
INTRODUCTION
Hedgehog (HH) signaling plays a critical role in normal cellular processes. It is pivotal in embryogenesis, tissue patterning, and differentiation [1-3]. The canonical HH pathway is critical to normal mammalian gastrointestinal development, where it is involved in the coordinate regulation of differentiation of normal intestinal villi [4-6]. The GLI genes, GLI1 and GLI2, are transcription factors that regulate target genes at the distal end of the canonical HH pathway (SHH->PTCH->SMO->GLI). Their expression in these processes is tightly regulated [1-3], with little expression detected in adult tissues [7]. GLI1 and GLI2 are transcriptional activators, binding to GACCACCCA-like consensus promoter sequences [1, 8, 9]. From genetic and biochemical studies, we and others suggest that GLI2 is the primary mediator of HH signaling, which activates GLI1 to transcriptionally regulate target genes and augment HH signaling quantitatively as well as qualitatively [1, 9-11]. Differences in the biological activities of GLI1 and GLI2 are evident, since GLI1-/- mice have no obvious phenotype [11], in contrast to homozygous GLI2-/- mice which die at birth [12, 13]. During development, GLI1 is strongly expressed along the midline and is a marker of the response to SHH. In contrast, GLI2 is expressed in the lateral regions, suggesting regulation by alternate factors [14]. GLI1 and GLI2 possess both independent and overlapping functions [1, 9-12, 15].
Both GLI1 and GLI2 are oncogenes, induce transformation and tumorigenesis [16-18], and are constitutively activated in many types of human cancers [1, 15]. Failure to terminate HH/GLI signaling, which occurs in cancer, leads to an amplified and persistent increase in GLI1 and GLI2 activity (reviewed in [15]). Amplification of GLI1 or GLI2, mutations in PTCH or SMO, upregulated expression of HH ligands, and activating mutations that initiate transformation can dysregulate HH signaling [1, 15]. Small molecule inhibitors of SMO upstream of GLI have probed the canonical, HH-SMO-GLI axis in preclinical models [19-25] and in human cancers [24, 26-28]. SMO inhibitors have limited or no clinical activity (GDC-0449, IPI-926, LDE225; reviewed in [24, 26]), except in a small number of HH-GLI-dependent tumors (e.g. basal cell carcinoma [29, 30], medulloblastoma [26, 31]). Acquired resistance to SMO antagonists also occurs [32].
Constitutive GLI activation progresses during colon carcinogenesis and in metastatic disease [21, 33, 34] by ligand-dependent (canonical) and ligand–independent (oncogenic) mechanisms [35-39]. Oncogenic pathways (KRAS/BRAF in colon cancer) circumvent the canonical HH-GLI axis by converging on and further driving GLI to a higher activating state in tumor cells, promoting cellular proliferation, tumor progression and survival [8, 15, 19, 40-42 43, 44]. Thus, potential targets upstream of GLI are bypassed, including SMO. Activating mutations in both KRAS [15, 42, 45-49] and BRAF [19, 48, 50, 51] are prevalent, and occur in high frequency in colon cancers [47-49, 51-53]. We have demonstrated that oncogenic KRAS/BRAF signaling activates GLI independent of the HH-SMO-GLI axis [38], inhibited by pharmacologic inhibitors of MEK (U0126 [38], AZD6244), and by GANT61, which targets GLI and GLI-dependent transcription. We have demonstrated that MEK inhibitors reduce GLI-luciferase activity [38]. Thus, GANT61 is effective when GLI (GLI1+GLI2) serves as a common node of activation through which oncogenic signals converge (schema, Figure 1). Due to the extensive cytotoxicity induced by GANT61 in human models of colon cancer [36, 38, 39], data suggest that GLI is a critical target in colon cancer cell survival, and also in other cancers where GLI is constitutively activated and/or an oncogenic KRAS-GLI axis drives proliferation.
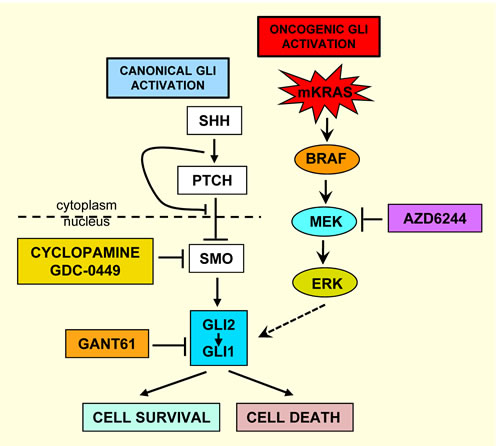
Figure 1: Schema of pathways for the aberrant activation of GLI in colon cancer
GANT61, an experimental agent in preclinical studies, was originally identified in a cell-based screen for small molecule inhibitors of GLI1-mediated transcription [54]. In this study GANT61 abrogated GLI function in the nucleus, blocked both GLI1- and GLI2- mediated transcription, inhibited GLI1-DNA binding, and demonstrated anti-tumor activity against human prostate cancer xenografts. We have demonstrated rapid inhibition of GLI1 and GLI2 binding to target gene promoters (1 hr), reduced reporter activity specific to GLI-luciferase, and rapid inhibition of gene transcription in human colon carcinoma cell lines [37]. Overexpression of GLI1 or GLI2 also protects cells from GANT61-mediated cell death [39]. Due to our findings of the critical role of GLI in colon cancer cell survival, and the importance of GANT61 as a unique small molecule inhibitor, we sought to determine the mechanism and specificity of GANT61 binding activity. We investigated whether GANT61 binds to the GLI protein or to DNA sequences, and further determined the exact sites of interaction.
GLI1 and GLI2 are zinc finger proteins, one of the most common DNA-binding motifs in eukaryotic transcription factors [7, 55]. The crystal structure of the five zinc finger GLI1-DNA complex is known (PDB ID 2GLI) [55]. Fingers 2 through 5 of GLI1 bind in the major groove and wrap around the DNA, with fingers 4 and 5 making the most extensive base contacts in a conserved 9-bp region. Fingers 2 and 3 only make a single base contact. Finger 1, which does not contact the DNA, makes extensive protein-protein interactions with Finger 2 [55]. To gain insight into the structural binding characteristics of GANT61, our findings from computational docking of GANT61 to PDB ID 2GLI, and experimentally by Surface Plasmon Resonance (SPR) technology, determined that docking of GANT61 to both the GLI1 protein and to the GLI1-DNA complex was identical; GANT61 did not bind to DNA or to other zinc finger transcription factors. The KD for dissociation (3.2-7.5 µM) and GANT61 concentrations that induce cell death in intact cells (10-20 µM) are in the same low µM range. These studies critically evaluate the mode of binding of GANT61, and identify that GANT61 specifically binds to GLI1 protein, which is strongly dependent on 2 amino acids. Overall these findings substantiate the importance of GLI as a target in cancers with activated GLI and/or oncogenic KRAS/BRAF signaling, and that inhibition of GLI-dependent transcription by a specific small molecule inhibitor can have profound effects on cell survival.
