
INTRODUCTION
The last decade has witnessed an unprecedented advance in the medical treatment of cancer thanks to the rapid development of targeted therapy and the recent revival of immunotherapy. Targeted agents can specifically inhibit oncogenic signaling in cancer cells and accomplish striking tumor responses in molecularly defined subsets of patients, although initial regression are commonly followed by the development of progressive diseases [1]. In parallel to these advances in targeting genetic drivers of tumorigenesis, another area of success in the treatment of cancer centers on the exploration of the mechanisms of tumor immunoevasion that has led to the development of several novel agents–most notably the immune checkpoint inhibitors which reverse T cell inhibition with antibodies against the programmed cell death protein-1 (PD-1) or its ligand programmed cell death-ligand 1 (PD-L1) and cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4)–that show durable efficacy in subsets of patients with diverse tumor types and could achieve disease control for extended periods [2]. In spite of these advantages, a deficiency of treatment with immune checkpoint inhibitor is the relatively low response rate in some tumor types because of the lack of definite predictive biomarkers. In addition, immunosuppressive effects of heavy baseline tumor burden may also limit efficacy of immunotherapy [3]. Given the dramatic tumor regression elicited by targeted therapy, as well as its immune potentiating effects [4], it is reasonable to explore the potential synergistic combination of targeted and immune therapy, which will hopefully yield a high and prolonged response. This review examines the immune-based mechanisms of targeted agents, presents available data related to the use of combined targeted- and immunotherapy, and discusses the challenges we face in the clinical application of the combination therapy.
Immune effects of targeted therapy
Targeted therapy has changed the treatment paradigm of cancer over the past decades. Apart from conventionally proposed mechanisms of inhibiting driver mutation in cancer cells, targeted agents can produce immunoactivating effects via bolstering tumor cells immunogenicity, enhancing immune cells effector function, and relieving tumor-mediated immunosuppression (Table 1). Several key steps are involved in an efficient anti-tumor response. First, tumor associated antigens released from immunogenic cell death are taken up processed by dendritic cells (DCs) in the tumor microenvironment. Then, DCs migrate to tumor-draining lymph nodes where they promote the priming and differentiation of naïve T cells. Subsequently, effector T cells travel from the lymph nodes through blood vessels to the tumor bed, and recognize and eliminate the tumor cell. As illustrated in Figure 1, targeted agents have a broad impact on the complex signals involved in the multistep process. Monoclonal antibodies (mAbs), which attach to and block the extracellular ligand binding domain of targeted kinase, play a direct immunoactivating role by antibody-dependent cellular cytotoxicity (ADCC). Thus, mAbs will not be further discussed here. In the following paragraph, we mainly discuss immune effect induced by small molecule inhibitors targeting different oncogenic signaling pathways.
Table 1: Immune-based mechanisms of targeted therapy
Pathway inhibition |
Immune Potentiating Effects |
Refs |
|---|---|---|
PI3K/AKT/mTOR pathway |
||
PI3K inhibition |
Heighten the antitumor properties of TLR ligands |
[7] |
Increase accumulation of effector T cells |
[7] |
|
Dampen Treg function |
[8] |
|
Inhibit myeloid cells |
[8] |
|
Inhibit immunosuppressive TAMs |
[8] |
|
AKT inhibition |
Sensitize tumor cells to immune destruction by disrupting Mcl-1 mediated anti-apoptotic signaling |
|
mTOR inhibition |
Generate memory CD8 + T cells |
|
Reduce Tregs |
||
MAPK pathway |
||
BRAF inhibition |
Directly enhance T cell function |
[31] |
Increase antigen expression |
||
Increase MHC class I expression |
[36] |
|
Restore IL-12 and TNF-α production by DCs |
[37] |
|
Restore CD80, CD83, and CD86 expression on DC |
[37] |
|
Reduce MDSCs |
[38] |
|
Reduce the expression of VEGF |
[39] |
|
Suppress the expression of IL-1 |
[40] |
|
Increase CD8 T cell and NK cells |
[41] |
|
MEK inhibition |
Protect effector CD8 T cells from death caused by chronic T cell receptor stimulation |
[32] |
Increase antigen expression |
[34] |
|
Restore IL-12 and TNF-α production by DCs |
[37] |
|
VEGF pathway |
||
VEGF/VEGFR inhibition |
Increase extravasation of T cell |
|
Augment DC maturation and function |
||
Multikinase inhibition |
Decrease the number and function of MDSCs and Tregs |
|
Increase cytotoxic lymphocyte infiltration and response |
||
Enhance IFN-gamma production |
||
Diminish expression of CTLA4, PD1, and PDL-1 |
||
C-kit pathway |
||
C-kit inhibition |
Facilitate production of Th1 cytokine |
|
Prompt NK cell activation |
||
Suppress IDO production |
[89] |
|
Inhibit Treg and MDSCs |
||
Epigenetic pathway |
||
Epigenetic inhibition |
Increase tumor antigen expression |
|
Increase MHC molecules expression |
||
Induce the expression of NKG2DL (MICA/B) |
||
Reduce Treg cells and MDSCs |
||
Others |
||
Proteasome inhibition |
Decrease expression of peptide–MHC class I complex (thereby sensitizing tumor to NK cells) |
[130] |
Increase expression of FAS and the TRAIL receptor DR5 |
[131] |
|
Induce NOXA-mediated enhancement of mitochondrial SMAC release (thereby increasing sensitivity to T cells) |
[132] |
|
HSP90 inhibition |
Increase tumor antigen presentation |
|
Augment the expression of NKG2DL (MICA/B) |
Abbreviations: TAMs, tumor-associated macrophages; TLR, toll-like receptor; Mcl-1, myeloid cell leukemia-1; Treg, regulatory T cell; MHC, major histocompatibility complex; IL, Interleukin; TNF--α, tumor necrosis factor-α; DCs, dendritic cells; NK, natural killer; MDSCs, myeloid-derived suppressor cells; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; PD-1, programmed death 1; PD-L1, programmed death-ligand 1; Th1, T helper 1; IDO, indoleamine 2, 3-dioxygenase; NKG2DL, natural killer group 2, member D ligands; MICA/B, major histocompatibility complex class I related-A and –B; TRAIL, TNF-related apoptosis-inducing ligand.
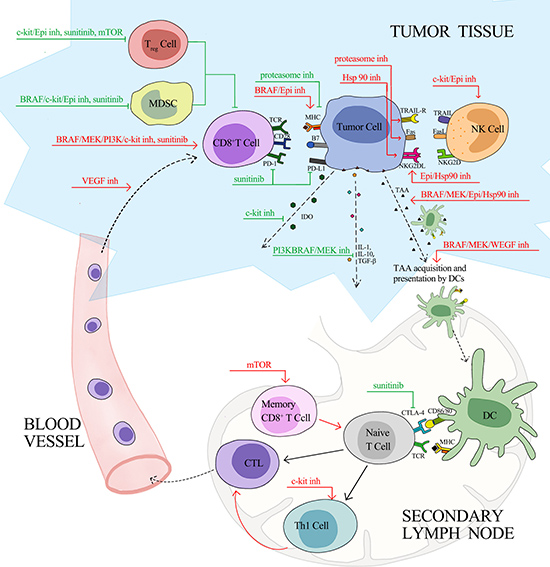
Figure 1: Effects of Targeted Therapy on Anti-Cancer Immunity. Red line, stimulatory effects; green line, inhibitory effects; inh, inhibitor; TAA, tumor associated antigen; TRAIL, TNF-related apoptosis-inducing ligand; TRAIL-R, TNF-related apoptosis-inducing ligand receptor; NK, natural killer; NKG2D, natural killer group 2, member D; NKG2DL, natural killer group 2, member D ligands; TCR, T-cell receptor; MHC, major histocompatibility complex; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; PD-1, programmed death 1; PD-L1, programmed death-ligand 1; IDO, indoleamine 2, 3-dioxygenase; IL, Interleukin; TGF, transforming growth factor; DC, dendritic cell; Th1, T helper 1; CTL, cytotoxic lymphocyte; Treg, regulatory T cell; MDSC, myeloid-derived suppressor cell.
PI3K/AKT/mTOR pathway inhibitors
The phosphatidylinositol 3-kinase (PI3K)/AKT/mechanistic target of rapamycin (mTOR) pathway is an intracellular signaling pathway that plays a crucial role in regulating cell survival and proliferation. Its dysregulation, which is implicated in a wide range of cancers, occurs via abnormal expression of pathway receptors and/or genetic mutations that lead to constitutive activation of key kinases in the pathway which could sever as targets for drug development [5].
PI3Ks are expressed in a broad range of cells, including all subtypes of leukocytes [6]. Therefore, PI3K inhibition could yield immune modulatory effects. Preclinical studies in different cancer models showed that PI3K inhibitor could heighten the antitumor properties of toll-like receptor (TLR) ligands and was associated with increased accumulation of polyfunctional T cells that secreted multiple effector cytokines, including interferon-γ (IFN-γ), interleukin-17 (IL-17), and IL-2 [7]. In addition, PI3K inhibition was shown to dampen Treg function, inhibit suppressive myeloid cells, and dampen immunosuppressive tumor-associated macrophages (TAMs) [8]. The activation of Akt was shown to increase programmed death ligand-1 (PD-L1) expression and lead to immunoresistance which could be reversed by inhibition of PI3K and mTOR [9]. In addition, AKT inhibition sensitized tumor cells to immune destruction by disrupting Mcl-1 mediated anti-apoptotic signaling [10, 11]. In an immune-resistant human papillomavirus type 16 (HPV-16) E7-expressing tumor cell line, AKT was shown to be over-activated. Consistently, retroviral transfer of a constitutively active form of AKT induced resistance against E7-specific CD8+ T-cell mediated apoptosis, which was associated with the upregulation of anti-apoptotic molecules. Besides, intratumoral injection of an AKT inhibitor enhanced the efficacy of immunotherapy [10]. Other studies also demonstrated that activation of AKT, by preventing apoptosis, induced resistance against the cytotoxicity of both antigen-specific cytotoxic T-cell lymphocytes (CTLs) in vitro and adoptively transferred cellular immune effectors in vivo [11].
