
INTRODUCTION
Non-small cell lung carcinoma (NSCLC) can be classified into distinct molecular subsets based on specific genomic alterations that drive tumorigenesis [1]. ALK rearrangement, most commonly EML4 -ALK, is detected in approximately 3–7% of unselected NSCLCs [2, 3]. EML4-ALK NSCLC is more frequently observed in patients with adenocarcinoma than with other diseases , in young adults than in older patients, and in non-smokers or light smokers (<15 packs/year) than in heavier smokers [4]. Crizotinib, a multiple tyrosine kinase inhibitor (TKI) of ALK, MET, and ROS1, is the only agent that has been approved for ALK-rearranged NSCLC. It shows dramatic clinical efficacy, with a response rate of about 60–80% and a progression free survival (PFS) of approximately 9–10 months in ALK-rearranged NSCLC patients [5]. However, almost all patients who strongly responded to crizotinib acquired resistance to these agents after varying periods of time [6].
Known mechanisms for resistance to crizotinib include the gatekeeper L1196M mutation [6], other secondary ALK gene mutations (F1174L, C1156Y, G1202R, S1206Y, 1151-T-ins, and G1269A) [7, 8, 9, 10], ALK amplification [7], and activation of bypass signals via activation of other receptors (KIT amplification and epidermal growth factor receptor (EGFR) autophosphorylation ) [8]. We recently reported that receptor ligands, such as epidermal growth factor (EGF), heparin binding-epidermal growth factor (HB-EGF), and transforming growth factor-α (TGF-α) , also activate EGFR as a bypass signal and induce crizotinib resistance in EML4-ALK NSCLC cells [11].
Alectinib is a highly selective, new generation ALK-TKI that also has inhibitory activity against EML4-ALK NSCLC cells with the gatekeeper L1196M mutation [12]. In a clinical trial for crizotinib-treatment naïve NSCLC patients with ALK rearrangement, there was a response rate of 93.5% to alectinib [13]. Moreover, alectinib demonstrated promising effects, even in the crizotinib-treated NSCLC patients with ALK rearrangement [14]. While it is clear that resistance may also develop against this class of inhibitor, the mechanisms of resistance to alectinib are largely unknown.
Heat shock protein 90 (Hsp90) is a molecular chaperone that plays a central role in regulating the correct folding, stability, and function of numerous “client proteins,” including human epidermal growth factor receptor 2 (HER2), BRAF , mutant EGFR, and EML4-ALK, Bcr-Abl, Raf-1, which are required for cancer cell survival [15, 16, 17, 18]. Hsp90 inhibition is therefore thought to be a promising strategy for controlling tumors, including those of EML4-ALK NSCLC. A natural product, geldanamycin, was found to directly bind to the ATP-binding pocket in the N-terminal domain of Hsp90 and block the binding of nucleotides to Hsp90; hence, geldanamycin was found to inhibit Hsp90 function. The first water-soluble, semi-synthetic derivative of geldanamycin is 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG) , which has shown excellent bioavailability and is quantitatively metabolized much less than other geldanamycin derivatives, such as 17-Allylamino 17-demethoxygeldanamycin (17-AAG) [19, 20].
In the present study, we examined whether receptor ligands would trigger resistance to a highly selective ALK-TKI, alectinib. Additionally, since we previously demonstrated that the Hsp90 inhibitor overcame EGFR-TKI resistance triggered by HGF, a ligand of MET, in EGFR-mutant lung cancer cells [21], we determined whether Hsp90 inhibition by 17-DMAG would overcome ligand-triggered alectinib resistance in ALK-rearranged NSCLC cells.
RESULTS
Exogenously added HGF and EGFR ligands induce resistance to alectinib in EML4-ALK NSCLC cells
Two EML4-ALK NSCLC cell lines, H2228 and H3122, were sensitive to crizotinib (IC50 0.3 µmol/L and 0.06 µmol/L, respectively). These cell lines were also sensitive to alectinib (IC50 0.24 µmol/L and 0.03 µmol/L, respectively). Exogenously added HGF and EGFR ligands (EGF, HB-EGF, and TGF-α) slightly stimulated cell growth, as determined by cell counting (Supplementary Fig. 1), and increased cell viability was determined by MTT assay (Fig. 1A-B). Under these experimental conditions, HGF and EGFR ligands remarkably reduced susceptibility of H2228 and H3122 cells to alectinib.