RESULTS
Critical role of GLI in cell survival
Following 72 hr drug exposure with equimolar concentrations of drugs (20 µM), inhibition of SMO in human colon carcinoma cell lines by GDC-0449 induced minimal cell death [36, 38, 39], while cell lines (HT29, SW480, HCT116, GC3/c1) with aberrant KRAS/BRAF signaling [51] demonstrated sensitivity to the MEK inhibitor AZD6244. GANT61 induced extensive cell death following termination of oncogenic signaling at the level of GLI in all of the 7 cell lines (Figure 2).
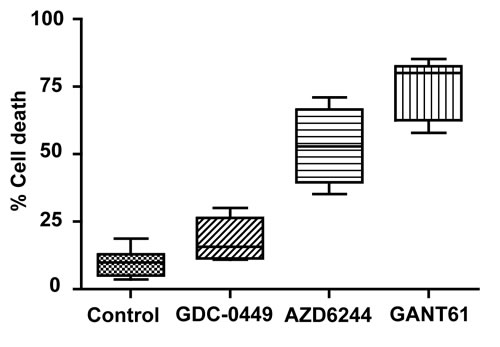
Figure 2: Human colon carcinoma cell lines were treated for 72 hr, in duplicate, to equimolar concentrations (20 µM) of GDC-0449 (n=5), AZD6244 (n=4), GANT61 (n=7), or were untreated (n=7). Cells were harvested by trypsinization, cell death analyzed by Annexin V/PI staining and FACS analysis, and raw data quantitated using CellQuest software, as described in Materials and Methods. Data represent the Mean, Range, and SD.
Computational docking of GANT61 to GLI1 or DNA
Computational docking analysis of GANT61 to GLI1, DNA, or the GLI1-DNA complex utilized GANT61-diamine, the active form of GANT61, in the analysis (Figure 3A). Docking predicted that GANT61-diamine binds to the GLI1 protein, but not to the DNA binding site, at amino acids E119 and E167, which lie within the groove between zinc fingers 2 and 3, on the opposite surface from, but in close proximity to, the GLI1-DNA binding region (Figure 3B). Predicted docking of GANT61-diamine is via two-way H bonds at E167 (2.2Å) involving the two imino protons of GANT61-diamine, and one H bond at E119 involving one imino proton (Figure 3C). No significant change in GANT61-diamine docking was observed when a complex of GLI1-DNA was used as the target; GANT61-diamine was not observed to bind with DNA. Alignment of GLI1 and GLI2 amino acid sequences, using Clustal 2.1, demonstrated that the residues within 3.5 Å of GANT61-diamine ligand atoms are conserved (Figure 3D). Computational modeling predicts the biological activity of GANT61 to be mediated by a direct GANT61-GLI1 interaction.
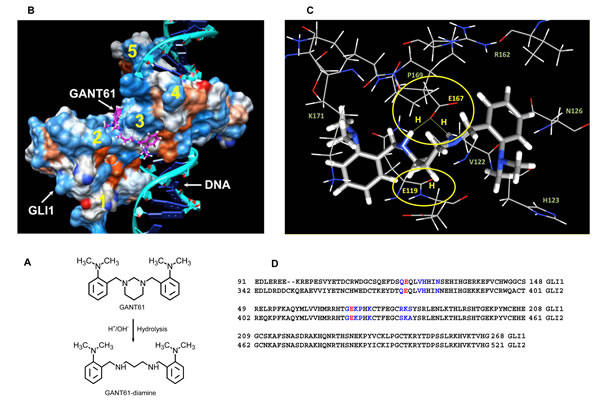
Figure 3: Computational docking of GANT61 to GLI1 using the known crystal structure of the five zinc finger GLI1-DNA complex (PDB ID 2GLI) [55]. A: Two dimensional chemical structures of GANT61 and GANT61-diamine drawn in ACD-ChemSketch (Advanced Chemistry Inc.); B: Computational modeling of GANT61-diamine binding predicts binding to GLI1 within the groove between fingers 2 and 3; zinc fingers 1-5 (yellow); C: Predicted GANT61-diamine bound to GLI1 at amino acids E119 (1 H bond) and E167 (2 H bonds), amino acids residues within 3.5 Å of any atom of GANT61 are shown as line. D: GLI1 and GLI2 alignment ; E119, E167 (red); residues within 3.5 Å of docked GANT61-diamine (blue).
GANT61 inhibits GLI1-DNA interactions by binding to GLI1 and not to DNA
To critically evaluate the structural characteristics of the GANT61-GLI-DNA binding interactions, we utilized SPR technology. To confirm the GLI-DNA interactions, biotinylated DNA (100 nM) was immobilized on a streptavidin pre-coated SA sensor chip (GE Healthcare) with varying concentrations of GLI1 (1–1,000 nM) as the analyte. Full length GLI1 binds to immobilized DNA on the sensor chip in a dose dependent manner (Figure 4A); the dissociation constant for binding (KD) was 5.5 µM (Figure 4C). To determine whether GANT61 binds directly to GLI1 as predicted from the docking model, GLI1 was immobilized on a CM5 sensor chip with varying concentrations of GANT61 (1–50 µM) as the analyte. A dose-dependent increase in the resonance response was observed (Figure 4B). Significant binding was determined at 10 µM GANT61 and was close to maximum at 25 µM GANT61. The KD for GANT61-GLI1 binding was 7.5 µM. To determine whether the binding of GANT61 was specific to GLI1 and not to other zinc finger transcription factors, KLF4 (Kruppel-like factor) or TFIIβ (general transcription factor) were each immobilized to sensor chips, and a supraphysiological concentration of GANT61 (40 µM) used as the analyte. Binding was compared to GANT61-∆GLI-WT binding. No significant binding of GANT61 to KLF4 or TFIIβ was detected (Figure 4D). To investigate whether GANT61 is able to disrupt the DNA-GLI1 interaction, and also to determine whether GANT61 binds to DNA, DNA (100 nM) was immobilized, and GANT61 (50 µM) in the absence or presence of GLI1 (100 nM) was employed as the analyte. In the absence of GLI1, there was no detectable binding of GANT61 to DNA. In contrast, in the absence of GANT61, significant binding of GLI1 to DNA was determined (Figure 5A). Subsequently, increasing concentrations of GANT61 (1–50 µM) were employed as the analyte in the presence of a fixed quantity of GLI1 (100 nM). A significant decrease in the binding of GLI1 to DNA was determined with GANT61 (5 µM), and close to maximal inhibition of binding at 10 µM GANT61 (Figure 5A). Maximum RU in the presence of GLI1 demonstrated a concentration-dependent decrease as the GANT61 concentration was increased and GLI1-DNA binding was inhibited (Figure 5B). The KD for GANT61 inhibition of GLI1-DNA binding was 3.2 µM (Figure 4C).