The role of mTOR in the regulation of immune cell function has been well characterized. MTOR inhibitors are widely used to suppress a rejection response by the immune system in organ transplantation [12]. Paradoxically, mTOR inhibitor was recently shown to potentiate immune response by generating memory CD8+ T-cells in a dose- and duration-dependent manner [13–16]. Li et al. [17] reported that a short course of high-dose, instead of low-dose rapamycin, generated memory CD8+ T-cell responses, and afforded more durable protection against tumor compared with persistent administration of either low or high dose. Besides, mTOR inhibitors may have a dual impact on FOXP3+ regulatory T (Treg) cells. Current evidence showed that transient mTOR inhibition with rapamycin promoted T cell receptor (TCR)-induced Treg cells proliferation before TCR stimulation [18], whereas prolonged inhibition led to significant Treg cells depletion [19]. Based on these findings, several investigations explored the potential synergy between mTOR inhibitors and cancer immunotherapy. In a murine model of metastatic renal cell carcinoma (mRCC), a combination of the adenosine triphosphate (ATP) -competitive mTOR kinase inhibitory agent AZD8055 and the alphaCD40 agonistic antibody yielded synergistic antitumor responses. The addition of AZD8055 increased the proliferation and activation of CD8 T-cells and natural killer cells, as well as matured macrophages and dendritic cells [20]. Another study on a murine model of RCC (RENCA) and melanoma (B16) showed that combined treatment with heat shock protein (HSP)-based cancer vaccines and temsirolimus augmented interferon-γ production and cytotoxic T-cell responses and enhanced generation of CD8 memory cells [21]. These preclinical results provide a strong rationale for further exploiting mechanisms by which PI3K/AKT/mTOR pathway inhibitors modulate antitumor immune response, thereby better guiding the clinical design, particularly in cancers harboring PI3K mutations, such as glioblastomas, breast, colon and endometrial cancers [22].
MAPK pathway inhibitors
The mitogen-activated protein kinase (MAPK) pathway includes the signaling molecules Ras, Raf, MEK, and ERK, and functions in the regulation of gene expression, cellular proliferation, and differentiation, and survival [23]. Abnormal MAPK signaling contributes to uncontrolled cell growth and resistance to apoptosis and is implicated in a wide range of cancer [24]. Besides, MAPK pathway also participates in the regulation of T cell expansion and differentiation, as well as T cell functions, including cytokine secretion and chemotaxis [25]. Two signals are the sine qua non for T cell activation: binding of the TCR to the antigen-major histocompatibility complex (MHC) and engagement of co-stimulatory molecules, which in turn, will activate downstream signal transduction cascades including MAPK pathway [25, 26]. Thus, MAPK pathway activation enhances T-cell proliferation and function. Conversely, its inhibition dampens T-cell response.
Besides acting on tumor cells carrying the RAF mutation, RAF inhibitors can also stimulate T cell-mediated immunity against tumors. Accumulating evidence suggests that RAF inhibitors have opposing effects on BRAF-mutant and BRAF wild-type cells. While RAF inhibitors block ERK signaling in BRAF-mutant cells, they paradoxically enhance ERK signaling in BRAF wild-type cells in preclinical studies [27, 28]. It is further validated by the clinical observation of treatment-induced squamous cell cancer and keratoakanthomas in patients receiving RAF inhibitors [29, 30]. In a BRAF V600E-driven murine model of melanoma, combined treatment with vemurafenib and TCR engineered adoptive cell therapy (ACT) resulted in superior antitumor responses compared with either therapy alone [31]. And vemurafenib increased intratumoral cytokine secretion and in vivo cytotoxic activity of adoptively transferred cells via MAPK activation [31]. Similarly, MEK inhibitors, a recent study in immune-competent mice found that MEK inhibitors protected effector CD8 T-cells from death caused by chronic T cell receptor stimulation while sparing cytotoxic activity [32].
Several studies have demonstrated that inhibition of MAPK pathway by BRAF and MEK inhibitors leads to increased expression of melanocyte differentiation antigens in both melanoma cell lines and clinical tumor samples from melanoma patients [33–35]. Further, loss of tumor antigen expression is observed when resistance to BRAF inhibition develops [35]. Besides, it has also been reported that BRAF inhibition induced upregulation of MHC class I expression in tumor cells and facilitated antigen presentation and recognition [36]. Besides, blockade of the MAPK pathway may enhance dendritic cell function. In vitro showed that MEK and BRAF inhibitors could restore the decreased production of IL-12 and tumor necrosis factor-α (TNF-α) by DCs co-cultured with BRAFV600E mutant cells [37]. Moreover, the CD80, CD83, and CD86 expression on DC was reduced upon co-culture and can be partially restored with BRAF inhibition.
Additional mechanisms by which MAPK pathway inhibitors bolster antitumor immunity involve improving the tumor microenvironment by reducing the suppressive immune cells and cytokines. In the blood sample of melanoma patients, vemurafenib, a BRAF inhibitor, decreased the frequency of monocytic myeloid-derived suppressor cells (MDSCs). And it inhibited MDSCs generation in an in vitro model of the melanoma microenvironment [38]. Furthermore, BRAF inhibitor can increase intratumoral infiltration and antitumor activity of TCR-engineered ACT by inhibiting tumor cell production of the vascular endothelial growth factor (VEGF) in xenograft mouse model [39]. In consistent, decreased VEGF expression was also observed in tumor biopsies of patients receiving BRAF inhibitor treatment [39]. BRAF inhibitors also reduce IL-1 expression in cell lines and tumor biopsies, which in turn may, as suggested by Khalili et al. [40], theoretically be able to relieve T cell suppression caused by IL-1 mediated upregulation of PD-1 ligands and COX-2 expression on tumor-associated fibroblasts (TAFs). Another study in BRAF V600E mouse melanoma transplants and in de novo melanomas demonstrated that BRAF inhibitor downregulated tumor expression of chemokine (C-C motif) ligand 2 (CCL2), and resulted in a robust increase in CD8+ T/FoxP3+CD4+ T cell ratio and natural killer (NK) cells [41].
Multiple studies have reported increased intratumoral T cells infiltration in BRAF mutant melanoma tumors treated with MAPK pathway inhibitors [35, 39, 42]. This may be due to the direct facilitation of T cell trafficking, or secondary to increases in tumor cell antigenicity and immunogenicity. As the aforementioned transient induction of melanoma tumor antigens by BRAF and MEK inhibitors, increase in intratumoral T cells resulted from MAPK pathway inhibition was also lost at the time of progression on therapy [35].
BRAF inhibitors were reported to be associated with an increase in immunosuppressive T cell exhaustion markers TIM-3 and PD1 and its ligand PD-L1 [35], suggesting that combining MAPK targeted therapy with PD1/PD-L1 inhibitors may improve responses. Subsequent studies have provided supporting evidence for the potential synergistic effect of this combination therapy [43, 44]. However, it is still unclear whether the enhanced expression of exhaustion markers resulted from the increase in T cell infiltration and cytotoxic activity or a direct stimulatory effect of MAPK inhibition on the expression of exhaustion markers. Following findings may shed light on this question. Elevated PD-L1 expression was observed in BRAF inhibitor-resistant melanoma cell lines and tumor samples [42, 45, 46]. Furthermore, induction of PD-L1 expression was shown to be limited to cell lines whose resistance to BRAF inhibitor did not depend on reactivation of the MAPK pathway [47]. Together, these studies implied that increased PD-L1 expression might be secondary to enhanced T cell infiltration facilitated by
In summary, BRAF inhibitors contribute to an intensified antitumor response via several different mechanisms: first, direct enhancing of T cell antitumor activity by a paradoxical activation of the MAPK pathway; second, promoting the tumor antigen expression, recognition, and presentation; third, altering the tumor microenvironment by reducing the suppressive immune cells and cytokines.
VEGF pathway inhibitors
The VEGF family in mammals is composed of five members: VEGF-A, VEGF-B, VEGF-C, VEGF-D and placenta growth factor (PGF), and binds to different receptors, including vascular endothelial growth factor receptor (VEGFR) and Neuropilin-1 (NRP1) [48]. The role of VEGF signaling pathway in tumorigenesis and progression is multifaceted. First, it is implicated in pathological angiogenesis, lymphangiogenesis and vascular permeability [49]. Second, it promotes tumor growth and inhibits apoptosis in an autocrine or paracrine manner [50]. Moreover, it facilitates epithelial–mesenchymal transition of tumor cells [51] and regulates the function of cancer stem cells, independent of angiogenesis [52–54]. Besides, VEGF signaling affects the function of immune cells and fibroblasts that are present in the tumor microenvironment [55]. Drugs that interfere with the VEGF signaling pathway include two types, one targeting VEGF, such as bevacizumab, an anti-VEGF-A antibody and aflibercept, a recombinant fusion protein containing the VEGF-A/B-binding domains of VEGFR1/2; the other targeting VEGFR, including small molecule multikinase inhibitors like sorafenib and sunitinib, as well as anti-VEGFR2 antibody ramucirumab.
