
INTRODUCTION
Colorectal cancer (CRC) is the third most commonly diagnosed malignancy in the World [1]. Despite advances in early detection and therapies, CRC still has a lethal outcome in nearly 40% of all diagnosed cases [2, 3]. CRC is an important contributor to cancer mortality [4]. Alterations in oncogenes, tumor suppressors, and DNA mismatch repair (MMR) genes lead to CRC development [5, 6]. The significant clinical heterogeneity within a given pathologic stage reflects the underlying molecular heterogeneity in CRC pathogenesis [3].
Molecularly, CRCs are categorized into those with microsatellite instability (MSI) which are primarily proximal and frequently associate with the CpG island methylator phenotype (CIMP) and hypermutation, and those that are microsatellite stable (MSS) but are chromosomally unstable [3, 7, 8]. MSI characterizes 10–15% of sporadic CRCs and has been related to a better patient prognosis compared with MSS colorectal cancer [7–12].
Tumors that demonstrate high MSI (MSI-H) as a result of alteration/loss of mismatch repair (MMR) protein(s) are referred to as MMR-deficient. MMR-proficient tumors include MSS and likely MSI-low tumors (MSI-L). The determination of MMR status is important for diagnosis and treatment.
DNA mismatch repair system is composed of 6 proteins (MLH1, MSH2, MSH3, MSH6, PMS1 and PMS2) whose function is to correct base-base mispairs introduced into short tandem repeats, termed microsatellites, during DNA synthesis [9, 13, 14]. We and others have previously reported the primary involvement of MLH1 and MSH2 alterations in MSI-H phenotype occurrence. However, alterations of MSH3 and MSH6 were also cited with proximal tumors that are poorly differentiated and mucine positive [14, 15].
APC is one of the key tumor suppressor genes (TSG) associated with early colon carcinogenesis [16, 17] in both familial adenomatous polyposis coli (FAP) and FAP-like sporadic CRCs [18]. Recent studies have shown mutations of APC in many cancers including CRC [7, 11, 17, 19–21]. APC’s impact on downstream effectors such as β-catenin, GSK and AXIN has been established in previous studies [17, 19, 21–24]. Altered WNT pathway signaling plays a significant role in CRC development and genetic variation in APC has been shown to correlate with increased predisposition to CRC [11].
We recently analyzed 12 pairs of African American CRC tumors and their matched normal tissue using whole exome sequencing and established a panel of novel variants that are potentially useful for better subtyping of CRC in this population [11]. We have reported APC, MSH3, and MSH6 among the top target genes for mutations. As such we further analyzed them through targeted sequencing in a larger sample size.
In this study, we examined APC, MSH3, and MSH6 sequence alterations and copy number variations (CNV) using targeted exome sequencing (TES) to determine specifics of these genes variants’ profiles in African American patients.
RESULTS
Clinico-pathological characteristics of patients
A: Discovery set (Ion Torrent sequencing [Thermo Fisher Scientific; Waltham, MA]): The study (discovery) set consisted of 123 patients (140 samples) (Figure 1 and Supplementary Table 1). Of these patients, 52 (43%) were females and 69 (57%) were males (for 2 samples, gender is missing). The age range of patients was 24 to 95 years, with a median of 62 years. With regards to cancer stage, 11% (6/56) were stage I, 34% (19/56) were stage II, 29% (16/56) were stage III, and 4% (2/56) were stage IV [23% (13/56) had no staging data].
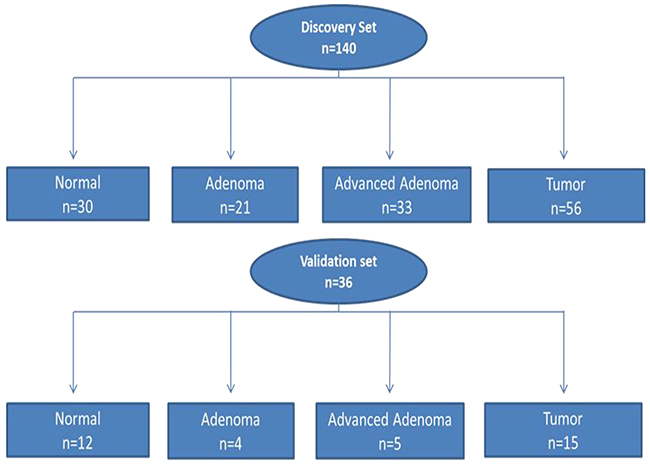
Figure 1: Flow chart of patient selection for both Discovery and Validation sets for somatic variant analysis. Discovery set: 140 samples (n=123 patients) and Validation set: 36 samples (n=26 patients).
B: Validation set (Illumina sequencing; San Diego, CA) was performed on a subset of the study group. This subset (Validation set) consisted of 36 samples from 26 patients. The main goal of this second sequencing is to rule-out technical sequencing artifacts and eliminate false variants. There were 11 (42%) females and 15 (58%) males. With regard to cancer stage, 60% (n = 9/15) were stage II, 26% (n = 4/15) were stage III and 13% (2/15) were stage IV. The age range was from 41 to 88 with a median age of 55 years (Table 1).