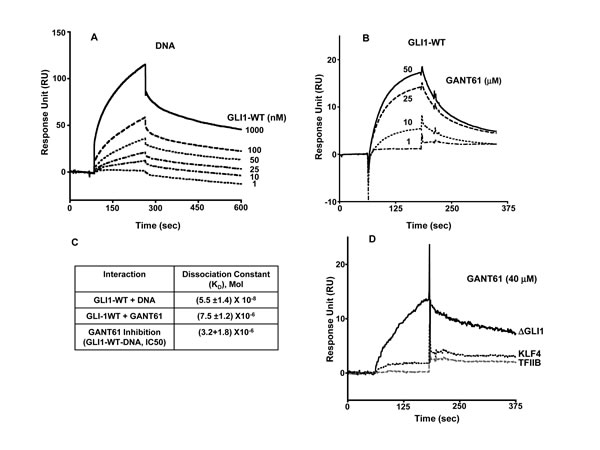
Figure 4: Surface Plasmon Resonance (SPR) of: A: Immobilized DNA with increasing concentrations of GLI1-WT as analyte: B: Immobilized GLI1-WT with determination of GANT61 binding in presence of increasing concentrations of GANT61 (1-50 µM); C: Dissociation Constants (KD) for the interaction between GLI1-WT and DNA, GLI1-WT and GANT61, or inhibition of GLI1-WT-DNA binding by GANT61; D: Binding of GANT61 (40 µM) to immobilized ∆GLI-WT in contrast to no binding of GANT61 (40 µM) to immobilized KLF4 or TFIIβ. The analyte was exposed to sensor chips for 3 min followed by 5 min in the absence of analyte as described in Materials and Methods. Response Units are shown vs time of incubation (sec).
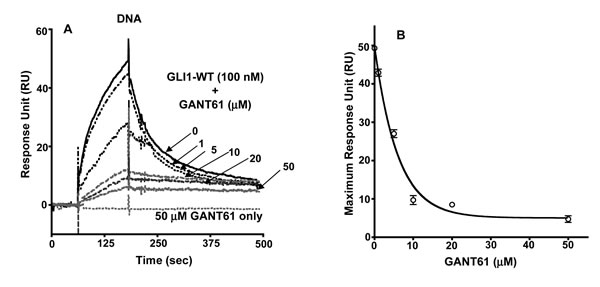
Figure 5: SPR of: A: Immobilized DNA +/- GLI1 (100 nM) +/- GANT61 at varied concentrations (1-50 µM); B: Maximum RU vs concentration of GANT61.
E119A and E167A mutations reduce GANT61 binding to GLI1
To determine the functional significance of the putative sites (E119, E167) on GLI1 in the GANT61-GLI1 interactions, fragments containing E119A and E167A mutations were amplified by PCR, cloned, expressed and purified using Ni-NTA affinity chromatography, as described in Materials and Methods. Purified proteins were immobilized on Ni-NTA sensor chips, and GANT61 (10-40 µM) was used as the analyte (∆GLI-WT, Figure 6A; ∆GLI-DM, Figure 6B). Significant inhibition of GANT61-GLI binding was determined following mutation of the two predicted binding sites, indicating the critical importance of these sites in the small molecule-protein interaction. Upon further analysis of a comparison of RUMax values (Figure 6C), mutagenesis of the two binding sites inhibited GANT61-GLI binding by ≈ 60%, yielding KD values for ∆GLI-WT and ∆GLI-DM of 11.2 µM and 26.1 µM, respectively (Figure 6D).
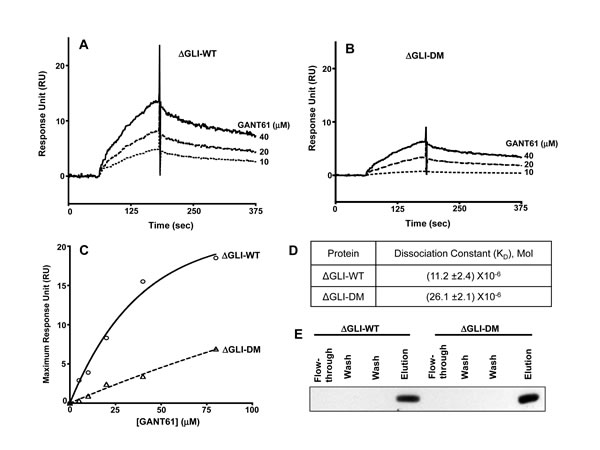
Figure 6: SPR conducted with immobilized ∆GLI-WT or ∆GLI-DM proteins following site directed mutagenesis of GLI1-WT, PCR amplification and protein purification of ∆GLI-WT without mutation, or ∆GLI-DM with both E119 and E167 sites mutated. Immobilized A: ∆GLI-WT or B: ∆GLI-DM with varied concentrations of GANT61 (10-40 µM) as analyte; C: Maximum Response for binding of GANT61 to ∆GLI-WT or ∆GLI-DM at varied concentrations of GANT61. D: Dissociation Constants (KD) for binding of GANT61 to ∆GLI-WT or ∆GLI-DM proteins; E: Purification of ∆GLI-WT or ∆GLI-DM proteins on Ni-NTA columns as described in Materials and Methods.
