
INTRODUCTION
Janus kinase 2 (JAK2) is a member of the non-receptor tyrosine kinase family and mediates intracellular signaling upon activation of cytokine receptors, which lack an intrinsic tyrosine kinase domain, such as cytokine receptor-like factor 2 (CRLF2). Ligand binding (e.g. TSLP for CLRF2) induces dimerization of cytokine receptors chains, resulting in activation of JAK proteins via cross-phosphorylation. JAKs activate signal transducers of transcription (STATs), which, upon dimerization, migrate to the nucleus and induce transcription of genes involved in differentiation and proliferation of hematopoietic cells [1].
JAK2 has seven homologous domains (JH1-JH7). The JH1 and JH2 domains are C-terminally located and comprise the catalytic kinase (JH1) and pseudokinase (JH2) domain. The JH2 domain has a dual regulatory function: exerting a negative regulatory effect on the kinase domain, and facilitating JAK2 activation upon receptor activation by ligand binding [2]. The JH3-JH4 domains share homology with Src homology 2 (SH2) domains and mediate protein-protein interactions. The N-terminal located JH6 and JH7 domains, also known as the FERM domain, are required for binding of JAK2 to cytokine receptors [1].
In pediatric BCP-ALL patients, gain of function mutations and translocations affecting JAK2 have been identified [3–16]. Genomic translocations of JAK2 have been observed in high-risk BCR-ABL1-like patients. For several of these fusion genes, constitutive JAK2 kinase activation has been demonstrated [12-14, 17]. Point mutations often occur in Down Syndrome ALL, mainly affect exon 16 (located in the pseudokinase domain), and functionally cooperate with overexpression of the type I cytokine receptor CRLF2 [6, 9, 10]. Indeed, requirement for the interaction of mutant JAK2 with a cytokine receptor was shown in cell lines models by several groups [6, 8, 9].
Mutations and translocations represent biologically distinct entities, but both are potential targets for precision medicines. JAK inhibitors were shown to be effective against mutant and translocated JAK2 in vitro [3, 5, 7, 8, 12, 13, 17, 18]. However, in vivo mouse studies show conflicting data and none has been reported to be curative [13, 18–22]. To date, clinical data with JAK inhibitors are scarce. The Children’s Oncology Group performed a phase 1 dosing study of the JAK inhibitor ruxolitinib, but no cases harboring JAK2 activating mutations or translocations were included [23].
Several papers have reported data with a focus on either fusion genes or mutations of JAK2, although often with small sample size or only in specific subtypes of BCP-ALL. Furthermore, most reports lack ex vivo efficacy data of JAK inhibitors in primary leukemic cells. To assess the clinical potential of JAK inhibitors in pediatric BCP-ALL, we performed a comprehensive study to determine the frequency and prognosis of JAK2 mutations and translocations among different subtypes of childhood BCP-ALL. Furthermore, the biological efficacy of the JAK inhibitors momelotinib and ruxolitinib was studied in primary leukemic cells harboring JAK2 aberrations, and the clonal stability of JAK2 mutations was investigated in ALL patient derived xenograft models. We show that JAK inhibitors are overall effective towards BCP-ALL cells, but also identified a number of limitations of JAK inhibitor therapy.
RESULTS
Frequency and type of JAK2 aberrations in BCP-ALL patients
JAK2 mutation status was analyzed in 461 newly diagnosed BCP-ALL cases representing all major subtypes seen in children, with a distribution that is comparable to the general pediatric BCP-ALL population. JAK2 exons 16, 20, 21 and 23 were examined by targeted amplicon sequencing at a median read depth of 673, 577, 711 and 944, respectively. Analyses revealed that 3.5% (16/461) of these BCP-ALL cases harbored JAK2 mutations, which were detected in 7.6% (6/79) of BCR-ABL1-like cases, 11.9% (8/67) of non-BCR-ABL1-like B-other cases, and 1.6% (2/124) of high hyperdiploid cases. No JAK2 mutations were detected in KMT2A-AFF1 (0/15), BCR-ABL1 (0/26), ETV6-RUNX1 (0/124) or TCF3-PBX1 (0/26) cases. The variant allele frequency (VAF) ranged from 1.0% to 56% (Figure 1A). Seven patients carried two different JAK2 mutations, and one patient even harbored three different JAK2 mutations. Mutations involved amino acid residue R683 in 13 of 16 mutated cases, which is an important amino acid for the JH2 domain mediated negative auto-regulation of JAK2 activity [24]. CRLF2 overexpression was detected in 87.5% (14/16) of these cases (Figure 1A, Supplementary Figure 1). One CRLF2 low expressing case harbored a subclonal JAK2 mutation, suggesting that CRLF2 overexpression might be subclonal as well. The other case harbored a JAK2R923H with a VAF of 50%, suggesting that this mutation in the kinase domain is not associated with CRLF2 overexpression.
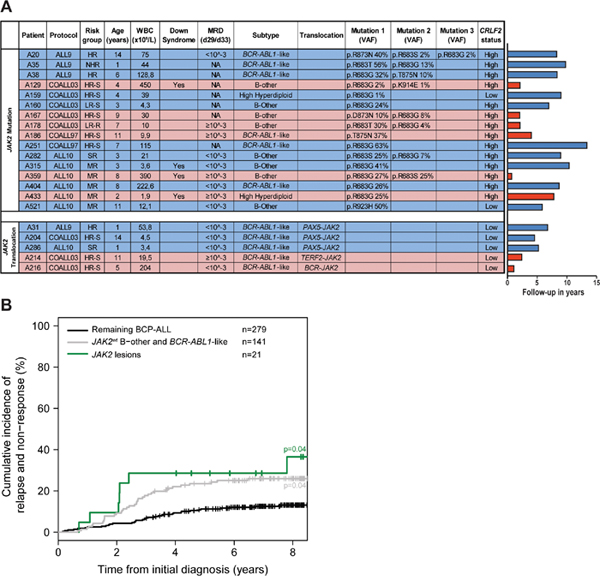
Figure 1: JAK2 aberrations in BCP-ALL patients. (A) Type of lesions, clinical characteristics and follow-up of JAK2 lesion positive patients. Treatment protocol and risk group assigned to each patient per protocol have been listed (HR-S: High Risk Standard. HR: High Risk. MR: Median Risk. SR: Standard Risk. LR-S: Low Risk Standard. LR-R: Low Risk Reduced. NHR: Non-High Risk). WBC indicates white blood cell count. Minimal residual disease (MRD) levels at day 29/33 of treatment of COALL and DCOG protocol, respectively. Type of translocation or mutation is listed. VAF indicates the variant allele frequency (%). CRLF2 status indicates gene expression below (low) or above (high) the 90th percentile levels. Right panel: Bar plot represents years from diagnosis to event or last contact. In blue: cases in complete clinical remission. In red: cases with an event (relapse or death). (B) Cumulative incidence of relapse curves for patients with JAK2 lesions (green line), JAK2 wildtype BCR-ABL1-like and B-other cases (grey line), and JAK2 wildtype remaining BCP-ALL cases (black line; ETV6-RUNX1, high hyperdiploid, TCF3-PBX1). Patients were treated according to ALL8, ALL9, ALL10, COALL97 or COALL03 protocol. Cumulative incidence of relapse (CIR) was estimated using a competing risk model. Relapse and non-response were considered as event, and death as competing event. Non-response was counted as event at day 79. The Gray’s test was applied to test for equality of CIRs (JAK2 lesion versus remaining BCP-ALL p=0.04; JAK2 wildtype B-other/BCR-ABL1-like versus remaining BCP-ALL p=0.04).
The screen for JAK2 fusion genes was confined to 153 BCP-ALL cases, negative for sentinel BCP-ALL associated lesions (KMT2A-rearranged, BCR-ABL1, ETV6-RUNX1, TCF-PBX1, high hyperdiploid), as JAK2 translocations were previously reported in this group of patients [12, 13, 25]. No JAK2 translocations were detected in 76 non-BCR-ABL1-like B-other cases, whereas in five of the 77 (6.5%) BCR-ABL1-like cases JAK2 tyrosine kinase activating fusion genes were identified. The cases involved three PAX5-JAK2 cases, one BCR-JAK2 case and one TERF2-JAK2 case (Figure 1A). The PAX5-JAK2 and BCR-JAK2 fusions contained identical exons as reported before [12, 13]. The TERF2-JAK2 case displayed an in frame fusion of TERF2 exon 10 to JAK2 exon 19. All JAK2 fusion genes harbored an intact JH1 kinase domain (Supplementary Figure 2). Gene expression data revealed high expression levels of JAK2 in these cases (Supplementary Figure 3). Absence of the cytokine receptor-binding FERM domain in JAK2 fusion protein suggests that they signal independent of a cytokine receptor.
