
INTRODUCTION
Ischemia/reperfusion (I/R) injury is a frequent event encountered in many pathophysiological situations, which results from complex interactions between reduced blood flow, production of reactive oxygen species (ROS) and activation of inflammatory processes that promote cell damage [1-3]. The initial sterile injury caused by I/R induces the activation of an innate immune response by pattern-recognition receptors. Among them, some members of the evolutionarily conserved family of Toll-like receptor (TLR) family, which recognize distinct microbial components and mediate innate immune responses for host defense [4,5], also play key roles in the initiation of inflammation and cell damages caused by I/R injury [2]. The immune hematopoietic cells have been considered to play predominant roles in the induction and control of the innate immune response. However, there is now growing lines of evidence that epithelial mucosal cells, including hepatocytes, intestinal cells, pulmonary cells, and renal tubule cells, or cardiomyocytes, exhibit some, but not all, of the TLRs, primarily identified in leukocytes [6-10]. In addition, epithelial cells stimulated by TLR agonists or pathogens may develop potent TLR-mediated inflammatory responses. These studies led to the emerging concept that epithelial cell, together with bone marrow-derived cells, are implicated in triggering an innate immune response to bacterial infection and/or ischemic stress [11,12].
TLR2 and TLR4 are constitutively expressed in bone marrow-derived cells and in a number of mucosal cells. A number of studies demonstrated that TLR4 play a key role in the mucosal inflammation caused by ischemia/reperfusion (I/R) in hepatic I/R [6,13], intestinal I/R [14,15], cardiac I/R [16], and hemorrhagic shock [17,18]. A number of studies have shown that liver, intestine or kidneys from TLR4 deficient mice are better protected than that of TLR4 expressing wild-type mice subjected to liver, intestinal or renal I/R injury [6,15,19]. TLR2 have also been reported to mediate inflammatory responses and apoptosis during renal I/R injury [20].
Like other TLRS (except TLR3), TLR2 and TLR4 exhibiting the co-adaptor MAL (or TIRAP) interact with adaptor myeloid differentiation factor 88 (MyD88). The recruitment of MyD88 facilitates the association of the Toll/IL-1R (TIR) domain with IL-1-receptor-associated kinases (IRAKs). Once phosphorylated, IRAKs become dissociated, and interact with TNF receptor-associated factor 6 (TRAF6) to activate TAK1, which then forms a complex with TK1 protein binding proteins, leading to the phosphorylation of the IκBs inhibitor proteins by IκB kinases (IKKs), their dissociation and subsequent degradation. This allows the transcription factor, NF-κB, to translocate into the nucleus [21,22]. Activation of TAK1 also stimulates mitogen-activated protein kinase (MAPK) pathways including extracellular signal-regulated kinase (ERK), p38 MAP kinase, and c-jun N-terminal kinase (JNK) [23-25]. MyD88 deficient mice were also shown to be better protected against renal I/R injury than wild-type mice [26]. Concerning the kidney, experiments performed on intact post-ichemic kidneys and primary cultures of renal tubule epithelial cells (RTEC) also revealed that TLR2 and TLR4 both drive the inflammatory response and apoptosis [19,20], suggesting that TLR2 and TLR4 are activated by endogenous ligands released by damaged cells to engage common or independent signaling pathways.
ROLE OF DAMAGE-ASSOCIATED MOLECULAR PATTERNS IN THE ACTIVATION OF TLR SIGNALING DURING I/R INJURY
The induction of non-bacterial, “sterile” inflammatory responses occurring during I/R results from the capacity of the so-called dammage-associated molecular patterns (DAMPs) to activate immune signaling pathways through TLR2 and/or 4. DAMPs can be classified as endogenous constituents released by damaged or necrotic cells, and as components of the extracellular matrix that are released by proteases [27]. A variety of DAMP constituents can activate TLR signaling. They include the high-mobility group box 1 (HMGB1) [28-30], heat shock proteins [31-33], fibrinogen [34], fibronectin [35], surfactant [36], β-defensin [37], uric acid [38], hyaluronan [39], and the matrix components biglycan and heparan sulfate [40,41]. Most of them were shown to activate TLR2 and TLR4. Genomic DNA from dying cells also induces the maturation of antigen-presenting cells, and self-mRNA released from necrotic cells activates TLR3 [42,43]. HMGB1, which is ubiquitously expressed in eukaryotic cells, is a member of the non-histone, chromatin-associated, high-mobility group (HMG) family of proteins [44]. HMGB1 is released by all cells in a context of necrotic cell death [45], and can also be secreted by macrophages or dendritic cells in response to LPS, interferon-γ, and TNF-α [46]. The role of HMGB1 in the activation of TLR-mediated inflammatory reponse has been extensively studied in the context of I/R. The release of HMGB1 from cultured hepatocytes was shown to require intact TLR4, and be regulated by reactive oxygen species (ROS). HMGB1 was also shown to activate downstream calcium-dependent signaling pathways, since inhibition of the calcium/calmodulin-dependent kinases (CMKs) reduces the release of HMGB1 induced by oxidative stress in cultured hepatocytes [6]. Conversely, inhibition of HMGB1 activity with a neutralising antibody markedly decreases the liver damage caused by I/R, whereas administration of recombinant HMGB1 worsened I/R-induced liver damages [6]. Similar observation have been made in experimental models of renal I/R: administration of blocking antibody to HMGB1 reduces renal tubule apoptosis and inflammatory responses caused by I/R, leading to the protection of renal function [47,48]. Interferon regulatory factor-1 (IRF-1) has been shown to contribute to the hepatocellular relase of HMGB1 by modulating the acetylation of HMGB1 through the nuclear histone acetyltransferase 330 [49]. Futhermore, the increase in the levels of acetyled HMGB1 is associated with concommitent decrease in nuclear histone deacetylase activity 1 and 4 [50], suggesting that the decrease in deacetylases 1 and 4 promotes the hyperacetylation and subsequent release of HMGB1 in hypoxic-stressed liver cells.