Table 1: Clinico-pathologic characteristics of the Validation set patients (n=26, 36 samples) with previously described and novel variants
Sample ID |
Age |
Sex |
Type of Tissue |
Location |
TNM |
STAGING |
APC Mut |
MSH6 |
|---|---|---|---|---|---|---|---|---|
CC1018N |
84 |
F |
N |
RIGHT |
N |
NA |
- |
- |
CC1018 |
84 |
F |
T |
LEFT |
T3N1b |
II |
- |
- |
CC1024 |
73 |
F |
T |
RIGHT |
T3N1MX |
III |
+ |
+ |
CC1028 |
42 |
M |
T |
LEFT |
T3N0MX |
II |
+ |
- |
CC1028N |
42 |
M |
N |
RIGHT |
N |
NA |
- |
- |
CC1029 |
51 |
F |
T |
RIGHT |
T2N0MX |
II |
+ |
+ |
CC1029N |
51 |
F |
N |
LEFT |
N |
NA |
- |
- |
CC1036 |
63 |
M |
T |
LEFT |
T3N2M1 |
IV |
+ |
- |
CC1036N |
63 |
M |
N |
RIGHT |
N |
NA |
- |
+ |
CC1038 |
54 |
M |
T |
LEFT |
T3N0Mx |
II |
+ |
- |
CC1038N |
54 |
M |
N |
RIGHT |
N |
NA |
- |
- |
CC1053 |
50 |
F |
T |
RIGHT |
T3N0M0 |
II |
+ |
- |
CC1053N |
50 |
F |
N |
LEFT |
N |
NA |
- |
- |
CC1054 |
53 |
M |
T |
RIGHT |
T3N0M0 |
II |
+ |
+ |
CC1054N |
53 |
M |
N |
LEFT |
N |
NA |
- |
- |
CC1055 |
79 |
F |
AA |
RIGHT |
AA |
NA |
+ |
|
CC1056 |
66 |
M |
T |
LEFT |
T1N1MX |
III |
+ |
- |
CC1056N |
66 |
M |
N |
RIGHT |
N |
NA |
- |
- |
CC1057 |
88 |
M |
T |
LEFT |
T3N2M0 |
III |
+ |
- |
CC1057N |
88 |
M |
N |
RIGHT |
N |
NA |
- |
- |
CC1059 |
60 |
F |
T |
RIGHT |
T3N1MX |
II |
+ |
+ |
CC1060 |
53 |
F |
T |
LEFT |
T3N2M1 |
IV |
+ |
|
CC1060N |
53 |
F |
N |
RIGHT |
N |
NA |
- |
+ |
CC1061N |
63 |
M |
N |
LEFT |
N |
NA |
+ |
- |
CC1065 |
41 |
M |
T |
LEFT |
T3N0M0 |
II |
+ |
- |
CC1109 |
62 |
F |
A |
RIGHT |
A |
NA |
+ |
+ |
CC1258 |
52 |
M |
T |
RIGHT |
T3N1bMx |
III |
+ |
- |
CC1386 |
70 |
M |
T |
LEFT |
T3N0Mx |
II |
- |
- |
CC1621N |
49 |
M |
N |
RIGHT |
N |
NA |
+ |
- |
CC1680 |
75 |
F |
AA |
RIGHT |
AA |
NA |
- |
- |
CC1681 |
75 |
M |
AA |
LEFT |
AA |
NA |
+ |
- |
CC1682 |
71 |
M |
AA |
RIGHT |
AA |
NA |
+ |
- |
CC1683 |
45 |
M |
A |
RIGHT |
AA |
NA |
+ |
- |
CC1698 |
54 |
M |
A |
RIGHT |
A |
NA |
+ |
- |
CC1720 |
70 |
F |
A |
RIGHT |
A |
NA |
+ |
- |
CC1721 |
54 |
M |
A |
Missing |
A |
NA |
+ |
+ |
N= Normal, T = CRC, A = Adenoma, AA = Advanced Adenoma, - =No variant. NA=Not Applicable
MMR genes
MSH3: We detected 315 MSH3 variants in the Discovery set, of which 298 were novel. Among these, 273 were non-synonymous, 23 were stopgain, and 2 were frameshift variants. Seventy variants altered the MutS_V domain, 21 altered the MutS_III domain, 37 altered the MutS_II domain and 32 altered the MutS_I domain (S.1).
Of the above variants, 14 previously identified variants were confirmed by Validation set using Illumina sequencing (Table 2). Six of these variants were non-synonymous altering exons 10, 21, and 23. Two were synonymous changes in exon 4 and 18. Variant at locus chr5:79966029 (HG19 reference) with a G to A change, had a frequency of 0.23 (7/30, 1 homozygous and 6 heterozygous) in normal, 0.52 (11/21, all heterozygous) in adenoma, 0.21 (7/33, 2 homozygous and 5 heterozygous) in advanced adenoma, and 0.29 (16/56, all heterozygous) in CRCs. Variant at locus chr5:80083459 with a G to A change, had a frequency of 0.03 (1/30, heterozygous) in normal, 0.06 (2/33, both heterozygous) in advanced adenoma, and 0.02 (1/56, heterozygous) in CRCs. The other variants were flanking intronic. The variants were mapped to the MSH3-MSH2-MSH6 region with 4 prior to EXO1, 3 in EXO1, 1 in MutS_I, 3 in MutS_II, 1 in MutS_III, and 3 in MutS_V domain (Figure 2A).