Clinical characteristics and prognosis of patients harboring JAK2 lesions
Ten out of sixteen (62.5%) JAK2-mutated patients remained in continuous complete remission at more than 5 years of follow up. The median time to relapse in the six other patients was 2.1 years [range 0.71-7.8 years]. Minimal residual disease (MRD) data were available for nine patients. The four patients with high MRD levels (≥10-3) at day 29/33 of treatment (time point 1 of COALL and DCOG protocol, respectively) relapsed, whereas the remaining five mutated patients with low MRD levels remained in continuous complete remission (p=0.008, Fisher exact test).
Three out of the five cases harboring JAK2 fusion genes remained in continuous complete remission at more than 5 years of follow up, whereas two cases suffered from a relapse within 2.4 years of diagnosis (Figure 1A). Both patients who relapsed were assigned to the High Risk arm of the COALL-03 study protocol because of unfavorable age (>10 years) or high white blood cell count at diagnosis (>50 WBC/nl).
Cumulative incidence of relapse in these JAK2 aberrant patients did not differ from JAK2 wildtype BCR-ABL1-like and B-other cases. Both displayed an unfavorable outcome compared to remaining BCP-ALL cases (p=0.04; Figure 1B). These findings underline the clinical relevance of JAK2 lesions. Mutations and translocations represent biologically distinct entities, but both may be targetable by JAK-inhibitors.
Leukemic cells with JAK2 lesions can be targeted by JAK inhibitors
Primary leukemic and patient-derived-xenograft (PDX) cells (Supplementary Figure 4, [26]) were exposed to momelotinib and ruxolitinib. JAK2 translocated cells were more sensitive to both momelotinib and ruxolitinib compared to JAK2 wildtype cases (P<0.05; Figure 2A and 2B, Supplementary Figure 5. JAK2 mutated cells were less sensitive to these inhibitors than JAK2 fusion positive cells, and were only marginally more sensitive than wildtype cells (P<0.05). Leukemic cells without genetic JAK2 aberrations were resistant to ruxolitinib, but showed some sensitivity to momelotinib. Importantly, normal bone marrow mononuclear cells were resistant to both inhibitors (Supplementary Figure 5). Both JAK inhibitors effectively reduced levels of phosphorylated STAT5Y694 and/or STAT1Y701 (Figure 2C and 2E, Supplementary Figure 6).
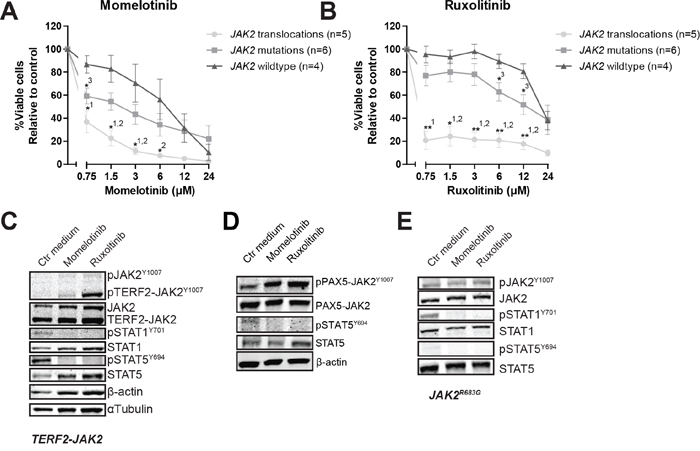
Figure 2: The efficacy of JAK inhibitors on JAK2 translocated and mutated cells. (A-B) Leukemic (PDX or primary patient) cells were incubated for four days with to an increasing concentration range of momelotinib or ruxolitinib, after which cell viability was measured using an MTT assay. Sensitivity of exposed cells was calculated relative to vehicle treated controls. Individual samples were tested in duplicate. Mean±SEM of JAK2 translocated cases (n=5), JAK2 mutated cases (n=6) and JAK2 wildtype cases (n=4) is shown. Cell viability of samples was compared using independent sample T-test. **p≤0.01, *p≤0.05, 1JAK2 translocations versus JAK2 wildtype,2JAK2 translocations versus JAK2 mutations,3JAK2 mutations versus JAK2 wildtype. (C-E) TERF2-JAK2, PAX5-JAK2 and JAK2R683G PDX cells were exposed for four hours to vehicle control medium, 1.5 μM momelotinib or 0.75 μM ruxolitinib, after which (phosphorylated) TERF2-JAK2, JAK2, STAT1 and STAT5 levels were analyzed using western blot (25 μg lysate).
The marginal sensitivity for both inhibitors and the low levels of phosphorylated STAT5 in JAK2 mutated cells may be explained by lack of human TSLP ligand to activate the CRLF2 pathway in our culture conditions. Addition of human TSLP sensitized JAK2 mutated cells to ruxolitinib, but not to momelotinib (P<0.01, Figure 3A and 3B, Supplementary Figures 7 and 8). TSLP exposure did not change the efficacy of JAK inhibitors in JAK2-fusion positive cells, confirming cytokine-independent signaling (Figure 3C and 3D, Supplementary Figure 7). JAK2 wildtype leukemic cells were not sensitized to JAK inhibitors by TSLP treatment (Figure 3E and 3F, Supplementary Figure 7). In the presence of TSLP both JAK2 translocated and JAK2 mutated cells were sensitive to JAK inhibitors (Figure 3G and 3H). At the protein level, TSLP exposure upregulated the levels of phosphorylated STAT1Y701 and STAT5Y694 in JAK2R683S mutated cells, whereas no effect was observed in JAK2 fusion positive and JAK2 wildtype leukemic cells (Figure 3I-3K, Supplementary Figure 9). Notably, TSLP triggered the phosphorylation and hence activation of the MEK/ERK pathway in JAK2R683S mutated cells, but not in JAK2R683G mutated cells (Figure 3I and 3L), suggesting that this activation is context-dependent. Phosphorylation of STAT1Y701 and STAT5Y694 was inhibited by momelotinib and ruxolitinib (Figure 3L).
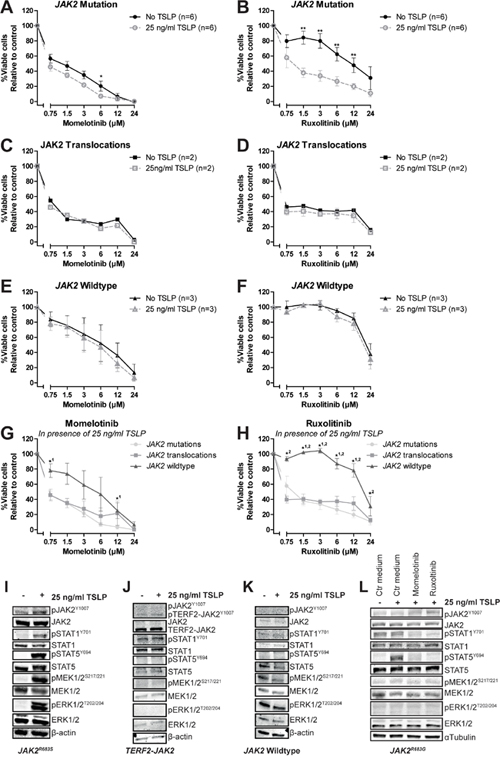
Figure 3: The effect of TSLP stimulation on the efficacy of JAK inhibitors. Cells (PDX or primary ALL) were pre-incubated for 1 hour with or without 25 ng/ml TSLP, after which cells were exposed for four days to indicated concentrations of momelotinib or ruxolitinib. Cell viability was measured using an MTT assay. Sensitivity was calculated relative to vehicle treated controls. Individual samples were tested in duplicate. (A-B) Efficacy of momelotinib and ruxolitinib on JAK2 mutated cells with or without TSLP pre-incubation. Mean±SEM of six independent samples is shown. (C-D) Efficacy of momelotinib and ruxolitinib on cells with JAK2 translocations. Mean±SEM of two independent samples is shown. (E-F) Efficacy of momelotinib and ruxolitinib on JAK2 wildtype PDX cells. Mean±SEM of three independent samples is shown. (G-H) Combined graph of the efficacy of momelotinib (G) and ruxolitinib (H) on TSLP stimulated cells with JAK2 mutations (n=6), JAK2 translocations (n=2), or JAK2 wildtype cells (n=3). Mean±SEM of independent samples is shown. Cell viability of samples was compared using the independent sample T-test. **p≤0.01, *p≤0.05. 1JAK2 translocations versus JAK2 wildtype,2JAK2 wildtype versus JAK2 mutations. (I-K) Western blot of JAK2R683S, TERF2-JAK2 and JAK2wt PDX cells with or without TSLP stimulation (25 ng/ml for 1 hour). (L) JAK2R683G cells were pre-incubated for 1 hour with or without 25 ng/ml TSLP, after which cells were exposed for four hours to vehicle control medium, 1.5 μM momelotinib or 0.75 μM ruxolitinib. Levels of (phosphorylated) JAK2, STAT1, STAT5, MEK1/2 and ERK1/2 were analyzed using western blot.
