
INTRODUCTION
Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) is an idiopathic focal epilepsy with a spectrum of clinical manifestations. The typical characteristics are clusters of brief motor seizures during light sleep that are often misdiagnosed as nightmares [1, 2, 3]. ADNFLE is a genetically heterogeneous condition, and the roles of genes encoding subunits of neuronal acetylcholine receptors (nAChRs), such as 4, 2, and 2 (CHRNA4, CHRNA2, and CHRNB2), have been clearly established in ADNFLE [4, 5]. Mutations in the KCNT1 gene, encoding a sodium-gated potassium channel subunit, have been identified in families with severe ADNFLE and accompanying psychiatric features and intellectual disability [6]. However, mutations in these genes only account for a small proportion of families affected with ADNFLE [7, 8], suggesting that other genes contributing to this disorder must exist. In this study, an ADNFLE pedigree in the South Chinese Han population was clinically characterized according to the typical disease phenotype, and whole-exome sequencing was performed in order to identify previously unknown genes linked to ADNFLE.
RESULTS
Description of the pedigree
The initial ADNFLE proband was a 37-year-old woman of the Southern Chinese Han population, who presented with brief, clustered, and involuntary motor actions that occurred during sleep. The involuntary motor actions lasted for 0.5 to 1 min and occurred 2 to 8 times per night, manifesting as head scratching, limb flexion, the sensation of being out of breath, tonic stiffening, and vocalization while the patient was conscious. No night blindness, visual disorders, or other abnormal genetic history was noted. The age at onset was 13 years. Examination of the nervous system, physical examination, and cerebral MRI showed no abnormality. The electroencephalogram showed sharp waves and spike and slow waves in bifrontal and bicentral areas during the non-rapid eye movement period. The other affected members of the ADNFLE pedigree showed similar symptoms, but differed in symptom severity (Table 1). All a patients of the pedigree responded well to Topamax and Levetiracetam treatment.
Table 1: Main clinical characteristics of seven patients of an ADNFLE pedigree in the Southern Chinese Han population
No. |
Sex |
Age at time of study (yrs.) |
Age at onset (yrs.) |
Diurnal seizures |
Intellectual disability |
Psychiatric disorder |
Interictal EEG |
MRI |
Drug resistance |
|---|---|---|---|---|---|---|---|---|---|
II4 |
F |
66 |
8 |
No |
No |
No |
NA |
N |
No |
III1 |
F |
45 |
15 |
Yes |
No |
No |
Rare frontal spikes (sleep recording) |
N |
No |
III3 |
F |
39 |
11 |
No |
No |
No |
NA |
N |
No |
III5 |
F |
37 |
13 |
No |
No |
No |
bifrontal and bicentral sharp waves and slow–sharp waves (sleep recording) |
N |
No |
III7 |
F |
34 |
11 |
No |
No |
No |
bifrontal sharp waves (sleep recording) |
N |
No |
IV1 |
F |
20 |
12 |
No |
No |
No |
Right frontotemporal spikes (sleep recording) |
N |
No |
IV6 |
M |
10 |
6 |
No |
No |
No |
Rare frontal spikes (sleep recording) |
N |
No |
F, female; M, male; N, normal; NA, not available.
Exome sequencing
Exome sequencing was performed on the genomic DNA of four affected individuals of the ADNFLE pedigree (Table 1) using NimbleGen Sequence Capture technology (SeqCap EZ library) according to the manufacturer's instructions. The enriched libraries underwent 90 base pair, paired-end sequencing on a HiSeq2000 next-generation sequencing platform (Illumina, San Diego, CA). The sequence data were aligned to the reference human genome (UCSC hg19) using SOAPaligner and variant calling used the SOAPsnp (v.1.03). After filtering PCR duplicates, previously reported variants (dbSNP135 as reported in the UCSC Genome Browser (http://genome.ucsc.edu/), the 1000 Genomes Project (http://www.1000genomes.org) and HapMap 8 (http://hapmap.ncbi.nlm.nih.gov/) databases, with frequency greater than 0.5 %,) and synonymous variants were removed.
Thirteen gene variants were found in the four patients of the ADNFLE pedigree (Table 2), and Sanger sequencing confirmed all the variants. Ten out of thirteen variants were proven to be false-positive. Only three gene mutations (CABP4, FAM72D, MKI67IP) were present in all four patients. However, except for the CABP4 mutation, the two other gene variants were not only found in affected individuals, but also in other unaffected pedigree members, thus, not separating according to the disease phenotype (Table 3).