GLI-luciferase activity
To evaluate the biological significance of the mutations introduced in the GLI1 protein (E119A, E167A), the effect of full length GLI1-DM on GLI-luc reporter activity was determined in HT29 cells. The cells were transiently cotransfected with pBabe-Puro, GLI1-WT cDNA, or GLI1-DM cDNA and both GLI-luc and pRLTK. Cells expressing GLI1-WT demonstrated a 7-fold increase in GLI-luc activity within 24 hr of transfection; in contrast, GLI1-DM increased GLI-luc activity by only 2-fold. Further, cells expressing GLI1-DM did not demonstrate decreased GLI-luc activity while cells expressing GLI1-WT demonstrated reduced GLI-luc activity in response to GANT61 (20 μM, 24 hr; Figure 7A). To further corroborate the biological effect of the GLI1-DM mutant, we employed an HT29-derived stable cell line that constitutively expresses GLI-luc. HT29-GLI-luc cells were transfected with pBabe-Puro, GLI1-WT or GLI1-DM plasmids and luciferase activity was visualized by live cell imaging. Similar to the findings with co-transfection, cells expressing GLI1-WT demonstrated increased GLI-luc activity while cells expressing GLI1-DM demonstrated reduced GLI-luc activity (Figure 7B). Administration of GANT61 (20 μM, 24 hr) reduced GLI-luc activity in both the untransfected and GLI1-WT-expressing cells but not in cells expressing GLI1-DM, which were insensitive (Figure 7B). Collectively, data demonstrate that mutating both GANT61 binding sites in the full length GLI1 cDNA significantly inhibits GLI1-DNA binding activity, resulting in reduced GLI-luc reporter activity, in addition to rendering resistance to GANT61.
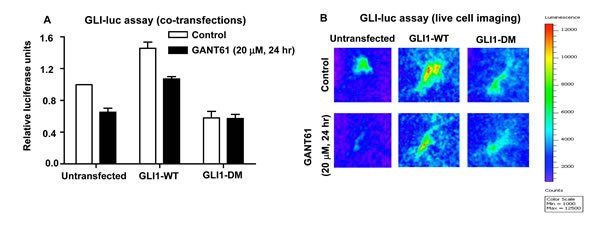
Figure 7: GLI-luciferase reporter assays in HT29 cells under 2 different conditions: A: Co-transfection of GLI-luc, pRLTK, and either pBabe-Puro, ∆GLI1-WT or ∆GLI1-DM cDNA into HT29 cells using Lipofectamine 2000; 24 hr post-transfection, cells were treated with GANT61 (20 µM) for 24 hr, and luciferase activity determined as described in Materials and Methods. B: An HT29-derived stable cell line expressing GLI-luc was exposed to GANT61 (20 µM) for 24 hr. Live cell imaging was performed using a Bruker optical and X-ray small animal imaging system, as described in Materials and Methods.
DISCUSSION
GLI is constitutively activated in a wide variety of human cancers, and we have demonstrated the critical role of GLI as a target in colon cancer cell survival using GANT61 as a probe [35-39]. In contrast we have demonstrated minimal effects on GLI targets in human colon carcinoma cells in response to physiologically relevant concentrations of SMO inhibitors [37-39]. Cells respond at higher concentrations of GDC-0449 (100 µM), used to select for drug resistance, and these cells remain sensitive to GANT61 [39]. The relative inactivity of SMO inhibitors in this model and others except in certain HH-GLI-dependent tumors [19-28], underscores the importance of activation of GLI by non-canonical pathways that drive cell proliferation and survival, in particular oncogenic KRAS/BRAF. We have shown decreased p-Erk and reduced expression of GLI targets and GLI-luciferase activity in human colon carcinoma cells following exposure to MEK inhibitors [38]. Cell line models with an activated KRAS pathway are found to be more sensitive to inhibition of MEK [56]. GLI therefore serves as a point of convergence and nodal activation by oncogenic signaling pathways, made evident by the extensive cytotoxicity induced by the small molecule inhibitor of GLI transcription, GANT61. However little is known regarding the mechanism by which GANT61 inhibits transcriptional activity of the GLI proteins.
The GLI genes are members of the GLI-Kruppel family of transcription factors [7, 55, 57]. Analysis of the GLI gene sequence indicates that it encodes a 118 kd protein that contains five repeats of a zinc finger DNA-binding motif. From the crystal structure of the 5-zinc finger GLI-DNA complex, it has been determined that finger 1 does not contact the DNA, while fingers 2-5 bind in the major groove and wrap around the DNA. Zinc fingers 4 and 5 of GLI1 make extensive base contacts in a conserved 9-base pair region [55]. Using this crystal structure of the GLI1-DNA complex, predicted docking of GANT61 to the GLI1 protein, the GLI1-DNA complex or to DNA itself, was determined. The predictions were that GANT61 binds between zinc fingers 2 and 3 of the GLI1 protein, at amino acids E119 and E167 by H bonds, in close proximity to, but independent of the DNA binding region. Close inspection revealed that the molecular surface where GANT61 binds is opposite from the groove where DNA binds. The sequence alignment showed that most of the residues within 3.5 Å of GANT61 are conserved between GLI1 and GLI2. BIAcore instrumentation using SPR-based biosensors has made it possible to routinely study the binding of small molecules (< 500 kD) to targets [58], and has been successfully employed to directly measure and quantitate the binding of small molecule inhibitors to target proteins, e.g. the p38-mitogen-activated protein kinase (MAPK) [59]. First, binding of GLI1 to immobilized DNA, or binding of GANT61 to immobilized GLI1, were demonstrated to be dependent on the concentration of GLI1 or GANT61, respectively, as the analyte. Second, the binding of GLI1 to DNA was inhibited by GANT61. No significant binding of GANT61 to DNA was determined even at the highest non-physiologic concentration (50 µM) of GANT61 employed. These data confirm the specificity of binding of GANT61 to the GLI1 protein, and negate any direct effect of GANT61 on binding to DNA. They also demonstrate inhibition of the binding of new GLI1 to DNA. We have demonstrated the appearance of γH2AX foci within 4 hr of exposure of cells to GANT61 [38, 39], which we now know to be independent of direct DNA binding. This is in contrast to other agents that induce DNA damage by direct DNA binding, including cisplatin, doxorubicin or etoposide. Our data are consistent with the introduction of DNA strand breaks and DNA damage due to inhibition of transcription, which has been shown to create R-loops (RNA:DNA hybrids) at sites of stalled RNA polymerase II, and has been associated with the mechanism of action of camptothecin. Thus, stabilized TopI cleavable complexes induced by camptothecin are potent transcription-blocking DNA lesions during elongation [60, 61]. GANT61-induced DNA damage is recognized at the initiation of S-phase with induction of a transient intra-S-phase checkpoint, where cells accumulate in early S, fail to progress, and undergo cell death [37-39]. Thus, DNA damage occurs prior to the induction of apoptosis. These mechanisms are currently being explored.