Table 2: Number of samples with confirmed variants in the targeted gene panel
Number of samples |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Loci |
Ref |
Var |
Gene |
Type |
Novel |
AA-Normal Het (30) |
AA-Normal Hom (30) |
AA-Adenoma Het (21) |
AA-Adenoma Hom (21) |
AA-Ad. Adenoma Het (33) |
AA-Ad. Adenoma Hom (33) |
AA-CRC Het (56) |
AA-CRC Hom (56) |
112043384 |
T |
G |
APC |
Intronic |
0 |
6 |
0 |
3 |
0 |
7 |
1 |
12 |
0 |
112103015 |
C |
A |
APC |
Stopgain |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
112116592 |
C |
T |
APC |
Stopgain |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
2 |
0 |
112128191 |
C |
T |
APC |
Stopgain |
0 |
0 |
0 |
1 |
0 |
2 |
0 |
2 |
0 |
112136947 |
A |
T |
APC |
Intronic |
0 |
3 |
0 |
2 |
0 |
3 |
0 |
3 |
0 |
112151261 |
C |
T |
APC |
Stopgain |
0 |
0 |
0 |
0 |
0 |
1 |
0 |
1 |
0 |
112154942 |
C |
T |
APC |
Stopgain |
0 |
0 |
0 |
1 |
0 |
1 |
0 |
1 |
0 |
112154980 |
T |
A |
APC |
Stopgain |
1 |
0 |
0 |
1 |
0 |
0 |
0 |
0 |
0 |
112157658 |
G |
T |
APC |
Stopgain |
1 |
0 |
0 |
1 |
0 |
0 |
0 |
0 |
0 |
112162854 |
T |
C |
APC |
Synonymous SNV |
0 |
11 |
1 |
7 |
2 |
14 |
2 |
17 |
6 |
112164561 |
G |
A |
APC |
Synonymous SNV |
0 |
16 |
5 |
14 |
6 |
20 |
7 |
27 |
16 |
112173553 |
T |
G |
APC |
Synonymous SNV |
0 |
1 |
0 |
1 |
0 |
0 |
0 |
0 |
0 |
112173899 |
C |
T |
APC |
Non-synonymous SNV |
0 |
1 |
0 |
1 |
0 |
2 |
0 |
3 |
0 |
112174096 |
C |
A |
APC |
Stopgain |
0 |
0 |
0 |
0 |
0 |
1 |
0 |
0 |
0 |
112174763 |
A |
T |
APC |
Stopgain |
1 |
0 |
0 |
0 |
0 |
0 |
0 |
1 |
0 |
112175023 |
A |
G |
APC |
Synonymous SNV |
0 |
3 |
0 |
2 |
0 |
4 |
0 |
3 |
0 |
112175030 |
G |
A |
APC |
Non-synonymous SNV |
0 |
1 |
0 |
0 |
0 |
0 |
0 |
2 |
0 |
112175069 |
C |
T |
APC |
Stopgain |
0 |
0 |
0 |
1 |
0 |
0 |
0 |
2 |
0 |
112175207 |
G |
T |
APC |
Stopgain |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
2 |
0 |
112175399 |
A |
T |
APC |
Stopgain |
0 |
0 |
0 |
0 |
0 |
2 |
0 |
1 |
0 |
112175576 |
C |
T |
APC |
Stopgain |
0 |
0 |
0 |
0 |
0 |
3 |
0 |
0 |
0 |
112175639 |
C |
T |
APC |
Stopgain |
0 |
0 |
0 |
2 |
0 |
4 |
0 |
2 |
0 |
112175770 |
G |
A |
APC |
Synonymous SNV |
0 |
17 |
5 |
13 |
7 |
20 |
8 |
26 |
14 |
112176325 |
G |
A |
APC |
Synonymous SNV |
0 |
19 |
4 |
14 |
6 |
21 |
6 |
26 |
17 |
112176541 |
C |
G |
APC |
Synonymous SNV |
0 |
1 |
0 |
1 |
0 |
0 |
0 |
2 |
0 |
112176559 |
T |
G |
APC |
Synonymous SNV |
0 |
18 |
4 |
13 |
7 |
19 |
8 |
26 |
17 |
112176756 |
T |
A |
APC |
Non-synonymous SNV |
0 |
3 |
26 |
4 |
17 |
8 |
24 |
4 |
50 |
112176918 |
G |
C |
APC |
Non-synonymous SNV |
1 |
0 |
0 |
0 |
0 |
1 |
0 |
0 |
0 |
112177171 |
G |
A |
APC |
Synonymous SNV |
0 |
16 |
5 |
14 |
6 |
21 |
7 |
31 |
15 |
112178492 |
C |
T |
APC |
Synonymous SNV |
0 |
0 |
0 |
0 |
0 |
1 |
0 |
1 |
0 |
112178795 |
G |
A |
APC |
Non-synonymous SNV |
0 |
1 |
0 |
0 |
0 |
0 |
0 |
2 |
0 |
112178995 |
A |
G |
APC |
Synonymous SNV |
0 |
7 |
0 |
3 |
0 |
6 |
0 |
6 |
2 |
112179909 |
C |
A |
APC |
Intronic |
0 |
9 |
1 |
9 |
1 |
12 |
0 |
20 |
3 |
79950497 |
C |
T |
DHFR, MSH3 |
Intronic |
0 |
6 |
4 |
6 |
2 |
3 |
2 |
14 |
5 |
79950508 |
C |
T |
DHFR, MSH3 |
Intronic |
0 |
4 |
0 |
1 |
0 |
3 |
0 |
5 |
0 |
79950512 |
A |
G |
DHFR, MSH3 |
Intronic |
0 |
5 |
14 |
1 |
8 |
3 |
8 |
13 |
26 |
79960955 |
G |
A |
MSH3 |
Intronic |
0 |
11 |
5 |
10 |
4 |
16 |
1 |
24 |
7 |
79966029 |
G |
A |
MSH3 |
Synonymous SNV |
0 |
6 |
1 |
11 |
0 |
5 |
2 |
16 |
0 |
79966197 |
G |
A |
MSH3 |
Intronic |
0 |
11 |
5 |
11 |
4 |
19 |
1 |
26 |
5 |
79968496 |
C |
T |
MSH3 |
Intronic |
0 |
5 |
3 |
3 |
3 |
11 |
1 |
13 |
3 |
80024685 |
C |
A |
MSH3 |
Non-synonymous SNV |
0 |
0 |
0 |
2 |
0 |
0 |