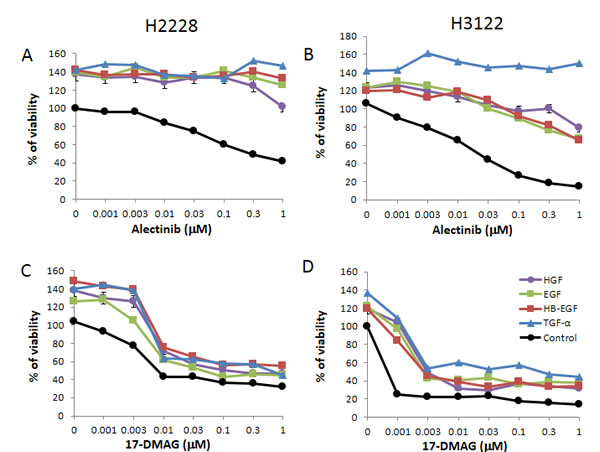
Figure 1: 17-DMAG suppresses the growth of EML4-ALK NSCLC cells in the presence of HGF and EGFR ligands. The EML4-ALK lung cancer cell lines human H2228 and human H3122 were treated with increasing concentrations of alectinib or 17-DMAG, with or without HGF (50 ng/mL), EGF (100 ng/mL), HB-EGF (10 ng/mL), and TGF-α (100 ng/mL), and cell viability was determined after 72 h by MTT assay. Data shown are representative of at least 3 independent experiments. Error bars indicate standard deviation (SD) of triplicate cultures.
17-DMAG inhibits the viability of EML4-ALK NSCLC cells, irrespective of the presence of exogenously added HGF or EGFR ligands
Only the Hsp90 inhibitor 17-DMAG inhibited the viability of H2228 (Fig. 1C) and H3122 (Fig. 1D) cells in a dose-dependent manner. Importantly, 17-DMAG inhibited the viability of H2228 and H3122 cells, even in the presence of HGF or EGFR ligands. These results suggest that 17-DMAG may overcome alectinib resistance triggered by HGF or EGFR ligands, such as EGF, HB-EGF, and TGF-α.
17-DMAG inhibits the viability of EML4-ALK NSCLC cells in the presence of endogenous HGF
Recently, HGF was reported to induce resistance to various molecular-targeted drugs in various types of cancers with oncogene drivers [22, 23]. Moreover, our previous study reported that HGF was overexpressed in the EGFR mutant cancer cells that acquired resistance to EGFR-TKIs, indicating endogenous HGF production by cancer cells [24]. These findings suggest that HGF can be overexpressed in EML4-ALK NSCLC cells that acquire resistance to ALK inhibitors.
Therefore, we next examined whether endogenously expressed HGF induced alectinib resistance in EML4-ALK NSCLC cells. To assess this question, we generated stable HGF-gene transfectants in H2228 cells (H2228/HGF); as a control, we generated H2228/Vec cells transfected with vector alone . H2228/HGF cells secreted high concentrations of HGF (16.0 ± 0.4 ng/mL), whereas the HGF concentrations secreted by H2228 and H2228/Vec cells were under the detection limit. Consistent with the results of exogenously added HGF, HGF-transfected H2228 (H2228/HGF) cells became insensitive to alectinib (Fig. 2A), indicating that endogenously-expressed HGF also induced resistance to alectinib in EML4-ALK NSCLC cells.
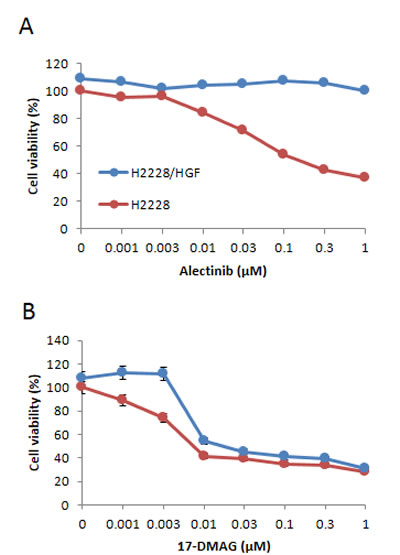
Figure 2: HGF-gene transfection resulted in reducing susceptibility of EML4-ALK NSCLC cells to alectinib but not 17-DMAG. H2228/Vec (A) or H2228/HGF (B) cells were treated with increasing concentrations of alectinib or 17-DMAG, and cell viability was determined after 72 h by MTT assay. Data shown are representative of at least 3 independent experiments. Error bars indicate SD of triplicate cultures.