The specificity of GANT61-GLI1 interactions were further evaluated by using 2 other zinc finger proteins/transcription factors, KLF4 and TFIIβ: KLF4 is a member of the GLI-Kruppel family with the classic Cys2His2 zinc finger structure [57, 62]; the general transcription factor TFIIβ, which has a zinc ribbon fold characterized by two beta-hairpins that form two structurally similar zinc-binding sub-sites [63]. GANT61 did not bind to KLF4 or TFIIβ, suggesting a binding mode unique to GLI1. Of interest, the predicted binding constants (KD) for GANT61-diamine-GLI1 (7.5 µM) or GANT61 inhibition of GLI-DNA binding (3.2 µM) are consistent with GANT61 concentrations of 10 µM-20 µM for 48 hr-72 hr exposures, required to induce cell death in human colon carcinoma cell lines [36-38].
To determine the importance of the predicted binding sites for GANT61 at E119 and E167 in the GLI1 protein, site-directed mutagenesis generated a double mutant (DM) with each of the two binding sites converted from E -> A. Following immobilization of the wild type or double mutant proteins on CM5 sensor chips and using GANT61 as the analyte, binding of GANT61 to ∆GLI-DM was decreased by 60%, with the KD for dissociation increased by 2.3-fold. This translated into inhibition of the ability of GANT61 to decrease GLI-luciferase activity in cells expressing GLI1-DM, in contrast to the inhibition of GLI-luc activity in untransfected cells or in cells transfected with GLI1-WT.
In summary, we have demonstrated that GLI1 is a specific target for the small molecule inhibitor, GANT61, which binds directly to the GLI1 protein and not to the DNA or to other zinc finger transcription factors. This provides a unique mechanism of induction of DNA damage following inhibition of GLI-dependent transcription, not related to direct DNA binding. These data underscore the critical importance of GLI as a target in survival of cancer cells. GLI1 and GLI2 are constitutively activated in colon cancer cells by oncogenic signaling pathways upstream of GLI. Targeting GLI terminates HH-SMO-GLI, KRAS-GLI, and HH-signaling in these cells, and is a more effective targeting strategy than employing agents that function upstream in these pathways. Colorectal cancer is the second leading cause of cancer deaths in the United States with >150,000 new cases annually [64, 65], and remains refractory to standard treatment approaches. Further, GLI1 and/or GLI2 are oncogenes, and are constitutively activated in many types of human cancers including epithelial cancers of the GI tract, brain tumors, melanoma, pediatric solid tumors, liver, lung, breast, pancreatic and prostate cancers. KRAS is mutated in 30% of all human cancers, and in 50% of colon carcinomas. Targeting GLI for therapeutics therefore has the potential for high impact.
MATERIALS AND METHODS
Cell Culture
Human colon carcinoma cell lines HT29, SW480, HCT116, GC3/c1, VRC5/c1, HCT8, RKO, have been described previously [35-38, 51]. All cell lines including HT29 were cultured in 10% FBS-supplemented RPMI medium and maintained at 37oC with 5% CO2.
Analysis of cell death
Human colon carcinoma cell lines were treated, in duplicate, with equimolar concentrations (20 µM) of each agent that inhibits SMO (GDC-0449; JS Research), MEK (AZD6244; Selleckchem) or GLI (GANT61; Calbiochem). Following 72 hr exposure, cells were collected by trypsinization and incubated with Annexin V FITC (BD Biosciences, CA) and propidium iodide (Sigma, MO) prior to analysis using a FACSCalibur flow cytometer. Raw data were analyzed by CellQuest software [36-39].
Molecular Docking
Using the known crystal structure of the five zinc finger GLI1-DNA complex (PDB ID 2GLI) [55], a prediction of how GANT61 may bind to GLI1, to DNA, or to the GLI1-DNA complex, was obtained. An unbiased docking of GANT61 was performed using AutoDock 3.0.5 with Autodock tools (ADT), as described [66]. The docking procedure allows GANT61 flexibility and the exploration of a large number of binding modes. GANT61 is a hexahydropyrimidine that hyrolyzes in solution at all pHs to an active diamine, which is the biologically active form of GANT61 [67]. The two-dimensional structure of GANT61-diamine was obtained from Lauth et al [67] and converted into the three-dimensional structure by using Accelrys Discovery Studio 1.7; energy minimization was achieved using CHARM (Accelrys Inc). The file for GLI1 was downloaded from the protein data bank (www.rcsb.org) and imported to ADT, adding polar hydrogen, charge, and solvation parameters; data for GANT61-diamine were also imported to ADT. For initial docking, the grid volume was set to accommodate the entire GLI1 molecule, with refinement performed using a grid volume consistent with the molar volume of GANT61 with spacing of 0.4 Å. A composite file of all possible conformations of GANT61-diamine with GLI1, DNA, or the GLI1-DNA complex was compiled. All three-dimensional docked complexes were analyzed and visualized by UCSF Chimera [68] and pymol (www.pymol.com).
Site Directed Mutagenesis of GLI1
Full length GLI1 cDNA was a gift from Dr. Graham Neill, Queen Mary University of London, UK. Two complimentary oligonucleotides for the double mutation (A50C/A65C) to convert E -> A at amino acids E119 and E167, were purchased from Integrated DNA Technology (Coralville, IA). The mutation reaction was set up using the QuikChange II Site-Directed Mutagenesis kit (Agilent Technologies Inc.). The reaction mixture contained 50 ng of template DNA (pBabe-GLI1), 125 ng each of the oligonucleotide primer, 200 µM dNTP mix, 2.5 U of PfuUltra HF DNA polymerase and I x reaction buffer in a total volume of 50 µl. The samples were denatured at 95oC for 30 sec and then cycled 16 times at 95oC for 30 sec., 55oC for 1 min followed by 68oC for 9 min. The parental plasmid DNA was digested by adding 10 U of Dpn I restriction enzyme to the amplification reaction and incubated at 37oC for 1 hr. DH5α competent cells (Agilent Technologies) were transformed with 1 µl Dpn I-treated DNA and plated on LB-agar plate containing 100µg/mL ampicillin. Plates were incubated at 37oC for 16 hr. Single colonies were isolated and plasmid DNA extracted and sequenced for mutation verification.
Sub-cloning of ∆GLI-WT and ∆GLI-DM mutant (A50C/A65C) fragments
Based on 1) the published crystal structure of the GLI1-DNA complex (PDB ID 2GLI) [55], and 2) the molecular modeling of GANT61-GLI1 binding, the GLI1 Zn-finger domain with DNA binding sequence was amplified by PCR to generate ∆GLI-WT and ∆GLI-DM fragments, which were subcloned into BamH1 and SalI sites of the pHIS-II-1 plasmid using the Infusion cloning kit (Clontech). The reaction mixture contained 1x infusion HD enzyme premix, 40 ng of pHISII-1 [69] linearized with BamH1 and Sal1 and 50 ng of ∆GLI-WT or ∆GLI-DM fragment in a total volume of 10 µl. The reaction was incubated at 50oC for 15 min and then placed on ice. DH5α competent cells were transformed with 2 µl of the cloning reaction and plated on LB-agar plate with 100 µg/ml ampicillin. The plates were incubated at 37oC overnight and single colonies were grown in a 5 ml culture. Plasmid DNA was extracted and sequenced for verification of the presence of the ∆GLI-WT or ∆GLI-DM fragment.