The signaling pathways activated downstream TLR2 and/or TLR4 which triggers pro-inflammatory responses and apoptosis are however less well known. The oxidative stress injury caused by I/R injury has been attributed to the activation of MAP kinase pathways, including those of ERK, JNK and p38 [50,51]. The activation of JNK/p38, via the activation of the Ser/Thr MAP kinase kinase, apoptosis signal-regulating kinase 1 (ASK1), plays a key role in cytokine- and stress-induced apoptosis [52-54]. The activation of ERK is thought to be involved in cell survival, although its activation has also been shown to be associated with drug-induced apoptosis [55-57]. Immunoblot analysis of total and phosphorylated ERK1/2, p38 and JNK MAP kinases in kidneys from wild-type, Tlr2-/- mice and Tlr4-/- mice subjected to I/R injury has revealed that I/R had almost no effect on p38 (not shown). I/R failed to activate JNK in both Tlr2-/- mice and Tlr4-/- mice, but in sharp contrast, differentially activated ERK1/2 (Fig. 1A), since I/R injury stimulated the expression of phosphorylated (p-) ERK1/2 in post-ischemic WT and Tlr4-/- kidneys, but not in Tlr2-/- kidneys [58]. IRI increased the expression level of TRAF2, and p-ASK1 and p-JNK, and a to much lesser extent p-p38 (not shown) in day-1 post-ischemic wild-type kidneys, but not in post-ischemic Tlr2-/- and Tlr4-/- kidneys (Fig. 1A). In addition, the pro-apoptotic Bcl-2 homologue BAX (but not Bcl-2) also significantly increased in wild-type, but not in Tlr2-/- (not shown) and Tlr4-/-post-ischemic kidneys (Fig. 1B). Fig. 1C provides a schematic representation of the differential activation of TLR2- and TLR4-mediated ERK1/2 and JNK signaling pathways activated during I/R. As a result, the number of TUNEL-positive apoptotic cells was significanltly less in in post-ischemic Tlr2-/-and Tlr4-/- kidneys than in wild-type kidneys (Fig. 1D) [19,20,58]. Overall, these findings have demonstrated that TLR2 selectively controls ERK1/2, and that both TLR2 and TLR4 mediates the activation of p-ASK1 and p-JNK, two key signal events responsible for apoptosis during IRI.
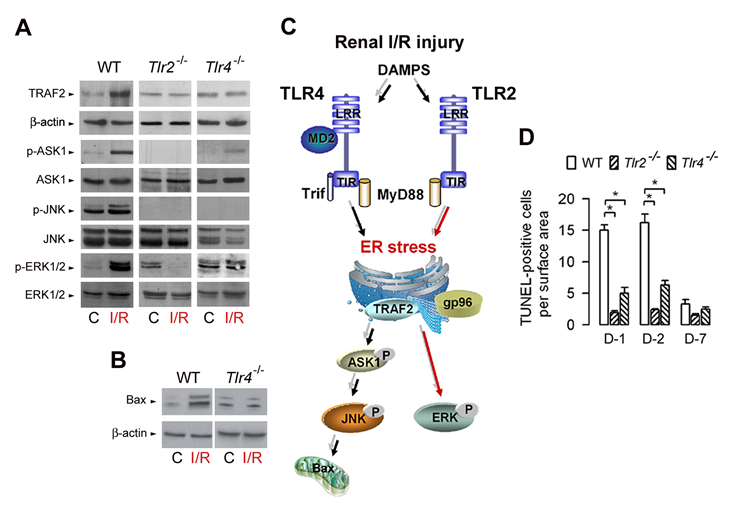
Figure 1: TLR2- and TLR4-mediated activation of ERK and JNK MAP kinases during renal ischemia/reperfusion injury. (A) Immunoblot analysis of TRAF2 and β-actin, phospho (p-) and total ASK1, JNK, and ERK1/2 in control (C), and day-1 post-ischemic (I/R) wild-type, Tlr2-/-, and Tlr4-/- kidneys. (B) Immunoblot analysis of BAX and β-actin in control (C), and day-1 post-ischemic (I/R) wild-type (WT) and Tlr4-/- kidneys. (C) Diagram depicting TLR2- and TLR4-mediated activation of ERK and JNK in kidneys subjected to I/R injury. (D) Number of apoptotic TUNEL-positive cells in non-ischemic (C) and day (D)-1, D-2, and D-7 post-ischemic WT, Tlr2-/- and Tlr4-/- kidneys. Values are means ± SE. * < p < 0.05. Redrawn from refs 58 and 78.
