
INTRODUCTION
Most frequently occurring cancer in women, breast cancer also presents the highest death rate. Cancer mortality is not correlated to tumor growth (due to proliferation mechanisms) but, in 90% of cases, to formation of metastasis [1] by migrating and invading cells. One of the mechanisms behind this invasion and metastasis process in breast cancer is the epithelial–mesenchymal transition (EMT) that allows epithelial cancer cells to dedifferentiate and undergo the rear-to-front polarization and to acquire high migratory capacity, invasiveness, enhanced resistance to apoptosis, and stem cell properties [2, 3]. Thus, EMT provides an explanation for why epithelial cancers with poor differentiation status are generally more aggressive and prone to metastasize than more differentiated cancers [4, 5]. Cell migration is a complex multistep process that involves protrusions of the leading edge of the cell, formation of adhesion complexes and the release of adhesions at the cell rear. Calcium (Ca2+) is a key effector of these migratory mechanisms by modulating the focal adhesion turnover or the cytoskeletal organization [6]. Ca2+ increases can occur in the form of waves, spikes or oscillations with various impact on cell migration progression [7]. Several plasma membrane channels that increase Ca2+ into the cytosol, such as the transient receptor potential (TRP) channels [8] and Orai/STIM channels [9, 10] have been described in cancer cell migration, but implication of inositol-(1,4,5)-trisphosphate (IP3) receptors (IP3Rs) [11] and ryanodine receptors (RyRs) [12] in such process remain fragmented.
IP3R protein subtypes (IP3R1, IP3R2 and IP3R3) are encoded by three different genes in mammals, however the resulting proteins share high similarity in their primary sequences and are expressed to varying degrees in different cell types [13]. Interestingly, Miyakawa et al. [14] demonstrated that IP3Rs subtypes differ by a specific Ca2+ signature, which is associated to various sensitivity to endogenous modulators such as IP3, Ca2+ and ATP. Thus, activation of IP3R1 generates very rapidly damped Ca2+ oscillations, IP3R2 stimulation produces regular and robust Ca2+ oscillations, whereas IP3R3 functions as an anti-Ca2+ oscillatory units with a Ca2+ transient signature [14, 15] and is able to modulate the spatiotemporal pattern of intracellular Ca2+ signals induced by ATP [16, 17].
Overexpression of IP3Rs has been demonstrated in various cancer types where a pro-apoptotic [18, 19] and pro-invasive [20, 21] roles of IP3Rs have been established. Concerning IP3R3 subtype, it appears as a key actor of carcinogenesis as its expression level is correlated with colorectal carcinoma aggressiveness [22], whereas its inhibition reduces breast cancer cell proliferation [17], migration, invasion and survival of glioblastoma cells [20]. Furthermore, we recently demonstrated that IP3R3 co-localizes and interacts, both at molecular and functional levels, with voltage- and Ca2+-dependent K+ channels (BKCa). This interaction appears to specifically occur in cancerous cells and increases cancer cell proliferation [23]. Based on our previous works [17, 23], we investigated the role of IP3R3 dependent Ca2+ signaling in the migratory process of three human breast cancer cell lines with different migration capacities. Our results clearly show that silencing of IP3R3 (siR3) reveals an oscillating Ca2+ signature and significantly decreases migration of invasive breast cancer cells (MDA-MB-231 and MDA-MB-435S). Conversely, IP3R3 overexpression in MCF-7 cells modifies the ATP-induced Ca2+ response from an oscillatory into a sustained signal and significantly increases their migration capability. This effect is completely reversed by siR3. Thus, our results strengthen the involvement of IP3R3 as a key player in the migration of breast cancer cells through modulation of their Ca2+ signature.
