
INTRODUCTION
Lung cancer is the most frequent cause of cancer-related death world-wide [1]. Non-small cell lung cancer (NSCLC) accounts for around 85% of all lung cancers. NSCLC is comprised of two subtypes, adenocarcinoma and SCC, based on histology. Recent progress in next generation sequencing (NGS) has allowed investigators to examine the various genetic mutations within malignant tumors. The discovery of driver mutations such as epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) has led to remarkable improvement in personalized therapy for pulmonary adenocarcinoma [2]. Further driver oncogenes have been identified in pulmonary adenocarcinoma. The identification of these mutations and amplifications can be used to predict sensitivity to clinical inhibitors of pulmonary adenocarcinoma [3]. Recently, genetic alterations in lung SCC have also been investigated broadly and genetic alterations related to lung SCC have been reported [4]. Molecular targeted therapies have taken a step forward with the discovery of these driver oncogenes for the treatment of lung SCC and several agents are expected to be effective for the treatment of the disease [5]. SCC is the predominant form of esophageal carcinoma. More than 450,000 people worldwide are suffering from esophageal cancer and the incidence is rapidly increasing. The overall 5-year survival rate for esophageal cancer is 15% to 30% and patients diagnosed at earlier stages have better outcomes than those diagnosed at later stages. In order to improve the efficacy of treatment of esophageal cancer multidisciplinary treatments have been conducted [6, 7]. In addition to the technical advance of minimally invasive surgery and endoscopic treatment, molecularly targeted agents have a key role in improving outcome. Molecular targeting agents containing small molecules and antibodies developed on the basis of molecular biology are being incorporated into multimodal therapies [8, 9], however effective molecular targeting therapies for esophageal cancer have not been established yet.
SOX2 is a transcription factor that is fundamental for early development and for the maintenance of stem cells in multiple adult tissues and also plays an important role in squamous cell differentiation [10, 11]. Amplification of chromosome 3q26 is the most common of the genetic alterations found in lung SCC [12]. SOX2 is a candidate oncogene present in this locus and amplification of SOX2 has been reported in lung and esophageal SCC [13]. Bass et al. showed that inhibition of SOX2 suppresses cell growth. There is a data that supports a role for this gene as a “lineage survival oncogene” [14]. In a previous study, we demonstrated that silencing SOX2 by siRNA induced G1 cell cycle arrest mediated by upregulation of CDKN1A expression resulting in an anti tumor effect in SOX2-expressing lung SCC cells both in vitro and in vivo [15]. As shown in Figure 1A, SOX2 expression was detected in 3 of 6 kinds of lung SCC cells, in 2 of 4 kinds of pulmonary adenocarcinoma cells and in all kinds of esophageal SCC cells, while expression of SOX2 was not detected in normal human foreskin fibroblast HFF1 and normal human lung fibroblast NHLF and human umbilical vein endothelial cells (HUVEC). In human lung SCC and esophageal SCC sections, SOX2 expression was detected in more than 87.5% of sections (Figure 1B and 1C) suggesting that molecular targeting of SOX2 might be useful for treating SCC.
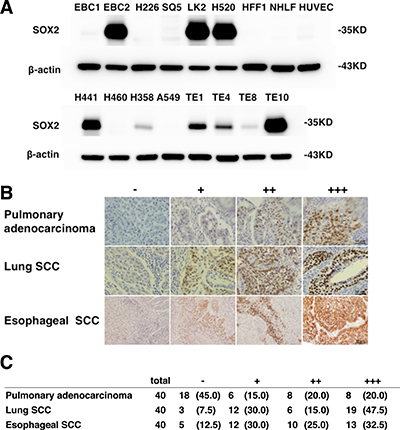
Figure 1: SOX2 expression in lung, esophageal SCC and pulmonary adenocarcinoma. (A) Immunoblot analysis of SOX2 in indicated cells. The expression level of β-actin is shown as a control. (B) The intensity of SOX2 staining was assigned the following scores: none = -, weak = +, moderate = ++, and strong = +++ expression. Examples of representative immunohistochemistry results are shown. Bars, 50 μm. (C) SOX2 expression in primary pulmonary adenocarcinoma samples of 40 patients, lung SCC samples of 40 patients and esophageal SCC samples of 40 patients who underwent surgical tumor resection at the Kawasaki Hospital Attached to Kawasaki Medical School between 2007 and 2012. Percentage values are given in parentheses.
As we reviewed previously, zinc finger -based artificial transcription factors (ATFs) [16] can be designed to regulate the expression of target genes and can provide powerful biotechnological tools for the investigation and treatment of disease [17, 18]. We demonstrated that a simple mode of DNA recognition by zinc finger domains makes it possible to design ATFs with novel sequence specificities [19]. The designed ATFs can perform DNA-binding activity and conduct natural transcription and they have been used to either activate or repress miscellaneous endogenous target genes [20–22]. We have also demonstrated that designed regulatory proteins (DRPs), in which artificial transcription factors (ATFs) are fused to cell-penetrating peptides (CPPs) effectively activate or repress target genes [23]. In a previous study, Stolzenburg et al. developed zinc finger-based artificial transcription factors (ATFs) to target SOX2 and showed their possible therapeutic use against breast cancer [24]. In order to target SOX2 as a candidate oncogene of lung SCC and esophageal SCC, we have developed a zinc finger-based artificial transcription factor (ATF) to selectively suppress SOX2 expression in cancer cells. We termed this system ATF/SOX2 (Figure 2A and 2B). Ad-ATF/SOX2 (Figure 2C) up-regulated CDKN1A mRNA and protein expression more significantly than did Ad-shSOX2 in SOX2-expressing lung and esophageal SCC cells in vitro. Furthermore, Ad-ATF/SOX2 induced an antitumor effect in SOX2-expressing SCC more effectively than did Ad-shSOX2 both in vitro and in vivo. These results indicate that the transcriptional SOX2 inhibition achieved by ATF/SOX2 activated CDKN1A and showed a greater antitumor effect more strongly than post-transcriptional SOX2 inhibition by shRNA. Here we report a novel SOX2-targeting therapy using an ATF for the treatment of lung and esophageal SCC.