Table 2: Exome sequencing identified thirteen gene variants in four affected individuals of an ADNFLE pedigree in the Southern Chinese Han population
Chromosome |
Gene name |
Mutation type |
Substitution |
Score from SIFT |
Prediction from SIFT |
|---|---|---|---|---|---|
Chr11 |
CABP4 |
Missense |
G155D |
0 |
DaMaging |
Chr1 |
FAM72D |
Missense |
G82V |
0.04 |
DaMaging |
Chr20 |
FRG1B |
Missense |
V12A |
0.03 |
DaMaging |
Chr20 |
FRG1B |
Missense |
D24N |
0.03 |
DaMaging |
Chr7 |
IGF2BP3 |
Missense |
I503T |
0 |
DaMaging |
Chr2 |
MKI67IP |
Missense |
V228M |
0.03 |
DaMaging |
Chr7 |
MLL3 |
Missense |
C988F |
0 |
DaMaging |
Chr11 |
MUC6 |
Missense |
T1911M |
0.04 |
DaMaging |
Chr4 |
NHEDC1 |
Missense |
A473V |
0.01 |
DaMaging |
Chr4 |
UGT2B11 |
Missense |
H481R |
0.02 |
DaMaging |
Chr19 |
ZNF285 |
Missense |
P455Q |
0.03 |
DaMaging |
Chr12 |
ZNF705A |
Missense |
R67W |
0.04 |
DaMaging |
Chr11 |
ZNHIT2 |
Missense |
R384W |
0.02 |
DaMaging |
Table 3: Three out of 13 identified gene mutations (CABP4, FAM72D, MK167IP) in the ADNFLE pedigree were confirmed by Sanger sequencing
Individual |
CaBP4 |
FAM72D |
MKI67IP |
|---|---|---|---|
II4 |
+ |
+ |
+ |
III1 |
+ |
- |
- |
III3 |
+ |
+ |
+ |
III5 |
+ |
+ |
+ |
III7 |
+ |
+ |
+ |
III9 |
- |
+ |
+ |
IV1 |
+ |
+ |
+ |
IV2 |
- |
+ |
- |
IV3 |
- |
- |
+ |
IV4 |
- |
+ |
+ |
IV5 |
+ |
+ |
+ |
IV6 |
+ |
+ |
+ |
IV7 |
- |
+ |
+ |
IV9 |
- |
+ |
+ |
Finally, only one mutation was present in all seven affected members in this family: the c.464G>A (p.G155D) mutation in the Ca2+-binding protein 4 (CABP4) gene (OMIM: 608965) (Figure 1). Although, the mutation existed in an unaffected, one year old individual, the member of the pedigree was too young to meet the average age of onset of ADNFLE. The c.464G>A mutant in the CABP4 gene was absent from the other unaffected individuals of the pedigree, and from 300 control cases.
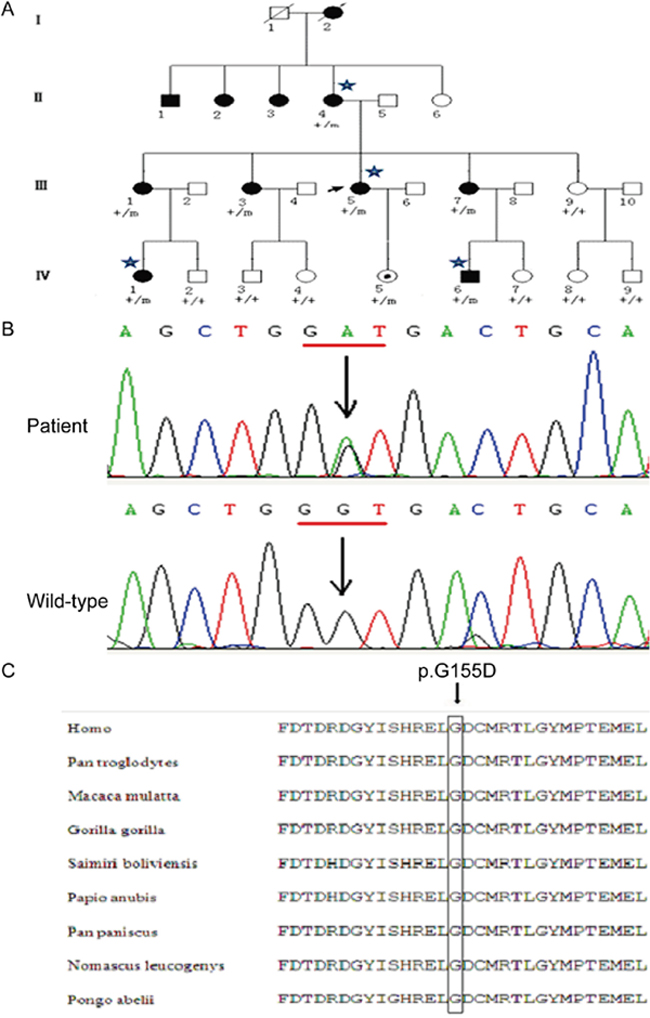
Figure 1: A Chinese ADNFLE pedigre with a mutation in the CABP4 gene. (A) ADNFLE pedigree with c.464G>A mutation in the CABP4 gene. Filled symbols indicate affected individuals and clear symbols unaffected individuals; squares: males; circles: females; arrow represents the proband, and asterisks mark the individuals for whom whole-exome sequencing was performed. Genotypes are indicated for each individual. “+” means wild type; “m” means mutant. (B) The c.464G>A mutation (↓) replaces D (glycine) with G (aspartic acid). (C) Evolutionary conservation of glycine G155. Black arrows indicate the positions of the missense mutations.