The immune modulating effects of VEGF come down to two aspects. On one side, VEGF-A reduces expression of endothelial cell adhesion molecules, which prompts an abnormal tumor vasculature and inhibits the infiltration of T cells and other immune cells [56–58]. On the other side, VEGF acts on receptors expressed on immune cells, thereby directly modulating the immune response. Preclinical studies demonstrated that VEGF-A suppressed dendritic cell differentiation and activity [59–61], promotes the expansion of MDSCs [59], upregulates the checkpoint molecules expression on CD8 T-cells [62], and modulates the proliferation of regulatory T cells [63, 64].
In accordance with the immune biology of VEGF, VEGF signaling blockade treatment shows multifaceted immune stimulatory effects. Studies in the murine model showed that modulation or normalization of tumor vasculature by anti-angiogenic therapy increased extravasation of T cell into tumors [65, 66], and exhibited a synergistic effect with anti-PD1 therapy [67]. Besides, the inhibitory effect of VEGF on DC maturation and function can be reversed by bevacizumab and sorafenib [59, 60]. Bevacizumab administration to patients with lung, breast, and colorectal carcinoma was associated with enhancement of DC maturation and antigen presentation [68]. In contrast, broader immune modulating effects were demonstrated in anti-angiogenic treatment with multikinase inhibitors. Preclinical and clinical studies have shown that sunitinib or sorafenib could decrease the number and function of MDSCs and Treg cells, enhance cytotoxic lymphocyte infiltration and response, increase IFN-gamma production, and diminish expression of CTLA4, PD1, and PDL-1 [69–71]. However, discordant results were reported. Liu et al. described an enhanced infiltration of Treg cells and an up-regulated expression of PD-L1 [72], while Guislain et al reported an augmented expression of PD-1 and an unvaried Treg cells infiltration [73]. Apart from inhibiting VEGFR, the multikinase inhibitor sunitinib also stimulates T cell response by interfering with signal transducer and activator of transcription 3 (STAT3) activation [70, 74].
Clinical trials combining VEGF-A or VEGFR inhibitors with immunotherapies showed enhanced antitumor activity. In a study of advanced metastatic melanoma treated with bevacizumab and ipilimumab combination therapy versus ipilimumab monotherapy, combination therapy increased expression of E-selectin on vessel endothelium, as well as intratumoral immune cell infiltration that was associated with clinical responses [75]. In patients with mRCC treated with atezolizumab (anti-PD-L1) in combination with bevacizumab, intratumoral T cells were increased by bevacizumab treatment alone, and were further increased upon combination with MPDL3280A [76]. Nonetheless, it should be noted that seemingly related small molecule inhibitors might not have identical activities; sorafenib, a multi-kinase inhibitor that inhibits angiogenesis, was shown to be immunosuppressive in vitro [77, 78].
C-kit inhibitors
Imatinib, a selective inhibitor of BCR-Abl and c-kit receptor tyrosine kinase, is a clear example of the success of targeted therapy for chronic myelogenous leukemia (CML) and gastrointestinal stromal tumors (GIST). Besides directly curbing tumor growth, imatinib also exerts both suppressive and stimulatory effects on host immune system.
The imatinib-induced immunosuppression is mediated by the following mechanisms. First, imatinib can impair the generation of dendritic cells (DCs) via reduced phosphorylation of AKT/ PKB Protein kinase B and nuclear accumulation of NFκB, thereby resulting in less efficient priming of CTLs [79]. Clinical observation of incomplete recovery of circulating DC numbers in CML patients treated with imatinib further provided supporting evidence for imatinib-induced dendritopoiesis [80]. Second, imatinib facilitates the conversion of the TAMs from anti-tumor M1 phenotype to pro-tumor M2 phenotype. In addition, imatinib restrains TCR induced T cell proliferation and activation [81, 82], as well as hampers cytokine synthesis by activated CD4 T cells [83].
Despite the above-mentioned immune inhibitory effects, the overall effect of imatinib on host anti-tumor immune response is stimulatory. The immune potentiating activities are mediated by hindering the activity of protein tyrosine kinases (PTKs) expressed on distinct cell types in host immune system. First, by acting on KIT in DCs, imatinib skews immune responses toward the production of T helper 1 (Th1) cytokines which will potentially contribute to tumor elimination [84, 85]. Besides, in vitro and in vivo studies of GIST showed that imatinib, by blocking KIT signaling in DCs, prompted DC-mediated NK cell activation, and stimulated the production of IFNα from NK cells [86]. Similar findings have been observed in clinical studies that imatinib treatment enhanced IFNγ secretion in GIST patients [86, 87]. In addition to the activation of favorable cross-talk between DC and NK cells, imatinib also activates CD8+ T-cells, induces Treg cells apoptosis, and reduces the expression of the immune suppressive enzyme indoleamine 2, 3-dioxygenase (IDO) through inhibition of oncogenic KIT signaling [88]. Imatinib was shown to have direct inhibitory effects on Treg cells by downregulating expression of the Treg cells master transcription factor FoxP3 and decreasing their number and suppressive capacity [89]. It also reduced MDSCs and restored effector lymphocyte responses [88]. Dasatinib, another KIT tyrosine kinase inhibitor, also could reduce the suppressive functions of MDSC and restore a Th1 response [90], and its antitumor activity can be strongly potentiated by immune stimulation with agonist anti-OX40 antibody therapy [91].
On the basis of the aforementioned preclinical findings, multiple investigations have studied the combined use of c-Kit inhibitor and various immunotherapeutic approaches. In several phase I and phase II trials, concomitant treatment with imatinib and vaccines against BCR–ABL1 epitopes showed an enhanced anti-tumor activity in patients with CML [92, 93]. Other studies explored the combination of imatinib and IFNα or IL-2 in the treatment of CML. Although several studies showed that the addition of IFNα to imatinib led to higher rates of molecular response in CML patients [94, 95], others yielded conflicting results [96, 97]. The difference among these studies might be ascribed to the high discontinuation rates resulted from IFNα related adverse events [97–99]. The combination of imatinib and pegylated IFNα 2b has also been investigated in patients with GIST, and interim analysis have shown promising results [100]. As to the combined use of imatinib and immune checkpoint inhibitor, it showed synergistic anticancer effects [88].
Epigenetic therapies and immunotherapy
Epigenetic regulation refers to the functional change in the genome without altering underlying nucleotide sequence, which is controlled through modifications on DNA and histone that includes methylation, acetylation, phosphorylation, ubiquitination, sumoylation, proline isomerization and ADP ribosylation [101, 102]. Multiple enzymes participate in the process, including DNA methyltransferases (DNMTs), histone deacetylases (HDACs), histone methyltransferases (HMTs), histone demethylases, histone acetyltransferases, ubiquitin ligases and deubiquitinases [102]. Epigenetic downregulation of tumor suppressor genes and upregulation of oncogenes are involved in cancer development, progression and drug resistance [103, 104]. Recent studies have revealed a dynamic interplay between epigenetic modulation and host antitumor immunity, laying the groundwork for the combination of epigenetic therapies and immunotherapy.
Epigenetic regulation of cancer immunity, which concerns both cancer cells and immune cells, includes downregulation of genes expression involved in recognition and eliminations of malignant cells by immune system, such as cytokine gene, MHC genes, and costimulatory genes [105, 106], as well as upregulation of genes involved in immunosuppressive pathways [107, 108]. Ergo, epigenetic therapies have the potential to render malignant cells more sensitive to immunosurveillance, as well as prime the host immune systems to respond to the immunotherapy.
A growing body of evidence suggests that epigenetic agents can heighten tumor immunogenicity by up-regulating the expression of tumor antigens, MHC molecules, and other molecules involved in antigen processing and presentation [109–111], sensitize tumor cells to NK group 2 member D (NKG2D)-mediated cytotoxicity of NK and T cells by inducing expression of MHC class I–related chain A and B (MICA/B) [112, 113], and reduce Treg cells and MDSCs [114, 115]. Correspondingly, HDAC inhibitors can boost the antitumor activity of immunotherapies in both lung cancer cell line [110] and murine model of various solid tumors [116–119]. However, it is noteworthy that epigenetic agents also exert immunosuppressive effects by augmenting Treg cell function, altering the Th1 and Th2 balance, and inhibiting proliferation and viability of lymphocytes [116, 120–122].
As to the effects on the expression of immune checkpoint molecules, it varies among different epigenetic agents. Preclinical studies have demonstrated that class I HDAC inhibitors, which target deacetylases mainly expressed in the nucleus, lead to upregulation of PD-L1 and PD-L2 in human and murine melanoma cell lines [123]. In contrast, the selective HDAC6 inhibitor rocilinostat (which belongs to class II HDAC inhibitors) or HDAC6-specific silencing is shown to cause the downregulation of PD-L1 in CLL patients [123]. In vitro and in vivo studies have shown that treatment with DNMT inhibitor decitabine gave rise to a dose-dependent upregulation of PD-1, PD-L1, PD-L2 and CTLA-4 expression [124]. Moreover, elevated expression of immune checkpoint genes was observed in patients resistant to DNMT inhibitor, suggesting its potential role in the development of resistance to demethylating agent [124]. A similar result was found that azacytidine (5-aza)-induced demethylation of PD-1 promoter was associated with heightened PD-1 expression and a significantly worse overall response rate [125]. Another study analyzing gene signatures in lung cancer samples has shown that 5-aza up-regulates genes involved in both tumor immune elimination and evasion. In particular, 5-aza up-regulates PD-L1 transcripts and protein expression, suggesting a superior combinatorial effect of the combined use of DNMT inhibitor and PD-L1 inhibitor [126].