The following chapters will summarises the recent findings concerning the mechanism governing the selective activation TLR2-mediqted ERK1/2 signaling and the mechanism controlling TLR4-mediated activation of pro-apoptotic pathways in post-ischemic kineys and primary cultures of renal tubule cells subjected to transient hypoxia.
SELECTIVE TLR2-MEDIATED ACTIVATION OF ERK1/2 IN POST-ISHEMIC KIDNEYS AND POST-HYPOXIC RENAL TUBULE CELLS: A ROLE FOR THE PROTEIN PHOSPHATASE 5
The activation of ERK has also been shown to be implicated in cell survival following oxidant injury or induction of endoplasmic reticulum (ER) stress-induced cell death signaling [59-61]. A variety of protein phosphatases and protein kinases are involved in the regulation of signaling pathways by a phosphorylation/dephosphorylation mechanism [62]. The protein phosphatase 5 (PP5), which is a member of the family of serine/threonine phosphatases is ubiquitously expressed in mammalian cells [63]. PP5 has been shown to act as a negative regulator of the apoptosis signal-regulating kinase 1 (ASK1)-JNK/p38 pathway that facilitates apoptosis in hypoxic and H2O2 stressed cell [64,65]. PP5 has been identified as an inactivator of the MEK-ERK pathway through its interaction with the kinase Raf-1 initiating this pathway [66]. PP5 physically was also shown to interact with the heat shock protein (Hsp) 90 which is complexed with the transcription factor heat shock factor 1 (HSF1), a key regulatory protein involved in Hsp synthesis. This complex present in unstressed cells dissociates in stressed cells, and inhibition of complexed Hsp90 or dissociation of the complex in condition of stress leads to an activation of HSF1 [67-69]. We have shown that benzoquinone ansamycin geldanamycin (GA), which binds to Hsp90 and destabilizes kinase complexes does not affect the phosphorylation state of p-ERK1/2 in control or post-hypoxic wild-type and Tlr4-/- renal tubule cells, but in contrast, restores the phosphorylation of ERK1/2 in post-hypoxic Tlr2-/- renal tubule cells [58]. On the other hand, GA had no effect on activated p-JNK in post-hypoxic wild-type RTECs, and did not restore the activation of phosphorylated JNK post-hypoxic Tlr2-/- RTECs as well as in post-hypoxic Tlr4-/- RTECs [58]. These findings have provided the first indication that gp96 play a key role in the induction of the TLR2-mediated activation of ERK1/2 in post-hypoxic renal tubule cells.
gp96 (also called grp94), one of the most abundant ER-residing Hsp proteins, homologue of cytosolic Hsp90, plays a key role in TLR-mediated innate immunity [70,71]. Co-Immunoprecipiation studies revealed that gp96 co-immunoprecipaited with PP5 in renal tubule cells The expression of gp96 increased significantly in renal tubule cells subjected to hypoxia [58]. Interestingly, PP5 does no longer physically interact with gp96 in post-hypoxic renal cells derived from wild-type kidneys, while it remains associated with gp96 in the protected, post-hypoxic Tlr2-/- renal cells [58] (Fig. 2A and 2B), suggesting that the disruption of the gp96-PP5 interaction leads to the inactivation of PP5. We reported that knock-down the gp96 protein by gp96 siRNA fully restored the activation of p-ERK1/2, but not of p-ASK1 and p-JNK, in the post-hypoxic Tlr2-/- renal tubule cells (Fig. 2C), indicating that gp96 plays a key role in controlling TLR2-mediated ERK activation in post-hypoxic renal tubule cells [58]. Furthermore, inhibition of PP5 by okadaic acid or silencing Pp5 siRNA restores the activation of p-ERK1/2 in Tlr2-/-renal cells [58]. In a similar way, extinction of PP5 by Pp5 siRNA also reactivated ERK1/2 in post-hypoxic Tlr2-/- renal cells, indicating that the selective TLR2-dependent activation of the ERK1/2 induced by hypoxia appears highly dependent on Pp5 activity in renal tubule cells [58]. PP5 associates with and inactivates ASK1 and JNK under redox stress [64,65]. Consistantly, the expression of p-JNK, as well as p-ASK1 dramatically increased in okadaic acid-pretreated and in in Pp5 siRNA-transfected Tlr2-/- renal cells subjected to hypoxia (Fig. 2D). As a result, silencing Pp5 mRNA expression also reactivates apoptosis in post-hypoxic Tlr2-/- renal tubule cells (Fig. 2E). Fig. 2F summarizes the possible mechanism of PP5-dependent inactivation/activation of ERK1/2 in non-hypoxic and post-hypoxic renal tubule cells.