0 |
0 |
0 |
80024738 |
A |
G |
MSH3 |
Non-synonymous SNV |
0 |
0 |
0 |
0 |
0 |
2 |
0 |
0 |
0 |
80024783 |
G |
A |
MSH3 |
Non-synonymous SNV |
0 |
1 |
0 |
0 |
0 |
5 |
0 |
2 |
0 |
80083459 |
G |
A |
MSH3 |
Synonymous SNV |
0 |
1 |
0 |
0 |
0 |
2 |
0 |
1 |
0 |
80149981 |
A |
G |
MSH3 |
Non-synonymous SNV |
0 |
2 |
27 |
4 |
16 |
7 |
23 |
5 |
48 |
80160610 |
T |
A |
MSH3 |
Intronic |
0 |
5 |
2 |
4 |
0 |
4 |
1 |
12 |
1 |
80168937 |
G |
A |
MSH3 |
Non-synonymous SNV |
0 |
11 |
15 |
10 |
8 |
19 |
10 |
22 |
30 |
48022981 |
G |
T |
MSH6 |
Intronic |
0 |
10 |
3 |
12 |
1 |
11 |
1 |
23 |
4 |
48023115 |
T |
C |
MSH6 |
Synonymous SNV |
0 |
9 |
1 |
5 |
1 |
7 |
1 |
14 |
1 |
48026286 |
C |
T |
MSH6 |
Synonymous SNV |
0 |
4 |
0 |
2 |
0 |
4 |
0 |
7 |
0 |
48027375 |
T |
C |
MSH6 |
Synonymous SNV |
0 |
4 |
0 |
2 |
0 |
7 |
0 |
5 |
1 |
48030692 |
T |
A |
MSH6 |
Synonymous SNV |
0 |
0 |
0 |
0 |
0 |
1 |
0 |
0 |
0 |
48030838 |
A |
T |
MSH6 |
Intronic |
0 |
10 |
2 |
12 |
0 |
8 |
1 |
17 |
2 |
48032908 |
A |
G |
MSH6 |
Intronic |
1 |
1 |
0 |
1 |
0 |
0 |
0 |
2 |
0 |
48032937 |
T |
C |
MSH6 |
Intronic |
0 |
4 |
25 |
4 |
17 |
9 |
21 |
12 |
40 |
48033514 |
T |
C |
MSH6 |
Intronic |
0 |
0 |
0 |
2 |
0 |
2 |
0 |
1 |
0 |
48033545 |
A |
C |
MSH6 |
Intronic |
1 |
0 |
0 |
0 |
0 |
0 |
0 |
1 |
0 |
48033551 |
C |
G |
MSH6 |
Intronic |
0 |
4 |
20 |
3 |
11 |
8 |
9 |
13 |
34 |
48033700 |
G |
A |
MSH6 |
Non-synonymous SNV |
0 |
1 |
0 |
2 |
0 |
0 |
0 |
2 |
0 |
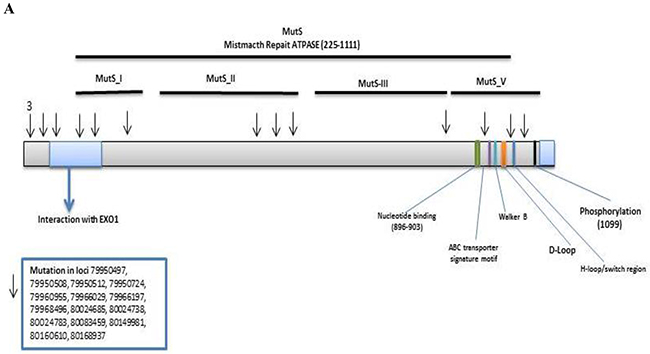
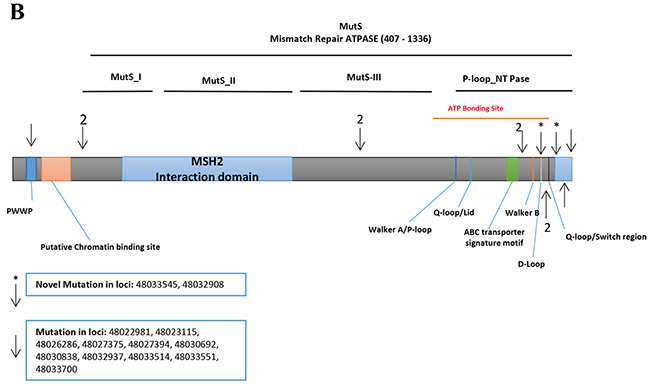
Figure 2: Distribution of confirmed and novel variants in proteins encoded by target genes. (A) MSH3 (B) MSH6, (Arrows show confirmed and asterisks the novel variants, respectively).
MSH6: We detected 434 variants in the Discovery set, of which 396 were novel (Supplementary table 2). Of these, 371 were non-synonymous, 22 were stopgain and 3 were frameshifts. Seventy-six variants altered the MutS_V domain, 29 altered the MutS_IV domain, 31 altered the MutS_III domain, 52 altered the MutS_II domain and 24 altered the MutS_I domain (S.1).
The Validation set sequencing confirmed 12 variants (by Illumina). Two of these were novel (Table 2). The A>G and A>C variants are flanking the MutS_V domain and the MSH2 binding site coding regions, respectively. An A>C intronic variant (IVS8-45) at locus chr2:48033545 was observed in 1 CRC sample with a frequency of 0.02 (1/56, heterozygous). The A>G variant at locus chr2:48032908 was intronic (IVS7-50) and was observed with a frequency of 0.03 (1/30, heterozygous) in normal, 0.05 (1/21, heterozygous) in adenoma, and 0.04 (2/56, both heterozygous) in CRC (Table 2). One variant was mapped in the PWWP domain, 2 prior to the MutS_I, 1 in MutS_III, and 8 in P-loop_NTPase (Figure 2B).
APC variants
We detected 944 variants in the Discovery set, of which 822 were novel. The Validation set sequencing led to the confirmation of 33 variants of which 4 were novel. The variant at locus chr5:112176918 with a G to C change, had a frequency of 0.03 (1/33, heterozygous) in advanced adenoma. The variant at locus chr5:112174763 with an A to T change, had a frequency of 0.02 (1/56, heterozygous) in CRCs. The variant at locus chr5:112154980 with a T to A change, had a frequency of 0.05 (1/21, heterozygous) in adenoma. The variant at locus chr5:112157658 with a G to T change, had a frequency of 0.05 (1/21, heterozygous) in adenoma. One variant was mapped in the 5’ UTR, 9 prior to ARM, 4 on the ARM domain, 3 prior to β-catenin interacting region, 12 in the β-catenin interacting region, 3 in the basic region, and 1 in the 3’ UTR (Table 2 and Figure 3).