The c. 464G>A substitution in the CABP4 gene replaces a glycine residue at position 155 with an aspartic acid residue (p.G155D). This missense mutation is predicted to affect the CABP4 protein function according to evaluation with the bioinformatics tools PolyPhen 2 and SIFT (Prediction score 0.00). Furthermore, the wild-type glycine residue which is affected by the p.G155D mutation is highly conserved between primate mammal species (Figure 1C). The three dimensional structure of the CaBP4 protein with the novel missense mutation was modeled using the ESyPred3D web server (Figure 2). The c.464G>A substitution replaces a glycine residue with an aspartic acid residue, additionally containing an acidic side chain, at position 155, which is within the first Ca2+ binding EF-hand motif (Figure 2).
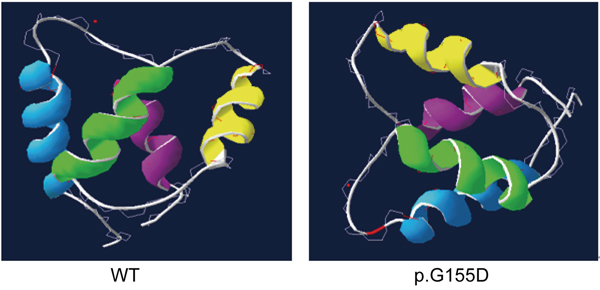
Figure 2: The three dimensional model of the CaBP4 protein containing the four EF-hand motifs (EF1-blue, EF2-green, EF3-yellow, and EF4-cyan) as revealed by the automated homology modeling program ESyPred3D. Left: wild-type (WT) protein; right: p.G155D mutant protein.
DISCUSSION
In the present study, a novel missense mutation c.464G>A (p.G155D) was identified in the neuronal Ca2+ binding protein CABP4 using whole-exome sequencing, and was found in all seven affected individuals from a 4-generation ADNFLE pedigree. This mutation was also found in one young, pre-symptomatic family member, but was absent from 300 unrelated control subjects. The mutation was localized in the EF hand Ca2+ binding domain and predicted to affect CABP4 protein function according to the bioinformatics tools PolyPhen 2 and SIFT.
CaBP4, which consists of 275 amino acids, is encoded by six exons on chromosome 11q11.3. CaBP4 belongs to the family of neuronal Ca2+-binding proteins (CaBPs) and shares structural homology with calmodulin. It has been shown to modulate voltage-dependent Ca2+ channels [9, 10]. CaBP4 contains four EF-hand Ca2+-binding motifs, but the lysine residue at position 1 of EF-hand 2 is not suitable for Ca2+ coordination; thus, the second EF-hand motif cannot coordinate Ca2+ [11].