JAK2 inhibition results in accumulation of phosphorylated JAK2
Exposure of primary leukemic cells, harboring TERF2-JAK2 or PAX5-JAK2 translocations, to momelotinib and ruxolitinib resulted in accumulation of phosphorylated JAK2Y1007 fusion proteins (Figure 2C and 2D). Wash out of both inhibitors induced a slight rebound effect with upregulation of pSTAT1Y701 and pSTAT5Y694 in TERF2-JAK2 cells (Figure 4A, Supplementary Figure 10). This rebound effect was also observed in the JAK2V617F-positive leukemic cell line HEL (Supplementary Figures 11 and 12). Phosphorylated JAK2Y1007 accumulated upon exposure to ruxolitinib. Removal of the inhibitor resulted in reactivation of JAK2 signaling, observed by a clear increase in phosphorylated STAT5Y694 levels within 4 hours (time point 100 hours; Supplementary Figure 11A and 11B). The inhibitory effect of momelotinib was more transient compared to ruxolitinib, resulting in an earlier reactivation of JAK2, observed by high levels of phosphorylated STAT5Y694 after 48 hours of momelotinib exposure (Supplementary Figure 11A).
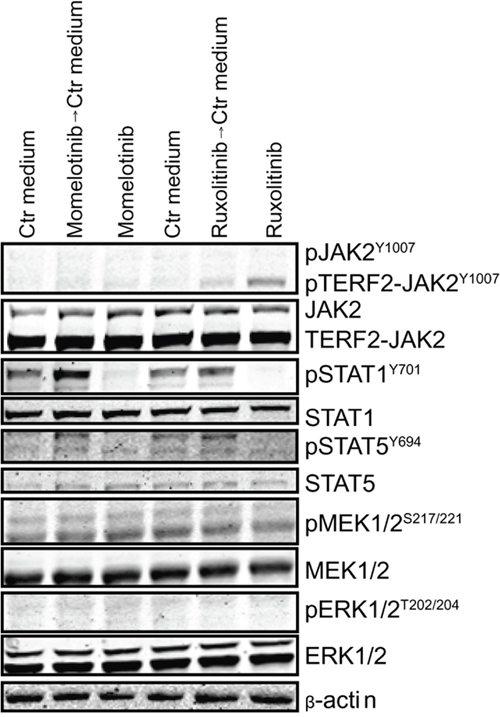
Figure 4: Accumulation of pJAK2Y1007 results in a rebound effect of JAK2. TERF2-JAK2 PDX cells were incubated for four hours with or without 1.5 μM momelotinib or 0.75 μM ruxolitinib, after which cells were washed to remove the JAK inhibitors. Half of the cells were exposed for another 1.5 hours to 1.5 μM momelotinib or 0.75 μM ruxolitinib, whereas the other cells were incubated in vehicle control (Ctr) medium. Protein expression levels were examined by western blot (25 μg lysate).
Mesenchymal stromal cells protect against JAK inhibitors
Leukemic cells reside in the bone marrow microenvironment, where they disrupt normal hematopoietic stem cell niches [27]. This abnormal niche protects ALL cells against chemotherapy [28, 29]. To study whether the bone marrow microenvironment protects against JAK inhibitors, we mimicked this niche by co-culturing PDX cells with bone marrow mesenchymal stromal cells (MSCs) derived from a leukemia patient. Survival of leukemic cells was improved in co-cultures together with MSCs compared to leukemic cells cultured without MSCs (Figure 5A). In these PDX/MSC co-cultures, JAK inhibitors decreased leukemic cell survival (Supplementary Figure 13A-13G). However, leukemic cells were more resistant to ruxolitinib in PDX/MSC co-culture compared to culture without MSCs (P<0.05). A similar trend was observed for momelotinib (Figure 5B and 5C, Supplementary Figure 13H and 13I).
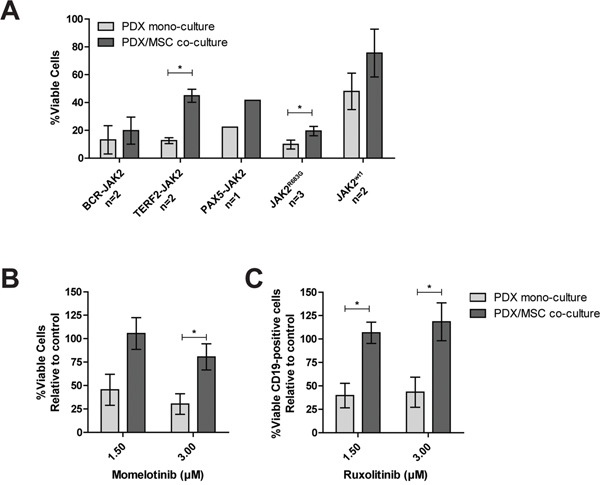
Figure 5: Efficacy of JAK inhibitors in co-culture. The response of CD19+ PDX cells, co-cultured with MSCs (hCD19-), to increasing concentrations of momelotinib and ruxolitinib, was assessed after four days of culture using flow cytometry. Cells were stained with Brilliant Violet 421 anti-human CD19 antibody, FITC Annexin V, and PI. Viability was calculated relative to vehicle controls. (A) Survival of PDX cells in mono-culture, or PDX/MSC co-culture. Individual samples were tested in duplicate. Bars represent the mean±SEM of two BCR-JAK2, two TERF2-JAK2, one PAX5-JAK2, three JAK2R683G and two JAK2wt samples. Cell viability of samples in mono-culture versus co-culture was compared using the independent sample T-test. *p≤0.05. (B-C) The effect of 1.5 μM and 3.0 μM momelotinib or ruxolitinib on the viability of PDX cells with JAK2 translocations in mono-culture, or in co-culture with primary MSCs. Mean±SEM of three JAK2 translocated samples is shown. Cell viability of samples was compared using independent sample T-test. *p≤0.05.
Different outgrowth pattern in xenografts
The outgrowth patterns of primary leukemic cells (>90% blast purity) in three NSG mice per patient was determined by paired-end deep-sequencing of JAK2 hot spot regions (exon 16, 20, 21 and 23; median read depth 554, 465, 411 and 593, respectively). PDX cells originating from a JAK2R863G mutated case had a different VAF profile compared to the original patient sample (Figure 6A, Supplementary Figure 14A). The primary sample contained a major JAK2R683G clone at a VAF of 63% and a minor KRASG12D clone at VAF 14%. In two out of three PDX samples generated, the VAF of the JAK2R683G mutation increased to 98% (PDX1) and 99% (PDX3), whereas in the remaining PDX sample the VAF decreased to 49% (PDX2). In contrast, the KRASG12D mutation increased to a VAF 23% in this PDX2 sample, whereas this mutation was not detected in PDX1 and PDX3. The reduced VAF of the JAK2 clone in PDX2 did not result in a decreased efficacy of momelotinib or ruxolitinib (Figure 6B). However, levels of pMEK1/2S217/221 and pERK1/2T202/204 in this sample were increased compared to the other two PDX samples (Figure 6C). Exposure to both JAK inhibitors did not decrease the levels of phosphorylated MEK and ERK.