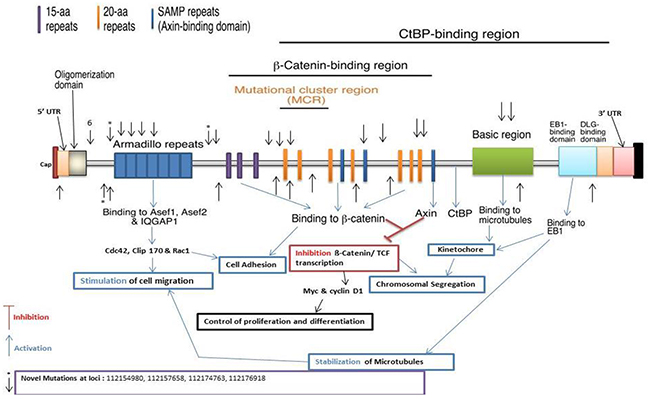
Figure 3: Distribution of APC confirmed and novel variants, (Arrows show confirmed and asterisks the novel variants, respectively).
We analyzed copy number variation (CNV) and loss of heterozygosity (LOH) in 16 matched tumor and normal samples in a blinded fashion by examining the sequencing read counts of 8978 variants arranged by patient, gene, and genomic location, as previously described [25, 26]. An NGS read counts imbalance of ≥0.6 (variant reads / reference + variant reads) across a minimum of 3 consecutive variants was used as criteria for a CNV/LOH alteration.
Fifteen of 16 pairs of samples were successfully analyzed, and 39 CNV/LOH were detected with an average of 1.6 alterations per tumor. Potential germline CNV/LOH (30/39, 77%) were observed, reflecting allele imbalance in both normal and tumor tissue in 11/15 (73%) pairs, that may be also attributed to PCR bias. Somatic CNV/LOH (9/39, 23%), heterozygous in normal tissue and homozygous in tumors, were observed in 6 pairs of tissues (40%). Two pairs of samples showed evidence of both germline and somatic CNV/LOH in different genes. MSH6 was most frequently altered, in 11 pairs (73%) by 8 germline and 3 somatic alterations; APC was altered in 8 pairs (53%) by 3 germline and 5 somatic alterations, and MSH3 in 5 pairs (33%) by 4 germline and 1 somatic alteration. CNV/LOH were examined in an additional 93 tumors using the criteria above and 83 samples (89%) were successfully analyzed to detect 117 alterations in 71/83 samples (86%) with an average of 1.7 alterations per sample for the 3 analyzed genes. Similar to the frequencies of gene sequence alterations observed in the matched T-N pairs, MSH6 was most frequently altered in 48 samples (68%), APC in 41 (58%), and MSH3 in 28 (39%). In total, the T-N pairs and the additional samples exhibited 156 CNV/LOH in 86/98 (88%) samples which altered MSH6 in 59/98 cases (60%), APC in 49 cases (50%), and MSH3 in 33 cases (34%).
DISCUSSION
Targeted exome sequencing can lead to the discovery of new targets for prevention, treatment, and diagnostic value through the definition of specific driver mutations, especially in African Americans, a population at high risk of CRC that generally presents aggressive and advanced tumors at diagnosis. In this study, we determined the frequencies of novel and known variants in neoplastic tissues in African Americans with CRC. We examined APC, MSH3 and MSH6 variants using TES and in-silico analysis of novel variants.
Platform usage in NGS is known to contribute to sequencing outcome and artifacts. We recently reported NGS outcome differences within and across different sequencing platforms which mandate the confirmation of predicted NGS variants [27].
While the frequency of many MSH3 variants was high, the level of homozygosity is low, which likely points to a deactivation of MSH3 through different variants. Indeed, several samples displayed two or more MSH3 variants at once (Table 2). It is noteworthy that many of the validated variants were present in normal matched specimens as well. These specimens were isolated from adjacent areas of the colonic lesions, and as such might correspond to a field effect of the neoplastic lesion, but are most likely germline. One limitation of confirming the somatic or germline nature of these variants is the lack of peripheral blood sample DNA.
For MSH6, two novel variants (Table 2) were flanking the region coding for the MutS-V domain and the MSH2 binding site, respectively. One variant was mapped in the PWWP domain, 2 prior to the MutS_I domain, 1 in MutS_III domain, and 8 in P-loop_NTPase domain (Figure 2B).
The majority of sequencing data deposited in public databases are from Caucasians. Minorities are generally underrepresented in such databases including TCGA and ClinVar [28]. This might be one of the reasons of the novelty of some of the variants we describe here. As such, there is a need to characterize the variant profile of all MMR genes associated with genomic instability in ethnically-diverse CRC patients.
APC mutations were associated with increased predisposition to colorectal neoplasia [17, 29]. While none of the APC variants presented here was homozygous, most specimens displayed 2 or more variants of which the cumulative effect is likely to impair APC function (Table 2). The confirmed APC variants are likely to lead to major changes in the activity of the APC protein since they are located in exons 5 and 15. Since stopgain and frameshift variants have major effects on APC protein structure, they will have drastic effects on the protein functions and involved pathways [11, 30, 31]. We report here 4 novel variants; three stopgain and one non-synonymous. One variant was mapped prior to the Armadillo repeat, one to the Armadillio repeat, one to the 15-amino acid repeat region, and one at the 20-amino acid repeat region (Figure 3). The variant observed in the 15-amino acid repeat (15R) region of the APC protein will affect the binding of β-catenin to the 15R of APC that is necessary and essential to target β-catenin for degradation. The first 15R displays the highest affinity for β-catenin in the 15R-20R module. Guda et al. recently reported 4 APC variants similar to ours [32, 33]. Sixteen confirmed APC previously described variants are described in our earlier CRC whole exome study [10, 11].
It is worth noting that the analyzed genes act as Tumor Suppressor Genes (TSGs), and as such their variants will only be effective if they lead to a complete loss of function. Our CNV analysis revealed that the 3 genes displayed a high level of LOH in favor of variant alleles. Indeed, MSH6 displayed the highest level of LOH (60%) in the analyzed samples, followed by the APC (50%) and MSH3 (34%). It is also important to note that some of the detected variants were found in normal tissue samples as well. The heterozygous presence of such variants in normal matched colon tissue samples may contribute to a higher genetic predisposition to CRC in AAs as well as early age onset of the disease in this population.