To date, three mutations have been identified in CaBP4 (Figure 3). All of them were detected in patients with retinal diseases, such as congenital stationary night blindness [12, 13, 14]. CaBP4 co-localizes with the voltage-gated L-type Ca2+ channel Cav1.4 in retinal synaptic terminals and regulates Ca2+ influx through the Ca2+ channel by binding to the ICDI (inhibitor of Ca2+-dependent inactivation) of CaV1.4. Thereby CaBP4 inhibits Cav1.4 inactivation and increases Ca2+ concentrations in synapses leading to enhanced neurotransmitter release [15]. Functional research of the CaBP4 mutations in congenital stationary night blindness have shown that the regulatory function of the mutant proteins is weaker compared to the wild type proteins [13, 16].
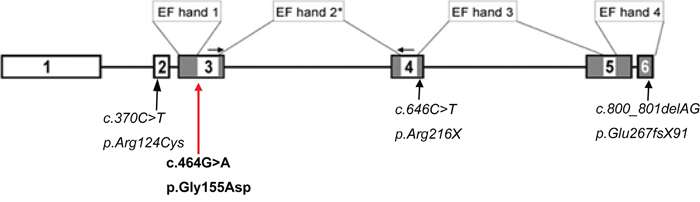
Figure 3: Schematic drawing of the CABP4 gene. Boxes: exons; lines: introns; gray boxes: parts of the gene that encode the EF hands EF1-4, the Ca2+-binding elements in CaBP4; In bold is the c.464G>A mutation and in italic are the previously identified mutations in CABP4. *EF hand 2 is not functional [10].
Interestingly, the N- and C-lobes of Ca2+-bound CaBP4 bind cooperatively to the IQ peptide of Cav1.4 [17]. The p.G155D mutation is located in the N-lobe of CaBP4 within EF1, which forms hydrophobic contacts and a salt bridge with the IQ domain of Cav1.4. Specifically, the CaBP4 residues Phe137 and Glu168 interact with Phe1586 and Arg1597, respectively, in Cav1.4.
Since the mutated p.G155D protein is predicted to bend and fold less easily if an aspartic acid exists at the G155 position, because of the additional acidic side chain, it is likely that the mutation induces structural changes in the Cav1.4 binding domain of CaBP4 between Phe137 and Glu168. These changes may interfere with cooperative binding to Cav1.4 or affect Ca2+ binding to the first EF-hand motif, which both may result in a weaker regulatory function. Therefore, it is tempting to speculate that the mutant protein decreases ion channel activation, leading to reduced Ca2+ concentrations, and thereby, for instance, interferes with the release of inhibitory neurotransmitters, thus, contributing to the etiology of ADNFLE function.
In summary, a novel missense mutation in CaBP4 was identified in patients of a Chinese Han ADNFLE pedigree by means of whole-exome sequencing. The c.464G>A mutation is the first CaBP4 mutation reported relating to ADNFLE, may provide a new insight into the pathology of ADNFLE. Further studies are necessary to identify CaBP4 mutations in other ADNFLE families, and functional investigations are necessary to predict the impact of the mutation in protein function and for the etiology of ADNFLE.
MATERIALS AND METHODS
Patients
In strict accordance with the classification criteria for the diagnosis of epilepsy and epilepsy syndrome by the 2010 International League Against Epilepsy (ILAE), we recruited 42 patients from 11 families with epilepsy and epilepsy syndrome. Diagnosis of these patients was based on the clinical history, EEG/ictal VEEG, video polysomnographic analysis, neurological examination and cranial magnetic resonance imaging (MRI). Among these families, a 4-generation pedigree from the Southern Chinese Han population, consisting of 27 members and 11 affected individuals, was diagnosed with ADNFLE (Figure 1). Fifteen family members participated in this study, including seven affected individuals and eight unaffected individuals. None of these patients had night blindness or visual disorders. The average age at onset was 11 years, ranging from 6 - 15 years. Three hundred healthy volunteers without epilepsy and related diseases were recruited as controls from the Southern Chinese Han population. Written informed consent was obtained from all study subjects and from healthy control individuals after explanation of the nature and possible consequences of the study. The study protocol was approved by the Ethics Committee for Human Research of the Guangdong General Hospital, and was in accordance with the tenets of the Declaration of Helsinki. Information was also obtained retrospectively from medical records.