Compatible with the aforementioned immune biology of epigenetic agents, combinatorial therapies of epigenetic agents and immunotherapy have shown promising results in preclinical studies. Combinations of CTLA-4 blocking antibody 9H10 with either 5-aza-2’-deoxycytidine (5-aza-CdR) [127] or the DNMT inhibitor guadecitabine [128] was shown to exert robust antitumor activity in syngeneic mouse models of mammary carcinoma TS/A and mesothelioma AB1. In another study on murine models of mammary and colorectal carcinoma, the addition of epigenetic-modulating drugs (5-aza and entinostat) to checkpoint inhibitors (anti-PD-1 and anti-CTLA-4 antibodies) remarkably improved treatment outcomes, curing more than 80% of the tumor-bearing mice. And function studies revealed that the improved antitumor activity might be partially due to the reduction of MDSCs [114].
Other targeted agents
Bortezomib is the first therapeutic proteasome inhibitor used in multiple myeloma [129]. While multiple mechanisms are likely to be involved, proteasome inhibitors are viewed as functioning mainly by inhibiting the degradation of proteins critical to growth inhibition and apoptosis, thereby allowing for activation of programmed cell death in malignant cells. Aside from the pro-apoptotic effect on tumor cells, bortezomib can sensitize tumor cells to NK cells by decreasing expression of peptide–MHC class I complexes [130] and increasing tumor cell surface expression of FAS and the TNF-related apoptosis-inducing ligand (TRAIL) receptor DR5 [131]. Furthermore, bortezomib may render tumor cells more susceptible to T cell-mediated cytolysis by inducing mitochondrial accumulation of NOXA and in turn potentiating the release of mitochondrial second mitochondria-derived activator of caspase (SMAC) in response to caspase-8 and granzyme B [132]. Accordingly, enhanced specific CTL response was described when combinatorial treatment with bortezomib and vaccine was administered [133].
Heat shock protein 90 (HSP90) is a major chaperone involved in maintaining correct folding of multiple client proteins, including several oncoproteins that regulate cell growth. HSP90 inhibitor disrupts the association between HSP90 and its client proteins, resulting in the degradation of client proteins [134]. As to the immunomodulatory effects, HSP90 inhibitor induces degradation of client proteins, which may be digested to short peptides and then be presented with MHC class I on cancer cells, thereby increasing tumor sensitivity to immune elimination [135, 136]. In addition, HSP90 inhibitors also augment the expression of natural killer group 2D (NKG2D) ligands, facilitating tumor recognition and elimination by NK cells [137, 138].
Considerations for the clinical development of combined therapy
Combination therapy may integrate the benefits of the high frequency of rapid tumor regressions achievable with targeted therapies with the durable responses induced by immunotherapy. However, some key considerations for such combinations include identifying predictive biomarkers, optimizing dosing regimen and schedule, and minimizing treatment-related toxicities.
First, the top of any treatment algorithms will be the selection of patients for initial therapy. Though it is well known that patients harboring specific genetic alterations respond to corresponding targeted agents, the predictive biomarkers for immunotherapy remain elusive. Moreover, as the effect on host anticancer immunity induced by targeted agents is still less characterized, combining or sequential use of targeted and immune therapy will further complicate the identification of biomarkers for determining patients who will benefit.
Then, optimization of the dosing regimen and schedule is needed to potentiate benefits. Would combining bring a greater survival benefit compared with sequencing of targeted and immune therapy? If sequencing administration is better, then what is the optimal sequence? Will immunotherapy before targeted agents be more beneficial or vice versa? Within the limited data concerning melanoma, ipilimumab given prior to a BRAF inhibitor appears to be more effective in BRAF mutation–positive melanoma. In a retrospective analysis, longer OS was observed when ipilimumab was given prior to a BRAF inhibitor (BRAFi) compared with BRAF inhibitor followed by ipilimumab, or with either agent alone [139]. However, there is a caveat: different criteria were used to select patients for initial ipilimumab or BRAFi therapy. Patients with a poorer prognosis, e.g., rapid disease progression or brain metastases, are more likely to receive upfront BRAFi treatment. The author suggested that the lower efficacy of ipilimumab after BRAFi might be due to the reduction of the tumor melanocytic antigen expression and T-cell infiltration at progression. Another explanation for the worse prognosis of patients receiving initial BRAFi followed by ipilimumab might be that the group of patients, who generally have several unfavorable prognostic features prior to therapy, experience aggressive tumor growth and short survival after progressing on BRAFi therapy and therefore are hardly able to benefit from ipilimumab, which typically requires weeks or months to show response. Notwithstanding these limitations, the data suggested that first-line ipilimumab might be a preferred option for BRAF-mutant metastatic melanoma patients with low risk of rapid progressing disease. However, this is not the case for PD1 inhibitors. PD1 blockade therapy was shown to be associated with high response rates and a rapid tumor regression. Moreover, BRAFi treatment increases T-cell infiltration into the tumor and may upregulate PD-L1 expression, which may result in a tumor microenvironment more predisposed to respond to subsequent anti-PD-1 therapy following initial BRAFi [140, 141]. And recent studies lend support to this hypothesis. In an open-label, randomized phase III study, patients who progressed after both ipilimumab and a BRAFi treatment still had a higher response rate with nivolumab compared with chemotherapy [142]. Similar results were reported in a randomized phase II trial of pembrolizumab [143]. Hence, awareness should be raised that immunotherapeutic agents acting on different immune signaling pathway may have distinct biological and clinical implications when it comes to sequential using with targeted therapy.
Most of the data regarding the sequence of the use of immunotherapy and targeted agents hitherto is derived from studies in melanoma. However, the scenario could vary wildly in other cancer types. For instance, current evidence showed that ipilimumab is effective in both BRAF mutation–positive or wild patients. In contrast, non-small-cell lung carcinoma (NSCLC) harboring EGFR mutations or ALK rearrangements, as found in a recent study, are associated with low rates of concurrent PD-L1 expression and CD8+ T-cells pre- and post-targeted treatment, as well as low response rates to PD-1/PD-L1 inhibitors, which argues against both combinational and sequential use of targeted agents and immune checkpoint inhibitors. In spite of discrepancies among various cancer types, it is anticipated that learnings obtained from melanoma will somehow apply to other tumor types as well.
Last but not least, avoiding toxicity associated with combinatorial therapy is crucial. A phase I trial evaluating concurrent vemurafenib and ipilimumab treatment in patients with BRAF V600 mutation–positive melanoma demonstrated dose-limiting hepatotoxicity and was terminated [144]. And it remains to be further investigated that if the hepatotoxicity resulted from an autoimmune damage caused by the combination of vemurafenib and ipilimumab-induced hyper-immunity, or a direct liver injury inflicted by the combing use of vemurafenib and ipilimumab. The result of this study highlights the importance of rigorous clinical trial aimed at evaluation and optimization of combined use of targeted and immune therapy in order to optimize the overall risk-benefit ratio.
CONCLUSIONS
It is increasingly clear that targeted agents, which are initially developed to inhibit tumor-intrinsic drivers of growth, have underappreciated but highly relevant effects on antitumor immunity. The aim of combined approaches is to bring the durable clinical benefit of immunotherapy along with the high response rates and rapid remission elicited by targeted therapy. To this end, future research should continue focusing on investigating the complex interplay between targeted agents and immunotherapy and optimizing parameters such as administration timing, dosage, and sequence that may maximize therapeutic index.
ACKNOWLEDGMENTS
None
CONFLICTS OF INTEREST
None
FUNDING
None
REFERENCES
1. Asić K. Dominant mechanisms of primary resistance differ from dominant mechanisms of secondary resistance to targeted therapies. Crit Rev in Oncol Hematol. 2016; 97:178–96. https://doi.org/10.1016/j.critrevonc.2015.08.004.
2. Park J, Kwon M, Shin EC. Immune checkpoint inhibitors for cancer treatment. Arch Pharm Res. 2016; 39:1577–87. https://doi.org/10.1007/s12272-016-0850-5.
3. Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, Joshua AM, Patnaik A, Hwu WJ, Weber JS, Gangadhar TC, Hersey P, Dronca R, et al. Association of Pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016; 315:1600-1609. https://doi.org/10.1001/jama.2016.4059.
4. Wargo JA, Reuben A, Cooper ZA, Oh KS, Sullivan RJ. Immune effects of chemotherapy, radiation, and targeted therapy and opportunities for combination with immunotherapy. Semin Oncol. 2015; 42:601–16. https://doi.org/10.1053/j.seminoncol.2015.05.007.
5. Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs. 2005; 16:797–803. https://doi.org/10.1097/01.cad.0000173476.67239.3b.
6. Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2014; 15:7–24. https://doi.org/10.1038/nrc3860.
7. Marshall NA, Galvin KC, Corcoran AM, Boon L, Higgs R, Mills KH. Immunotherapy with PI3K inhibitor and toll-like receptor agonist induces IFN-γ+IL-17+ polyfunctional T cells that mediate rejection of murine tumors. Cancer Res. 2012; 72:581–91. https://doi.org/10.1158/0008-5472.CAN-11-0307.
8. Okkenhaug K, Graupera M, Vanhaesebroeck B. Targeting PI3K in cancer: Impact on tumor cells, their protective stroma, angiogenesis, and immunotherapy. Cancer Discovery. 2016; 6:1090–1105. https://doi.org/10.1158/2159-8290.CD-16-0716.
9. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, Mischel PS, Stokoe D, Pieper RO. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007; 13:84–8. https://doi.org/10.1038/nm1517.
10. Noh KH, Kang TH, Kim JH, Pai SI, Lin KY, Hung CF, Wu TC, Kim TW. Activation of Akt as a mechanism for tumor immune evasion. Mol Ther. 2009; 17:439–47. https://doi.org/10.1038/mt.2008.255.