We understand that CRC incidence in African Americans has many other confounding factors, like utilization, access, and equality of treatment, as well as other socio-epidemiological factors that play a strong role in the development and progression of CRC. Our data, however, shows that this population has many novel variants of potentially deleterious nature that might account for part of the clinical phenotype. Indeed, the in-silica analysis showed that the novel variants have far-reaching consequences. The detected MSH3 and MSH6 variants seem to disrupt the whole DNA MMR system which will increase the rate of mutation within the whole genome while the variants within the APC gene will impact major pathways, including the WNT pathway
We were among the first to report on the MSI status in African Americans that was predominantly linked to MLH1 and MSH2 genes’ alterations. We here report the presence of distinctive variants in other DNA MMR genes that might play a role in the MSI status as well [10]. More importantly, alterations within the MSH3 gene are associated with the EMAST phenotype characterized by instability in tetranucleotide repeats and poor prognosis, even in MSI-H background patients. This phenotype is more prevalent in African Americans [34, 35]. Studies showed that MSI-H patients with EMAST colorectal cancers show diminished prognosis compared to patients without an EMAST phenotype. Since the distribution of EMAST relates to MSH3 gene mutations and it is more prevalent in African Americans this is potentially a reason why some African American MSI-H CRC patients have poor prognosis [34-36].
In conclusion, we defined novel deleterious variants for APC, MSH3 and MSH6 genes that might serve as novel markers for neoplastic lesions subtyping and CRC risk assessment in African Americans. These and other known variants might serve as targets for personalized and targeted treatment in the future.
MATERIALS AND METHODS
Patients and samples
A total of 140 colon samples (Supplementary Table 1), including 30 normal tissues, 21 adenomas, 33 advanced adenomas, and 56 cancers collected from 123 African American patients were used to establish variant profiles by targeted exome sequencing, using a Discovery set of patients analyzed using an Ion Torrent sequencing platform (Figure 1). Patients’ DNA was quantitated using PicoGreen double-stranded DNA binding assay (Thermo-Fisher Scientific, Waltham, MA). DNA metrics were used to confirm the quality of the library preps. Only samples that passed quality control for the NGS, as a part of standard NGS library preparation, were further processed.
To discriminate technical artifacts from real variants, we used a second sequencing platform (Illumina) in a Validation set consisting of 36 samples from 26 patients including 12 normal tissue samples, 4 adenomas, 5 advanced adenomas and 15 cancers. The Validation set samples are a subset of the Discovery set (n=140; Figure 1).
Discovery set sequencing
A targeted, multiplex PCR primer panel was designed using the custom Ion Ampliseq Designer v1.2 (Thermo Fisher Scientific). The primer panel covered 56.9 kb and included the coding regions of APC, MSH3 and MSH6 genes, with an average coverage of 96.9% of the protein coding regions and splice junctions. The panel was designed to amplify PCR products appropriate for use with DNA from formalin-fixed paraffin embedded (FFPE) tissue with an average amplicon size of 150 base pairs (bp). Sample DNA (20 ng/primer pool) was amplified using the primer panel, and libraries were prepared using the Ion Ampliseq Library Preparation kit following the manufacturer’s protocol (Thermo-Fisher Scientific, Grand Island, NY). Individual samples were barcoded, pooled, and sequenced on an Ion Torrent Proton Sequencer using the Ion PI Template OT2 200v3 and Ion PI Sequencing 200v2 kits per manufacturer’s instructions. Raw sequencing reads were filtered for high-quality reads, and the adaptors were removed using the Ion Torrent Suite 4.0.4, then reads were aligned to the hg19 reference sequence by TMAP (https://github.com/iontorrent/TS/tree/master/Analysis/TMAP) using default parameters. Resulting BAM files were processed through an in-house quality control (QC) filter and coverage analysis pipeline. BAM files were aligned using GATK LeftAlignIndels module. Amplicon primers were trimmed from aligned reads by Torrent Suite. Variant calls were made by Torrent Variant Caller 4.0 (http://mendel.iontorrent.com/ion-docs/Torrent-Variant-Caller-Plugin.html).
Validation set sequencing
Details regarding DNA quantification and quality assessment, MiSeq platform sequencing (Illumina, San Diego, CA), SNV and indel detection, and assessment of copy number alterations were performed as previously described [10-12, 37].
Copy number variation and loss of heterozygosity
Variants were organized by genomic coordinates and used to assess regions likely to be altered by a CNV selected from a minimum of 3 grouped or neighboring variants showing allelic imbalance. Cancer and normal sample sequencing data were analyzed in a blinded fashion as described previously [25, 26]. In optimal cases, multiple consecutive variants showed read count imbalance including ≥1 novel variant not in dbSNP likely to be somatic or a rare germline variant.
Bioinformatics
Comparison of African American CRC data to public databases
We used R software (version 2.15.2, http://www.r-project.org/) to compare the variants in the normal and tumor samples with those in the 1000Genomes database, which represents a nominally noncancerous population and dbSNP database. All samples displayed more or less an equal number of single nucleotide variants (SNVs) in their tumors compared with their matched normal samples. In addition, we compared our results with the recently published African American CRC Whole Exome study by Guda et al [32].
An average sequencing depth of >1000x was achieved and >98% of targeted bases (coding and within 5 bp of intron-exon boundaries) were examined by >10 reads required for variant identification. Variants were annotated using ANNOVAR [38] and filtered using the 1000 Genomes database, which represents a nominally noncancerous population, and dbSNP build 138. In addition, variants were filtered using the COSMIC database, a database of cancer somatic mutations. Variants present in any of those three datasets were marked as non-novel. All samples displayed more or less an equal number of SNVs in their tumors compared with their matched normal samples.