Exome sequencing and analysis
DNA was extracted from subjects’ peripheral blood using a QIAamp Blood DNA Kit (Sango Inc., Shanghai, China). Four affected individuals from the ADNFLE pedigree (II-4, III-5, IV-1 and IV-6, Figure 1) were selected for exome sequencing. Exome capture was performed with a SureSelect Human All Exon Kit. Exon-enriched DNA was sequenced on an Illumina Genome Analyzer II platform in accordance with the manufacturer’s instructions (BGI Inc., Shenzhen, China). The resulting sequences were further analyzed using the SOAP (Short OligonucleotideAnalysis Package) software, which allows the fast and accurate alignment for huge amounts of short reads generated by the Illumina Genome Analyzer (http://soap.genomics.org.cn/).
Sequencing analyses and polymerase chain reaction (PCR)
To confirm the presence of gene variants identified via exome sequencing, and to screen for these variants in relatives and control subjects, genomic DNA derived from blood samples from 15 individuals from the ADNFLE pedigree and 300 control subjects was used for PCR amplification of the CaBP4 gene comprising the c.464G>A mutant. PCR primers for the CaBP4 gene comprising the c.464G>A mutant were ‘shown in Table 4) and were purchased from Sigma Inc, USA. PCR was performed using standard protocols in the 9700 PCR cycler (ABI Inc., USA). Sequencing of PCR amplification products was performed using an Applied Biosystems 3730XL DNA Analyzer (ABI Inc., Carlsbad, USA).
Table 4: Primers sequence and annealing temperature of exons of CABP4 gene
Exon |
5’– 3’ |
3’– 5’ |
Temperature (°C) |
|---|---|---|---|
1 |
GCAGAGGACTGGGATTAGGG |
GGGAGGGTTATTGCTGCTC |
20 |
1 |
ATGACCACAGAGCAGGCAAG |
TCAGTCTGGGGCAGAAGAG |
20 |
3 |
CGGGTGTTTCTTCCTAGGTG |
CTGGACGGATCCTCTAGCTG |
20 |
4 |
CAGCCTGGTGACAGAGCA |
ACAAACTCTGCGGGAGAGAA |
18 |
5 |
GAGTCTCCTTCCCGGAAAAC |
CAGCCATCCTCTCCATTCAT |
20 |
Bioinformatics tools
PolyPhen-2 (Polymorphism Phenotyping, v2.2.2) is a bioinformatics tool that predicts possible impact of an amino acid substitution on the structure and function of a human protein using straightforward physical and comparative considerations [18]. The software uses protein sequences from the UniProtKB database (UniRef100), structures from PDB/DSSP Snapshot (78,304 entries), and UCSC MultiZ multiple alignments of 45 vertebrate genomes with the hg19/GRCh37 human genome (http://genetics.bwh.harvard.edu/pph2/). SIFT (Sorting Intolerant From Tolerant, v 5.2.2) predicts whether an amino acid substitution affects protein function based on sequence homology and the physical properties of amino acids [19]. Three dimensional model of CaBP4 was simulated with ESyPred3D (Lambert et al., 2002), a new automated homology modeling program (http://www.unamur.be/sciences/biologie/urbm/bioinfo/esypred/) [20].
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
We express special thanks to BGI Genomics (BGI-Shenzhen, China) for providing us with experimental guidance. This work was funded by the 2010 Chinese National Natural Science Foundation Project (81071046); and Guangdong Provincial Science and Technology Program (2013B0220000084, 2014A020212242, 2017A020215052); and Guangzhou Science and Technology Foundation Project (201510010189); and Guangzhou traditional Chinese medicine Fund (20171005); and Special Fund of Guangdong General Hospital (Y012015010).
REFERENCES
1. Scheffer IE, Bhatia KP, Lopes-Cendes I, Fish DR, Marsden CD, Andermann E, Andermann F, Desbiens R, Keene D, Cendes F, Manson JI, Constantinou JE, Mclntosh A, Berkovic SF. Autosomal dominant nocturnal frontal lobe epilepsy. A distinctive clinical disorder. Brain. 1995; 118:61–73.
2. Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, Scheffer IE, Berkovic SF. A missense mutation in the neuronal nicotinic acetylcholine receptor α4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995; 11:201-203.
3. Oldani A, Zucconi M, Asselta R, Modugno M, Bonati MT, Dalprà L, Malcovati M, Tenchini ML, Smirne S, Ferini-Strambi L. Autosomal dominant nocturnal frontal lobe epilepsy. A video-polysomnographic and genetic appraisal of 40 patients and delineation of the epileptic syndrome. Brain. 1998; 121:205-223.