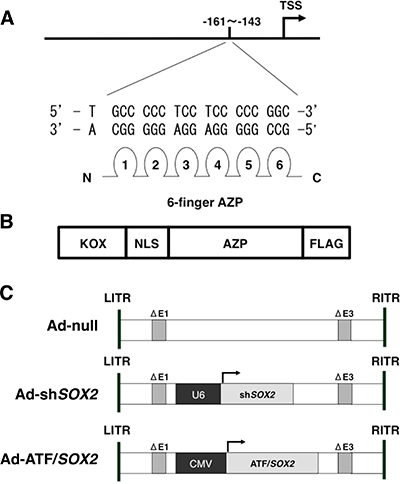
Figure 2: Schematic representation of ATF/SOX2. (A) Design of Artificial zinc finger protein (AZP) to target a 19 bp sequence (-161: TGCCCCCTCCTCCCCCGGC:-143) in human SOX2 distal promoter region. TSS; transcription start site. (B) The Artificial Transcription Factor (ATF) contains the KOX suppressor domain, a nuclear localization signal (NLS), the Artificial zinc finger protein (AZP) and a FLAG tag. We termed this ATF as ATF/SOX2. (C) Schematic representation of Ad-null, Ad-shSOX2 and Ad-ATF/SOX2. The PCR-generated expression cassette of ATF/SOX2 from pcDNA3.1 ATF/SOX2 or shSOX2 from pBAsi-mU6 shSOX2 (described in Materials and Methods section) were subcloned into the linearized E1 deleted adenovirus type 5 genome. E1; Adenovirus early region 1, E3; Adenovirus early region 3, LITR; Left Inverted Terminal Repeat, RITR; Right Inverted Terminal Repeat.
RESULTS
ATF/SOX2 suppressed SOX2 transcriptional activity and protein expression in lung and esophageal SCC cells
In order to suppress SOX2 expression in lung SCC, we have generated a zinc finger-based artificial repressor for SOX2 termed ATF/SOX2. We engineered ATF/SOX2 using six zinc finger arrays designed to bind a 19 bp site in the SOX2 distal and proximal promoter region [24]. The six zinc finger domains are linked to the nuclear localization signal (NLS) and KOX (zinc finger 10) repressor domain (Figure 2A, 2B). To test the ability of ATF/SOX2 to suppress SOX2 expression we measured SOX2 transcriptional activity in lung SCC cells in the presence or absence of ATF/SOX2 cloned into the mammalian expression vector pcDNA3.1. As shown in Figure 3A and 3B, in the control, which lacked ATF/SOX2, the SOX2 distal and proximal promoter and 5’UTR region –1990/+436 exhibited significant transcriptional activity in EBC2 SCC cells (52 -fold) 24 hours after transfection. On the other hand, in the presence of pcDNA3.1 ATF/SOX2, transcriptional activity of the SOX2 was significantly decreased in EBC2 cells (1.9-fold). In esophageal SCC cells, the region –1990/+436 exhibited significant transcriptional activity in TE1 cells (32 -fold) and in TE4 cells (65 -fold). ATF/SOX2 suppressed significantly the transcriptional activity in TE1 cells (1.9 -fold) and in TE4 cells (1.9 -fold). These results indicate that ATF/SOX2 significantly suppresses SOX2 transcriptional activity in lung and esophageal SCC cells.
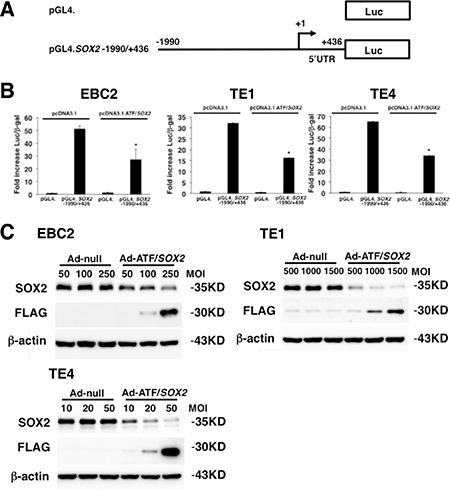
Figure 3: Repression of SOX2 transcriptional activity and protein expression by ATF/SOX2 in lung and esophageal SCC cells. (A) Schematic representation of SOX2 distal and proximal promoter reporter constructs. TSS; transcription start site. Luc; Luciferase. (B) Transient transfection reporter assays in lung SCC cells and esophageal squamous carcinoma cells with the indicated luciferase reporter constructs (2 μg, pGL4), effector constructs (2 μg, pcDNA3.1) and pCMV. β-gal (1 μg). Results are presented as fold induction of relative light units normalized to β-galactosidase activity relative to that observed for control constructs. Statistical analysis was performed using Student’s t test (two-tailed, unpaired). Statistical significance was defined as *p < 0.01 vs pcDNA3.1 transfected group. (C) Ad-ATF/SOX2 dose dependently increased ATF/SOX2 fused to the FLAG epitope and suppressed SOX2 expression in EBC2 cells, TE1 and TE4 cells 48 hours after infection. SOX2 expression was not changed after Ad-null infection in these kinds of cells.
Next, we evaluated whether SOX2 expression could be suppressed by adenoviral mediated ATF/SOX2 (Ad-ATF/SOX2, Figure 2C) induction in lung and esophageal SCC cells. Immunoblot analysis was performed using anti-FLAG antibody in order to detect ATF/SOX2 expression. As shown in Figure 3C, Ad-ATF/SOX2 dose dependently increased FLAG tagged ATF/SOX2 expression in EBC2 lung SCC cells, TE1 and TE4 esophageal SCC cells 48 hours after infection. Ad-ATF/SOX2 suppressed SOX2 expression in these kinds of cells in a dose dependent manner. FLAG-tagged ATF/SOX2 was not induced and SOX2 expression was not changed after Ad-null (Figure 3C) infection in the cells.
ATF/SOX2 induced CDKN1A expression more strongly than shSOX2 in SOX2-expressing lung and esophageal SCC cells
We previously reported that CDKN1A is a SOX2 downstream gene and that silencing of SOX2 increases the expression of CDKN1A which induces cell cycle arrest in lung SCC cells [15]. In order to analyze the antitumor effect induced by ATF/SOX2 we transfected SCC cells with Ad-ATF/SOX2. We also used shSOX2 in an adenoviral vector (Ad-shSOX2) to compare the efficacy of ATF/SOX2 and shSOX2 inhibition of SOX2 expression. We then determined SOX2 and CDKN1A expression in lung and esophageal SCC cells after Ad-shSOX2 or Ad-ATF/SOX2 infection. The optimal multiplicity of infection (MOI) was determined by infecting each cell line with Ad-CMV/GFP and choosing the MOI in which over 80% of the cells were infected [25]. As shown in Figure 4, both Ad-shSOX2 and Ad-ATF/SOX2 effectively suppressed SOX2 expression in EBC2 lung SCC cells, TE1 and TE4 esophageal SCC cells 48 hours after adenoviral infections. Importantly, in EBC2 cells and TE4 cells, SOX2 protein expression was more robustly suppressed by Ad-shSOX2 than by Ad-ATF/SOX2, however, CDKN1A expression was greater in all kinds of cells in the Ad-ATF/SOX2 treated cells than in Ad-shSOX2 treated cells. qPCR analysis also showed that Ad-ATF/SOX2 induced CDKN1A mRNA more significantly than Ad-shSOX2 in these kinds of cells (Supplementary Figure 1). Moreover, as shown in Supplementary Figure 2, G1 cell cycle arrest was induced in EBC2 cells and TE4 cells 36 hours after Ad-ATF/SOX2 infection, whereas the G0/G1 cell population was very weakly increased after Ad-shSOX2 infection. CDKN1A is known as one of the downstream genes of TP53. In this study, EBC2 lung SCC cells, TE1 and TE4 esophageal SCC cells harbor mutant TP53. TP53 expression was not altered after Ad-ATF/SOX2 infection in lung and esophageal squamous SCC cells, suggesting that ATF/SOX2 induces CDKN1A in TP53 independent manner (Figure 4) [15].