11. Hähnel PS, Thaler S, Antunes E, Huber C, Theobald M, Schuler M. Targeting AKT signaling sensitizes cancer to cellular immunotherapy. Cancer Res. 2008; 68:3899–906. https://doi.org/10.1158/0008-5472.CAN-07-6286.
12. Kahan BD. The limitations of calcineurin and mTOR inhibitors: New directions for immunosuppressive strategies. Transplant Proc. 2002; 34:130–3. https://doi.org/10.1016/S0041-1345(01)02702-6.
13. Srivastava RK, Utley A, Shrikant PA. Rapamycin: A rheostat for CD8(+) T-cell-mediated tumor therapy. Oncoimmunology. 2012; 1:1189–90. https://doi.org/10.4161/onci.20663.
14. Li Q, Rao RR, Araki K, Pollizzi K, Odunsi K, Powell JD, Shrikant PA. A central role for mTOR kinase in homeostatic proliferation induced CD8+ T cell memory and tumor immunity. Immunity. 2011; 34:541–53. https://doi.org/10.1016/j.immuni.2011.04.006.
15. Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase eetermines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and eomesodermin. Immunity. 2010; 32:67–78. https://doi.org/10.1016/j.immuni.2009.10.010.
16. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009; 460:108–12. https://doi.org/10.1038/nature08155.
17. Li Q, Rao R, Vazzana J, Goedegebuure P, Odunsi K, Gillanders W, Shrikant PA. Regulating mammalian target of Rapamycin to tune vaccination-induced CD8+ T cell responses for tumor immunity. J Immunol. 2012; 188:3080–7. https://doi.org/10.4049/jimmunol.1103365.
18. Procaccini C, De Rosa V, Galgani M, Abanni L, Calì G, Porcellini A, Carbone F, Fontana S, Horvath TL, La Cava A, Matarese G. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010; 33:929–41. https://doi.org/10.1016/j.immuni.2010.11.024.
19. Wang Y, Camirand G, Lin Y, Froicu M, Deng S, Shlomchik WD, Lakkis FG, Rothstein DM. Regulatory T cells require mammalian target of Rapamycin signaling to maintain both homeostasis and alloantigen-driven proliferation in lymphocyte-replete mice. J Immunol. 2011; 186:2809–18. https://doi.org/10.4049/jimmunol.0903805.
20. Jiang Q, Weiss JM, Back T, Chan T, Ortaldo JR, Guichard S, Wiltrout RH. mTOR kinase inhibitor AZD8055 enhances the immunotherapeutic activity of an agonist CD40 antibody in cancer treatment. Cancer Res. 2011; 71:4074–84. https://doi.org/10.1158/0008-5472.CAN-10-3968.
21. Wang Y, Wang XY, Subjeck JR, Shrikant PA, Kim HL. Temsirolimus, an mTOR inhibitor, enhances anti-tumour effects of heat shock protein cancer vaccines. Br J Cancer. 2011; 104:643–52. https://doi.org/10.1038/bjc.2011.15.
22. Vogt PK, Kang S, Elsliger MA, Gymnopoulos M. Cancer-specific mutations in phosphatidylinositol 3-kinase. Trends Biochem Sci. 2007; 342–9. https://doi.org/10.1016/j.tibs.2007.05.005.
23. Sowa Y, Sakai T. [Ras/Raf/MEK/ERK cascade]. [Article in Japanese]. Nihon Rinsho. 2012; 70:63–6.
24. Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012; 16:103–19. https://doi.org/10.1517/14728222.2011.645805.
25. Tsukamoto H, Irie A, Senju S, Hatzopoulos AK, Wojnowski L, Nishimura Y. B-Raf-mediated signaling pathway regulates T cell development. Eur J Immunol. 2008; 38:518–27. https://doi.org/10.1002/eji.200737430.
26. Janardhan SV, Praveen K, Marks R, Gajewski TF. Evidence implicating the Ras pathway in multiple CD28 costimulatory functions in CD4 + T cells. PLoS One. Department of Pathology, The University of Chicago, Chicago, Illinois, United States of America. 2011; 6: e24931. https://doi.org/10.1371/journal.pone.0024931.
27. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010; 464:427–30. https://doi.org/10.1038/nature08902.
28. Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, Morales T, Aliagas I, Liu B, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010; 464:431–5. https://doi.org/10.1038/nature08833.
29. Robert C, Arnault JP, Mateus C. RAF inhibition and induction of cutaneous squamous cell carcinoma. Curr Opin Oncol. 2011; 23:177–82. https://doi.org/10.1097/CCO.0b013e3283436e8c.
30. Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, Kong X, Koya RC, Flaherty KT, Chapman PB, Kim MJ, Hayward R, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012; 366:207–15. https://doi.org/10.1056/NEJMoa1105358.
31. Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, Minasyan A, Graham NA, Graeber TG, Chodon T, Ribas A. BRAF inhibitor vemurafenib improves the antitimour activity of adoptive cell immunotherapy. Cancer Res. 2012; 72:3928–37. https://doi.org/10.1158/0008-5472.CAN-11-2837.
32. Ebert PJ, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, Gould SE, Maecker H, Irving BA, Kim JM, Belvin M, Mellman I. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity. 2016; 44:609–21. https://doi.org/10.1016/j.immuni.2016.01.024.
33. Boni A, Cogdill AP, Dang P, Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CJ, Sloss CM. Selective BRAF V600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010; 70:5213–9. https://doi.org/10.1158/0008-5472.CAN-10-0118.
34. Kono M, Dunn IS, Durda PJ, Butera D, Rose LB, Haggerty TJ, Benson EM, Kurnick JT. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol Cancer Res. 2006; 4:779–92. https://doi.org/10.1158/1541-7786.MCR-06-0077.
35. Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, Peng W, Sullivan RJ, Lawrence DP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013; 19:1225–31. https://doi.org/10.1158/1078-0432.CCR-12-1630.
36. Sapkota B, Hill CE, Pollack BP. Vemurafenib enhances MHC induction in BRAF(V600E) homozygous melanoma cells. Oncoimmunology. 2013; 2: e22890. https://doi.org/10.4161/onci.22890.
37. Ott PA, Henry T, Baranda SJ, Frleta D, Manches O, Bogunovic D, Bhardwaj N. Inhibition of both BRAF and MEK in BRAFV600E mutant melanoma restores compromised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol Immunother. 2013; 62:811–22. https://doi.org/10.1007/s00262-012-1389-z.
38. Schilling B, Sucker A, Griewank K, Zhao F, Weide B, Görgens A, Giebel B, Schadendorf D, Paschen A. Vemurafenib reverses immunosuppression by myeloid derived suppressor cells. Int J Cancer. 2013; 133:1653–63. https://doi.org/10.1002/ijc.28168.
39. Liu C, Peng W, Xu C. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013; 19:393–403. https://doi.org/10.1158/1078-0432.CCR-12-1626.
40. Khalili JS, Liu S, Rodríguez-Cruz TG, Whittington M, Wardell S, Liu C, Zhang M, Cooper ZA, Frederick DT, Li Y, Zhang M, Joseph RW, Bernatchez C, et al. Oncogenic Braf(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012; 18:5329–40. https://doi.org/10.1158/1078-0432.CCR-12-1632.
41. Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, Haynes NM, Kinross K, Yagita H, Koya RC, Graeber TG, Ribas A, McArthur GA, Smyth MJ. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J Clin Invest. 2013; 123:1371–81. https://doi.org/10.1172/JCI66236DS1.
42. Kakavand H, Wilmott JS, Menzies AM, Vilain R, Haydu LE, Yearley JH, Thompson JF, Kefford RF, Hersey P, Long GV, Scolyer RA. PD-L1 expression and tumor-infiltrating lymphocytes define different subsets of MAPK inhibitor-treated melanoma patients. Clin Cancer Res. 2015; 21:3140–9. https://doi.org/10.1158/1078-0432.CCR-14-2023.
43. Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, Pinheiro EM, Koya RC, Graeber TG, Comin-Anduix B, Ribas A. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med. 2015; 7: 279ra41. https://doi.org/10.1126/scitranslmed.aaa4691.
44. Homet Moreno B, Mok S, Comin-Anduix B, Hu-Lieskovan S, Ribas A. Combined treatment with dabrafenib and trametinib with immune-stimulating antibodies for BRAF mutant melanoma. Oncoimmunology. 2016; 5: e1052212. https://doi.org/10.1080/2162402X.2015.1052212.
45. Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. 2013; 19:598–609. https://doi.org/10.1158/1078-0432.CCR-12-2731.
46. Pd- T, Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, Yang J, Seestaller-wehr L, Zhang S, Hopson C, Tsvetkov L, Jing J, et al. The BRAF and MEK inhibitors Dabrafenib and Trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res. 2015; 21:1639–51. https://doi.org/10.1158/1078-0432.CCR-14-2339.
47. Atefi M, Avramis E, Lassen A, Wong DJ, Robert L, Foulad D, Cerniglia M, Titz B, Chodon T, Graeber TG, Comin-Anduix B, Ribas A. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. 2014; 20:3446–57. https://doi.org/10.1158/1078-0432.CCR-13-2797.
48. Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011; 437:169–83.
49. Senger DR. Vascular endothelial growth factor: much more than an angiogenesis factor. Mol Biol Cell. 2010; 21:377–9. https://doi.org/10.1091/mbc.E09-07-0591.
50. Schoeffner DJ, Matheny SL, Akahane T, Factor V, Berry A, Merlino G, Thorgeirsson UP. VEGF contributes to mammary tumor growth in transgenic mice through paracrine and autocrine mechanisms. Lab Invest. 2005; 85:608–23. https://doi.org/10.1038/labinvest.3700258.