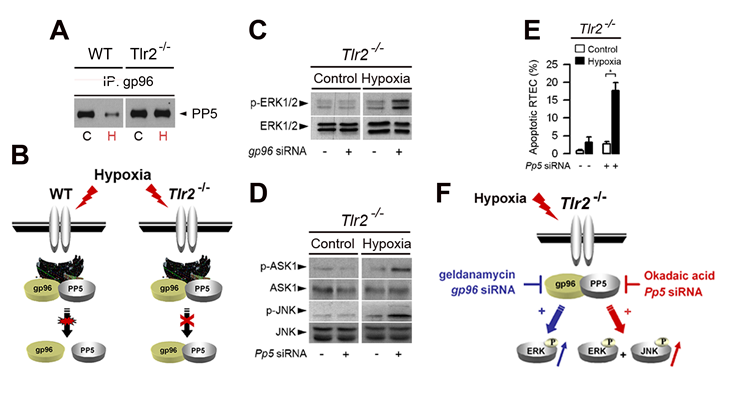
Figure 2: Role of the endoplasmic reticulum-resident gp96 and protein phosphatase 5 in the control of TLR2-mediated activation of ERK1/2 in hypoxic renal tubule cells. (A) Non-hypoxic (C) and day-1 post-hypoxic (H) wild-type (WT) or Tlr2-/- renal tubule cells were subjected to immunoprecipitation (IP) using an antibody against gp96, and proteins were detected using an anti-protein phosphatase 5 (PP5) antibody. (B) Schematic representation of the PP5-dependent activation of ERK1/2 in non-hypoxic and post-hypoxic WT and Tlr2-/- renal tubule cells. (C, D) Immunoblot analyses of p- and total ERK1/2 (C) or total and p-ASK1 and -JNK (D) in non-hypoxic (Control) and 24 h post-hypoxic (Hypoxia) Tlr2-/- renal tubule cells transfected (+) or not (-) with specific gp96 siRNA (C) or Pp5 siRNA. (E) Percentage of apoptotic cells measured in non-hypoxic (Control) or day-1 post-hypoxic (Hypoxia) Tlr2-/- RTECs transfected or not with a Pp5 siRNA or negative control siRNA. Values are means ± SE. * p < 0.05 between groups. (F) Schematic representation of the TLR2-mediated ERK activation in post-hypoxic renal tubule cells. Hypoxia stimulates gp96, but does not induce the dissociation of gp96 bound to PP5 in Tlr2-/- deficient renal tubule cells. Inhibition of gp96 activity by geldanamycin, or knockdown gp96 mRNA induces the reactivation of ERK1/2, but not of JNK or ASK1 in Tlr2-/- deficient RTECs. Extinction of Pp5 mRNA expression also induces the reactivation of p-ERK1/2 and p-JNK. Redrawn from ref 58.
TLR4-MEDIATED APOPTOSIS IN POST-HYPOXIC RENAL TUBULE CELLS: A ROLE FOR THE NADPH OXIDASE 4 PRODUCING ROS
ROS-induced cell injury has been attributed in part to activation of mitogen-activated protein kinase (MAPK) pathways [72]. Members of the NAD(P)H oxidase (NOX) family producing ROS, which are homologues of the gp91phox catalytic subunit of phagocytic NAD(P)H oxidase (NOX2), have been shown to be involved in cellular functions related to innate immunity, signal transduction, proliferation, and/or apoptosis [73]. Among the NOX isoforms, NOX4 was shown to be required for LPS-induced H2O2 generation in TLR4-expressing HEK 293T cells, and to interact directly with TLR4 in regulating NF-κB activation [74]. NOX4, which is sthough to constituvely produce ROS [73,75] is the most abundantly NOX isform expressed within kidneys [76,77]. Recently, we have shown that the mouse kidney express both the full-lenght 65-kDa NOX4 protein, and also a 28-kDa NOX4 isoform [78], similar to a the human NOX4 D variant lacking exons 3-11 with a predicted molecular weight of 28-kDa [79]. We also showed that hypoxia stimulates the expression of the 28 kDa isoform in wild-type renal tubule cells (Fig. 3A) and that silencing both the full length 65-kDa and 28-kDa Nox4 mRNAs or only the 28-kDa Nox4 mRNA isoform prevented both the induction of ROS in post-hypoxic wild-type cells (Fig. 3B), suggesting that the 28-kDa NOX4 isoform play a role in the induction of TLR4-mediated apoptosis caused by I/R injury.