4. Xu J, Cohen BN, Zhu Y, Dziewczapolski G, Panda S, Lester HA, Heinemann SF, Contractor A. Altered activity-rest patterns in mice with a human autosomal-dominant nocturnal frontal lobe epilepsy mutation in the β2 nicotinic receptor. Mol Psychiatry. 2011; 16:1048–61.
5. O'Neill HC, Laverty DC, Patzlaff NE, Cohen BN, Fonck C, McKinney S, McIntosh JM, Lindstrom JM, Lester HA, Grady SR, Marks MJ. Mice expressing the ADNFLE valine 287 leucine mutation of the Β2 nicotinic acetylcholine receptor subunit display increased sensitivity to acute nicotine administration and altered presynaptic nicotinic receptor function. Pharmacol Biochem Behav. 2013; 103:603-621.
6. Heron SE, Smith KR, Bahlo M, Nobili L, Kahana E, Licchetta L, Oliver KL, Mazarib A, Afawi Z, Korczyn A, Plazzi G, Petrou S, Berkovic SF, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 2012; 44:1188-1190.
7. Steinlein OK, Hoda JC, Bertrand S, Bertrand D. Mutations in familial nocturnal frontal lobe epilepsy might be associated with distinct neurological phenotypes. Seizure. 2012; 21:118-123.
8. Rozycka A, Skorupska E, Kostyrko A, Trzeciak WH. Evidence for S284L mutation of the CHRNA4 in a white family with autosomal dominant nocturnal frontal lobe epilepsy. Epilepsia. 2003; 44:1113-1117.
9. Haeseleer F, Sokal I, Verlinde CL, Erdjument-Bromage H, Tempst P, Pronin AN, Benovic JL, Fariss RN, Palczewski K. Five members of a novel Ca2+-binding protein (CABP) subfamily with similarity to calmodulin. J Biol Chem. 2000; 275:1247-1260.
10. Sokal I, Li N, Verlinde CL, Haeseleer F, Baehr W, Palczewski K. Ca2+-binding proteins in the retina: from discovery to etiology of human disease. Biochim Biophys Acta. 2000; 1498:233-251.
11. Haeseleer F, Imanishi Y, Maeda T, Possin DE, Maeda A, Lee A, Rieke F, Palczewski K. Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat Neurosci. 2004; 7:1079-1087.
12. Zeitz C, Kloeckener-Gruissem B, Forster U, Kohl S, Magyar I, Wissinger B, Mátyás G, Borruat FX, Schorderet DF, Zrenner E, Munier FL, Berger W. Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am J Hum Genet. 2006; 79:657-667.
13. Littink KW, van Genderen MM, Collin RW, Roosing S, de Brouwer AP, Riemslag FC, Venselaar H, Thiadens AA, Hoyng CB, Rohrschneider K, den Hollander AI, Cremers FP, van den Born LI. A novel homozygous nonsense mutation in CABP4 causes congenital cone-rod synaptic disorder. Invest Ophthalmol Vis Sci. 2009; 50:2344-2350.
14. Aldahmesh MA, Al-Owain M, Alqahtani F, Hazzaa S, Alkuraya FS. A null mutation in CABP4 causes Leber’s congenital amaurosis-like phenotype. Mol Vis. 2010; 16:207.
15. Maeda T, Lem J, Palczewski K, Haeseleer F. A critical role of CaBP4 in the cone synapse. Invest Ophthalmol Vis Sci. 2005; 46:4320-4327.
16. Shaltiel L, Paparizos C, Fenske S, Hassan S, Gruner C, Rötzer K, Biel M, Wahl-Schott CA. Complex regulation of voltage-dependent activation and inactivation properties of retinal voltage-gated Cav1. 4 L-type Ca2+ channels by Ca2+-binding protein 4 (CaBP4). J Biol Chem. 2012; 287:36312-36321.
17. Park S, Li C, Haeseleer F, Palczewski K, Ames JB. Structural Insights into Activation of the Retinal L-type Ca2+ Channel (Cav1.4) by Ca2+-binding Protein 4 (CaBP4). J Biol Chem. 2014; 289:31262-31273.
18. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010; 7:248-249.
19. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009; 4:1073-81.
20. Lambert C, Leonard N, De Bolle X, Depiereux E. ESyPred3D: Prediction of proteins 3D structures. Bioinformatics. 2002; 18:1250-1256.