We further found that 17-DMAG inhibited the growth of both H2228/Vec and H2228/HGF cells, because each had an IC50 of 0.01 µmol/L (Fig. 2B). These findings indicate that 17-DMAG may overcome alectinib resistance triggered by endogenously-produced HGF.
HGF reduces alectinib susceptibility via MET phosphorylation, and 17-DMAG reduces expression of ALK and MET
To explore the molecular mechanism by which HGF reduced susceptibility to alectinib and 17-DMAG inhibited cell growth, even in the presence of HGF, we examined the protein expression and phosphorylation status of MET, ALK, and their downstream molecules (PI3K/AKT, ERK1/2, and STAT3) by Western blotting (Fig. 3). Since HGF reduced alectinib susceptibility more potently in H2228 compared with H3122 cells (Fig. 1A-B), we mainly used H2228 cells in the following experiments. H2228 (data not shown) and H2228/Vec (Fig. 3) cells expressed ALK and MET proteins (ALK were phosphorylated but MET were not), as well as the downstream molecules AKT, ERK1/2, and STAT3. In the absence of HGF, alectinib inhibited ALK phosphorylation, thereby inhibiting AKT, ERK1/2, and STAT3 phosphorylation.
In the presence of HGF, alectinib failed to inhibit MET, AKT, and ERK1/2 phosphorylation, although it inhibited ALK and STAT3 phosphorylation. These results suggest that HGF reduced susceptibility to alectinib by mainly restoring AKT and ERK1/2 pathways via MET activation.
In parallel experiments, 17-DMAG decreased the expression of ALK and MET proteins and inhibited their phosphorylation and AKT, ERK1/2, and STAT3 phosphorylation, irrespective of HGF presence. Similar results were observed in H2228/HGF (Fig. 3) and H3122 cells (Supplementary Fig. 2). These results indicate that 17-DMAG decreases protein expression of ALK and MET, thereby suppressing downstream signaling and overcoming alectinib resistance caused by HGF.
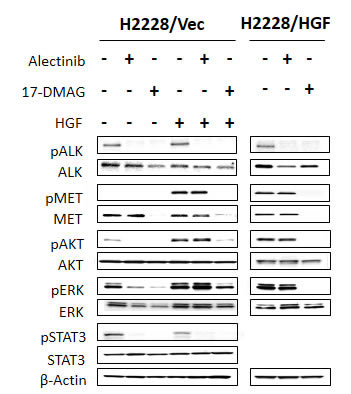
Figure 3: 17-DMAG reduced MET protein expression and inhibited downstream pathways, even in the presence of HGF. H2228/Vec or H2228/HGF cells were treated with or without alectinib (0.3 μmol/L) for 2 h or 17-DMAG (0.3 μmol/L) for 24 h and then stimulated with or without HGF (50 ng/mL) for 10 minutes. The resultant cells were lysed, and the indicated proteins were detected by immunoblotting. Data shown are representative of at least 3 independent experiments.
17-DMAG reduces EGFR and AKT protein expression and inhibits downstream pathways, even in the presence of EGFR ligands
We also examined the protein expression and phosphorylation status of EGFR and its downstream molecules in H2228 cells stimulated with EGFR ligands (Fig. 4). H2228 expressed EGFR, but EGFR was not constitutively phosphorylated in our experimental conditions. The EGFR ligands EGF, HB-EGF, and TGF-α remarkably induced EGFR phosphorylation. In these experimental conditions, alectinib failed to inhibit phosphorylation of EGFR or downstream AKT and ERK1/2, while it inhibited STAT3 phosphorylation. These results suggest that EGFR ligands reduced susceptibility to alectinib mainly by restoring AKT and ERK1/2 pathways via EGFR activation.
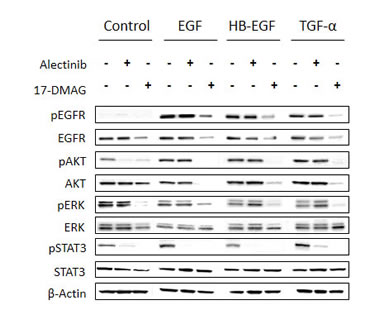
Figure 4: 17-DMAG reduced MET protein expression and inhibited downstream pathways, even in the presence of EGFR ligands. H2228 cells were treated with or without alectinib (0.3 μmol/L) for 2 h or 17-DMAG (0.3 μmol/L) for 24 h, and then stimulated with or without EGF (100 ng/mL), HB-EGF (10 ng/mL), and TGF-α (100 ng/mL) for 10 min. The resultant cells were lysed, and the indicated proteins were detected by immunoblotting. Data shown are representative of at least 3 independent experiments.