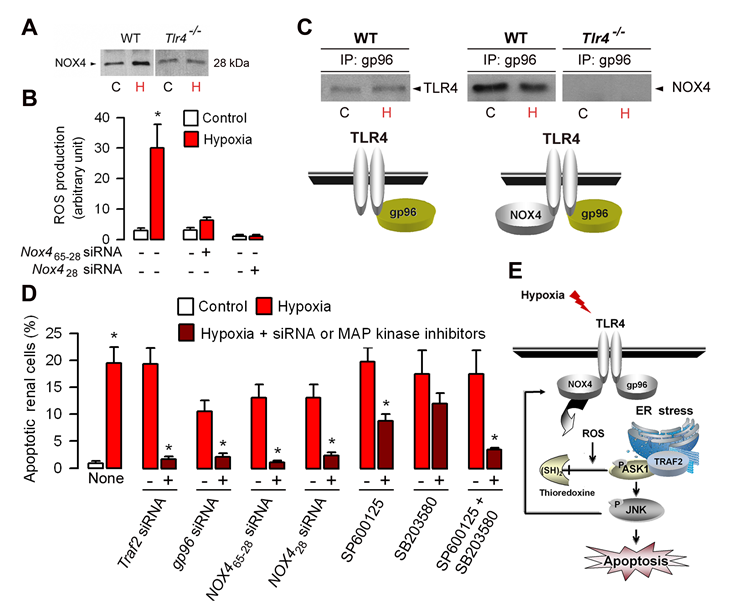
Figure 3: Role of gp96 and NOX4 in the activation of TLR4-mediated apoptosis in hypoxic renal tubule cells. (A) Immunoblot analysis of the 28 kDa NOX4 isoform in control (C) and day-1 post-hypoxic (H) wild-type (WT) and Tlr4-/- renal tubule cells. (B) ROS production in non-hypoxic (Control) and post-hypoxic (Hypoxia) wild-type RTECs transfected (+) or not (-) with specific Nox4 siRNAs, which target sequences encoding both the full length 65-kDa and the 28-kDa isoform (Nox465-28) or only the 28-kDa NOX4 isoform (Nox428). (C) ) Immunoprecipitated lysates from non-hypoxic (C) or 24 h post-hypoxic (H) wild-type renal tubule cells using an anti-gp96 antibody were subjected to Western blot analysis using anti-TLR4 or NOX4 antibodies. (D) Percentage of apoptotic cells in non-hypoxic (Control) and day-1 post-hypoxic (Hypoxia) wild-type renal tubule cells and in post-hypoxic cells transfected (+) ot not (-) with specific Traf2, gp96, or the two Nox465-28 or Nox428 siRNAs, or incubated with (+) or without (-) the JNK inhibitor SP600125 or the ERK inhibitor SB203580. Values are means ± SE. * p < 0.05 between groups. (E) Schematic representation of the TLR4-mediated activation of ASK1/JNK inducing apoptosis. Hypoxia activates NOX4 producing ROS which induces the dissociation of thioredoxine from endogenous ASK1. Redrawn from ref 78.
INTERPLAYS BETWEEN TLR4, GP96, AND NOX4 IN TLR4-MEDIATED ACTIVATION OF PRO-APOPTOTIC SIGNALING INDUCED BY HYPOXIA
Tlr4-/- post-ischemic kidneys and post-hypoxic renal tubule cells exhibit significantly less active caspase-3 positive apoptotic cells and produce less ROS than in the corresponding wild-type post-ischemic kidneys and post-hypoxic renal tubule cells [19,78]. Like in TLR4-transfected HEK 293 cells, TLR4 interacts NOX4 [79] and with gp96 in non-hypoxic renal tubule cells, but TLR4 does not binds with gp96 and hypoxia did not alter the physical interaction between TLR4 and NOX4 [78]. These findings are summarized in Fig. 3C. However, silencing gp96 mRNA expression prevented the increase in TRAF2, p-ASK1 and p-JNK in post-hypoxic wild-type renal tubule cells, a well as ROS production and induction of apoptosis [78]. Co-immunoprecipitation experiments also revealed that TRAF2, which is not associated with ASK1 in non-hypoxic renal tubule cells, co-immunoprecipitates with ASK1 upon hypoxia [78]. These data have confirmed that endogenous TLR4 binds with NOX4 in intact renal tubule cells, and also provided strong evidence that the ER-resident gp96 play a key role in the activation of TRAF2-mediated signaling pathway downstream TLR4. Silencing Traf2 mRNA expression almost fully impaired the activation of NOX4, p-ASK1 and p-JNK and ROS production in post-hypoxic wild-type renal tubule cells [78]. Consistantly, the induction of apoptosis was no longer detected in Traf2 siRNA-transfected WT renal tubule cells subjected to hypoxia (Fig. 3D). Silencing Nox428 siRNA expression does not prevent the activation of TRAF2 in post-hypoxic, wild-type renal tubule cells, but impairs the hypoxia-induced activation of p-ASK1 and p-JNK [78]. Silencing NOX4 RNAs encoding the the full lengt 65 kDa or the 28 kDa isoform using specific NOX465-28 and Nox428 siRNAs, respectively, also fully impaired the induction of apoptosis and ROS production in post-hypxic wWT renal tubule cells (Fig. 3D) [78]. Using JNK and p38 inhibitors, we also showed that JNK can reciprocally regulate NOX4 and prevent the induction of ROS production and cell apoptosis caused by hypoxia [78]. Inhibition of JNK appears to be more efficient that the inhibition of p-p38 to reduce apoptosis induced by hypoxia (Fig. 3D). Moreover, concomittent inhibition of JNK and p38 had additive effects on apoptosis inhibition (Fg. 3D), suggesting that both JNK, and to a lesser extend p38, are involved in the induction of apoptotic signaling activated by hypoxia. Fig. 3E provides a schematic representation of the proposed mechanism of the TLR4/NOX4-mediated activation of the TRAF2/ASK1/JNK apoptotic pathway induced by hypoxia.