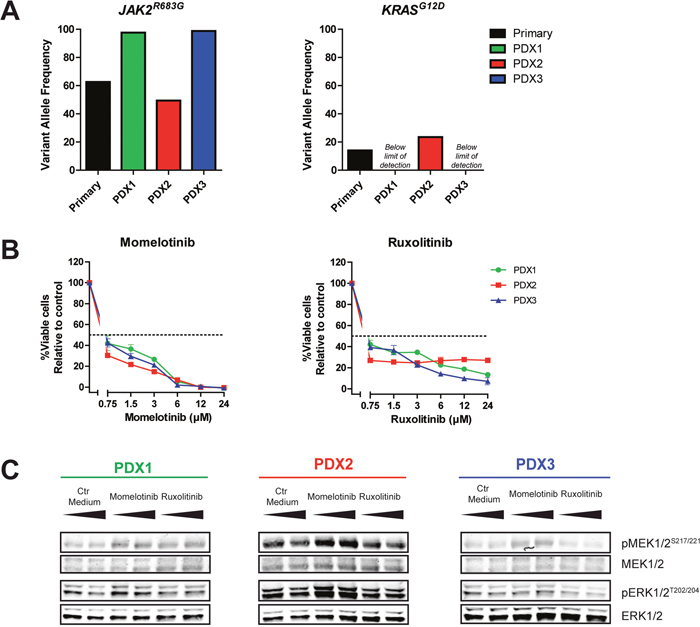
Figure 6: Outgrowth pattern of JAK2 mutated samples in xenografts. (A) VAF of JAK2R683G and KRASG12D in primary leukemic cells and three PDX samples of the same patient was determined using targeted amplicon sequencing. (B) PDX cells were pre-incubated for 1 hour with 25 ng/ml TSLP, after which they were exposed for four days to indicated concentrations of momelotinib or ruxolitinib in the presence of TSLP. Cell viability was measured using an MTT assay. Cell survival was calculated relative to vehicle treated controls. Samples were tested in duplicate. Mean±SD of each sample is shown. (C) Protein expression levels of PDX samples, exposed to 1.5 μM momelotinib or 0.75 μM ruxolitinib for four hours.
Sanger sequencing of JAK2R683S and JAK2R683T PDX models also indicated a change in the VAF of PDX cells compared to the primary sample. In PDX cells from three NSG mice injected with JAK2R683S cells, the A/T peak ratio at nucleotide position 2049 differed, suggesting heterogeneity in frequency of JAK2 mutations between samples (Supplementary Figure 14B). Strikingly, two mice injected with JAK2R683T mutated leukemic cells developed JAK2 wildtype leukemia (Supplementary Figure 14C). Although the JAK2 mutation was lost, CRLF2 expression levels remained high (Supplementary Figure 14D and 14E). TSLP stimulation activated the JAK2 pathway signaling (Supplementary Figure 14F), but cells were not sensitive for JAK2 inhibition (Supplementary Figure 14G and 14H).
DISCUSSION
This study aimed to evaluate the clinical need (frequency of lesions and prognostic value) and potential of JAK inhibitors in pediatric BCP-ALL. For this purpose pediatric BCP-ALL patients were screened for JAK2 lesions. JAK2 point mutations were found in 3.6% of our BCP-ALL patients, of which the majority were JAK2R683 mutations. These mutations were solely detected in BCR-ABL1-like, B-other and high hyperdiploid patients, but not in KMT2A-AFF1, BCR-ABL1, ETV6-RUNX1 or TCF3-PBX1 patients. JAK2 translocations were detected in the poor prognostic BCR-ABL1-like group, but not in non-BCR-ABL1-like B-other cases. The prognosis of patients with JAK2 aberrations was as poor as JAK2 wildtype BCR-ABL1-like and non-BCR-ABL1-like B-other patients.
JAK2 mutations were not detected as frequently in our DCOG/COALL sequencing cohort as reported for COG high-risk cohorts [5, 10]. Two independent classifiers are used to describe BCR-ABL1-like BCP-ALL [30–32]. Differences in genetic ancestry between the American COG and European DCOG/COALL cohorts likely affected the signatures to classify patients as BCR-ABL1-like. Hence, genetic differences, or more specifically the lack of Hispanic/Latino cases [10, 33], might explain the lower frequencies of JAK2 mutations, as well as the difference in treatment outcome.
A targeted approach was used to detect JAK2 translocations. All cases with high JAK2 expression levels harbored one of the known fusion genes, making it therefore unlikely that novel fusion genes were missed.
The poor outcome of cases harboring JAK2 lesions underline the clinical relevance of activated JAK2. The therapeutic potential of JAK2 was demonstrated by efficacy of JAK inhibitors in JAK2 mutated and JAK2 translocated primary BCP-ALL cells. JAK2 translocated cells were sensitive to both JAK inhibitors, independent of cytokine activation. In contrast, the efficacy of JAK inhibitors in JAK2 mutated cells highly depended on CRLF2 activation by TSLP, which is in concordance with previous reports [3, 5, 7, 8, 18]. These results highlight the importance of TSLP in in vitro and in vivo studies involving JAK2 mutations. Since human CRLF2 cannot be activated by mouse TSLP, conventional mouse xenograft models are not suitable to test the efficacy of ruxolitinib and other JAK inhibitors on JAK2 mutated cells. Absence of human TSLP in ALL xenograft mouse models might therefore explain the disappointing efficacy of JAK inhibitors in in vivo models [19–22]. The recently engineered xenograft model which synthesizes human TSLP may overcome this limitation [34].
Despite the need for human TSLP, we and others observed engraftment of primary JAK2 mutated cells in NSG mice, giving rise to leukemia within months after intra-femoral injection [19, 20, 22]. This implies that the proliferation of JAK2 mutated leukemic cells in the mouse bone marrow does not (solely) depend on CRLF2 signaling, but may be supported by activation of alternative pathways [35, 36]. This argues against targeted monotherapy, as cells might escape via an alternative pathway, such as the RAS pathway [36, 37]. This is further strengthened by a study in Down syndrome ALL, in which a switch was observed from a JAK2 mutation at initial diagnosis to a RAS-mutation at relapse [38]. In the current study, we also observed co-occurrence of JAK2 and KRAS lesions in non-Down syndrome BCP-ALL cells. More importantly, these JAK2 and KRAS mutations showed different outgrowth patterns in NSG xenograft models. Co-occurrence of these mutations suggests that a combination of JAK and RAS pathway inhibitors may be more effective for JAK2-aberrant cases. Moreover our results indicate that JAK2 mutations by itself might not be drivers of leukemic outgrowth, which is also corroborated by the outgrowth of JAK2 wildtype leukemic cells in mice injected with JAK2R683T cells. Taken together, these data imply that 1) the expansion properties of JAK2 mutated leukemic cells vary, 2) other subclones (e.g. KRASG12D) can grow out at the expense of the major JAK2 mutated clone, 3) JAK2 mutations can get lost while leukemia progresses. Therefore, JAK2 mutations are most likely secondary lesions, in contrast to JAK2 translocation, which are primary lesions. The selection of alternative pathways indicates a potential risk for use of JAK inhibitors in JAK2 mutated cases without detailed monitoring of the mutational status of leukemic cells in time.
Interestingly, we observed a difference in the cytotoxicity of momelotinib and ruxolitinib. Both agents fully inhibited JAK/STAT signaling by downregulation of phosphorylation levels of STAT1 and STAT5. However, only momelotinib affected JAK2 wildtype and non-TSLP stimulated JAK2 mutated cells, regardless of activation of STAT5. Therefore, it is likely that momelotinib also affects alternative targets [39, 40]. In addition, our data revealed that exposure to JAK inhibitors, especially ruxolitinib, resulted into accumulation of phosphorylated JAK2Y1007, which consequently induced a profound re-activation of downstream STAT signaling. Accumulation of phosphorylated JAK2Y1007 is in concordance with other reports [21, 41–43]. The rebound effect observed in the present study identifies an important limitation of these agents. As type I inhibitors bind JAK2 in its active conformation, reactivation of JAK2 may be overcome by type II inhibitors, which bind JAK2 in its inactive conformation [18, 44]. Preclinical in vitro and in vivo data indicate high efficacy of the type II inhibitor CHZ868. Clinical data of this inhibitor and other novel inhibitors are warranted.