51. Mak P, Leav I, Pursell B, Bae D, Yang X, Taglienti CA, Gouvin LM, Sharma VM, Mercurio AM. ERbeta impedes prostate cancer EMT by destabilizing HIF-1alpha and inhibiting VEGF-mediated snail nuclear localization: implications for Gleason grading. Cancer Cell. 2010; 17:319–32. https://doi.org/10.1016/j.ccr.2010.02.030.
52. Beck B, Driessens G, Goossens S, Youssef KK, Kuchnio A, Caauwe A, Sotiropoulou PA, Loges S, Lapouge G, Candi A, Mascre G, Drogat B, Dekoninck S, et al. A vascular niche and a VEGF–Nrp1 loop regulate the initiation and stemness of skin tumours. Nature. 2011; 478:399–403. https://doi.org/10.1038/nature10525.
53. Cao Y, E G, Wang E, Pal K, Dutta SK, Bar-Sagi D, Mukhopadhyay D. VEGF exerts an angiogenesis-independent function in cancer cells to promote their malignant progression. Cancer Res. 2012; 72:3912–8. https://doi.org/10.1158/0008-5472.CAN-11-4058.
54. Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer. 2013; 13:871–82. https://doi.org/10.1038/nrc3627.
55. Yaqoob U, Cao S, Shergill U, Jagavelu K, Geng Z, Yin M, De Assuncao TM, Cao Y, Szabolcs A, Thorgeirsson S, Schwartz M, Yang JD, Ehman R, et al. Neuropilin-1 stimulates tumor growth by increasing fibronectin fibril assembly in the tumor microenvironment. Cancer Res. 2012; 72:4047–59. https://doi.org/10.1158/0008-5472.CAN-11-3907.
56. Peske JD, Woods AB, Engelhard VH. Control of CD8 T-cell infiltration into tumors by vasculature and microenvironment. Adv Cancer Res. 2015; 128:263–307. https://doi.org/10.1016/bs.acr.2015.05.001.
57. Bouma-Ter Steege JC, Baeten CI, Thijssen VL, Satijn SA, Verhoeven IC, Hillen HF, Wagstaff J, Griffioen AW. Angiogenic profile of breast carcinoma determines leukocyte infiltration. Clin Cancer Res. 2004; 10:7171–8. https://doi.org/10.1158/1078-0432.CCR-04-0742.
58. Dirkx AE, Oude Egbrink MG, Kuijpers MJ, van der Niet ST, Heijnen VV, Bouma-ter Steege JC, Wagstaff J, Griffioen AW. Tumor angiogenesis modulates leukocyte-vessel wall interactions in Vivo by reducing endothelial adhesion molecule expression. Cancer Res. 2003; 63:2322–9.
59. Alfaro C, Suarez N, Gonzalez A, Solano S, Erro L, Dubrot J, Palazon A, Hervas-Stubbs S, Gurpide A, Lopez-Picazo JM, Grande-Pulido E, Melero I, Perez-Gracia JL. Influence of bevacizumab, sunitinib and sorafenib as single agents or in combination on the inhibitory effects of VEGF on human dendritic cell differentiation from monocytes. Br J Cancer. 2009; 100:1111–9. https://doi.org/10.1038/sj.bjc.6604965.
60. Yang DH, Park JS, Jin CJ, Kang HK, Nam JH, Rhee JH, Kim YK, Chung SY, Choi SJ, Kim HJ, Chung IJ, Lee JJ. The dysfunction and abnormal signaling pathway of dendritic cells loaded by tumor antigen can be overcome by neutralizing VEGF in multiple myeloma. Leuk Res. 2009; 33:665–70. https://doi.org/10.1016/j.leukres.2008.09.006.
61. Gabrilovich D, Chen H, Girgis K. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996; 2:1096–103.
62. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, Latreche S, Bergaya S, Benhamouda N, Tanchot C, Stockmann C, Combe P, Berger A, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8 + T cells in tumors. J Exp Med. 2015; 212:139–48. https://doi.org/10.1084/jem.20140559.
63. Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, Dubreuil O, Carpentier AF, Tartour E, Taieb J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013; 73:539–49. https://doi.org/10.1158/0008-5472.CAN-12-2325.
64. Hansen W, Hutzler M, Abel S, Alter C, Stockmann C, Kliche S, Albert J, Sparwasser T, Sakaguchi S, Westendorf AM, Schadendorf D, Buer J, Helfrich I. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J Exp Med. 2012; 209:2001–16. https://doi.org/10.1084/jem.20111497.
65. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyteinfiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010; 70:6171–80.
66. Dirkx AE, oude Egbrink MG, Castermans K, van der Schaft DW, Thijssen VL, Dings RP, Kwee L, Mayo KH, Wagstaff J, Bouma-ter Steege JC, Griffioen AW. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J. 2006; 20:621–30. https://doi.org/10.1096/fj.05-4493com.
67. Yasuda S, Sho M, Yamato I, Yoshiji H, Wakatsuki K, Nishiwada S, Yagita H, Nakajima Y. Simultaneous blockade of programmed death 1 and vascular endothelial growth factor receptor 2 (VEGFR2) induces synergistic anti-tumour effect in vivo. Clin Exp Immunol. 2013; 172:500–6. https://doi.org/10.1111/cei.12069.
68. Osada T, Chong G, Tansik R, Hong T, Spector N, Kumar R, Hurwitz HI, Dev I, Nixon AB, Lyerly HK, Clay T, Morse MA. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol Immunother. 2008; 57:1115–24. https://doi.org/10.1007/s00262-007-0441-x.
69. Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, Schwartz M, Divino CM, Pan PY, Chen SH. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009; 69:2514–22. https://doi.org/10.1158/0008-5472.CAN-08-4709.
70. Avella DM, Li G, Schell TD, Liu D, Zhang SS, Lou X, Berg A, Kimchi ET, Tagaram HR, Yang Q, Shereef S, Garcia LS, Kester M, et al. Regression of established hepatocellular carcinoma is induced by chemoimmunotherapy in an orthotopic murine model. Hepatology. 2012; 55:141–52. https://doi.org/10.1002/hep.24652.
71. Bose A, Taylor JL, Alber S, Watkins SC, Garcia JA, Rini BI, Ko JS, Cohen PA, Finke JH, Storkus WJ. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int J Cancer. 2011; 129:2158–70. https://doi.org/10.1002/ijc.25863.
72. Liu XD, Hoang A, Zhou L, Kalra S, Yetil A, Sun M, Ding Z, Zhang X, Bai S, German P, Tamboli P, Rao P, Karam J a., et al. Resistance to antiangiogenic therapy is associated with an immunosuppressive tumor microenvironment in metastatic renal cell carcinoma. Cancer Immunol Res. 2015; 3:1017–29. https://doi.org/10.1158/2326-6066.CIR-14-0244.
73. Guislain A, Gadiot J, Kaiser A, Jordanova ES, Broeks A, Sanders J, van Boven H, de Gruijl TD, Haanen JB, Bex A, Blank CU. Sunitinib pretreatment improves tumor - infiltrating lymphocyte expansion by reduction in intratumoral content of myeloid - derived suppressor cells in human renal cell carcinoma. Cancer Immunol Immunother. 2015; 64:1241–50. https://doi.org/10.1007/s00262-015-1735-z.
74. Kujawski M, Zhang C, Herrmann A, Reckamp K, Scuto A, Jensen M, Deng J, Forman S, Figlin R, Yu H. Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res. 2010; 70:9599–610. https://doi.org/10.1158/0008-5472.CAN-10-1293.
75. Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, Zeng W, Giobbie-Hurder A, Atkins MB, Ibrahim N, Friedlander P, Flaherty KT, Murphy GF, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014; 2:632–42. https://doi.org/10.1158/2326-6066.CIR-14-0053.
76. Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, Hernandez G, Mier J, He X, Hodi FS, Denker M, Leveque V, Cañamero M, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016; 7: 12624. https://doi.org/10.1038/ncomms12624.
77. Hipp MM, Hilf N, Walter S, Werth D, Brauer KM, Radsak MP, Weinschenk T, Singh-Jasuja H, Brossart P. induction of primary immune responses Sorafenib , but not sunitinib , affects function of dendritic cells and induction of primary immune responses. Blood. 2012; 111:5610–20. https://doi.org/10.1182/blood-2007-02-075945.
78. Krusch M, Salih J, Schlicke M, Baessler T, Kampa KM, Mayer F, Salih HR. The kinase inhibitors sunitinib and sorafenib differentially affect NK cell antitumor reactivity in vitro. J Immunol. 2009; 183:8286–94. https://doi.org/10.4049/jimmunol.0902404.
79. Appel S, Rupf A, Weck MM, Schoor O, Bru TH, Weinschenk T, Gru F. Effects of imatinib on monocyte-derived dendritic cells are mediated by inhibition of nuclear factor- κ B and Akt signaling pathway. Clin Cancer Res. 2005; 11:1928–40.
80. Boissel N, Rousselot P, Raffoux E, Cayuela JM, Maarek O, Charron D, Degos L, Dombret H, Toubert A, Rea D. Defective blood dendritic cells in chronic myeloid leukemia correlate with high plasmatic VEGF and are not normalized by imatinib mesylate. Leukemia. 2004; 18:1656–61. https://doi.org/10.1038/sj.leu.2403474.
81. Dietz AB, Souan L, Knutson GJ, Bulur PA, Litzow MR, Vuk-Pavlovic S. Imatinib mesylate inhibits T-cell proliferation in vitro and delayed-type hypersensitivity in vivo. Blood. 2004; 104:1094–9. https://doi.org/10.1182/blood-2003-12-4266.