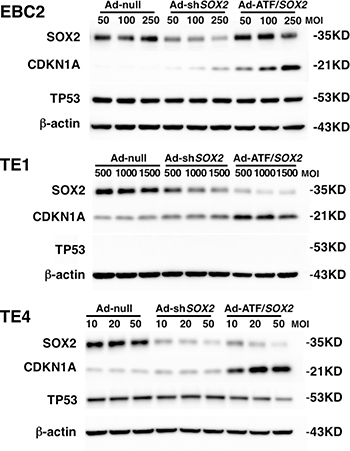
Figure 4: Ad-ATF/SOX2 up-regulated CDKN1A in lung and esophageal SCC cells more robustly than Ad-shSOX2. Immunoblot analysis shows Ad-ATF/SOX2 increased CDKN1A expression more than Ad-shSOX2 48 hours after adenoviral infections in EBC2 lung SCC cells, TE1 and TE4 esophageal SCC cells.
Inhibition of SOX2 by Ad-ATF/SOX2 suppressed cell viability and colony formation of SOX2-expressing lung and esophageal SCC cells
In order to elucidate the antitumor effect of the ATF for SOX2, we analyzed cell viability and colony formation of lung SCC cells after Ad-ATF/SOX2 infection. 48 hours after infection Ad-ATF/SOX2 significantly suppressed the cell proliferation of EBC2 lung SCC cells, TE1 and TE4 esophageal SCC cells compared to Ad-null and Ad-shSOX2 (Figure 5A). Furthermore, as shown in Figure 5B and 5C, Ad-ATF/SOX2 significantly decreased colony formation more than Ad-null and Ad-shSOX2 in EBC2, TE1 and TE4 cells. These results indicate that Ad-ATF/SOX2 shows an antitumor effect more strongly than Ad-shSOX2 in all the SOX2 positive lung and esophageal SCC cells indicated. In this experiment, Ad-shSOX2 inhibited colony formation of EBC2 cells but not of TE1 cells and TE4 cells. This is concordant with the results shown in Figure 4. There was little difference in CDKN1A expression after Ad-null and Ad-shSOX2 infection in TE1 cells and TE4 cells. On the other hand, CDKN1A expression was clearly different after Ad-null and Ad-shSOX2 infection in EBC2 cells. It is possible that Ad-shSOX2 could not significantly inhibit cell viability in EBC2 lung SCC cells just 48 hours after infection but that it could show anti tumor effect in colony formation of EBC cells during a longer time incubation after treatment but not in TE1 and TE4 cells.
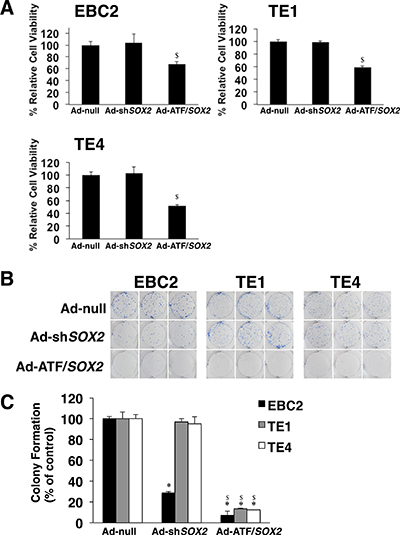
Figure 5: Ad-ATF/SOX2 significantly decreases cell viability and colony formation of SOX2-expressing lung and esophageal SCC cells. (A) Ad-ATF/SOX2 more significantly inhibited cell growth of SOX2-expressing EBC2 lung SCC cells and TE1, TE4 esophageal SCC cells than Ad-shSOX2. Cell viability was assessed 48 hours after adenoviral infection with a TC20 automated cell counter. Results represent the mean ± SD (n = 3). Statistical analysis was performed using Student’s t test (two-tailed, unpaired). Statistical significance was defined as $p < 0.01 vs Ad-shSOX2 treated group. (B) Colony formation of EBC2 lung SCC cells, TE1 and TE4 esophageal SCC cells treated with Ad-null, Ad-shSOX2 or Ad-ATF/SOX2. 7 to 14 days after treatment, cells were fixed and stained with Diff-Quik. Representative pictures of experiments performed in triplicate are shown. (C) Mean colony number was derived from quantitation of triplicate dishes for each treatment and was arbitrarily set to 100%. Data are shown relative to the control group. Results represent the mean ± SD (n = 3). Statistical analysis was performed using Student’s t test (two-tailed, unpaired). Statistical significance was defined as *p < 0.01 vs Ad-null treated group at the same MOI; $p < 0.01 vs Ad-shSOX2 treated group at the same MOI.
Down-regulation of SOX2 suppresses lung SCC growth in a xenograft mouse model
In addition to the cell-based experiments, we used an EBC2 lung SCC xenograft nude mice tumor model to determine whether ATF/SOX2 suppresses tumor growth in vivo. The tumor volume in the mice group treated with Ad-shSOX2 was approximately 37% of those in the mice group treated with Ad-null (p = 0.0026). Importantly, Ad-ATF/SOX2 completely inhibited the tumor growth (p = 0.00039 vs Ad-shSOX2 treated group, Figure 6A and 6B). These results indicate that Ad-ATF/SOX2 significantly induced an antitumor effect against SCC more than Ad-shSOX2 in vivo.
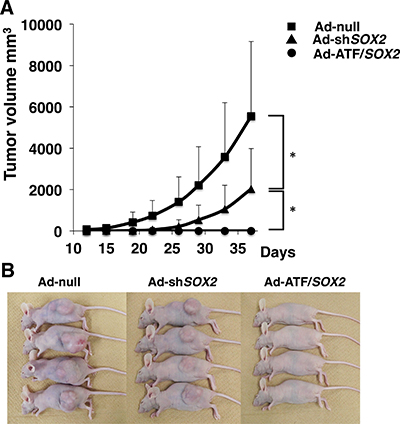
Figure 6: Effect of EBC2 xenograft tumor volumes as a function of time and treatment with adenoviral vectors. (A) Volume of the tumors derived from EBC2 lung SCC cells treated with Ad-null, Ad-shSOX2 or Ad-ATF/SOX2 at a MOI of 250 is shown. The volume was monitored over time (days) after inoculation of tumor cells. Fifteen mice were studied in each group. Tumor growth is expressed as mean tumor volume; bars represent SD. Statistical analysis was performed using Student’s t test (two-tailed, unpaired). Statistical significance was defined as *p < 0.01. (B) Macroscopic appearance of EBC2 lung SCC tumors in xenograft mice at 39 days after inoculation.