Besides studying the mechanism of action, we showed influences of the bone marrow microenvironment on the efficacy of JAK inhibitors. Microenvironment-induced resistance of leukemic cells has been reported for conventional drugs, e.g. prednisolone and asparaginase, for imatinib in BCR-ABL1-positive cells, and for JAK inhibitors in the present study [45–49]. These data imply that the local tumor environment can stimulate a survival program, which may provide leukemic cells a way to escape from JAK inhibitors.
Taken together, JAK2 lesions are infrequently found in children with newly diagnosed BCP-ALL and are mainly restricted to BCR-ABL1-like and non-BCR-ABL1-like B-other cases (negative for sentinel cytogenetic lesions). Although mutations and translocations represent biologically distinct entities, both may be targetable by JAK inhibitors. Indeed, we showed efficacy of momelotinib and ruxolitinib in both cell types. As both momelotinib and ruxolitinib have a short half-life (less than half a day), the observed rebound effect may be therapeutically of risk [50, 51]. Although it should be confirmed by functional in vivo studies, our data suggest that the effect of JAK inhibition may be compromised by microenvironment-induced resistance and induction of alternative survival pathways. In conclusion, JAK inhibitors may be considered to be added to, but not substitute, chemotherapy for patients with JAK2 aberrations, especially for those at high risk of relapse due to high MRD levels upon induction therapy. Therapeutic monitoring for activation of alternative pathways (e.g. RAS) is highly recommended.
materials and METHODS
Primary patient-derived material
Bone marrow and/or peripheral blood samples were obtained from children (1-18 years) with newly diagnosed ALL. Written informed consent was obtained from parents or guardians to use excess diagnostic material for research purposes, as approved by the institutional review board. These studies were conducted in accordance with the Declaration of Helsinki. Mononuclear cells were isolated and processed as described previously [52]. Samples were enriched towards > 90% leukemic cells by depletion of non-leukemic cells using immunomagnetic beads. The major cytogenetic subtypes were determined using fluorescent in situ hybridization, (RT-) PCR, and the 110-probeset gene expression classifier [31]. Patients were treated according to the DCOG ALL8, ALL9 or ALL10 protocol, or the COALL-06-97 and COALL-07-03 study protocols [53–57]. Patient characteristics were provided by the central study centers of DCOG (The Hague, the Netherlands) and COALL (Hamburg, Germany).
Mesenchymal stromal cells (MSCs) were isolated from the bone marrow aspirate of a BCR-ABL1-like patient as described previously [58]. Purity of MSCs was assessed by negativity for the hematopoietic markers CD34, CD45, and CD19, and positivity for the mesenchymal markers CD13, CD29, CD44, CD54, CD73, CD90, CD105, CD146 and CD166, as detected by flow cytometry (MACS Quant). Expression was measured using the human mesenchymal stem cell marker antibody panel (R&D Systems, Minneapolis, Minnesota, USA), and CD13-APC, CD29-APC, CD34-PE, CD54-PE, CD73-PE, IgG1-PE, and IgG1-APC (BD Biosciences, San Jose, California, USA).
Cell lines
The human erythroleukemia cell line HEL was obtained from the German Collection of Microorganisms and Cell lines (DSMZ, Braunschweig, Germany) and cultured in RPMI-1640 medium, supplemented with 10% fetal calf serum (Bodinco BV, Alkmaar, Netherlands), 100 units/ml penicillin, 100 μg/ml streptomycin and 0.125 μg/mL fungizone (Life Technologies, Bleiswijk, Netherlands). The identity of the cell line was routinely verified by DNA fingerprinting. Additionally, presence of Mycoplasma was excluded every 25 passages by PCR.
Patient-derived-xenograft cells
Primary leukemic cells were transplanted by intra-femoral injection in 7-12 weeks old female NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (Charles River, Wilmington, Massachusetts, USA; three NSG mice per patient), as approved by the animal ethics committee (EMC 2863). Mice were sacrificed upon overt leukemia or 6 months after injection. Leukemic cells were isolated from bone marrow and spleen. The percentage of human leukemic cells was analyzed by flow cytometry (APC anti-human CD19, PE anti-human CD45 and FITC anti-mouse CD45; Biolegend, London, UK) and May-Grünwald-Giemsa staining. Patient-derived-xenograft cells (PDX) were enriched for > 90% human leukemic cells using anti-human-CD19 immunomagnetic beads (MACS Miltenyi, Leiden, Netherlands).
JAK2 mutations
JAK2 mutations status was analyzed in 461 BCP-ALL cases, representing the major cytogenetic subtypes (6% BCR-ABL1, 17% BCR-ABL1-like, 15% non-BCR-ABL1-like B-other, 27% ETV6-RUNX1, 27% high hyperdiploid, 3% KMT2A-AFF1, 6% TCF3-PBX1). Genomic DNA was isolated using Trizol reagent (Life Technologies), or in some cases using the DNeasy blood and tissue kit (Qiagen) according to manufacturer’s instructions, and quantified using the Qubit dsDNA Broad Range Assay Kit (Life Technologies). 100-250 ng genomic DNA was used to prepare sequencing libraries according to manufacturer’s instructions. Successful library preparation, correct amplicon length and concentration were assessed using the Labchip GX genomic analyzer (Caliper Life Sciences Benelux N.V.) with the HT DNA 12K Reagent Kit, Version 2. Samples were pooled equi-molarly and sequenced on an Illumina MiSeq in paired-end reads of 250bp each. The custom amplicons covered the exons 16, 20, 21 and 23 and represent the mutational hotspot region of JAK2.
Sequence reads were exported in fastq format and, aligned to the standard 1000 genomes human reference sequences (version b37, from the GATK resource bundle provided by the Broad institute, USA), using BWA version 0.7.10 and the GATK indel realigner version 3.3-0. Freebayes version 0.9.18-24, Varscan version 2.3.7, Bcftools version 1.0, and GATK version 3.3-0 were used to call single nucleotide variants. The resulting variant call format files were annotated using snpEff and snpSift version 4.1a and the dbNSFP version 2.7 database. Variants were combined and filtered based on several criteria using an in-house developed pipeline: variants were excluded if they were not located in targeted regions, were reported by only one caller, had coverage of < 100 reads, or had < 10 reads supporting the variant allele. Furthermore, variants had to occur at least once with a VAF above 2% in any sample and to be distributed equally between runs according to a chi-square test. Additionally only variants were taken into account if they were reported in the COSMIC V73 GRCh37 database, lead to an amino acid change, were unlikely to be a germline variant and not a known SNP. SNPs were defined as having a mean population frequency of ≥ 5% across the 1000Gp1 complete human population, 1000Gp1 population of European decent, and the ESP-6500 population of European decent allele frequency databases. Variants found in ≥ 10 samples with a mean and median VAF > 40% were labelled as possible germline. Sufficient coverage was crucial to detect subclonal mutations, which could be reliably detected ≥ 5% VAF in on average 91% of the cases. Detection of variants with a frequency of 1% was limited to a smaller group (26%), and is therefore a conservative estimate in the present study.
Genomic DNA of PDX cells was used to identify JAK2 mutations in exon 16. Samples were analyzed by Baseclear B.V. using Sanger sequencing (forward primer 5’-ATGCCTCCAAATTATTATACTATCA-3’, reverse primer 5’-ATCACCTCACAGTCCATGGTTAT-3’).
JAK2 translocations
Presence of ten recurrently reported JAK2 translocations was examined by RT-PCR in 153 BCP-ALL cases, negative for sentinel BCP-ALL associated lesions (n=77 BCR-ABL1-like, n=76 non-BCR-ABL1-like B-other). Total RNA was extracted from leukemic cells using Trizol reagents (Life Technologies). cDNA was synthesized using random hexamers and oligodT primers, and M-MLV reverse transcriptase (Promega, Leiden, Netherlands). RT-PCR was performed in a final volume of 25 μl, containing 0.3 μM forward primer, 0.3 μM reverse primer, 200 μM dNTPs, 1x PCR buffer II, 4 mM MgCl2, 0.125 μl AmpliTaq DNA polymerase (Promega) and 2.5 μl cDNA. Primer sequences are shown in Supplementary Table 1. PCR products were Sanger sequenced by Baseclear B.V. (Leiden, Netherlands).