On the other hand, 17-DMAG decreased EGFR protein expression, resulting in inhibition of AKT, ERK1/2, and STAT3 phosphorylation, irrespective of the presence of EGFR ligands. These results suggest that 17-DMAG decreases EGFR protein expression, thereby suppressing downstream signaling and overcoming alectinib resistance triggered by EGFR ligands.
17-DMAG induces apoptosis of EML4-ALK lung cancer cells, even in the presence of HGF
We next assessed whether alectinib and 17-DMAG induced H2228/Vec cell apoptosis in the absence or presence of HGF. Alectinib induced apoptosis of H2228/Vec cells in the absence, but not presence, of HGF (Fig. 5). In contrast, 17-DMAG induced apoptosis in both the presence and absence of HGF. In a similar fashion, 17-DMAG, but not alectinib, induced H2228/HGF cell apoptosis.
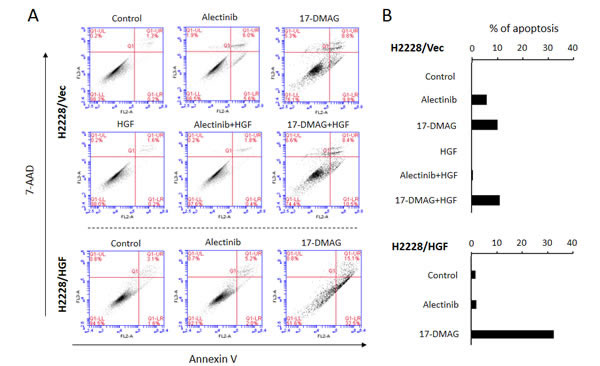
Figure 5: 17-DMAG induced apoptosis of EML4-ALK NSCLC cells, even in the presence of HGF. A. Apoptotic cells were evaluated by the 7-AAD cell viability assay , as described in the Materials and Methods. B. Quantification of apoptotic cells.
17-DMAG inhibits H2228 cell viability, even in the presence of both of HGF and EGFR ligands
Since several growth factors can be simultaneously produced in cancer microenvironments [25, 26], it is possible that HGF and EGFR ligands are co-expressed in EML4-ALK NSCLC cells . Crizotinib inhibits MET, ALK, and ROS1, and it is supposed to overcome alectinib resistance caused by HGF alone. We therefore examined the effect of 17-DMAG compared with crizotinib in the presence of HGF plus EGFR ligands. H2228 and H3122 cells became insensitive to alectinib in the presence of HGF, TGF-α, and HGF with TGF-α (Fig. 6). These cells were sensitive to crizotinib in the presence of HGF, but they became much less sensitive to crizotinib in the presence of TGF-α with or without HGF. However, H2228 and H3122 were sensitive to 17-DMAG in the presence of HGF, TGF-α, or HGF with TGF-α. These results suggest that 17-DMAG may overcome alectinib resistance, even in the presence of ligands for two different receptors.
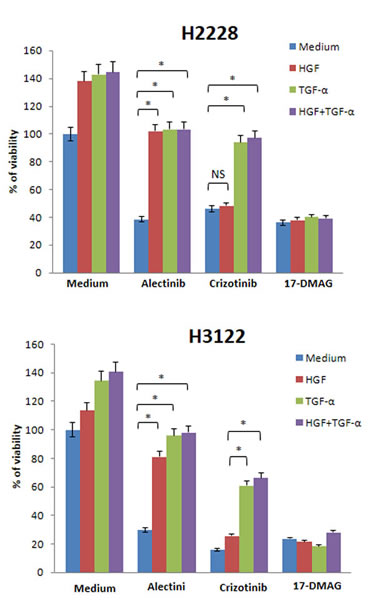
Figure 6: 17-DMAG reduced viability of EML4-ALK NSCLC cells, even in the presence of both HGF and TGF-α. H2228 and H3122 cells were incubated with or without alectinib (0.1 μmol/L), crizotinib (0.1 μmol/L), and/or HGF (50 ng/mL) and TGF-α (100 ng/mL), and cell viability was determined after 72 h by MTT assay. The percentage of cell viability is shown relative to controls without HGF or TGF-α treatment. * , P < 0.001 (one-way ANOVA). NS, not significant. Data shown are representative of at least 3 independent experiments. Error bars indicate SD of triplicate cultures.