CONCLUSIONS
Activation of both TLR2 and TLR4 signaling during I/R injury induces potent inflammatory response and induction of cell apoptosis. Studies on renal I/R injury have revealed that TLR2 selectively stimulated the ERK1/2 through specific interactions between the ER-resident gp96 chaperone and the protein phosphatase 5. Activation of TLR4 signaling promotes the activation of the TRAF2/ASK1/JNK pro-apoptotic pathway. In the kidney, the non-phagocytic NOX4 which physically interacts with NOX4 appears to play a key role in the induction of aptoptosis. However, a number of question still remains to be solved. There are multiple source of ROS production during hypoxia, including the mitochondrial respiratory chain, xanthine oxidase, xanthine-oxygenase, lipoxygenase, and NOX enzymes in stressed cells [80]. Although the 28-kDa NOX4 isform appears to play a key role in the control of apoptosis in post-hypoxic renal tubule cells, the question arises wether this Nox4 isoform lacking most transmembrane domains [79], is also directly involved in the ROS production induced by hypoxia. NOX4 has been shown to be involved in the up-regulation of the hypoxia-induced transcription factor HIF-1α [81], a key regulator of oxygen-regulated gene expression [82]. Conversely, the induction of NOX4 by HIF-1α also shown to maintain sustain levels of ROS after hypoxia [83]. The question arises to know as to whether NOX4 interacts with TLR4 and whether NOX4 plays a role a in the control of cell apoptosis in other tissue² than the kidney. The identification of NOX4 as a sensor in the ER stress-induced, TLR4-mediated, pro-apoptotic signaling cascade should has implications for selective anti-superoxide-generating enzyme strategies intended to prevent the the induction of apoptosis in hypoxic tissues.
Acknowledgments
This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM) and in part by a grant from the Agence Nationale de la Recherche (ANR-08-MIEN-030-01). A.Vandewalle was in receipt of a contrat hospitalier de recherche translationnelle (CHRP) INSERM-APHP.
Conflict of interest
The authors declare no conflict of interest
Reference List
1. Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol. 2003; 14: 2199-2210.
2. Boros P, Bromberg JS. New cellular and molecular immune pathways in ischemia/reperfusion injury. Am J Transplant. 2006; 6: 652-658.
3. Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002; 82: 47-95.
4. Akira S, Takeda K. Toll-like receptor signalling. Nature Rev Immunol. 2004; 4: 499-511.
5. Barton GM, edzhitov R. Toll-like receptor signaling pathways. Science. 2003; 300: 1424-1425.
6. Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005; 201: 1135-1143.
7. Fallach R, Shainberg A, Avlas O, Fainblut M, Chepurko Y, Porat E, Hochhauser EJ. Cardiomyocyte Toll-like receptor 4 is involved in heart dysfunction following septic shock or myocardial ischemia. Mol Cell Cardiol. 2010; 48: 1236-1244.
8. Hornef MW, Frisan T, Vandewalle A, Normark S, Richter-Dahlfors A. Toll-like receptor 4 resides in the Golgi apparatus and colocalizes with internalized lipopolysaccharide in intestinal epithelial cells. J Exp Med. 2002; 195: 559-570.
9. Guillot L, Medjane S, Le-Barillec K, Balloy V, Danel C, Chignard M, Si-Tahar M. Response of human pulmonary epithelial cells to lipopolysaccharide involves Toll-like receptor 4 (TLR4)-dependent signaling pathways: evidence for an intracellular compartmentalization of TLR4. J Biol Chem. 2004; 279: 2712-2718.
10. Tsuboi N, Yoshikai Y, Matsuo S, Kikuchi T, Iwami K, Nagai Y, Takeuchi O, Akira S, Matsuguchi T. Roles of toll-like receptors in C-C chemokine production by renal tubular epithelial cells. J Immunol. 2002; 169: 2026-2033.
11. Gribar SC, Anand RJ, Sodhi CP, Hackam DJ. The role of epithelial Toll-like receptor signaling in the pathogenesis of intestinal inflammation. J Leukoc Biol. 2008; 83: 493-498.
12. Vandewalle A. Toll-like receptors and renal bacterial infections. Chang Gung Med J. 2008; 31: 525-537.
13. Zhai Y, Shen XD, O’Connell R, Gao F, Lassman C, Busuttil RW, Cheng G, Kupiec-Weglinski JW. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004; 173: 7115-7119.
14. Watson MJ, Ke B, Shen XD, Gao F, Busuttil RW, Kupiec-Weglinski JW, Farmer DG. Intestinal ischemia/reperfusion injury triggers activation of innate toll-like receptor 4 and adaptive chemokine programs. Transplant Proc. 2008; 40: 3339-3341.