DISCUSSION
In this study, we have shown both in vitro and in vivo that the targeted down-regulation of SOX2 using ATF based technologies can be used as an effective tool for the treatment of SCC in lung and esophageal cancers that express SOX2. We showed that ATF/SOX2 up-regulated CDKN1A, one of the target genes of SOX2, and induced cell G1 cycle arrest more effectively than shSOX2 in SOX2-expressing EBC2 lung cells and TE4 esophageal SCC cells (Figure 4, Supplementary Figure 1, Supplementary Figure 2). In addition, the ATF more significantly suppressed cell viability and colony formation of SOX2-expressing lung and esophageal SCC cells compared to shSOX2, whereas little CDKN1A expression was induced after Ad-ATF/SOX2 infection in SOX2 negative lung SCC cells and normal human cells HUVEC and NHLF (Supplementary Figure 3A). Cell viability was not significantly suppressed in these kinds of normal cells after Ad-ATF/SOX2 treatment, compared to control vector (Supplementary Figure 3B). These results indicate that Ad-ATF/SOX2 could induce antitumor effect in SCC cells expressing SOX2 but not in normal cells. Each zinc finger domain specifically recognizes 3 or 4 bp of DNA and ATF/SOX2 recognizes a 19 bp sequence in close proximity to the transcriptional start of the human SOX2 distal and proximal promoter region, providing a high degree of specificity. It is theoretically necessary to recognize at least 16 bp of DNA for specific recognition of one genomic region in human cells based on a genome size of 3 × 109 bp. The Basic Local Alignment Search Tool (BLAST) on the human genome revealed that the 19 bp target sequence in the human SOX2 distal promoter region is unique. Ad-ATF/SOX2 induced CDKN1A expression in all three kinds of SOX2-expressing lung and esophageal SCC cells but not in all five kinds of SOX2 negative cells (Figure 4, Supplementary Figure 3A). Moreover, as shown in Supplementary Figure 4, Ad-ATF/SOX2 did not change mRNA expression of ABL1, RTN4 and KMT2B that contained a highly similar sequence to the 19 bp target sequence of ATF/SOX2 in EBC2 cells. In addition to this, the other kinds of zinc finger based ATFs previously reported [26] alter neither SOX2 nor CDKN1A expression in EBC2 cells (Supplementary Figure 5). These results indicate that induction of CDKN1A, the downstream targets of SOX2, after Ad-ATF/SOX2 infection was not the result of off-target action of ATF/SOX2 (was on-target effect of ATF/SOX2). In this study, SOX2 expression was more robustly suppressed by Ad-shSOX2 than by Ad-ATF/SOX2 in EBC2 cells and TE4 cells. However, Ad-ATF/SOX2 induced CDKN1A and the other SOX2 related genes [15] more than Ad-shSOX2 in these kinds of cells (Figure 4, Supplementary Figure 6). We believe that this discrepancy is not the result of off-target action of ATF/SOX2 but could possibly be due to kinetics-dependent differences of ATF and shRNA. There could also be another possibility in that the distal or proximal promoter region of SOX2 could suppress the expression of unidentified noncoding RNA, which shRNA for SOX2 does not target, leading to induction of CDKN1A. Recently, a noncoding RNA, SOX2 overlapping transcript (SOX2OT), has been identified as having a role in lung and esophageal cancer [27]. SOX2OT is a long noncoding RNA (lncRNA) mapped to human chromosome 3q26.3-q27 where the SOX2 gene is embedded within its third intron [28]. Shahryari et al. reported that suppression of a novel splice variant of SOX2OT (SOX2OT-S1) increased the number of cells in G1 phase in human embryonal carcinoma NTERA2 cells [29]. Furthermore, the KOX transcriptional repressor domain used in the current study works as an effective repressor of distal and proximal gene regulatory elements including enhancers [30, 31], therefore ATF/SOX2 might inhibit the expression of not only SOX2 but also an unidentified distal noncoding RNA. Further investigation of the loci around the SOX2 gene is needed in order to elucidate this discrepancy.
In our previous investigation, we identified 15 SOX2 related genes (4 negatively-correlated and 11 positively-correlated with SOX2) in lung SCC cells using RNA-seq data from 178 lung SCC specimens [32] and another RNA-seq dataset from 105 non-small cell lung cancer cell lines [4]. As shown in Supplementary Figure 6, the ATF suppressed MSH6 mRNA and protein expression in EBC2 cells, TE4 cells and TE10 cells suggesting that these genes might be downstream genes of SOX2 in lung and esophageal SCC. On the other hand, the PI3 Kinase inhibitor wortmannin suppressed SOX2 expression in EBC2 lung SCC cells and in TE1, TE10 esophageal SCC cells whereas ATF/SOX2 did not alter phosphorylated AKT (pAKT) expression, suggesting that pAKT might be an upstream regulator of SOX2 in these kinds of cells (Supplementary Figure 7) [33, 34]. Importantly, AKT and β-actin expression were not altered after wortmannin treatment. However, further investigation is needed to clarify whether pAKT directly regulates SOX2 or not. In this experiment, we used TE10 esophageal SCC cells because TE4 esophageal SCC cells have little PI3 Kinase expression (data not shown). Thus a combination treatment of the ATF targeting SOX2 and PI3 Kinase inhibitor might prove to be an effective antitumor treatment for esophageal and lung SCC (Supplementary Figure 8).
The technology of gene activation / repression with zinc finger (ZF)-based artificial transcription factors (ATFs), TAL Effector or CRISPR a/i are evolving rapidly [35–37]. RNAi is known as the most universally used gene repression technology and is categorized as either small interfering RNAs (siRNA) or short hairpin RNAs (shRNA) [38]. To achieve repression, RNAi requires only a single small component, while other systems need 2 or 3 components (e.g.: dCas9, sgRNA and repression module) that are difficult to deliver [36, 37]. On the other hand, there is concern that RNAi may have many off-target effects and reproducibility issues [39]. Compared to RNAi, off-target effects with the use of ATFs, TALE and CRISPR intervention is considered to be minimal [40]. Furthermore, the molecular weight of ATFs is much smaller than TAL effector and CRISPR a/i thus ATFs may have an advantage in delivery to target cells over the other systems.
Delivery of therapeutic viral vectors into the target organ and tumor is required for the treatment of lung cancer or esophageal cancer [41–43]. One of the drawbacks of this approach is the lack of means to effectively deliver the therapeutic virus to target different kinds of cells that reside within an intricate lung and esophageal structure. However, we have previously demonstrated that the ATFs fuse to cell-penetrating peptides (CPPs) and effectively activate or repress target genes in vitro [23]. Importantly, this approach does not depend on cell types and could prevent the risk of insertional mutagenesis and cause relatively fewer off-target effects than ATF gene delivery systems that rely on expression from DNA and mRNA [44]. Thus, CPPs fused to ATF might be feasible treatments to target lung and esophageal SCC in the near future.
