, Dobreva G. The Isl1/Ldb1 Complex Orchestrates Genome-wide Chromatin Organization to Instruct Differentiation of Multipotent Cardiac Progenitors. Cell Stem Cell. 2015; 17: 287-99.
36. Costello I, Nowotschin S, Sun X, Mould AW, Hadjantonakis AK, Bikoff EK, Robertson EJ. Lhx1 functions together with Otx2, Foxa2, and Ldb1 to govern anterior mesendoderm, node, and midline development. Genes Dev. 2015; 29: 2108-22.
37. Li L, Jothi R, Cui K, Lee JY, Cohen T, Gorivodsky M, Tzchori I, Zhao Y, Hayes SM, Bresnick EH, Zhao K, Westphal H, Love PE. Nuclear adaptor Ldb1 regulates a transcriptional program essential for the maintenance of hematopoietic stem cells. Nat Immunol. 2011; 12: 129-36.
38. Dawid IB, Breen JJ, Toyama R. LIM domains: multiple roles as adapters and functional modifiers in protein interactions. Trends Genet. 1998; 14: 156-62.
39. Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999; 399: 491-6.
40. Drost J, Mantovani F, Tocco F, Elkon R, Comel A, Holstege H, Kerkhoven R, Jonkers J, Voorhoeve PM, Agami R, Del Sal G. BRD7 is a candidate tumour suppressor gene required for p53 function. Nat Cell Biol. 2010; 12: 380-9.
41. Tae S, Karkhanis V, Velasco K, Yaneva M, Erdjument-Bromage H, Tempst P, Sif S. Bromodomain protein 7 interacts with PRMT5 and PRC2, and is involved in transcriptional repression of their target genes. Nucleic Acids Res. 2011; 39: 5424-38.
42. Alaña L, Sesé M, Cánovas V, Punyal Y, Fernández Y, Abasolo I, de Torres I, Ruiz C, Espinosa L, Bigas A, Y Cajal SR, Fernández PL, Serras F, et al. Prostate tumor OVerexpressed-1 (PTOV1) down-regulates HES1 and HEY1 notch targets genes and promotes prostate cancer progression. Mol Cancer. 2014; 13: 74.
43. Zhu P, Wang Y, Du Y, He L, Huang G, Zhang G, Yan X, Fan Z. C8orf4 negatively regulates self-renewal of liver cancer stem cells via suppression of NOTCH2 signalling. Nat Commun. 2015; 6: 7122.