82. Seggewiss R, Loré K, Greiner E, Magnusson MK, Price DA, Douek DC, Dunbar CE, Wiestner A. Imatinib inhibits T-cell receptor – mediated T-cell proliferation and activation in a dose-dependent manner. Blood. 2005; 105:2473–9. https://doi.org/10.1182/blood-2004-07-2527.
83. Gao H, Lee BN, Talpaz M, Donato NJ, Cortes JE, Kantarjian HM, Reuben JM. Imatinib mesylate suppresses cytokine synthesis by activated CD4 T cells of patients with chronic myelogenous leukemia. Leukemia. 2005; 19:1905–11.
84. Oriss TB, Krishnamoorthy N, Ray P, Ray A. Dendritic cell c-kit signaling and adaptive immunity: implications for the upper airways. Curr Opin Allergy Clin Immuno. 2014; 14:7–12. https://doi.org/10.1097/ACI.0000000000000019.
85. Ray P, Krishnamoorthy N, Oriss TB, Ray A. Signaling of c-kit in dendritic cells influences adaptive immunity. Ann N Y Acad Sci. 2010; 1183:104–22. https://doi.org/10.1111/j.1749-6632.2009.05122.x.
86. Borg C, Terme M, Taïeb J, Ménard C, Flament C, Robert C, Maruyama K, Wakasugi H, Angevin E, Thielemans K, Le Cesne A, Chung-Scott V, Lazar V, et al. Novel mode of action of c-KIT tyrosin kinase inhibitors leading to NK cell dependent antitumor effects. J Clin Invest. 2004; 114:379–88.
87. Ménard C, Blay JY, Borg C, Michiels S, Ghiringhelli F, Robert C, Nonn C, Chaput N, Taïeb J, Delahaye NF, Flament C, Emile JF, Le Cesne A, Zitvogel L. Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res. 2009; 69:3563–9. https://doi.org/10.1158/0008-5472.CAN-08-3807.
88. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, Sorenson EC, Popow R, Ariyan C, Rossi F, Besmer P, Guo T, Antonescu CR, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011; 17:1094–100. https://doi.org/10.1038/nm.2438.
89. Larmonier N, Janikashvili N, LaCasse CJ, Larmonier CB, Cantrell J, Situ E, Lundeen T, Bonnotte B, Katsanis E. Imatinib mesylate inhibits CD4+ CD25+ regulatory T cell activity and enhances active immunotherapy against BCR-ABL- tumors. J Immunol. 2008; 181:6955–63. https://doi.org/181/10/6955.
90. Christiansson L, Söderlund S, Mangsbo S, Hjorth-Hansen H, Höglund M, Markevärn B, Richter J, Stenke L, Mustjoki S, Loskog A, Olsson-Strömberg U. The tyrosine kinase inhibitors imatinib and dasatinib reduce myeloid suppressor cells and release effector lymphocyte responses. Mol Cancer Ther. 2015; 14:1181–91. https://doi.org/10.1158/1535-7163.mct-14-0849.
91. Yang Y, Liu C, Peng W, Lizée G, Overwijk WW, Liu Y, Woodman SE, Hwu P. Antitumor T-cell responses contribute to the effects of dasatinib on c-KIT mutant murine mastocytoma and are potentiated by anti-OX40. Blood. 2012; 120:4533–43. https://doi.org/10.1182/blood-2012-02-407163.
92. Bocchia M, Gentili S, Abruzzese E, Fanelli A, Iuliano F, Tabilio A, Amabile M, Forconi F, Gozzetti A, Raspadori D, Amadori S, Lauria F. Effect of a p210 multipeptide vaccine associated with imatinib or interferon in patients with chronic myeloid leukaemia and persistent residual disease: A multicentre observational trial. Lancet. 2005; 365:657–62. https://doi.org/10.1016/S0140-6736(05)17945-8.
93. Cathcart K, Pinilla-Ibarz J, Korontsvit T, Schwartz J, Zakhaleva V, Papadopoulos EB, Scheinberg DA. A multivalent bcr-abl fusion peptide vaccination trial in patients with chronic myeloid leukemia. Blood. 2003; 103:1037–42.
94. Preudhomme C, Guilhot J, Nicolini FE, Guerci-Bresler A, Rigal-Huguet F, Maloisel F, Coiteux V, Gardembas M, Berthou C, Vekhoff A, Rea D, Jourdan E, Allard C, et al. Imatinib plus peginterferon alfa-2a in chronic myeloid leukemia. N Engl J Med. 2010; 363:2511–21. https://doi.org/10.1056/NEJMoa1004095.
95. Simonsson B, Gedde-Dahl T, Markevärn B, Remes K, Stentoft J, Almqvist A, Björeman M, Flogegård M, Koskenvesa P, Lindblom A, Malm C, Mustjoki S, Myhr-Eriksson K, et al. Combination of pegylated interferon-α2b with imatinib increases molecular response rates in patients with low or intermediate risk chronic myeloid leukemia. Blood. 2011; 118:3228–36. https://doi.org/10.1182/blood-2011-02-336685.
96. Hehlmann R, Lauseker M, Jung-Munkwitz S, Leitner A, Muller MC, Pletsch N, Proetel U, Haferlach C, Schlegelberger B, Balleisen L, Hanel M, Pfirrmann M, Krause SW, et al. Tolerability-adapted imatinib 800 mg/d versus 400 mg/d versus 400 mg/d plus interferon-alpha in newly diagnosed chronic myeloid leukemia. J Clin Oncol. 2011; 29:1634–42. https://doi.org/10.1200/JCO.2010.32.0598.
97. Cortes J, Quintas-Cardama A, Jones D, Ravandi F, Garcia-Manero G, Verstovsek S, Koller C, Hiteshew J, Shan J, O’Brien S, Kantarjian H. Immune modulation of minimal residual disease in early chronic phase chronic myelogenous leukemia: a randomized trial of frontline high-dose imatinib mesylate with or without pegylated interferon alpha-2b and granulocyte-macrophage colony-stimulating fact. Cancer. 2011; 117:572–80. https://doi.org/10.1002/cncr.25438.
98. Baccarani M, Martinelli G, Rosti G, Trabacchi E, Testoni N, Bassi S, Amabile M, Soverini S, Castagnetti F, Cilloni D, Izzo B, De Vivo A, Messa E, et al. Imatinib and pegylated human recombinant interferon- 2b in early chronic-phase chronic myeloid leukemia. Blood. 2004; 104:4245–51. https://doi.org/10.1182/blood-2004-03-0826.
99. Palandri F, Iacobucci I, Castagnetti F, Testoni N, Poerio A, Amabile M, Breccia M, Intermesoli T, Iuliano F, Rege-Cambrin G, Tiribelli M, Miglino M, Pane F, et al. Front-line treatment of Philadelphia positive chronic myeloid leukemia with imatinib and interferon-α: 5-year outcome. Haematologica. 2008; 93:770–4. https://doi.org/10.3324/haematol.12265.
100. Chen LL, Chen X, Choi H, Sang H, Chen LC, Zhang H, Gouw L, Andtbacka RH, Chan BK, Rodesch CK, Jimenez A, Cano P, Jones KA, et al. Exploiting antitumor immunity to overcome relapse and improve remission duration. Cancer Immunol Immunother. 2012; 61:1113–24. https://doi.org/10.1007/s00262-011-1185-1.
101. Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. 2014; 15:703–8. https://doi.org/10.1038/nrm3890.
102. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011; 21:381–95. https://doi.org/10.1038/cr.2011.22.
103. Sarkar S, Horn G, Moulton K, Oza A, Byler S. Cancer development, progression, and therapy: an epigenetic overview. Int J Mol Sci. 2013; 14:21087–113. https://doi.org/10.3390/ijms141021087.
104. Glasspool RM, Teodoridis JM, Brown R. Epigenetics as a mechanism driving polygenic clinical drug resistance. Br J Cancer. 2006; 94:1087–92. https://doi.org/10.1038/sj.bjc.6603024.
105. Tomasi TB, Magner WJ, Khan AN. Epigenetic regulation of immune escape genes in cancer. Cancer Immol Immother. 2006; 55:1159–84. https://doi.org/10.1007/s00262-006-0164-4.
106. Fonsatti E, Nicolay HJ, Sigalotti L, Calabrò L, Pezzani L, Colizzi F, Altomonte M, Guidoboni M, Marincola FM, Maio M. Functional up-regulation of human leukocyte antigen class I antigens expression by 5-aza-2'-deoxycytidine in cutaneous melanoma: immunotherapeutic implications. Clin Cancer Res. 2007; 13:3333–8. https://doi.org/10.1158/1078-0432.CCR-06-3091.
107. Sadun RE, Sachsman SM, Chen X, Christenson KW, Morris WZ, Hu P, Epstein AL. Immune signatures of murine and human cancers reveal unique mechanisms of tumor escape and new targets for cancer immunotherapy. Clin Cancer Res. 2007; 13:4016–25. https://doi.org/10.1158/1078-0432.CCR-07-0016.
108. Bhatia A, Kumar Y. Cellular and molecular mechanisms in cancer immune escape: a comprehensive review. Expert Rev Clin Immunol. 2014; 10:41–62. https://doi.org/10.1586/1744666X.2014.865519.
109. Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, Keiser N, Santaniello F, Tomasi TB. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000; 165:7017–24. https://doi.org/10.4049/jimmunol.165.12.7017.
110. Setiadi AF, Omilusik K, David MD, Seipp RP, Hartikainen J, Gopaul R, Choi KB, Jefferies WA. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008; 68:9601–7. https://doi.org/10.1158/0008-5472.CAN-07-5270.