DISCUSSION
We demonstrated that ligands of MET (HGF) and EGFR (EGF, HB-EGF, and TGF-α) triggered resistance to alectinib in ALK-rearranged NSCLC cells, and that the Hsp90 inhibitor 17-DMAG overcame the resistance triggered by these receptor ligands. 17-DMAG inhibited protein levels of ALK, EGFR, and MET, even in the presence of ligand activation, and suppressed of AKT and ERK1/2 phosphorylation, thereby inducing apoptosis of ALK-rearranged NSCLC cells, irrespective of the presence of HGF or EGFR ligands. Since the Hsp90 inhibitor by itself could inhibit both driver (from rearranged ALK) and resistance signals (from activated receptors; MET and EGFR), it may be an ideal agent for overcoming ligand-triggered alectinib resistance in ALK-rearranged NSCLC.
Activation of bypass signals is a common resistance mechanism for targeted drugs. For example, EGFR-TKI resistance could be caused by MET amplification [27], HGF-triggered MET activation [23], Gas6-triggered AXL activation [28], and HER2 amplification [29] in EGFR mutant lung cancer. BRAF inhibitor resistance could be caused by HGF-triggered MET activation [30], and IGF-1 triggered its receptor activation [31] in BRAF mutant melanoma. Crizotinib resistance could be caused by EGFR ligand-triggered EGFR activation [11], and stem cell factor (SCF)-triggered amplified cKIT activation [8] in EML4-ALK NSCLC. Therefore, HGF and EGFR ligands may be common resistance triggers that activate bypass survival signal via their receptor activation. The results in the present study are consistent with previous research indicating that alectinib resistance was induced by HGF and EGFR ligands.
Previous studies reported that several signaling pathways, including PI3K/AKT, MEK/ERK, and STAT3, are essential for survival and/or resistance to ALK inhibitors in ALK-rearranged NSCLC cells [12, 32]. Accordingly, we found that alectinib inhibited STAT3 and ALK phosphorylation. In the presence of alectinib, HGF or EGFR ligands restored AKT and ERK1/2, but not STAT3, phosphorylation and thereby made EML4-ALK cells insensitive to alectinib. These observations indicate that, when activated by their ligands, AKT and ERK signals from MET or EGFR play pivotal roles in alectinib resistance of EML4-ALK NSCLC cells.
It is of interest in the present study that HGF and EGFR ligands induced not only ALK-TKI resistance but also increased cell growth of EML4-ALK NSCLC cells. HGF and EGFR ligands also induced morphological change of H2228 cells (Supplementary Fig. 3). Therefore, these receptor ligands may modulate various cancer phenotypes of EML4-ALK NSCLC cells. HGF-MET and EGFR-ligands-EGFR axises play pivotal roles in progression of various types of tumors (33, 34). We are planning further studies to explore the molecular mechanisms of this morphological change and co-relation between the expression of receptor ligands in patient specimens and clinical characteristics in ALK-rearranged NSCLC.
Inter- and/or intra-tumor heterogeneity is a critical obstacle in cancer therapy with targeted drugs [35]. This is also the case in ALK-TKI resistance. Intra-tumor heterogeneity caused by crizotinib resistance results from the L1196M gatekeeper ALK mutation, and other ALK secondary C1156Y mutations co-existed in malignant pleural effusion of a patient who acquired crizotinib resistance [6]. Moreover, activation of different two receptors, EGFR and amplified KIT (both of which could induce crizotinib resistance), also co-existed in one crizotinib-resistant tumor [8].
Hsp90 inhibitors have been reported to overcome crizotinib resistance caused by several mechanisms, including ALK amplification, L1196M gatekeeper ALK mutation, other secondary ALK mutations (including F1174L), and epithelial to mesenchymal transition. Furthermore, we demonstrated that the Hsp90 inhibitor may overcome alectinib resistance, even when ligands of MET and EGFR co-exist . A new generation of Hsp90 inhibitors, including ganetespib, has recently been developed, and remarkable efficacy has been reported in a co-clinical model and early phase clinical studies [36]. Therefore, Hsp90 inhibition using new generation inhibitors may be a promising strategy to treat ALK-rearranged NSCLCs that acquire resistance to alectinib.
MATERIALS AND METHODS
Cell culture
The H2228 human lung adenocarcinoma cell line, with EML4-ALK fusion protein variant3 (E6;A20), was purchased from the American Type Culture Collection (Manassas, VA ). The H3122 human lung adenocarcinoma cell line, with EML4-ALK fusion protein variant1 (E13;A20), was kindly provided by Dr. Jeffrey A. Engelman of the Massachusetts General Hospital Cancer Center (Boston, MA) [37]. H2228 and H3122 cells were cultured in RPMI-1640 medium supplemented with 5% fetal bovine serum (FBS), penicillin (100 U/mL), and streptomycin (50 μg/mL) in a humidified CO2 incubator at 37°C. All cells were passaged for less than 3 months before renewal from frozen, early-passage stocks obtained from the indicated sources. Cells were regularly screened for Mycoplasma using a MycoAlert Mycoplasma Detection Kit (Lonza , Basel, Swiss).