Author contributions
HAs, HB and MN study design and manuscript writing and critical evaluation of data; SV, PT, HAz collecting data/literature review, bioinformatics and statistical analysis.
ACKNOWLEDGMENTS
This project was supported (in part) by the National Institute on Minority Health and Health Disparities of the National Institutes of Health under Award Number G12MD007597.
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65: 87-108. https://doi.org/10.3322/caac.21262.
2. Clark ME, Smith RR. Liver-directed therapies in metastatic colorectal cancer. J Gastrointest Oncol. 2014; 5: 374-87. https://doi.org/10.3978/j.issn.2078-6891.2014.064.
3. Jass JR. Molecular heterogeneity of colorectal cancer: Implications for cancer control. Surg Oncol. 2007; 16: S7-9. https://doi.org/10.1016/j.suronc.2007.10.039.
4. Siegel R, Desantis C, Virgo K, Stein K, Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, Lin C, Leach C, Cannady RS, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012; 62: 220-41. https://doi.org/10.3322/caac.21149.
5. Karahan B, Argon A, Yildirim M, Vardar E. Relationship between MLH-1, MSH-2, PMS-2,MSH-6 expression and clinicopathological features in colorectal cancer. Int J Clin Exp Pathol. 2015; 8: 4044-53.
6. Coates R, Williams M, Melillo S, Gudgeon J. Genetic testing for lynch syndrome in individuals newly diagnosed with colorectal cancer to reduce morbidity and mortality from colorectal cancer in their relatives. PLoS Curr. 2011; 3: RRN1246. https://doi.org/10.1371/currents.RRN1246.
7. Ashktorab H, Rahi H, Wansley D, Varma S, Shokrani B, Lee E, Daremipouran M, Laiyemo A, Goel A, Carethers JM, Brim H. Toward a comprehensive and systematic methylome signature in colorectal cancers. Epigenetics. 2013; 8: 807-15. https://doi.org/10.4161/epi.25497.
8. Park JM, Huang S, Tougeron D, Sinicrope FA. MSH3 mismatch repair protein regulates sensitivity to cytotoxic drugs and a histone deacetylase inhibitor in human colon carcinoma cells. PLoS One. 2013; 8: e65369. https://doi.org/10.1371/journal.pone.0065369.
9. Haugen AC, Goel A, Yamada K, Marra G, Nguyen TP, Nagasaka T, Kanazawa S, Koike J, Kikuchi Y, Zhong X, Arita M, Shibuya K, Oshimura M, et al. Genetic instability caused by loss of MutS homologue 3 in human colorectal cancer. Cancer Res. 2008; 68: 8465-72. https://doi.org/10.1158/0008-5472.CAN-08-0002.
10. Ashktorab H, Varma S, Brim H. Next-generation sequencing in African Americans with colorectal cancer. Proc Natl Acad Sci U S A. 2015; 112: E2852. https://doi.org/10.1073/pnas.1503760112.
11. Ashktorab H, Daremipouran M, Devaney J, Varma S, Rahi H, Lee E, Shokrani B, Schwartz R, Nickerson ML, Brim H. Identification of novel mutations by exome sequencing in African American colorectal cancer patients. Cancer. 2015; 121: 34-42. https://doi.org/10.1002/cncr.28922.
12. Guda K, Veigl ML, Varadan V, Nosrati A, Ravi L, Lutterbaugh J, Beard L, Willson JK, Sedwick WD, Wang ZJ, Molyneaux N, Miron A, Adams MD, et al. Reply to Ashktorab et al.: Mutational landscape of colon cancers in African Americans. Proc Natl Acad Sci U S A. 2015; 112: E2853. https://doi.org/10.1073/pnas.1505059112.
13. Woerner SM, Tosti E, Yuan YP, Kloor M, Bork P, Edelmann W, Gebert J. Detection of coding microsatellite frameshift mutations in DNA mismatch repair-deficient mouse intestinal tumors. Mol Carcinog. 2014; 54:1376-86. https://doi.org/10.1002/mc.22213.
14. Mello JA, Acharya S, Fishel R, Essigmann JM. The mismatch-repair protein hMSH2 binds selectively to DNA adducts of the anticancer drug cisplatin. Chem Biol. 1996; 3: 579-89.
15. Acharya S. Mutations in the signature motif in MutS affect ATP-induced clamp formation and mismatch repair. Mol Microbiol. 2008; 69: 1544-59. https://doi.org/10.1111/j.1365-2958.2008.06386.x.
16. Cierna Z, Adamcikova Z, Zajac V, Wachsmannova L, Stevurkova V, Hainova K, Holec V, Janega P, Janegova A, Klincova E, Babal P. The role of adenomatous polyposis coli protein in prevention of colorectal carcinoma in the APC+/APC1638N mouse model. Virchows Archiv. 2011; 459: S162-S.
17. Segditsas S, Rowan AJ, Howarth K, Jones A, Leedham S, Wright NA, Gorman P, Chambers W, Domingo E, Roylance RR, Sawyer EJ, Sieber OM, Tomlinson IP. APC and the three-hit hypothesis. Oncogene. 2009; 28: 146-55.
18. Leoz ML, Carballal S, Moreira L, Ocana T, Balaguer F. The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management. Appl Clin Genet. 2015; 8: 95-107. https://doi.org/10.2147/TACG.S51484.
19. Femia AP, Dolara P, Giannini A, Salvadori M, Biggeri A, Caderni G. Frequent mutation of Apc gene in rat colon tumors and mucin-depleted foci, preneoplastic lesions in experimental colon carcinogenesis. Cancer Res. 2007; 67: 445-9.
20. Ashktorab H, Hassanzadeh Namin H, Taylor T, Williams C, Brim H, Mellman T, Shokrani B, Holt CL, Laiyemo AO, Nouraie M. Role of life events in the presence of colon polyps among African Americans. BMC Gastroenterol. 2013; 13: 101. https://doi.org/10.1186/1471-230X-13-101.