CRLF2 status
CRLF2 expression levels were determined by Affymetrix gene expression microarrays (U133 Plus 2.0; Santa Clara, California, USA), of a previously published cohort of pediatric BCP-ALL patients at initial diagnosis [59]. High CRLF2 expression levels are indicative P2RY8-CRLF2 and IGH-CRLF2 translocations in different studies, though not all cases with high CRLF2 mRNA expression levels harbor CRLF2 rearrangements [10, 16, 59–61]. Of 406 cases, both CRLF2 gene expression data and JAK2 mutation status was available. Signal intensity of probeset 208303_s_at above the 90th percentile levels was classified as CRLF2 high, as described previously [59].
In addition, CRLF2 expression in primary leukemic and PDX samples was determined using RT-qPCR and SYBR green. CRLF2 expression (forward primer 5’-ACGGGGATCTCCTCTATG-3’, reverse primer: 5’-GAGGCGTTGG TGTCTCT-3’) was calculated relative to RSP20 expression (forward primer 5’-AAGGGCTGAGGATTTT TG-3’, reverse primer 5’-CGTTGCGGCTTGTTAG-3’), using the comparative cycle time (Ct) method; 2-DCt x 100%, whereby DCt = CtCRLF2 – CTRPS20. RT-qPCR expression values were correlated to Affymetrix microarray data of probeset 208303_s_at.
JAK2 expression levels
JAK2 expression levels were analyzed using Affymetrix gene expression microarrays, in a previously published pediatric BCP-ALL cohort [59]. Signal intensity of probeset 205841_at was used to quantify JAK2 gene expression.
Ex vivo drug cytotoxicity assays
Cell-intrinsic resistance towards momelotinib and ruxolitinib (Selleck Chemicals, Kirby Drive, Houston, USA) was evaluated as described previously [52]. Briefly, leukemic cells were exposed to a concentration range (24μM to 750 nM) of these compounds for four days and cytotoxicity was quantified using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). For TSLP stimulation, cells were pre-incubated for 1 hour with 25 ng/ml TSLP (R&D systems, Oxon, UK). In addition to single cell cultures, leukemic cells (1*106 cells) were co-cultured with primary MSCs (5*104 cells) for four days in a 24 well plate in the presence of a dilution series of momelotinib and ruxolitinib. Cell survival was quantified using flow cytometry (MACSQuant, FlowJo 10.0.8r1), and cells were stained with Brilliant Violet 421 anti-human CD19 antibody (Biolegend), FITC Annexin V (Biolegend), and Propidium Iodide (PI; Invitrogen, Bleiswijk, Netherlands), as described previously [45].
Western blotting
Leukemic cells were lysed in lysis buffer supplemented with freshly added protease and phosphatase inhibitors. 25μg (BCA method; Thermo Scientific) lysate was loaded on 10% mini protean precast gels (BioRad, Veenendaal, Netherlands), and transferred to a nitrocellulose membrane (Biorad). Primary antibody incubation was performed according to manufacturer’s protocol. Anti-JAK2 (#3230), anti-phospho-JAK2-Tyr1007 (#4406), anti-phospho-STAT5-Tyr694 (#9351), anti-phospho-STAT1-Tyr701 (#9167), anti-Stat1 (#9175), anti-phospho-MEK1/2-Ser217/221 (#9154), anti-MEK1/2 (#4694), anti-phospho-Erk1/2-T202/204 (#4370), anti-Erk1/2 (#91078), and anti-αTubulin (#2144) were obtained from Cell Signaling Technology (Danvers, Massachusetts, USA). Anti-β-actin (ab6276) was obtained from Abcam (Cambridge, UK), and anti-STAT5 (sc-835) from Santa Cruz (Heidelberg, Germany). Blots were stained with secondary antibodies (IRDye 680RD- or 800CW-labelled anti-rabbit and IRDye 680RD- or 800CW-labelled anti-mouse; Li-Cor Biosciences, Leusden, Netherlands) and scanned using the Odyssey infrared imaging system (Li-Cor Biosciences). To reprobe membranes, they were stripped in NewBlot Nitrocellulose stripping buffer (Li-Cor Biosciences) according to manufacturer’s protocol. BCR-JAK2, PAX5-JAK2 and TERF2-JAK2 proteins were separated from wildtype JAK2 based on size (~94, 57, 95 and 125 kDa, respectively).
Statistics
Cumulative incidence of relapse (CIR) was estimated using a competing risk model. Relapse and non-response (counted at day 79) were considered as event, and death as competing event. The Gray’s test was applied to test for equality of CIRs [62]. Outcome analyses were performed in R 3.0.1, using the packages cmprsk version 2.2-7 [63], mstate version 0.2.7 [64], and survival version 2.37-4 [65].
Abbreviations
BCP-ALL: B-cell precursor acute lymphoblastic leukemia; CIR: cumulative incidence of relapse; CRLF2: cytokine receptor-like factor 2; JAK2: Janus kinase 2; MSCs: mesenchymal stromal cells; MRD: minimal residual disease; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NSG: NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ; PDX: patient-derived-xenograft; STATs: signal transducers of transcription; VAF: variant allele frequency.
Author contributions
EMPS and ISJ designed and performed experiments, and analyzed and interpreted data. MJK designed deep sequencing primers. ISJ and NJMB performed deep sequencing. ISJ, AQH and JMB analyzed and interpreted deep sequence data. EMPS and WGN performed ex vivo experiments and analyzed data. EMPS, AB and CvdV collected PDX samples. EMPS and AB performed PCR reactions for Sanger sequencing analyses. RP, HAdGK, CMZ and MH provided clinical characteristics and clinical outcome data, and interpreted data. MLdB designed the study and interpreted data. EMPS, ISJ and MLdB drafted the manuscript. The manuscript was revised and approved by all authors.
ACKNOWLEDGMENTS AND FUNDING
We thank Ies Nijman, Annelies Smouters and Edwin Cuppen (University Medical Centre Utrecht) for their help in setting up the sequencing experiments and analysis pipeline.
This work was supported by the VICI program grant 016.126.612 from Netherlands Organization for Scientific Research (NWO), the Dutch Cancer Society grants AMC 2008-4265 and EMCR 2014-6998, the Kika Foundation (grant 132 and 161), the Pediatric Oncology Foundation Rotterdam (KOCR), and by the Centre for Personalized Cancer Treatment, which is a collaboration between all University Medical Cancer Centres and the Netherlands Cancer Institute Amsterdam and is supported by grants from KWF/Alpe d’Huzes and Nuts/Ohra.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Smith CA, Fan G. The saga of JAK2 mutations and translocations in hematologic disorders: pathogenesis, diagnostic and therapeutic prospects, and revised World Health Organization diagnostic criteria for myeloproliferative neoplasms. Hum Pathol. 2008; 39:795-810.
2. Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem. 2002; 277:47954-47963.
3. Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, Shochat C, Cazzaniga G, Biondi A, Basso G, Cario G, Schrappe M, Stanulla M, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet. 2008; 372:1484-1492.
4. Russell LJ, Capasso M, Vater I, Akasaka T, Bernard OA, Calasanz MJ, Chandrasekaran T, Chapiro E, Gesk S, Griffiths M, Guttery DS, Haferlach C, Harder L, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009; 114:2688-2698.
5. Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA, Tasian SK, Loh ML, Su X, Liu W, Devidas M, Atlas SR, Chen IM, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2009; 106:9414-9418.
6. Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, Ma J, Coustan-Smith E, Harvey RC, Willman CL, Mikhail FM, Meyer J, Carroll AJ, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009; 41:1243-1246.
7. Kearney L, Gonzalez De Castro D, Yeung J, Procter J, Horsley SW, Eguchi-Ishimae M, Bateman CM, Anderson K, Chaplin T, Young BD, Harrison CJ, Kempski H, So CW, et al. Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood. 2009; 113:646-648.