111. Maeda T, Towatari M, Kosugi H, Saito H. Up-regulation of costimulatory/adhesion molecules by histone deacetylase inhibitors in acute myeloid leukemia cells. Blood. 2000; 96:3847–56.
112. Skov S, Pedersen MT, Andresen L, Straten PT, Woetmann A, Odum N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 2005; 65:11136–45. https://doi.org/10.1158/0008-5472.CAN-05-0599.
113. Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, Kaiser S, Smirnow I, Wagner A, Steinle A, Salih HR. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005; 65:6321–9.
114. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, Diaz LA Jr, Papadopoulos N, Kinzler KW, Vogelstein B, Zhou S. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014; 111:11774–9. https://doi.org/10.1073/pnas.1410626111.
115. Shen L, Ciesielski M, Ramakrishnan S, Miles KM, Ellis L, Sotomayor P, Shrikant P, Fenstermaker R, Pili R. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PLoS One. 2012; 7: e30815. https://doi.org/10.1371/journal.pone.0030815.
116. Kato Y, Yoshimura K, Shin T, Verheul H, Hammers H, Sanni TB, Salumbides BC, Van Erp K, Schulick R, Pili R. Synergistic in vivo antitumor effect of the histone deacetylase inhibitor MS-275 in combination with interleukin 2 in a murine model of renal cell carcinoma. Cell. 2007; 13:4538–46. https://doi.org/10.1158/1078-0432.CCR-07-0014.
117. Murakami T, Sato A, Chun NA, Hara M, Naito Y, Kobayashi Y, Kano Y, Ohtsuki M, Furukawa Y, Kobayashi E. Transcriptional modulation using HDACi depsipeptide promotes immune cell-mediated tumor destruction of murine B16 melanoma. J Invest Dermatol. 2008; 128:1506–16. https://doi.org/10.1038/sj.jid.5701216.
118. Vo DD1, Prins RM, Begley JL, Donahue TR, Morris LF, Bruhn KW, de la Rocha P, Yang MY, Mok S, Garban HJ, Craft N, Economou JS, Marincola FM, et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. 2009; 69:8693–9. https://doi.org/10.1158/0008-5472.CAN-09-1456.
119. Christiansen AJ, West A, Banks KM, Haynes NM, Teng MW, Smyth MJ, Johnstone RW. Eradication of solid tumors using histone deacetylase inhibitors combined with immune-stimulating antibodies. Proc Natl Acad Sci U S A. 2011; 108:4141–6. https://doi.org/10.1073/pnas.1011037108.
120. Johnson J, Pahuja A, Graham M, Hering B, Hancock WW, Pratima BP. Effects of histone deacetylase inhibitor SAHA on effector and FOXP3+regulatory T cells in rhesus macaques. Transplant Proc. 2008; 40:459–61. https://doi.org/10.1016/j.transproceed.2008.01.039.
121. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, Li B, Turka LA, Olson EN, Greene MI, Wells AD, Hancock WW. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007; 13:1299–307. https://doi.org/10.1038/nm1652.
122. Brogdon JL, Xu Y, Szabo SJ, An S, Buxton F, Cohen D, Huang Q. Histone deacetylase activities are required for innate immune cell control of Th1 but not Th2 effector cell function. Blood. 2007; 109:1123–30. https://doi.org/10.1182/blood-2006-04-019711.
123. Woods DM, Sodré AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy with PD-1 Blockade. Cancer Immunol Res. 2015; 3:1375–85. https://doi.org/10.1158/2326-6066.CIR-15-0077-T.
124. Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, Fang Z, Nguyen M, Pierce S, Wei Y, Parmar S, Cortes J, Kantarjian H, Garcia-Manero G. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014; 28:1280–1288. https://doi.org/10.1038/leu.2013.355.
125. Ørskov AD, Treppendahl MB, Skovbo A, Holm MS, Friis LS, Hokland M, Grønbæk K. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS / AML patients: A rationale for combined targeting of PD-1 and DNA methylation. Oncotarget. 2015; 6:9612–26. https://doi.org/10.18632/oncotarget.3324.
126. Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, Vancriekinge W, Demeyer T, Du Z, Parsana P, Rodgers K, Yen RW, Zahnow CA, et al. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget. 2013; 4:2067–79. https://doi.org/10.18632/oncotarget.1542.
127. Covre A, Coral S, Nicolay H, Parisi G, Fazio C, Colizzi F, Fratta E, Di Giacomo AM, Sigalotti L, Natali PG, Maio M. Antitumor activity of epigenetic immunomodulation combined with CTLA-4 blockade in syngeneic mouse models. Oncoimmunology. 2015; 4: e1019978. https://doi.org/10.1080/2162402X.2015.1019978.
128. Covre A, Coral S, Di Giacomo AM, Taverna P, Azab M, Maio M. Epigenetics meets immune checkpoints. Semin Oncol. 2015; 42:506–13.
129. Richardson PG, Mitsiades C, Hideshima T, Anderson KC. Bortezomib: proteasome inhibition as an effective anticancer therapy. Annu Rev Med. 2006; 57:33–47. https://doi.org/10.1146/annurev.med.57.042905.122625.
130. Shi J, Tricot GJ, Garg TK, Malaviarachchi PA, Szmania SM, Kellum RE, Storrie B, Mulder A, Shaughnessy JD Jr, Barlogie B, van Rhee F. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood. 2008; 111:1309–17. https://doi.org/10.1182/blood-2007-03-078535.
131. Hallett WH, Ames E, Motarjemi M, Barao I, Shanker A, Tamang DL, Sayers TJ, Hudig D, Murphy WJ. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol. 2008; 180:163–70. https://doi.org/10.4049/jimmunol.180.1.163.
132. Seeger JM, Schmidt P, Brinkmann K, Hombach AA, Coutelle O, Zigrino P, Wagner-Stippich D, Mauch C, Abken H, Krönke M, Kashkar H. The proteasome inhibitor bortezomib sensitizes melanoma cells toward adoptive CTL attack. Cancer Res. 2010; 70:1825–34. https://doi.org/10.1158/0008-5472.CAN-09-3175.
133. Tseng CW, Monie A, Wu CY, Huang B, Wang MC, Hung CF, Wu TC. Treatment with proteasome inhibitor bortezomib enhances antigen-specific CD8+ T-cell-mediated antitumor immunity induced by DNA vaccination. J Mol Med. 2008; 86:899–908. https://doi.org/10.1007/s00109-008-0370-y.
134. Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol [Internet]. Whitehead Institute for Biomedical Research, 9 Cambridge Center, Cambridge, Massachusetts 02142, USA.; 2010; 11:515–28. https://doi.org/10.1038/nrm2918.
135. Lin CC, Tu CF, Yen MC, Chen MC, Hsieh WJ, Chang WC, Chang WT, Lai MD. Inhibitor of heat-shock protein 90 enhances the antitumor effect of DNA vaccine targeting clients of heat-shock protein. Mol Ther. 2007; 15:404–10. https://doi.org/10.1038/sj.mt.6300014.
136. Kawabe M, Mandic M, Taylor JL, Vasquez CA, Wesa AK, Neckers LM, Storkus WJ. Heat shock protein 90 inhibitor 17-dimethylaminoethylamino-17-demethoxygeldanamycin enhances EphA2+ tumor cell recognition by specific CD8+ T cells. Cancer Res. 2009; 69:6995–7003. https://doi.org/10.1158/0008-5472.CAN-08-4511.
137. Böll B, Eltaib F, Reiners KS, von Tresckow B, Tawadros S, Simhadri VR, Burrows FJ, Lundgren K, Hansen HP, Engert A, von Strandmann EP. Heat shock protein 90 inhibitor BIIB021 (CNF2024) depletes NF-kappaB and sensitizes Hodgkin's lymphoma cells for natural killer cell-mediated cytotoxicity. Clin Cancer Res. 2009; 21:5108–16. https://doi.org/10.1158/1078-0432.CCR-09-0213.
138. Fionda C, Soriani A, Malgarini G, Iannitto ML, Santoni A, Cippitelli M. Heat shock protein-90 inhibitors increase MHC class I-related chain A and B ligand expression on multiple myeloma cells and their ability to trigger NK cell degranulation. J Immunol. 2009; 183:4385–94. https://doi.org/10.4049/jimmunol.0901797.
139. Ascierto PA, Simeone E, Giannarelli D, Grimaldi AM, Romano A, Mozzillo N. Sequencing of BRAF inhibitors and ipilimumab in patients with metastatic melanoma: a possible algorithm for clinical use. J Transl Med. 2012; 10: 107. https://doi.org/10.1186/1479-5876-10-107.
140. Callahan MK, Masters G, Pratilas CA, Ariyan C, Katz J, Kitano S, Russell V, Gordon RA, Vyas S, Yuan J, Gupta A, Wigginton JM, Rosen N, et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol Res. 2014; 2:70–9. https://doi.org/10.1158/2326-6066.CIR-13-0160.
141. Hersey P, Gallagher S. A focus on PD-L1 in human melanoma. Clin Cancer Res. 2013; 19:514–6. https://doi.org/10.1158/1078-0432.CCR-12-3312.
142. Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr, Lao CD, Linette GP, Thomas L, Lorigan P, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial.. Lancet Oncol. 2015; 2045:1–2.
143. Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, Hodi FS, Schachter J, Pavlick AC, Lewis KD, Cranmer LD, Blank CU, O'Day SJ, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015; 16:908–18.
144. Ribas A, Hodi FS, Callahan MK, Konto C, Wolchok JD. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013; 368:1365–6. https://doi.org/10.1056/NEJMc1301264.