Although several technical challenges and uncertainties remain, further advances in understanding and improvements in ATF technology will open the next era of cancer therapy. In this study we have used ATF technology to successfully suppress SCC in vitro and in vivo in SOX2-expressing SCC lung and esophageal cancer cells as a first step in the search for an effective treatment for SCC lung and esophageal cancers.
MATERIALS AND METHODS
Cell lines and culture conditions
The human lung SCC cells H520, H226, the human pulmonary adenocarcinoma cells H358, H441, H460 and A549, the human esophageal SCC cells TE1, TE4, TE8, TE10 and human foreskin fibroblast HFF1 obtained from the American Type Culture Collection (Manassas, VA) and the normal human lung fibroblast cells NHLF obtained from Lonza (Portsmouth, NH) were grown in RPMI 1640 (H226, H358, H460, H520, TE1, TE4, TE8, TE10) or high glucose Dulbecco’s modified Eagle medium (H441, A549, HFF1, NHLF) supplemented with 10% heat-inactivated fetal bovine serum. Human umbilical vein endothelial cells (HUVEC) were purchased and grown in Endothelial Cells Growth Medium (Medium 200) supplemented with Low Serum Growth Supplement kit (Thermo Fisher Scientific, Rockford, IL). The lung squamous cell carcinoma cells EBC1, EBC2, SQ5, LK2 were kindly provided by Dr. Kiura Katsuyuki (Department of Respiratory Medicine, Okayama University Graduate School of Medicine and Dentistry, Okayama, Japan) and grown in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum. All cell lines were cultured in 5% CO2 at 37°C.
Immunohistochemistry
Sections were sequentially deparaffinized through a series of xylene, graded ethanol, and water immersion steps. After being boiled in target retrieval solution (Dako, Carpinteria, CA, USA) for 15 minutes, sections were incubated with 3% hydrogen peroxide for 5 minutes to block endogenous peroxidase activity. A primary antibody specific for human SOX2 was obtained from Cell Signaling Technology (Beverly, MA). Specimens were incubated overnight at 4°C with a 1:100 dilution of antibody followed by three washes with TBS. The slides were treated with streptavidin-biotin complex (Envision System labeled polymer, horseradish peroxidase [HRP], Dako, Carpinteria, CA) for 60 minutes at room temperature. Immunoreactions were visualized using a 3,3′-diaminobenzidine (DAB) substrate-chromogen solution (Dako Cytomation Liquid DAB Substrate Chromogen System, Dako) and counterstained with hematoxylin. Sections were immersed in an ethanol and xylene bath and mounted for examination. For immunohistochemistry analysis, 40 lung SCC tissue samples, 40 pulmonary adenocarcinoma tissues samples and 40 esophageal SCC samples in tissue sections were obtained from patients diagnosed with lung SCC and pulmonary adenocarcinoma and who underwent surgical resection at Kawasaki Hospital, Okayama, Japan. The experimental protocol was approved by the Ethics Review Committee of Kawasaki Medical School (Ethics Committee reference number: 1310).
Design and construction of ATF/SOX2
We designed and constructed an artificial zinc finger protein (AZP) targeting –161 to –143 in the human SOX2 gene, where +1 is the transcription start site, by using our recognition code table as described [19]. The DNA encoding the AZP was cloned into pcDNA3.1+ (Thermo Fisher Scientific) containing a Krüppel-associated box domain of KOX1; a nuclear localization signal from the simian virus 40 large T antigen; and a FLAG epitope tag to construct the ATF-expression vector pcDNA3.1 ATF/SOX2.
Adenoviral vectors
A plasmid vector expressing shSOX2: pBAsi-mU6 shSOX2 was constructed by ligating a shRNA sequence for SOX2 (GCTCTTGGCTCCATGGGTT) into pBAsi-mU6 Pur (Takara Bio Inc. Otsu. Japan). The recombinant adenoviral vector Ad-shSOX2 was generated by subcloning the expression cassette of mU6 shSOX2 from pBAsi-mU6 shSOX2 using PCR primers (5′- GTAACTATAACGG TCATGTGGTATGGCTGATTATGATCGAATCG and 5′- ATTACCTCTTTCTCCTAAAACGACGGCCAGTG CCAAGC). Ad-ATF/SOX2 was generated by subcloning the expression cassette of ATF/SOX2 from pcDNA3.1 ATF/SOX2 using PCR primers (5′- GTAACTATAACG GTCGCGATGTACGGGCCAGATATAC and 5′- ATTA CCTCTTTCTCCCTGGTTCTTTCCGCCTCAGA). These PCR-generated fragments were directly subcloned into the linearized pAdenoX vector using Adeno-X Adenoviral System 3 Universal according to manufacture’s protocol (Takara Bio Inc.). The viral titer for each vector was determined by the Adeno-X™ Rapid Titer Kit (Takara Bio Inc.) and the optimal multiplicity of infection (MOI) was determined by infecting each cell line with Ad-CMV/GFP and assessing the expression of GFP [25].
Luciferase reporter construct and transient transfection reporter assay
The SOX2 proximal and distal promoter region of was obtained from human genomic DNA (Invitrogen, Life Technologies, Carlsbad, CA). The position of the transcription initiation site (+1) was determined by the Ensembl Human Genome browser. The luciferase reporter construct: pGL4. SOX2 –1990/+436 was generated by subcloning the SOX2 promoter region –1990/+436 amplified from the genomic DNA using PCR primers (5′-tttNhe1GCTAGC-1990acaagccataacttgagagaaaaaggagaaccttc and 5′-aaaHindIIIaagctt +436gcgg gcgctgtgcgcgggcccggcccgccggcggc), digested with NheI and HindIII and subcloned into pGL4.23 luciferase reporter construct (Promega. Madison. WI.). All of the transfections were carried out in 6-well plates. Cells were seeded 1 day before transfection at a density of 2 × 105 per well. Transfections were carried out with Lipofectamine 3000 (Thermo Fisher Scientific) in accordance with the manufacturer’s protocol as indicated. Transfected cells were harvested at 24 hours. Results of one representative experiment are presented as fold induction of relative light units normalized to β-galactosidase activity relative to that observed for the control vectors. Each experiment was repeated at least three times. Error bars indicate the SD from the average of the triplicate samples in one experiment.