Protein Expression and purification
BL21(DE3) bacterial cells (Agilent Technologies) were transformed with either pHIS-II-1-∆GLI-WT or pHIS-II-1-∆GLI-DM. Single colonies were grown at 37oC and shaking at 225 rpm overnight in 5 ml LB medium containing 100 µg/ml ampicillin. The culture was diluted 1:10 and shaken at 37oC at 225 rpm until the optical density at 600 nm reached 0.7. The culture was cooled to 4oC and IPTG was added to a final concentration of 0.3 mM. The culture was then grown at 20oC for 18 hr. Cells were harvested and washed with 50 mM Tris pH 8.5 buffer containing 150 mM NaCl. Cell pellets were resuspended in 10 ml of lysis buffer (50 mM Tris, pH 8.5, 5 mM MgCl2, 50 mM KCl, I mM EDTA, 5% glycerol, 14 mM 2-mercaptoethanol, 50 µM PMSF) containing 1x protease inhibitor cocktail (Roche). The cells were sonicated for 15 cycles (10 sec ‘on’ and 15 sec ‘off’, each cycle). The sonicated fraction was centrifuged and the supernatant was bound to Ni-NTA. The Ni-NTA-bound protein was washed x 3 with washing buffer (50 mM Tris, pH 8.5, 5 mM MgCl2, 500 mM NaCl, I mM EDTA, 5% glycerol, 7 mM 2-mercaptoethanol, 50 µM PMSF) followed by washing x 2 with washing buffer containing 10 mM imidazole. ∆GLI-WT and ∆GLI-DM proteins were eluted with wash buffer containing 200 mM imidazole, and detected by SDS-PAGE/coomassie blue staining and by Western analysis using the His-probe antibody H15 (Santa Cruz Biotechnology).
Surface Plasmon Resonance
The full length GLI1 protein was purchased from OriGene, KLF4 from Peprotech, and TFIIβ from Abcam. Amine group covalent coupling chemistry was used to immobilize GLI1, KLF4 or TFIIβ proteins on a CM5 sensor chip (GE Health Care) via free primary amine groups (lysine residues), which are present in GLI1, and also present in KLF4 and TFIIβ. The analyte was GANT61. To perform the reverse binding, the biotin-labeled synthetic GLI1 DNA-binding sequence (21-mer; purchased from Integrated DNA Technologies [IDT]), was captured on a streptavidin pre-coated SA sensor chip (GE Healthcare). The analyte was either full-length GLI1 protein and/or GANT61. Purified recombinant (His)6-tagged ∆GLI-WT or ∆GLI-DM fragments were immobilized on a Ni-NTA chip [70]; the analyte was GANT61. Varying concentrations of GLI1 and/or GANT61, were passed over the DNA-biotin-SA, GLI1-CM5, or ∆GLI sensor chips at a flow rate of 20 µl/min for 3 min followed by 5 min dissociation in HBS-P buffer alone. Sensograms were recorded and the response units (RU) and maximum resonance units (Rmax) at equilibrium determined. All experiments were carried out on a Biacore Model 3000 and analyzed in bia evalution 4.0.1 (GE LifeSciences). The data were imported to Prizm to generate fit curves.
GLI-luciferase Assay
The 12 GLI-binding site driven luciferase reporter (2 µg, GLI-luc, gift from Dr. Rune Toftgard, Karolinska Institutet [71]) and Renilla luciferase (0.2 µg, pRLTK) were cotransfected with either pBabe-Puro (2 µg, empty vector), full length GLI1 cDNA (2 µg, GLI1-WT), or full length GLI1-DM into HT29 cells using Lipofectamine 2000 (Invitrogen); 24 hr post-transfection, cells were treated with GANT61 (20 M) and allowed to grow for another 24 hr. Cells were subsequently harvested using the Dual luciferase reporter assay system (Promega Corporation) according to the manufacturer’s protocol. Luciferase activity was detected by a Victor2 multilabel counter and normalized to Renilla luciferase activity as a control for transfection efficiency.
HT29-derived stable cell lines expressing the GLI-luciferase reporter were generated by transducing HT29 cells with pCignal Lenti-TRE-GLI-luciferase viral particles (SABiosciences) in the presence of polybrene (to a final concentration of 8 μg/ml) as flocculation agent to increase infectivity. The plate was gently swirled to mix. The cells were incubated overnight at 37°C in a humidified incubator with 5% CO2. Cells were tested after two passages for GLI-luciferase reporter activity. HT29-GLI-luc cells was transfected with either pBabe-Puro, GLI1-WT or GLI1-DM cDNA, and exposed to GANT61 (20 µM) for 24 hr. Live cell imaging was performed using a Bruker optical and X-ray small animal imaging system (Bruker Corporation) before or following treatment with GANT61.
REFERENCES
1. Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med 2009; 9: 873-886.
2. Yu M, Gipp J, Yoon JW, Iannaccone P, Walterhouse D, Bushman W. Sonic hedgehog-responsive genes in the fetal prostate. J Biol Chem 2009; 284:5620-5629.
3. Hooper JE, Scott MP. Communicating with Hedgehogs. Nat Rev Mol Cell Biol 2005; 6:306-317.
4. Varnat F, Zacchetti G, Ruiz i Altaba A. Hedgehog pathway activity is required for the lethality and intestinal phenotypes of mice with hyperactive Wnt signaling. Mech Dev 2005: 127:73-81.
5. Alinger B, Kiesslich T, Datz C, Aberger F, Strasser F, Berr F, Dietze O, Kaserer K, Hauser-Kronberger C. Hedgehog signaling is involved in differentiation of normal colonic tissue rather than in tumor proliferation. Virchows Arch 2009; 454:369-379.
6. Van den Brink, G.R. Linking pathways in colorectal cancer. Nat Genet 36, 1038-1039 (2004).
7. Kinzler KW, Ruppert JM, Bigner SH, Vogelstein B. The GLI gene is a member of the Kruppel family of zinc finger proteins. Nature 1988; 332:371-374.
8. Kasper M, Schnidar H, Neill GW, Hanneder M, Klingler S, Blaas L, Schmid C, Hauser-Kronberger C, Regl G, Philpott MP, Aberger F. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol Cell Biol 2006; 26:6283-6298.