15. Moses T, Wagner L, Fleming SD. TLR4-mediated Cox-2 expression increases intestinal ischemia/reperfusion-induced damage. J Leukoc Biol. 2009; 86: 971-980.
16. Oyama J, Blais Jr C, Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004; 109: 784-789.
17. Barsness KA, Arcaroli J, Harken AH, Abraham E, Banerjee A, Reznikov L, McIntyre RC. Hemorrhage-induced acute lung injury is TLR-4 dependent. Am J Physiol Regul Integr Comp Physiol. 2004; 287: R592-R599.
18. Prince JM, Levy RM, Yang R, Mollen KP, Fink MP, Vodovotz Y, Billiar TR. Toll-like receptor-4 signaling mediates hepatic injury and systemic inflammation in hemorrhagic shock. J Am Coll Surg. 2006; 202: 407-417.
19. Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007; 117: 2847-2859.
20. Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, Akira S, van der Poll T, Weening JJ, Florquin S. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest. 2005; 115: 2894-2903.
21. Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988; 242: 540-546.
22. Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995; 267: 1485-1488.
23. Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999; 398: 252-256.
24. An H, Yu Y, Zhang M, Xu H, Qi R, Yan X, Liu S, Wang W, Guo Z, Guo J, Qin Z, Cao X. Involvement of ERK, p38 and NF-kappaB signal transduction in regulation of TLR2, TLR4 and TLR9 gene expression induced by lipopolysaccharide in mouse dendritic cells. Immunology. 2002; 106: 38-45.
25. Schieven GL. The biology of p38 kinase: a central role in inflammation. Curr Top Med Chem. 2005; 5: 921-928.
26. Shigeoka AA, Holscher TD, King AJ, Hall FW, Kiosses WB, Tobias PS, Mackman N, McKay DB. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J Immunol. 2007; 178: 6252-6258.
27. Srikrishna G, Freeze HH. Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia. 2009; 11: 615-628.
28. Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004; 279: 7370-7377.
29. Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005; 201: 1135-1143.
30. Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006; 26: 174-179.
31. Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002; 277: 15107-15112.
32. Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002; 277: 15028-15034.
33. Ohashi K, Burkart V, Flohé S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000; 164: 558-561.
34. Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001; 167: 2887-2894.
35. Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF 3rd. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001; 276: 10229-10233.
36. Guillot L, Balloy V, McCormack FX, Golenbock DT, Chignard M, Si-Tahar M. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J Immunol. 2002; 168: 5989-5992.
37. Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, Shirakawa AK, Farber JM, Segal DM, Oppenheim JJ, Kwak LW. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 2002; 298: 1025-1029.
38. Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003; 425: 516-521.
39. Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002; 195: 99-111.
40. Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, Marsche G, Young MF, Mihalik D, Götte M, Malle E, Schaefer RM, Gröne HJ. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest. 2005; 115: 2223-2233.
41. Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 2002; 168: 5233-5239.
42. Ishii KJ, Suzuki K, Coban C, Takeshita F, Itoh Y, Matoba H, Kohn LD, Klinman DM. Genomic DNA released by dying cells induces the maturation of APCs. J Immunol. 2001; 167: 2602-2607.
43. Karikó K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004; 279: 12542-12550.
44. Thomas JO, Travers AA. HMG1 and 2, and related ‘architectural’ DNA-binding proteins. Trends Biochem Sci. 2001; 26: 167-174.
45. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002; 418: 191-195.
46. Rucci A, Palumbo R, Bianchi ME. HMGB1: a signal of necrosis. Autoimmunity. 2007; 40: 285-289.
47. Li J, Gong Q, Zhong S, Wang L, Guo H, Xiang Y, Ichim TE, Wang CY, Chen S, Gong F, Chen G.Neutralization of the extracellular HMGB1 released by ischaemic damaged renal cells protects against renal ischaemia-reperfusion injury. Nephrol Dial Transplant. 2010 [Epub ahead of print].
48. Wu H, Ma J, Wang P, Corpuz TM, Panchapakesan U, Wyburn KR, Chadban SJ. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol. 2010; 21: 1878-1890.
49. Dhupar R, Klune JR, Evankovich J, Cardinal J, Zhang M, Ross M, Murase N, Geller DA, Billiar TR, Tsung A. Interferon regulatory factor-1 mediates acetylation and release of high mobility group Box-1 from hepatocytes during liver ischemia-reperfusion injury. Shock. 2010 [Epub ahead of print].
50. Evankovich J, Cho SW, Zhang R, Cardinal J, Dhupar R, Zhang L, Klune JR, Zlotnicki J, Billiar TR, Tsung A.High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem. 2010 [Epub ahead of print].
51. Nakano H, Nakajima A, Sakon-Komazawa S, Piao JH, Xue X, Okumura K. Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ. 2006; 13: 730-737.
52. Matsuzawa A, Ichijo H. Stress-responsive protein kinases in redox-regulated apoptosis signaling. Antioxid Redox Signal. 2005; 7: 472-481.
53. Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997; 275: 90-94.
54. Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001; 2: 222-228.