Reagents
Alectinib, crizotinib, and 17-DMAG were purchased from Seleck Chemicals (Houston, TX). Recombinant EGF, TGF-α, and HB-EGF were purchased from R&D Systems (Minneapolis, MN). Recombinant HGF was prepared as described in a previous study [38].
Cell growth assay
Cell proliferation was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl terazolium bromide (MTT) dye reduction method [39]. Tumor cells were harvested at 80% confluence, seeded at 2 × 103 cells per well in 96-well plates, and incubated in appropriate medium for 24 h. Several concentrations of alectinib, crizotinib, 17-DMAG, and/or EGF, TGF-α, HB-EGF, and HGF were added to each well, and incubation continued for another 72 h. Fifty μL MTT (2 mg/mL; Sigma, St.Louis, MO) was added to each well, followed by incubation for 2 h at 37°C. The media were removed and the dark blue crystals in each well were dissolved in 100 μL of dimethyl sulfoxide (DMSO). Absorbance was measured with an MTP-120 Microplate reader (Corona Electric , Hitachinaka, Ibaraki, Japan) at test and reference wavelengths of 550 and 630 nm, respectively. The percentage growth was calculated relative to untreated controls. Each assay was carried out at least in triplicate, with results based on 3 independent experiments.
HGF-gene transfection
One day before transfection, aliquots of 1 × 105 H2228 cells in 1 mL of antibiotic-free medium were plated on 6-well plates. The full-length HGF cDNA cloned into the BCMGSneo expression vector [40] was transfected using Lipofectamine 2000 according to the manufacturer’s instructions. After incubation for 24 h, the cells were washed with phosphate buffered saline (PBS) and incubated for an additional 72 h in antibiotic-containing medium. Then, the cells were selected in G418 sulfate (Calbiochem, Jolla, CA). After limiting dilution, the HGF-producing cells, H2228/HGF, were established. HGF production by H2228/HGF was confirmed by enzyme linked immunosolvent assay (ELISA) .
HGF production
Cells (2 × 105) were cultured in RPMI-1640 medium with 10% FBS for 24 h. The cells were washed with PBS and incubated for 48 h in 2 mL of RPMI-1640 medium with 10% FBS. Then, culture medium was harvested and centrifuged, and the supernatant was stored at –70°C until analysis. HGF concentrations were determined by IMMUNIS HGF EIA (Institute of Immunology, Tokyo) according to the manufacturer’s protocols. All samples were run in duplicate. Color intensity was measured at 450 nm using a spectrophotometric plate reader. Growth factor concentrations were determined by comparison with standard curves, and the HGF detection limit was 100 pg/mL.
Apoptosis assay
Cell apoptosis induced by alectinib and 17-DMAG was measured by the PE Annexin V Apoptosis Detection Kit I (BD Biosciences, San Jose, CA) which detects and quantifies apoptotic cells with phycoerythrin (PE) Annexin V and 7-amino-actinomycin (7-AAD) staining. Cells were analyzed on a FACSCalibur flow cytometer with CellQuest software (Becton Dickinson, Franklin Lakes, NJ).
Western blotting
Sodium dodesyl sulfate (SDS) polyacrylamide gels (Bio-Rad , Hercules, CA) were loaded with 40 μg total protein per lane; following electrophoresis, the proteins were transferred onto polyvinylidene difluoride membranes (Bio-Rad ), which were incubated with Blocking One (Nacalai Tesque, Kyoto, Japan ) for 1 h at room temperature, followed by overnight incubation at 4°C with anti-ALK (C26G7), anti-phospho-ALK (Tyr1604), anti-phospho-EGFR (Tyr1068), anti-STAT3 (79D7), anti-phospho-STAT3 (Y705), anti-AKT, anti-phospho-AKT (Ser473), anti-ErbB4 (111B2), anti-phospho-ErbB4 (Tyr1284), anti-MET (25H2), anti-phospho-MET (Y1234/Y1235) (3D7), or anti-β-actin (13E5) antibodies (1:1,000 dilution each; Cell Signaling Technology , Danvers, MA), or with anti-human EGFR (1 μg/mL), anti-human/mouse/rat extracellular signal-regulated kinase (Erk)1/Erk2 (0.2 μg/mL), or anti-phospho-Erk1/Erk2 (T202/Y204) (0.1 μg/mL) antibodies (R&D Systems ). After washing 3 times, the membranes were incubated for 1 h at room temperature with secondary antibodies (horseradish peroxidase-conjugated species-specific antibodies).