9. Thiyagarajan S, Bhatia N, Reagan-Shaw S, Cozma D, Thomas-Tikhonenko A, Ahmad N, Spiegelman VS. Role of GLI2 transcription factor in growth and tumorigenicity of prostate cells. Cancer Res 2007; 67:10642-10646.
10. Ikram MS, Neill GW, Regl G, Eichberger T, Frischauf AM, Aberger F, Quinn A, Philpott M. GLI2 is expressed in normal human epidermis and BCC and induces GLI1 expression by binding to its promoter. J Invest Dermatol 2004: 122:1503-1509.
11. Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 2002; 129:4753-4761.
12. Eichberger T, Sander V, Schnidar H, Regl G, Kasper M, Schmid C, Plamberger S, Kaser A, Aberger F, Frischauf AM. Overlapping and distinct transcriptional regulator properties of the GLI1 and GLI2 oncogenes. Genomics 2006; 87:616-632.
13. Mill P, Mo R, Fu H, Grachtchouk M, Kim PC, Dlugosz AA, Hui CC. Sonic hedgehog-dependent activation of Gli2 is essential for embryonic hair follicle development. Genes Dev 2003; 17:282-294.
14. Roessler E, Du YZ, Mullor JL, Casas E, Allen WP, Gillessen-Kaesbach G, Roeder ER, Ming JE, Ruiz i Altaba A, Muenke M. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc Natl Acad Sci U S A 2003; 100:13424-13429.
15. Ruiz i Altaba A, Mas C, Stecca B. The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol 2007; 17:438-447.
16. Kinzler KW, Bigner SH, Trent JM, Law ML, O’Brien SJ, Wong AJ, Vogelstein B. Identification of an amplified, highly expressed gene in a human glioma. Science 1987; 236:70-73.
17. Kasper M, Regl G, Frischauf AM, Aberger F. GLI transcription factors: mediators of oncogenic Hedgehog signalling. Eur J Cancer 2006; 42:437-445.
18. Zhang D, Cao L, Li Y, Lu H, Yang X, Xue P. Expression of glioma-associated oncogene 2 (Gli 2) is correlated with poor prognosis in patients with hepatocellular carcinoma undergoing hepatectomy. World J Surg Oncol 2013; 11-25.
19. Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, Beermann F, Ruiz I Altaba A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci U S A 2007; 104:5895-5900.
20. Sanchez P, Hernández AM, Stecca B, Kahler AJ, DeGueme AM, Barrett A, Beyna M, Datta MW, Datta S, Ruiz i Altaba A. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc Natl Acad Sci U S A 2004; 101:12561-12566.
21. Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, Ruiz i Altaba A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med 2009; 1:338-351.
22. Ecke I, Rosenberger A, Obenauer S, Dullin C, Aberger F, Kimmina S, Schweyer S, Hahn H. Cyclopamine treatment of full-blown Hh/Ptch-associated RMS partially inhibits Hh/Ptch signaling, but not tumor growth. Mol Carcinog 2008; 47:361-372.
23. Sarangi A, Valadez JG, Rush S, Abel TW, Thompson RC, Cooper MK. Targeted inhibition of the Hedgehog pathway in established malignant glioma xenografts enhances survival. Oncogene 2009; 28:3468-3476.
24. Mas C, Ruiz i Altaba A. Small molecule modulation of HH-GLI signaling: current leads, trials and tribulations. Biochem Pharmacol 2010; 80:712-723.
25. Robarge KD, Brunton SA, Castanedo GM, Cui Y, Dina MS, Goldsmith R, Gould SE, Guichert O, Gunzner JL, Halladay J, Jia W, Khojasteh C, Koehler MF, Kotkow K, La H, Lalonde RL, Lau K, Lee L, Marshall D, Marsters JC Jr, Murray LJ, Qian C, Rubin LL, Salphati L, Stanley MS, Stibbard JH, Sutherlin DP, Ubhayaker S, Wang S, Wong S, Xie M. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg Med Chem Lett 2009; 19:5576-5581.
26. Low JA, de Sauvage FJ. Clinical experience with Hedgehog pathway inhibitors. J Clin Oncol 2010; 28:5321-5326.
27. De Smaele E, Ferretti E, Gulino A. Vismodegib, a small-molecule inhibitor of the hedgehog pathway for the treatment of advanced cancers. Curr Opin Investig Drugs 2010; 11:707-718.
28. Lorusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad M., Hann CL, Brahmer JR, Chang I, Darbonne WC, Graham RA, Zerivitz KL, Low JA, Von Hoff DD. Phase I Trial of Hedgehog Pathway Inhibitor Vismodegib (GDC-0449) in Patients with Refractory, Locally Advanced or Metastatic Solid Tumors. Clin Cancer Res 2011; 17:2502-2511.
29. Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Mackey HM, Lum BL, Darbonne WC, Marsters JC, de Sauvage FJ, Low JA. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009; 361:1164-1172.
30. Dierks C. GDC-0449--targeting the hedgehog signaling pathway. Recent Results Cancer Res 2010; 184:235-238.
31. Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, LoRusso PM, Von Hoff DD, de Sauvage FJ, Low JA. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med 2009; 361:1173-1178.
32. Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T, Bazan JF, Kan Z, Seshagiri S, Hann CL, Gould SE, Low JA, Rudin CM, de Sauvage FJ. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009; 326:572-574.
33. Yoshikawa K, Shimada M, Miyamoto H, Higashijima J, Miyatani T, Nishioka M, Kurita N, Iwata T, Uehara H. Sonic hedgehog relates to colorectal carcinogenesis. J Gastroenterol 2009; 44:1113-1117.
34. Bian YH, Huang SH, Yang L, Ma XL, Xie JW, Zhang HW. Sonic hedgehog-Gli1 pathway in colorectal adenocarcinomas. World J Gastroenterol 2007; 13:1659-1665.
35. Shi T, Mazumdar T, DeVecchio J, Duan ZH, Agyeman A, Aziz M, Houghton JA. cDNA microarray gene expression profiling of hedgehog signaling pathway inhibition in human colon cancer cells. PloS one 2010; 5, e13054.
36. Mazumdar T, DeVecchio J, Shi T, Jones J, Agyeman A, Houghton JA. Hedgehog signaling drives cellular survival in human colon carcinoma cells. Cancer Res 2010; 71: 1092-1102.
37. Mazumdar T, Devecchio J, Agyeman A, Shi T, Houghton J.A. Blocking Hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res 2011; 71:5904-5914.