Cell viability assay
EBC2 lung SCC cells were plated at a density of 1 × 105 cells per well, TE1 esophageal SCC cells were plated at a density of 2 × 105 cells and TE4 esophageal SCC cells were plated at a density of 4 × 105 cells in a 6-well plates and cultured overnight at 37°C. The following day Ad-null, Ad-shSOX2 or Ad-ATF/SOX2 was infected at a MOI of 250 for EBC2 cells, at a MOI of 1500 for TE1 and at a MOI of 50 for TE4. Cells were harvested 48 hours after adenoviral infection and viable cells were assessed by a TC20 automated cell counter (Bio-Rad, Hercules, CA). [45].
Co-transfection and puromycin selection
Cells were transfected using Lipofectamine 3000 (Thermo Fisher Scientific) according to manufacturer’s protocol. Cells were seeded into 6-well plates at a density of 2.5 × 105 per well 1 day before transfection. To enrich for transfected cells, plasmid with puromycin resistant cassette pPUR (0.5 μg, Takara Bio, Inc.) was co-transfected with pcDNA3.1, pGFPZFN1.4-B2H, pGFPZFN2-B2H, pST1374, pPIGAZFN-L1 and pPIGAZFN-R2 (2 μg, obtained from Addgene, Cambridge, MA) [26]. After 24 hours, cells were selected in 2 μg/ml puromycin for 48 hours, then cells were harvested and the protein was isolated according to the manufacturer’s instructions.
Colony formation assay
EBC2 cells were first plated at a density of 1 × 105 cells, TE1 cells were plated at a density of 2 × 105 cells and TE4 cells were plated at a density of 4 × 105 cells per well in 6-well plates 24 hours before virus infection. The following day Ad-null, Ad-shSOX2 or Ad-ATF/SOX2 were infected at a MOI of 250 for EBC2 cells at a MOI of 1500 for TE1and at a MOI of 50 for TE4 cells for 24 hours. EBC2 cells were released from the dish by incubation with trypsin/EDTA, counted, plated in triplicate at a density of 1 × 103 cells in 6-well plates for 7 days. TE1 cells were plated in triplicate at a density of 2 × 103 cells in 6-well plates for 8 days. TE4 cells were plated in triplicate at a density of 3 × 103 cells in 6-well plates for 14 days. The cells were fixed and stained with Diff-Quik (Sysmex, Kobe, Japan) [46]. Colonies (a group of aggregated cells numbering at least 50) were then counted [47]. The mean number of the control group was arbitrarily set to 100%, and all other numbers were normalized and percentage-specific cytotoxicity compared to colony formation in the control group was calculated.
Immunoblot analysis
Cells were lysed in ice-cold M-PER lysis buffer purchased from Thermo Fisher Scientific. Cell lysates were clarified by centrifugation (20 min at 15,000 rpm at 4°C) and protein concentration determined using the BCA Protein Assay (Thermo Fisher Scientific). Equal amounts of protein were separated on an SDS-PAGE gel. The gel was electrophoretically transferred to a Hybond PVDF transfer membrane (Millipore, Bedford, Massachusetts) and incubated with primary and secondary antibodies according to the Supersignal® West Pico chemiluminescence protocol (Pierce, Rockford, IL). Antibody specific for FLAG (DYKDDDDK epitope) tag was obtained from Takara Bio, Inc. (Shiga, Japan) and antibody specific for SOX2, CDKN1A, VIM, AKT and phosphorylated AKT (Ser473) were purchased from Cell Signaling Technology (Beverly, MA). Antibody specific for TP53 and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and antibody specific for MSH6 and BMP2 were purchased from Proteintech Japan (Tokyo, Japan). Secondary horseradish peroxidase-conjugated antibodies were obtained from Jackson Immunoresearch Laboratories (West Grove, PA). Each experiment was repeated at least three times and the representative data is displayed.
Real time PCR
Total RNA from the cultured cells was obtained by using TRIzol Reagent (Thermo Fisher Scientific). 2 μg of total RNA was used for reverse transcription. Reverse transcription was performed at 37°C for 15 min using PrimeScript RT reagent Kit (Takara Bio, Inc.). The specific probe for CDKN1A (Hs00355782_m1), BMP2 (Hs00154192_m1), SNAI1 (Hs00195591_m1), VIM (Hs00958111_m1), ABL1 (Hs01104728_m1), RTN4 (Hs00199671_m1), KMT2B (Hs00207065_m1), MSH6 (Hs00943000_m1) and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Hs03929097_g1) were derived from the commercially available TaqMan Gene Expression Assays (Applied Biosystems, Life Technologies, CA). The real-time PCR reactions were carried out in a 48-well microtiter plate using the TaqMan Gene Expression Master Mix (TaqMan One-Step RT-PCR kit) (Applied Biosystems). All samples were analyzed in triplicate in three independent experiments. The fluorescence of the PCR products was detected by the same apparatus. The number of cycles for the amplification plot to reach the threshold limit (Ct value) was used for quantification. GAPDH was used as endogenous control.
Flow cytometric analysis for cell cycle
For cell cycle analysis, EBC2 cells were plated at a density of 1 × 105 cells, TE4 cells were plated at a density of 4 × 105 cells per well in 6-well plates and cultured overnight at 37°C. The following day Ad-null, Ad-shSOX2 or Ad-ATF/SOX2 was infected at a MOI of 250 for EBC2 cells and at a MOI of 50 for TE4. 36 hours after infection, cells were harvested and washed once with PBS. Cells were resuspended in PBS containing 0.1 % Triton X-100 and 1 mg/ml RNase for 5 min at room temperature and then stained with propidium iodide at 100 μg/ml to determine DNA cell cycle using a FACS Verse (BD Bioscience, San Jose, CA). Doublets, cell debris, and fixation artifacts were gated out, and DNA cell cycle was determined using FACSuite Version 1.0.2.2238.
Mouse experiments
The experimental protocol was approved by the Ethics Review Committee for Animal Experimentation of Kawasaki University Graduate School of Medicine and Dentistry (Ethics Committee reference number: 15–046). EBC2 lung SCC cells were plated in 15 cm dishes at a density of 4 × 106 per dish and cultured overnight at 37°C. The following day cells were infected with Ad-null, Ad-shSOX2 or Ad-ATF/SOX2 at a MOI of 250 for 24 hours. Cells were harvested and resuspended in culture medium. Human lung cancer xenografts were established in 6-wk-old female BALB/c nude mice (CLEA Japan, Inc., Tokyo, Japan) by subcutaneous (s.c.) inoculation of the adenoviral treated EBC2 cells (2 × 106 cells/0.1 ml) into the dorsal flank. Fifteen mice were studied in each group. Animals were then observed closely and survival studies were performed. Tumors were measured 2 times a week, and tumor volume was calculated as a × b2 × 0.5, where a and b were large and small diameters, respectively.
Statistical analysis
Statistically significant differences between means and medians of the study groups were evaluated using Student’s t-test (two-tailed, unpaired). Statistical significance was defined as *p < 0.01.