Immunoreactive bands were visualized with SuperSignal West Dura Extended Duration Substrate Enhanced Chemiluminescent Substrate (Pierce, Osaka, Japan ). Each experiment was independently carried out at least 3 times.
Statistical analysis
Differences were analyzed by one-way ANOVA. All statistical analyses were carried out using GraphPad StatMate 4 (GraphPad Software, Inc ., San Diego, CA). P < 0.05 was considered significant.
ACKNOWLEDGMENTS
The authors thank Dr. Jeffrey A. Engelman (Massachusetts General Hospital Cancer Center) for providing the H3122 cells.
Grant support:
This study was supported by Grants-in-Aid for Cancer Research (Yano, 21390256), Scientific Research on Innovative Areas “Integrative Research on Cancer Microenvironment Network” (S. Yano, 22112010A01), and P-DIRECT from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Disclosure statement
Seiji Yano received honoraria and research funding from Chugai Pharmaceutical Co., Ltd.
REFERENCES
1. Oxnard GR, Binder A, Janne PA. New targetable oncogenes in non-small-cell lung cancer. Journal of clinical oncology. 2013; 31(8):1097-1104.
2. Camidge DR, Doebele RC. Treating ALK-positive lung cancer--early successes and future challenges. Nature reviews. Clinical oncology. 2012; 9(5):268-277.
3. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007; 448(7153):561-566.
4. Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, Solomon B, Stubbs H, Admane S, McDermott U, Settleman J, Kobayashi S, Mark E.J, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. Journal of clinical oncology. 2009; 27(26):4247-4253.
5. Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, Riely G.J, Solomon B, Ou S.H, Kim D.W, Salgia R, Fidias P, Engelman J.A, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. The lancet oncology. 2012; 13(10):1011-1019.
6. Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, Yatabe Y, Takeuchi K, Hamada T, Haruta H, Ishikawa Y, Kimura H, Mitsudomi T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. The New England journal of medicine. 2010; 363(18):1734-1739.
7. Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo K.L, Linderman D.J, Heasley L.E, Franklin W.A, Varella-Garcia M, Camidge D.R, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clinical cancer research. 2012; 18(5):1472-1482.
8. Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop N.A, Wain J.C, Yeo A.T, Benes C, Drew L, Saeh J.C, Crosby K,et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Science translational medicine. 2012; 18(5):1472-1482.
9. Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer research. 2011; 71(18):6051-6060.
10. Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer research. 2010; 70(24):10038-10043.
11. Yamada T, Takeuchi S, Nakade J, Kita K, Nakagawa T, Nanjo S, et al. Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clinical cancer research. 2012; 18(13):3592-3602.
12. Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011; 19(5):679-690.
13. Seto T, Kiura K, Nishio M, Nakagawa K, Maemondo M, Inoue A, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1-2 study. The lancet oncology. 2013; 14(7):590-598.
14. Alexander W, 2013 European cancer congress. P & T. 2013; 38(11):709-711.
15. Normant E, Paez G, West KA, Lim AR, Slocum KL, Tunkey C, et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene. 2011; 30(22):2581-2586.
16. MV Blagosklonny. Hsp-90-associated oncoproteins: multiple targets of geldanamycin and its analogs. Leukemia. 2002; 16(4):455-62.
17. Demidenko ZN, An WG, Lee JT, Romanova LY, McCubrey JA, Blagosklonny MV et al. Kinase-addiction and bi-phasic sensitivity-resistance of Bcr-Abl- and Raf-1-expressing cells to imatinib and geldanamycin. Cancer Biol Ther. 2005; 4(4):484-90.
18. de Billy E, Travers J, Workman P. Shock about heat shock in cancer. Oncotarget. 2012; 3(8):741-3.
19. Egorin MJ, Lagattuta TF, Hamburger DR, Covey JM, White KD, Musser SM, Eiseman J.L. Pharmacokinetics, tissue distribution, and metabolism of 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (NSC 707545) in CD2F1 mice and Fischer 344 rats. Cancer Chemother Pharmacol. 2002; 49(1):7-19.