38. Mazumdar T, DeVecchio J, Agyeman A, Shi T, Houghton JA. The GLI genes as the molecular switch in disrupting Hedgehog signaling in colon cancer. Oncotarget 2011; 2:638-645.
39. Agyeman A, Mazumdar T, Houghton JA. Regulation of DNA Damage Following Termination of Hedgehog (HH) Survival Signaling at the level of the GLI Genes in Human Colon Cancer. Oncotarget 2010; 3:854-868.
40. Yoon JW, Kita Y, Frank DJ, Majewski RR, Konicek BA, Nobrega MA, Jacob H, Walterhouse D, Iannaccone P. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J Biol Chem 2002; 277:5548-5555.
41. Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C, Regl G, Kroismayr R, Moriggl R, Sibilia M, Aberger F. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res 2009; 69:1284-1292.
42. Lauth M, Toftgard R. Non-canonical activation of GLI transcription factors: implications for targeted anti-cancer therapy. Cell Cycle 2007; 6:2458-2463.
43. Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 2006; 24:21-44.
44. Little AS, Smith PD, Cook SJ. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene 2012; 32:1207-1215.
45. Ji Z, Mei FC, Xie J, Cheng X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J Biol Chem 2007; 282:14048-14055.
46. Sunaga N, Shames DS, Girard L, Peyton M, Larsen JE, Imai H, Soh J, Sato M, Yanagitani N, Kaira K, Xie Y, Gazdar AF, Mori M, Minna JD. Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol Cancer Ther 2011; 10:336-346.
47. Tentler JJ, Nallapareddy S, Tan AC, Spreafico A, Pitts TM, Morelli MP, Selby HM, Kachaeva MI, Flanigan SA, Kulikowski GN, Leong S, Arcaroli JJ, Messersmith WA, Eckhardt SG. Identification of predictive markers of response to the MEK1/2 inhibitor selumetinib (AZD6244) in K-ras-mutated colorectal cancer. Mol Cancer Ther 2010; 9:3351-3362.
48. Friday BB, Yu C, Dy GK, Smith PD, Wang L, Thibodeau SN, Adjei AA. BRAF V600E disrupts AZD6244-induced abrogation of negative feedback pathways between extracellular signal-regulated kinase and Raf proteins. Cancer Res 2008; 68:6145-6153.
49. Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359:1757-1765.
50. Hingorani SR, Jacobetz MA, Robertson GP., Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res 2003; 63:5198-5202.
51. Hinoue T, Weisenberger DJ, Pan F, Campan M, Kim M, Young J, Whitehall VL, Leggett BA, Laird PW. Analysis of the association between CIMP and BRAF in colorectal cancer by DNA methylation profiling. PloS one 2009; 4: e8357.
52. Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 2009; 27:2091-2096.
53. Mercer KE, Pritchard CA. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim Biophys Acta 2003; 1653:25-40.
54. Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A 2007; 104:8455-8460.
55. Pavletich NP, Pabo CO. Crystal structure of a five-finger GLI-DNA complex: new perspectives on zinc fingers. Science 1993; 261:1701-1707.
56. Hofmann I, Weiss A, Elain G, Schwaederle M, Sterker D, Romanet V, Schmelzle T, Lai A, Brachmann SM, Bentires-Alj M, Roberts TM, Sellers WR, Hofmann F, Maira S-M. K-RAS mutant pancreatic tumors show higher sensitivity to MEK than to PI3K inhibition in vivo. PloS One 2012; 7: e44146.
57. Ruppert JM, Kinzler,KW, Wong AJ, Bigner SH, Kao FT, Law ML, Seuanez HN, O’Brien SJ, Vogelstein B. The GLI-Kruppel family of human genes. Mol Cell Biol 1988; 8:3104-3113.
58. Day YS, Baird CL, Rich RL, Myszka DG. Direct comparison of binding equilibrium, thermodynamic, and rate constants determined by surface- and solution-based biophysical methods. Protein Science : A Publication of the Protein Society 2002; 11:1017-1025.
59. Thurmond RL, Wadsworth SA, Schafer PH, Zivin, RA, Siekierka JJ. Kinetics of small molecule inhibitor binding to p38 kinase. Eur J Biochem / FEBS 2001; 268:5747-5754.
60. Sordet O, Nakamura AJ, Redon CE, Pommier Y. DNA double-strand breaks and ATM activation by transcription-blocking DNA lesions. Cell Cycle 2010; 9:274-278.
61. Sordet O, Redon CE, Guirouilh-Barbat J, Smith S, Solier S, Douarre C, Conti C, Nakamura AJ, Das BB, Nicolas E, Kohn KW, Bonner WM, Pommier Y. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep 2009; 10:887-893.
62. Tiwari N, Meyer-Schaller N, Arnold P, Antoniadis H, Pachkov M, van Nimwegan E, Christofori G. Klf4 is a transcriptional regulator of genes critical for EMT, including Jnk1 (Mapk8). PloS one 2013; 8: e57329.
63. Elsby LM, Roberts SG. Interaction of the TFIIB zinc ribbon with RNA polymerase II. Biochem Soc Trans 2008; 36:595-598.
64. Saif MW, Chu E. Biology of colorectal cancer. Cancer J 2010; 16:196-201.
65. Siegel R, Naishadham D, Jemal A. Cancer Statistics, 2012. CA: A Cancer Journal For Clinicians 2012; 62:10-29.
66. Thakur CS, Jha BK, Dong B, Das Gupta J, Silverman KM, Mao HS, Sawai H, Nakamura AO, Banerjee AK, Godkov A, Silverman,RH. Small-molecule activators of RNase L with broad-spectrum antiviral activity. Proc Natl Acad Sci U S A 2007; 104:9585-9590.
67. Lauth M, Rohnalter V, Bergstrom A, Kooshesh M, Svenningsson P, Toftgard R. Antipsychotic drugs regulate hedgehog signaling by modulation of 7-dehydrocholesterol reductase levels. Mol Pharmacol 2010; 78:486-496.
68. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004; 25:1605-1612.
69. Sheffield P, Garrard S, Derewenda Z. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein expression and purification 1999; 15:34-39.
70. Nieba L, Nieba-Axmann SE, Persson A, Hämäläinen M, Edebratt F, Hansson A, Lidholm J, Magnusson K, Karlsson AF, Plückthun A. BIACORE analysis of histidine-tagged proteins using a chelating NTA sensor chip. Analytical Biochemistry 1997; 252:217-228.
71. Kogerman P, Grimm T, Kogerman L, Krause D, Undén AB, Sandstedt B, Toftgård R, Zaphiropoulos PG. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nature Cell Biol 1999; 1:312-319.