Abbreviations
SCC, squamous cell carcinoma; ATF, artificial transcription factor; bp, base pair; shSOX2, SOX2 short hairpin RNA; NSCLC, Non-small cell lung cancer; NGS, next generation sequencing; EGFR, epidermal growth factor receptor; ALK, anaplastic lymphoma kinase; HFF1, human foreskin fibroblast; NHLF, normal human lung fibroblast; HUVEC, human umbilical vein endothelial cells; ATFs, artificial transcription factors; DRPs, designed regulatory proteins; CPPs, cell-penetrating peptides; NLS, nuclear localization signal; UTR, untranslated region; MOI, multiplicity of infection; BLAST, Basic local alignment search tool; bp, base pair; EMT, epithelial-mesenchymal transition; RNAi, RNA interference; siRNA, small interfering RNAs; TALE, transcription activator-like effector; CRISPR, clustered regularly interspaced short palindromic repeat; CRISPR a/i, CRISPR activation and interference; GFP, green fluorescence protein.
Author contributions
E.Y., N.M., T.I., T.M., S.O. performed the experiments. T.F. and T.S. designed the study. T.F. and T.S. wrote the manuscript. T.Y., M.T., and M.H. provided technical help. N.T. and Y.N. provided scientific discussion. All authors contributed to the discussion and review of the manuscript.
ACKNOWLEDGMENTS
The authors thank M. Durbin, Y. Maeda and H. Matsuzaki for critical reading and discussion.
CONFLICTS OF INTEREST
The authors have declared that no conflicts of interest exists.
FUNDING
This study was supported by the Ministry of Education, Science, and Culture, Japan to T. Fukazawa, and Y. Naomoto.
REFERENCES
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136:E359–86. https://doi.org/10.1002/ijc.29210.
2. Hirsch FR, Scagliotti GV, Mulshine JL, Kwon R, Curran WJ Jr, Wu YL, Paz-Ares L. Lung cancer: current therapies and new targeted treatments. Lancet. 2017; 389:299–311. https://doi.org/10.1016/s0140-6736(16)30958-8.
3. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511:543–50. https://doi.org/10.1038/nature13385.
4. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012; 489:519–25. https://doi.org/10.1038/nature11404.
5. Gandara DR, Hammerman PS, Sos ML, Lara PN Jr, Hirsch FR. Squamous cell lung cancer: from tumor genomics to cancer therapeutics. Clin Cancer Res. 2015; 21:2236–43. https://doi.org/10.1158/1078-0432.ccr-14-3039.
6. Holmes RS, Vaughan TL. Epidemiology and pathogenesis of esophageal cancer. Semin Radiat Oncol. 2007; 17:2–9. https://doi.org/10.1016/j.semradonc.2006.09.003.
7. Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013; 381:400–12. https://doi.org/10.1016/s0140-6736(12)60643-6.
8. Shah MA, Ramanathan RK, Ilson DH, Levnor A, D’Adamo D, O’Reilly E, Tse A, Trocola R, Schwartz L, Capanu M, Schwartz GK, Kelsen DP. Multicenter phase II study of irinotecan, cisplatin, and bevacizumab in patients with metastatic gastric or gastroesophageal junction adenocarcinoma. J Clin Oncol. 2006; 24:5201–6. https://doi.org/10.1200/jco.2006.08.0887.
9. Gefitinib extends survival in some esophageal cancers. Cancer Discov. 2015; 5:OF4. https://doi.org/10.1158/2159-8290.cd-nb2014-174.
10. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131:861–72. https://doi.org/10.1016/j.cell.2007.11.019.
11. Que J, Okubo T, Goldenring JR, Nam KT, Kurotani R, Morrisey EE, Taranova O, Pevny LH, Hogan BL. Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development. 2007; 134:2521–31. https://doi.org/10.1242/dev.003855.
12. Choi YW, Choi JS, Zheng LT, Lim YJ, Yoon HK, Kim YH, Wang YP, Lim Y. Comparative genomic hybridization array analysis and real time PCR reveals genomic alterations in squamous cell carcinomas of the lung. Lung Cancer. 2007; 55:43–51. https://doi.org/10.1016/j.lungcan.2006.09.018.
13. Hussenet T, Dali S, Exinger J, Monga B, Jost B, Dembele D, Martinet N, Thibault C, Huelsken J, Brambilla E, du Manoir S. SOX2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas. PLoS One. 2010; 5:e8960. https://doi.org/10.1371/journal.pone.0008960.
14. Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, Ramos AH, Woo MS, Weir BA, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009; 41:1238–42. https://doi.org/10.1038/ng.465.
15. Fukazawa T, Guo M, Ishida N, Yamatsuji T, Takaoka M, Yokota E, Haisa M, Miyake N, Ikeda T, Okui T, Takigawa N, Maeda Y, Naomoto Y. SOX2 suppresses CDKN1A to sustain growth of lung squamous cell carcinoma. Sci Rep. 2016; 6: 20113. https://doi.org/10.1038/srep20113.
16. Sera T. Zinc-finger-based artificial transcription factors and their applications. Adv Drug Deliv Rev. 2009; 61:513–26. https://doi.org/10.1016/j.addr.2009.03.012.
17. Segal DJ, Goncalves J, Eberhardy S, Swan CH, Torbett BE, Li X, Barbas CF 3rd. Attenuation of HIV-1 replication in primary human cells with a designed zinc finger transcription factor. J Biol Chem. 2004; 279:14509–19. https://doi.org/10.1074/jbc.M400349200.
18. Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS, Malani N, Anguela XM, Sharma R, Ivanciu L, Murphy SL, Finn JD, Khazi FR, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011; 475:217–21. https://doi.org/10.1038/nature10177.
19. Sera T, Uranga C. Rational design of artificial zinc-finger proteins using a nondegenerate recognition code table. Biochemistry. 2002; 41:7074–81.
20. Tan S, Guschin D, Davalos A, Lee YL, Snowden AW, Jouvenot Y, Zhang HS, Howes K, McNamara AR, Lai A, Ullman C, Reynolds L, Moore M, et al. Zinc-finger protein-targeted gene regulation: genomewide single-gene specificity. Proc Natl Acad Sci U S A. 2003; 100:11997–2002. https://doi.org/10.1073/pnas.2035056100.
21. Rebar EJ, Huang Y, Hickey R, Nath AK, Meoli D, Nath S, Chen B, Xu L, Liang Y, Jamieson AC, Zhang L, Spratt SK, Case CC, et al. Induction of angiogenesis in a mouse model using engineered transcription factors. Nat Med. 2002; 8:1427–32. https://doi.org/10.1038/nm795.
22. Zhang B, Xiang S, Zhong Q, Yin Y, Gu L, Deng D. The p16-specific reactivation and inhibition of cell migration through demethylation of CpG islands by engineered transcription factors. Hum Gene Ther. 2012; 23:1071–81. https://doi.org/10.1089/hum.2012.070.