20. Eiseman JL, Lan J, Lagattuta TF, Hamburger DR, Joseph E, Covey JM, Egorin MJ. Pharmacokinetics and pharmacodynamics of 17-demethoxy 17-[[(2-dimethylamino)ethyl]amino]geldanamycin (17DMAG, NSC 707545) in C.B-17 SCID mice bearing MDA-MB-231 human breast cancer xenografts. Cancer Chemother Pharmacol. 2005; 55(1):21-32.
21. Koizumi H, Yamada T, Takeuchi S, Nakagawa T, Kita K, Nakamura T, et al. Hsp90 inhibition overcomes HGF-triggering resistance to EGFR-TKIs in EGFR-mutant lung cancer by decreasing client protein expression and angiogenesis. Journal of thoracic oncology. 2012; 7(7):1078-1085.
22. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nature reviews. Molecular cell biology. 2003; 4(12):915-925.
23. Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, Ogino H, Kakiuchi S, Hanibuchi M, Nishioka Y, Uehara H, Mitsudomi T, Yatabe Y, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer research. 2008; 68(22):9479-9487.
24. Yano S, Yamada T, Takeuchi S, Tachibana K, Minami Y, Yatabe Y, et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. Journal of thoracic oncology. 2011; 6(12):2011-2017.
25. Masuya D, Huang C, Liu D, Nakashima T, Kameyama K, Haba R, Ueno M, Yokomise H, et al. The tumour-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non-small-cell lung cancer patients. British journal of cancer. 2004; 90(8):1555-1562.
26. Meert AP, Martin B, Delmotte P, Berghmans T, Lafitte JJ, Mascaux C, et al. The role of EGF-R expression on patient survival in lung cancer: a systematic review with meta-analysis. The European respiratory journal. 2002; 20(4):975-981.
27. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316(5827):1039-1043.
28. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat genetics. 2012; 44(8):852-860.
29. Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, et al. HER2 Amplification: A Potential Mechanism of Acquired Resistance to EGFR Inhibition in EGFR-Mutant Lung Cancers That Lack the Second-Site EGFRT790M Mutation. Cancer discovery. 2012; 2(10):922-933.
30. Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012; 487(7408):500-504.
31. Hilmi C, Larribere L, Giuliano S, Bille K, Ortonne JP, Ballotti R, et al. IGF1 promotes resistance to apoptosis in melanoma cells through an increased expression of BCL2, BCL-X(L), and survivin. J Invest Dermatol. 2008; 128(6):1499-1505.
32. Takezawa K, Okamoto I, Nishio K, Janne PA, Nakagawa K. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clinical cancer research. 2011; 17(8):2140-2148.
33. Natan S, Tsarfaty G, Horev J, Haklai R, Kloog Y, Tsarfaty I, et al. Interplay between HGF/SF-Met-Ras signaling, tumor metabolism and blood flow as a potential target for breast cancer therapy. Oncoscience. 2014; 1: 30-38.
34 Tang C, Fontes Jardim DL, Falchook GS, Hess K, Fu S, Wheler JJ, et al. MET nucleotide variations and amplification in advanced ovarian cancer: characteristics and outcomes with c-Met inhibitors. Oncoscience. 2014; 11: 5-13.
35. Janku F. Tumor heterogeneity in the clinic: is it a real problem?. Therapeutic advances in medical oncology. 2014; 6(2):43-51.
36. Socinski MA, Goldman J, El-Hariry I, Koczywas M, Vukovic V, Horn L, Paschold E,Salgia R, West H, Sequist L.V, Bonomi P, Brahmer J, Chen L.C, et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clinical cancer research. 2013; 19(11):3068-3077.
37. Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clinical cancer research. 2008; 14(13):4275-4283.
38. Montesano R, Matsumoto K, Nakamura T, Orci L. Identification of a fibroblast-derived epithelial morphogen as hepatocyte growth factor. Cell. 1991; 67(5):901-908.
39. Yasumoto K, Yamada T, Kawashima A, Wang W, Li Q, Donev IS, et al. The EGFR ligands amphiregulin and heparin-binding egf-like growth factor promote peritoneal carcinomatosis in CXCR4-expressing gastric cancer. Clinical cancer research. 2011; 17(11):3619-3630.
40. Ueki T, Kaneda Y, Tsutsui H, Nakanishi K, Sawa Y, Morishita R, Matsumoto K, Nakamura T, Takahashi H, Okamoto E, Fujimoto J, et al. Hepatocyte growth factor gene therapy of liver cirrhosis in rats. Nature medicine. 1999; 5(2):226-230.