8. Yoda A, Yoda Y, Chiaretti S, Bar-Natan M, Mani K, Rodig SJ, West N, Xiao Y, Brown JR, Mitsiades C, Sattler M, Kutok JL, DeAngelo DJ, et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2010; 107:252-257.
9. Hertzberg L, Vendramini E, Ganmore I, Cazzaniga G, Schmitz M, Chalker J, Shiloh R, Iacobucci I, Shochat C, Zeligson S, Cario G, Stanulla M, Strehl S, et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood. 2010; 115:1006-1017.
10. Harvey RC, Mullighan CG, Chen IM, Wharton W, Mikhail FM, Carroll AJ, Kang H, Liu W, Dobbin KK, Smith MA, Carroll WL, Devidas M, Bowman WP, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010; 115:5312-5321.
11. Tirado CA, Chen W, Huang LJ, Laborde C, Hiemenz MC, Valdez F, Ho K, Winick N, Lou Z, Koduru P. Novel JAK2 rearrangement resulting from a t(9;22)(p24;q11.2) in B-acute lymphoblastic leukemia. Leuk Res. 2010; 34:1674-1676.
12. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, McCastlain K, Ding L, Lu C, Song G, Ma J, Becksfort J, Rusch M, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014; 371:1005-1015.
13. Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, Chen SC, Payne-Turner D, Churchman ML, Harvey RC, Chen X, Kasap C, Yan C, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012; 22:153-166.
14. Cuesta-Dominguez A, Ortega M, Ormazabal C, Santos-Roncero M, Galan-Diez M, Steegmann JL, Figuera A, Arranz E, Vizmanos JL, Bueren JA, Rio P, Fernandez-Ruiz E. Transforming and tumorigenic activity of JAK2 by fusion to BCR: molecular mechanisms of action of a novel BCR-JAK2 tyrosine-kinase. PLoS One. 2012; 7:e32451.
15. Boer JM, Steeghs EM, Marchante JR, Boeree A, Beaudoin JJ, Beverloo HB, Kuiper RP, Escherich G, van der Velden VH, van der Schoot CE, de Groot-Kruseman HA, Pieters R, den Boer ML. Tyrosine kinase fusion genes in pediatric BCR-ABL1-like acute lymphoblastic leukemia. Oncotarget. 2017; 8:4618-4628. https://doi.org/10.18632/oncotarget.13492.
16. Reshmi SC, Harvey RC, Roberts KG, Stonerock E, Smith A, Jenkins H, Chen IM, Valentine M, Liu Y, Li Y, Shao Y, Easton J, Payne-Turner D, et al. Targetable kinase gene fusions in high-risk B-ALL: a study from the Children's Oncology Group. Blood. 2017; 129:3352-3361.
17. Schinnerl D, Fortschegger K, Kauer M, Marchante JR, Kofler R, Den Boer ML, Strehl S. The role of the Janus-faced transcription factor PAX5-JAK2 in acute lymphoblastic leukemia. Blood. 2015; 125:1282-1291.
18. Wu SC, Li LS, Kopp N, Montero J, Chapuy B, Yoda A, Christie AL, Liu H, Christodoulou A, van Bodegom D, van der Zwet J, Layer JV, Tivey T, et al. Activity of the type II JAK2 inhibitor CHZ868 in B cell acute lymphoblastic leukemia. Cancer Cell. 2015; 28:29-41.
19. Suryani S, Bracken LS, Harvey RC, Sia KC, Carol H, Chen IM, Evans K, Dietrich PA, Roberts KG, Kurmasheva RT, Billups CA, Mullighan CG, Willman CL, et al. Evaluation of the in vitro and in vivo efficacy of the JAK inhibitor AZD1480 against JAK-mutated acute lymphoblastic leukemia. Mol Cancer Ther. 2015; 14:364-374.
20. Maude SL, Tasian SK, Vincent T, Hall JW, Sheen C, Roberts KG, Seif AE, Barrett DM, Chen IM, Collins JR, Mullighan CG, Hunger SP, Harvey RC, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2012; 120:3510-3518.
21. Weigert O, Lane AA, Bird L, Kopp N, Chapuy B, van Bodegom D, Toms AV, Marubayashi S, Christie AL, McKeown M, Paranal RM, Bradner JE, Yoda A, et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J Exp Med. 2012; 209:259-273.
22. Tasian SK, Teachey DT, Li Y, Shen F, Harvey RC, Chen IM, Ryan T, Vincent TL, Willman CL, Perl AE, Hunger SP, Loh ML, Carroll M, Grupp SA. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2017; 129:177-187.
23. Loh ML, Tasian SK, Rabin KR, Brown P, Magoon D, Reid JM, Chen X, Ahern CH, Weigel BJ, Blaney SM. A phase 1 dosing study of ruxolitinib in children with relapsed or refractory solid tumors, leukemias, or myeloproliferative neoplasms: a Children's Oncology Group phase 1 consortium study (ADVL1011). Pediatr Blood Cancer. 2015; 62:1717-1724.
24. Li F, Guo HY, Wang M, Geng HL, Bian MR, Cao J, Chen C, Zeng LY, Wang XY, Wu QY. The effects of R683S (G) genetic mutations on the JAK2 activity, structure and stability. Int J Biol Macromol. 2013; 60:186-195.
25. Imamura T, Kiyokawa N, Kato M, Imai C, Okamoto Y, Yano M, Ohki K, Yamashita Y, Kodama Y, Saito A, Mori M, Ishimaru S, Deguchi T, et al. Characterization of pediatric Philadelphia-negative B-cell precursor acute lymphoblastic leukemia with kinase fusions in Japan. Blood Cancer J. 2016; 6:e419.
26. Schmitz M, Breithaupt P, Scheidegger N, Cario G, Bonapace L, Meissner B, Mirkowska P, Tchinda J, Niggli FK, Stanulla M, Schrappe M, Schrauder A, Bornhauser BC, Bourquin JP. Xenografts of highly resistant leukemia recapitulate the clonal composition of the leukemogenic compartment. Blood. 2011; 118:1854-1864.
27. Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008; 322:1861-1865.
28. McMillin DW, Delmore J, Weisberg E, Negri JM, Geer DC, Klippel S, Mitsiades N, Schlossman RL, Munshi NC, Kung AL, Griffin JD, Richardson PG, Anderson KC, Mitsiades CS. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat Med. 2010; 16:483-489.
29. Lane SW, Scadden DT, Gilliland DG. The leukemic stem cell niche: current concepts and therapeutic opportunities. Blood. 2009; 114:1150-1157.
30. Boer JM, Marchante JR, Evans WE, Horstmann MA, Escherich G, Pieters R, Den Boer ML. BCR-ABL1-like cases in pediatric acute lymphoblastic leukemia: a comparison between DCOG/Erasmus MC and COG/St. Jude signatures. Haematologica. 2015; 100:e354-357.
31. Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, Van Zutven LJ, Beverloo HB, Van der Spek PJ, Escherich G, Horstmann MA, Janka-Schaub GE, Kamps WA, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009; 10:125-134.
32. Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, Ma J, Liu W, Cheng C, Schulman BA, Harvey RC, Chen IM, Clifford RJ, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009; 360:470-480.
33. Hicks C, Miele L, Koganti T, Young-Gaylor L, Rogers D, Vijayakumar V, Megason G. Analysis of patterns of gene expression variation within and between ethnic populations in pediatric B-ALL. Cancer Inform. 2013; 12:155-173.
34. Francis OL, Milford TA, Martinez SR, Baez I, Coats JS, Mayagoitia K, Concepcion KR, Ginelli E, Beldiman C, Benitez A, Weldon AJ, Arogyaswamy K, Shiraz P, et al. A novel xenograft model to study the role of TSLP-induced CRLF2 signals in normal and malignant human B lymphopoiesis. Haematologica. 2016; 101:417-426.
35. Ma X, Edmonson M, Yergeau D, Muzny DM, Hampton OA, Rusch M, Song G, Easton J, Harvey RC, Wheeler DA, Ma J, Doddapaneni H, Vadodaria B, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun. 2015; 6:6604.