23. Tachikawa K, Schroder O, Frey G, Briggs SP, Sera T. Regulation of the endogenous VEGF-A gene by exogenous designed regulatory proteins. Proc Natl Acad Sci U S A. 2004; 101:15225–30. https://doi.org/10.1073/pnas.0406473101.
24. Stolzenburg S, Rots MG, Beltran AS, Rivenbark AG, Yuan X, Qian H, Strahl BD, Blancafort P. Targeted silencing of the oncogenic transcription factor SOX2 in breast cancer. Nucleic Acids Res. 2012; 40:6725–40. https://doi.org/10.1093/nar/gks360.
25. Gu J, Zhang L, Huang X, Lin T, Yin M, Xu K, Ji L, Roth JA, Fang B. A novel single tetracycline-regulative adenoviral vector for tumor-specific Bax gene expression and cell killing in vitro and in vivo. Oncogene. 2002; 21:4757–64. https://doi.org/10.1038/sj.onc.1205582.
26. Zou J, Maeder ML, Mali P, Pruett-Miller SM, Thibodeau-Beganny S, Chou BK, Chen G, Ye Z, Park IH, Daley GQ, Porteus MH, Joung JK, Cheng L. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009; 5:97–110. https://doi.org/10.1016/j.stem.2009.05.023.
27. Khandelwal A, Bacolla A, Vasquez KM, Jain A. Long non-coding RNA: A new paradigm for lung cancer. Mol Carcinog. 2015; 54:1235–51. https://doi.org/10.1002/mc.22362.
28. Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, FitzPatrick DR. Mutations in SOX2 cause anophthalmia. Nat Genet. 2003; 33:461–3. https://doi.org/10.1038/ng1120.
29. Shahryari A, Rafiee MR, Fouani Y, Oliae NA, Samaei NM, Shafiee M, Semnani S, Vasei M, Mowla SJ. Two novel splice variants of SOX2OT, SOX2OT-S1, and SOX2OT-S2 are coupregulated with SOX2 and OCT4 in esophageal squamous cell carcinoma. Stem Cells. 2014; 32:126–34. https://doi.org/10.1002/stem.1542.
30. Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, Maehr R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods. 2015; 12:401–3. https://doi.org/10.1038/nmeth.3325.
31. Thakore PI, D’Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, Reddy TE, Crawford GE, Gersbach CA. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods. 2015; 12:1143–9. https://doi.org/10.1038/nmeth.3630.
32. Klijn C, Durinck S, Stawiski EW, Haverty PM, Jiang Z, Liu H, Degenhardt J, Mayba O, Gnad F, Liu J. A comprehensive transcriptional portrait of human cancer cell lines. Nature. 2015; 33:306–12. https://doi.org/10.1038/nbt.3080.
33. Schaefer T, Wang H, Mir P, Konantz M, Pereboom TC, Paczulla AM, Merz B, Fehm T, Perner S, Rothfuss OC, Kanz L, Schulze-Osthoff K, Lengerke C. Molecular and functional interactions between AKT and SOX2 in breast carcinoma. Oncotarget. 2015; 6:43540–56. https://doi.org/10.18632/oncotarget.6183.
34. Keysar SB, Le PN, Miller B, Jackson BC, Eagles JR, Nieto C, Kim J, Tang B, Glogowska MJ, Morton JJ, Padilla-Just N, Gomez K, Warnock E, et al. Regulation of Head and Neck Squamous Cancer Stem Cells by PI3K and SOX2. J Natl Cancer Inst. 2017; 109. https://doi.org/10.1093/jnci/djw189.
35. Beerli RR, Segal DJ, Dreier B, Barbas CF 3rd. Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc Natl Acad Sci U S A. 1998; 95:14628–33.
36. Zhang Z, Wu E, Qian Z, Wu WS. A multicolor panel of TALE-KRAB based transcriptional repressor vectors enabling knockdown of multiple gene targets. Sci Rep. 2014; 4: 7338. https://doi.org/10.1038/srep07338.
37. Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014; 159:647–61. https://doi.org/10.1016/j.cell.2014.09.029.
38. Karagiannis TC, El-Osta A. RNA interference and potential therapeutic applications of short interfering RNAs. Cancer Gene Ther. 2005; 12:787–95. https://doi.org/10.1038/sj.cgt.7700857.
39. Sigoillot FD, Lyman S, Huckins JF, Adamson B, Chung E, Quattrochi B, King RW. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat Methods. 2012; 9:363–6. https://doi.org/10.1038/nmeth.1898.
40. La Russa MF, Qi LS. The New State of the Art: Cas9 for Gene Activation and Repression. Mol Cell Biol. 2015; 35:3800–9. https://doi.org/10.1128/mcb.00512-15.
41. Li L, Li S, Cai T, Wang H, Xie X, Liu Z, Zhang Y. The targeted inhibitory effects of human amniotic fluid stem cells carrying CXCR4 promoter and DAL-1 on non-small cell lung carcinoma growth. Gene Ther. 2015; 23:214–22. https://doi.org/10.1038/gt.2015.90.
42. Fukazawa T, Maeda Y, Matsuoka J, Ono T, Mominoki K, Yamatsuji T, Shigemitsu K, Morita I, Murakami I, Tanaka H, Durbin ML, Naomoto Y. Targeting KRAS mutation-bearing lung cancer in vivo by pulmonary surfactant-adenovirus-mediated gene transfer. Anticancer Res. 2010; 30:4925–35.
43. Ma G, Kawamura K, Shan Y, Okamoto S, Li Q, Namba M, Shingyoji M, Tada Y, Tatsumi K, Hiroshima K, Shimada H. Combination of adenoviruses expressing melanoma differentiation-associated gene-7 and chemotherapeutic agents produces enhanced cytotoxicity on esophageal carcinoma. Gene Ther. 2014; 21:31–7. https://doi.org/10.1038/cgt.2013.79.
44. Gaj T, Gersbach CA, Barbas CF 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013; 31:397–405. https://doi.org/10.1016/j.tibtech.2013.04.004.
45. Kosmac K, Bantug GR, Pugel EP, Cekinovic D, Jonjic S, Britt WJ. Glucocorticoid treatment of MCMV infected newborn mice attenuates CNS inflammation and limits deficits in cerebellar development. PLoS Pathog. 2013; 9:e1003200. https://doi.org/10.1371/journal.ppat.1003200.
46. Menges CW, Sementino E, Talarchek J, Xu J, Chernoff J, Peterson JR, Testa JR. Group I p21-activated kinases (PAKs) promote tumor cell proliferation and survival through the AKT1 and Raf-MAPK pathways. Mol Cancer Res. 2012; 10:1178–88. https://doi.org/10.1158/1541-7786.mcr-12-0082.
47. Twentyman PR, Walls GA, Wright KA. The response of tumour cells to radiation and cytotoxic drugs--a comparison of clonogenic and isotope uptake assays. Br J Cancer. 1984; 50:625–31.