55. Wang X, McCullough KD, Franke TF, Holbrook NJ. Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J Biol Chem. 2000; 275: 14624-14631.
56. Stefanelli C, Tantini B, Fattori M, Stanic’ I, Pignatti C, Clo C, Guarnieri C, Caldarera CM, Mackintosh CA, Pegg AE, Flamigni F. Caspase activation in etoposide-treated fibroblasts is correlated to ERK phosphorylation and both events are blocked by polyamine depletion. FEBS Lett. 2002; 527: 223-228.
57. Sarró E, Tornavaca O, Plana M, Meseguer A, Itarte E. Phosphoinositide 3-kinase inhibitors protect mouse kidney cells from cyclosporine-induced cell death. Kidney Int. 2008; 73: 77-85.
58. Mkaddem SB, Werts C, Goujon JM, Bens M, Pedruzzi E, Ogier-Denis E, Vandewalle A. Heat shock protein gp96 interacts with protein phosphatase 5 and controls toll-like receptor 2 (TLR2)-mediated activation of extracellular signal-regulated kinase (ERK) 1/2 in post-hypoxic kidney cells. J Biol Chem. 2009; 284: 12541-12549.
59. Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem. 1996; 271: 4138-4142.
60. di Mari JF, Davis R, Safirstein RL. MAPK activation determines renal epithelial cell survival during oxidative injury. Am J Physiol. 1999; 277: F195-F203.
61. Hung CC, Ichimura T, Stevens JL, Bonventre JV. Protection of renal epithelial cells against oxidative injury by endoplasmic reticulum stress preconditioning is mediated by ERK1/2 activation. J Biol Chem. 2003; 278: 29317-29326.
62. Mumby MC, Walter G. Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiol Rev. 1993; 73: 673-699.
63. Cohen PT. Novel protein serine/threonine phosphatases: variety is the spice of life. Trends Biochem Sci.1997; 22: 245-251.
64. Morita K, Saitoh M, Tobiume K, Matsuura H, Enomoto S, Nishitoh H, Ichijo H. Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J. 2001; 20: 6028-6036.
65. Zhou G, Golden T, Aragon IV, Honkanen RE. Ser/Thr protein phosphatase 5 inactivates hypoxia-induced activation of an apoptosis signal-regulating kinase 1/MKK-4/JNK signaling cascade. J Biol Chem. 2004; 279: 46595-46605.
66. von Kriegsheim A, Pitt A, Grindlay GJ, Kolch W, Dhillon AS. Regulation of the Raf-MEK-ERK pathway by protein phosphatase 5. Nat Cell Biol. 2006; 8: 1011-1016.
67. Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998; 94: 471-480.
68. Bharadwaj S, Ali A, Ovsenek N. Multiple components of the HSP90 chaperone complex function in regulation of heat shock factor 1 In vivo. Mol Cell Biol. 1999; 19: 8033-8041.
69. Guo Y, Guettouche T, Fenna M, Boellmann F, Pratt WB, Toft DO, Smith DF, Voellmy R. Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J Biol Chem. 2001; 276: 45791-45799.
70. Randow F, Seed B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat Cell Biol. 2001; 3: 891-896.
71. Yang Y, Liu B, Dai J, Srivastava PK, Zammit DJ, Lefrançois L, Li Z. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity. 2007; 26: 215-226.
72. Nakano H, Nakajima A, Sakon-Komazawa S, Piao JH, Xue X, Okumura K. Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ. 2006; 13: 730-737.
73. Bedard K, Krause KH. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol Rev. 2007; 87: 245-313.
74. Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol. 2004; 173: 3589-3593.
75. Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O et al. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J. 2007; 406: 105-114.
76. Geiszt M, Kopp JB, Varnai P, Leto TL. Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci USA. 2000; 97: 8010-8014.
77. Shiose A, Kuroda J, Tsuruya K, Hirai M, Hirakata H, Naito S, Hattori M, Sakaki Y, Sumimoto H. A novel superoxide-producing NAD(P)H oxidase in kidney. J Biol Chem. 2001; 276: 1417-1423.
78. Ben Mkaddem S, Pedruzzi E, Werts C, Coant N, Bens M, Cluzeaud F, Goujon JM, Ogier-Denis E, Vandewalle A. Heat shock protein gp96 and NAD(P)H oxidase 4 play key roles in Toll-like receptor 4-activated apoptosis during renal ischemia/reperfusion injury. Cell Death Differ. 2010; 17: 1474-1485.
79. Goyal P, Weissmann N, Rose F, Grimminger F, Schäfers HJ, Seeger W et al. Identification of novel Nox4 splice variants with impact on ROS levels in A549 cells. Biochem Biophys Res Commun. 2005; 329: 32-39.
80. Kamata H, Hirata H. Redox regulation of cellular signalling. Cell Signal. 1999; 11: 1-14.
81. Bonello S, Zähringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, Kietzmann T, Görlach A. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol. 2007; 27: 755-761.
82. Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000; 88: 1474-1480.
83. Diebold I, Petry A, Hess J, Görlach A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol Biol Cell. 2010; 21: 2087-2096.