36. Vesely C, Frech C, Eckert C, Cario G, Mecklenbrauker A, Zur Stadt U, Nebral K, Kraler F, Fischer S, Attarbaschi A, Schuster M, Bock C, Cave H, et al. Genomic and transcriptional landscape of P2RY8-CRLF2-positive childhood acute lymphoblastic leukemia. Leukemia. 2017; 31:1491-1501.
37. Winter PS, Sarosiek KA, Lin KH, Meggendorfer M, Schnittger S, Letai A, Wood KC. RAS signaling promotes resistance to JAK inhibitors by suppressing BAD-mediated apoptosis. Sci Signal. 2014; 7:ra122.
38. Nikolaev SI, Garieri M, Santoni F, Falconnet E, Ribaux P, Guipponi M, Murray A, Groet J, Giarin E, Basso G, Nizetic D, Antonarakis SE. Frequent cases of RAS-mutated Down syndrome acute lymphoblastic leukaemia lack JAK2 mutations. Nat Commun. 2014; 5:4654.
39. Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S, Moody SE, Shen RR, Schinzel AC, Thai TC, Reibel JB, Tamayo P, Godfrey JT, et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014; 4:452-465.
40. Asshoff M, Petzer V, Warr MR, Haschka D, Tymoszuk P, Demetz E, Seifert M, Posch W, Nairz M, Maciejewski P, Fowles P, Burns CJ, Smith G, et al. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood. 2017; 129:1823-1830.
41. Haan S, Wuller S, Kaczor J, Rolvering C, Nocker T, Behrmann I, Haan C. SOCS-mediated downregulation of mutant Jak2 (V617F, T875N and K539L) counteracts cytokine-independent signaling. Oncogene. 2009; 28:3069-3080.
42. Hart S, Goh KC, Novotny-Diermayr V, Hu CY, Hentze H, Tan YC, Madan B, Amalini C, Loh YK, Ong LC, William AD, Lee A, Poulsen A, et al. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia. 2011; 25:1751-1759.
43. Grandage VL, Everington T, Linch DC, Khwaja A. Go6976 is a potent inhibitor of the JAK 2 and FLT3 tyrosine kinases with significant activity in primary acute myeloid leukaemia cells. Br J Haematol. 2006; 135:303-316.
44. Meyer SC, Levine RL. Molecular pathways: molecular basis for sensitivity and resistance to JAK kinase inhibitors. Clin Cancer Res. 2014; 20:2051-2059.
45. Polak R, de Rooij B, Pieters R, den Boer ML. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood. 2015; 126:2404-2414.
46. Iwamoto S, Mihara K, Downing JR, Pui CH, Campana D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J Clin Invest. 2007; 117:1049-1057.
47. Vianello F, Villanova F, Tisato V, Lymperi S, Ho KK, Gomes AR, Marin D, Bonnet D, Apperley J, Lam EW, Dazzi F. Bone marrow mesenchymal stromal cells non-selectively protect chronic myeloid leukemia cells from imatinib-induced apoptosis via the CXCR4/CXCL12 axis. Haematologica. 2010; 95:1081-1089.
48. Feldhahn N, Arutyunyan A, Stoddart S, Zhang B, Schmidhuber S, Yi SJ, Kim YM, Groffen J, Heisterkamp N. Environment-mediated drug resistance in Bcr/Abl-positive acute lymphoblastic leukemia. Oncoimmunology. 2012; 1:618-629.
49. Ehsanipour EA, Sheng X, Behan JW, Wang X, Butturini A, Avramis VI, Mittelman SD. Adipocytes cause leukemia cell resistance to L-asparaginase via release of glutamine. Cancer Res. 2013; 73:2998-3006.
50. Shilling AD, Nedza FM, Emm T, Diamond S, McKeever E, Punwani N, Williams W, Arvanitis A, Galya LG, Li M, Shepard S, Rodgers J, Yue TY, Yeleswaram S. Metabolism, excretion, and pharmacokinetics of [14C]INCB018424, a selective Janus tyrosine kinase 1/2 inhibitor, in humans. Drug Metab Dispos. 2010; 38:2023-2031.
51. Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM, Druker BJ, Burns CJ, Fantino E, Deininger MW. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood. 2010; 115:5232-5240.
52. Den Boer ML, Harms DO, Pieters R, Kazemier KM, Gobel U, Korholz D, Graubner U, Haas RJ, Jorch N, Spaar HJ, Kaspers GJ, Kamps WA, Van der Does-Van den Berg A, et al. Patient stratification based on prednisolone-vincristine-asparaginase resistance profiles in children with acute lymphoblastic leukemia. J Clin Oncol. 2003; 21:3262-3268.
53. Escherich G, Zimmermann M, Janka-Schaub G; CoALL study group. Doxorubicin or daunorubicin given upfront in a therapeutic window are equally effective in children with newly diagnosed acute lymphoblastic leukemia. A randomized comparison in trial CoALL 07-03. Pediatr Blood Cancer. 2013; 60:254-257.
54. Veerman AJ, Kamps WA, van den Berg H, van den Berg E, Bokkerink JP, Bruin MC, van den Heuvel-Eibrink MM, Korbijn CM, Korthof ET, van der Pal K, Stijnen T, van Weel Sipman MH, van Weerden JF, et al. Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997-2004). Lancet Oncol. 2009; 10:957-966.
55. Escherich G, Troger A, Gobel U, Graubner U, Pekrun A, Jorch N, Kaspers G, Zimmermann M, zur Stadt U, Kazemier K, Pieters R, Den Boer ML, Horstmann M, et al. The long-term impact of in vitro drug sensitivity on risk stratification and treatment outcome in acute lymphoblastic leukemia of childhood (CoALL 06-97). Haematologica. 2011; 96:854-862.
56. Kamps WA, Bokkerink JP, Hakvoort-Cammel FG, Veerman AJ, Weening RS, van Wering ER, van Weerden JF, Hermans J, Slater R, van den Berg E, Kroes WG, van der Does-van den Berg A. BFM-oriented treatment for children with acute lymphoblastic leukemia without cranial irradiation and treatment reduction for standard risk patients: results of DCLSG protocol ALL-8 (1991-1996). Leukemia. 2002; 16:1099-1111.
57. Pieters R, de Groot-Kruseman H, Van der Velden V, Fiocco M, van den Berg H, de Bont E, Egeler RM, Hoogerbrugge P, Kaspers G, Van der Schoot E, De Haas V, Van Dongen J. Successful therapy reduction and intensification for childhood acute lymphoblastic leukemia based on minimal residual disease monitoring: study ALL10 from the dutch childhood oncology group. J Clin Oncol. 2016; 34:2591-2601.
58. van den Berk LC, van der Veer A, Willemse ME, Theeuwes MJ, Luijendijk MW, Tong WH, van der Sluis IM, Pieters R, den Boer ML. Disturbed CXCR4/CXCL12 axis in paediatric precursor B-cell acute lymphoblastic leukaemia. Br J Haematol. 2014; 166:240-249.
59. van der Veer A, Waanders E, Pieters R, Willemse ME, Van Reijmersdal SV, Russell LJ, Harrison CJ, Evans WE, van der Velden VH, Hoogerbrugge PM, Van Leeuwen F, Escherich G, Horstmann MA, et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood. 2013; 122:2622-2629.
60. Cario G, Zimmermann M, Romey R, Gesk S, Vater I, Harbott J, Schrauder A, Moericke A, Izraeli S, Akasaka T, Dyer MJ, Siebert R, Schrappe M, Stanulla M. Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BFM 2000 protocol. Blood. 2010; 115:5393-5397.
61. Chen IM, Harvey RC, Mullighan CG, Gastier-Foster J, Wharton W, Kang H, Borowitz MJ, Camitta BM, Carroll AJ, Devidas M, Pullen DJ, Payne-Turner D, Tasian SK, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's Oncology Group study. Blood. 2012; 119:3512-3522.
62. Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999; 94:496-509.
63. Gray RJ. cmprsk: Subdistribution Analysis of Competing Risks. R package version 22-6 2013; http://CRAN.R-project.org/package=cmprsk.
64. De Wreede LC, Fiocco M, Putter H. mstate: An R Package for the analysis of competing risks and multi-state models. J Stat Softw. 2011; 38:1-30.
65. Therneau T. A Package for Survival Analysis in S. R package version 236-12. 2012.