21. Nagel R, le Sage C, Diosdado B, van der Waal M, Oude Vrielink JA, Bolijn A, Meijer GA, Agami R. Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res. 2008; 68: 5795-802. https://doi.org/10.1158/0008-5472.Can-08-0951.
22. Inra JA, Steyerberg EW, Grover S, McFarland A, Syngal S, Kastrinos F. Racial variation in frequency and phenotypes of APC and MUTYH mutations in 6,169 individuals undergoing genetic testing. Genet Med. 2015; 17:815-21. https://doi.org/10.1038/gim.2014.199.
23. Moreno-Bueno G, Hardisson D, Sanchez C, Sarrio D, Cassia R, Garcia-Rostan G, Prat J, Guo MZ, Herman JG, Matias-Guiu X, Esteller M, Palacios J. Abnormalities of the APC/beta-catenin pathway in endometrial cancer. Oncogene. 2002; 21: 7981-90. https://doi.org/10.1038/sj.onc.1205924.
24. Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, Kovar CL, Lewis LR, Morgan MB, Newsham IF, Reid JG, Santibanez J, Shinbrot E, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487: 330-7. https://doi.org/10.1038/Nature11252.
25. Nickerson ML, Dancik GM, Im KM, Edwards MG, Turan S, Brown J, Ruiz-Rodriguez C, Owens C, Costello JC, Guo G, Tsang SX, Li Y, Zhou Q, et al. Concurrent alterations in TERT, KDM6A, and the BRCA pathway in bladder cancer. Clin Cancer Res. 2014; 20: 4935-48. https://doi.org/10.1158/1078-0432.CCR-14-0330.
26. Nickerson ML, Im KM, Misner KJ, Tan W, Lou H, Gold B, Wells DW, Bravo HC, Fredrikson KM, Harkins TT, Milos P, Zbar B, Linehan WM, et al. Somatic alterations contributing to metastasis of a castration-resistant prostate cancer. Hum Mutat. 2013; 34: 1231-41. https://doi.org/10.1002/humu.22346.
27. Ashktorab H, Azimi H, Nickerson ML, Bass S, Varma S, Brim H. Targeted Exome Sequencing Outcome Variations of Colorectal Tumors within and across Two Sequencing Platforms. Next Gener Seq Appl. 2016; 3. https://doi.org/10.4172/2469-9853.1000123.
28. Kessler MD, Yerges-Armstrong L, Taub MA, Shetty AC, Maloney K, Jeng LJ, Ruczinski I, Levin AM, Williams LK, Beaty TH, Mathias RA, Barnes KC, Consortium on Asthma among African-ancestry Populations in the A, et al. Challenges and disparities in the application of personalized genomic medicine to populations with African ancestry. Nat Commun. 2016; 7: 12521. https://doi.org/10.1038/ncomms12521.
29. Schirosi L, Pellegrino M, Tarantino P, Mauro S, Tinelli A, Greco M. A new germline stop codon mutation in exon 15 of the APC gene predisposing to familial adenomatous polyposis. Int J Biol Markers. 2013; 28: e405-8. https://doi.org/10.5301/jbm.5000042.
30. Soravia C, DeLozier CD, Dobbie Z, Berthod CR, Arrigoni E, Brundler MA, Blouin JL, Foulkes WD, Hutter P. Double frameshift mutations in APC and MSH2 in the same individual. Int J Colorectal Dis. 2005; 20: 466-70. https://doi.org/10.1007/s00384-005-0764-z.
31. Varesco L, Groden J, Spirio L, Robertson M, Weiss R, Gismondi V, Ferrara GB, White R. A Rapid Screening Method to Detect Nonsense and Frameshift Mutations - Identification of Disease-Causing Apc Alleles. Cancer Res. 1993; 53: 5581-4.
32. Guda K, Veigl ML, Varadan V, Nosrati A, Ravi L, Lutterbaugh J, Beard L, Willson JK, Sedwick WD, Wang ZJ, Molyneaux N, Miron A, Adams MD, et al. Novel recurrently mutated genes in African American colon cancers. Proceedings of the National Academy of Sciences of the United States of America. 2015; 112: 1149-54. https://doi.org/10.1073/pnas.1417064112.
33. Guda K, Veigl ML, Varadan V, Nosrati A, Ravi L, Lutterbaugh J, Beard L, Willson JK, Sedwick WD, Wang ZJ, Molyneaux N, Miron A, Adams MD, et al. Reply to Ashktorab et al.: Mutational landscape of colon cancers in African Americans. Proceedings of the National Academy of Sciences of the United States of America. 2015; 1 12:E2853. https://doi.org/10.1073/pnas.1505059112.
34. Venderbosch S, van Lent-van Vliet S, de Haan AF, Ligtenberg MJ, Goossens M, Punt CJ, Koopman M, Nagtegaal ID. EMAST is associated with a poor prognosis in microsatellite instable metastatic colorectal cancer. PLoS One. 2015; 10: e0124538. https://doi.org/10.1371/journal.pone.0124538.
35. Ashktorab H, Ahuja S, Kannan L, Llor X, Nathan E, Xicola RM, Adeyinka LO, Carethers JM, Brim H, Nouraie M. A meta-analysis of MSI frequency and race in colorectal cancer. Oncotarget. 2016; 7:34546-57. https://doi.org/10.18632/oncotarget.8945.
36. Carethers JM, Koi M, Tseng-Rogenski SS. EMAST is a Form of Microsatellite Instability That is Initiated by Inflammation and Modulates Colorectal Cancer Progression. Genes (Basel). 2015; 6: 185-205. https://doi.org/10.3390/genes6020185.
37. Ashktorab H, Mokarram P, Azimi H, Olumi H, Varma S, Nickerson ML, Brim H. Targeted exome sequencing reveals distinct pathogenic variants in Iranians with colorectal cancer. Oncotarget. 2016; 8:7852-7866. https://doi.org/10.18632/oncotarget.13977.
38. Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015; 10: 1556-66. https://doi.org/10.1038/nprot.2015.105.