RESULTS
IP3R3 expression level is specifically correlated to migration capacity of breast cancer cell lines
We have previously reported that IP3R3 is the unique isoform to positively regulate the 17-beta estradiol-induced proliferation of the estrogen-dependent MCF-7 cell line [17]. Here, we investigated the expression levels of the three IP3Rs subtypes in three breast cancer cell lines with various degree of malignancy: a very low migrating- (MCF-7), a metastatic- (MDA-MB-231), and a highly metastatic (MDA-MB-435S) cell lines. We first realized cell migration measurements using the Boyden transwell chamber assays in order to characterize and compare the migration potential of each cell line. The relative rank order of cell migration capacity of MCF-7, MDA-MB-231 and MDA-MB-435S is reported in Figure 1A. Relative cell migration is 1 ± 0.66, 15.43 ± 1.33 and 25.48 ± 1.59 N = 3, for MCF-7, MDA-MB-231 and MDA-MB-435S, cells respectively. In parallel, we measured and compared the expression level of IP3R3 at the RNA (Figure 1B) and at the protein (Figure 1C) levels in each cell line. Interestingly, it appears that a higher RNA and protein IP3R3 expression level is correlated to a higher migration capacity of breast cancer cell lines. The relative IP3R3 RNA and protein expression levels are respectively in MCF-7 (1 ± 0.04 (N = 3) and 1 ± 0.06 (N = 3)); MDA-MB-231 (1.41 ± 0.08 (N = 3, p = 0.003) and 1.78 ± 0.18 (N = 3, p = 0.04)) and MDA-MB-435S (1.52 ± 0.06 (N = 3, p = 0.004) and 2.41 ± 0.28 (N = 3, p = 0.02)). Immunostaining with anti-IP3R3 antibody confirmed this marked labeling in highly migrating cells MDA-MB-435S compared to MDA-MB-231 and MCF-7 cells (Figure 1D). This correlation between the cell migration potential and the IP3R3 expression is specific to IP3R3 subtype, since it is not observed with the others IP3R1 and IP3R2 subtypes (Figure 2). To appreciate the IP3R3 expression compared to the two others IP3R subtypes, we also investigated RNA and protein expression levels of both IP3R1 (Figure 2Aa and 2Ab, Table 1) and IP3R2 (Figure 2Ba and 2Bb, Table 1) in the same batch of the three cell lines. Similarly to IP3R3, IP3R1 is predominantly expressed in MDA-MB435s (Table 1), whereas IP3R2 appeared as expressed in MCF-7 as in MDA-MB-435S cell lines (Table 1). Moreover, the rationalization of IP3R3 expression to IP3R1 (Figure 2Ac) or IP3R2 (Figure 2Bc) protein expression levels confirmed its overexpression in migrating cell lines compared to the low migrating MCF-7 cell line. The slight IP3R1 and IP3R2 levels measured in MDA-MB-231 cells accentuate the predominance of IP3R3 in these cells (Figure 2Ac and 2Bc), even if MDA-MB-435S cells show the highest IP3R3 expression level (Figure 1B). Taken together, our results establish, for the first time, a specific correlation between IP3R3 expression level and the cell migration capacities in breast cancer cell lines.
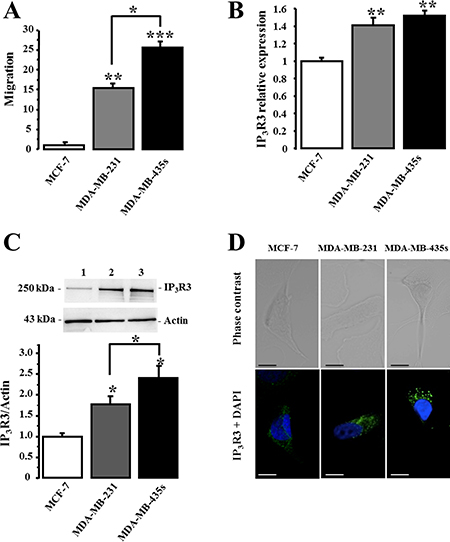
Figure 1: IP3R3 expression level is correlated to migration capacity of breast cancer cell lines. (A) Relative migration capacities of MCF-7, MDA-MB-231 and MDA-MB-435S breast cancer cell lines were evaluated using Boyden chamber migration assay. For each experiment, the number of migrating cells per area for each condition was normalized to MCF-7 cells. (B) IP3R3 mRNA relative expression detected in MCF-7, MDA-MB-231 and MDA-MB-435S cells was quantified using RT-qPCR and results are expressed as average ± SEM of IP3R3/β-actin mRNA ratio. (C) IP3R3 protein expression level was analyzed by Western-blot in MCF-7 (1), MDA-MB-231 (2) and MDA-MB-435S cells (3). Actin protein was used as loading control and quantitative analysis are the average of three independent experiments. Histogram summarizes quantification of IP3R3 expression level in the three cell lines. (D) Immunolabelling of IP3R3 in breast cancer cell lines MCF-7 (1), MDA-MB-231 (2) and MDA-MB-435S (3). Cells were immunostained with anti-IP3R3 antibody (green) and nuclei were stained with DAPI (blue). Scale bar = 20 μm. Values are reported as mean ± SEM normalized to the MCF-7 cells (N = 3). *p < 0.05, **p < 0.01, ***p < 0.001.
Table 1: RNA and protein expression levels of IP3R1 and IP3R2 in breast cancer cell lines
Cell type |
IP3R1 |
IP3R2 |
||
|---|---|---|---|---|
ARN value |
Protein value |
ARN value |
Protein value |
|
MCF-7 |
1 ± 0.36 |
1 ± 0.23 |
1 ± 0.32 |
1 ± 0.07 |
MDA-MB-231 |
0.19 ± 0.006 |
0.52 ± 0.19 |
0.31 ± 0.09 |
0.36 ± 0.08b |
MDA-MB-435S |
1.58 ± 0.11a |
1.15 ± 0.15 |
0.21 ± 0.05 |
0.85 ± 0.19c |
The values are mean of triplicate assays (N = 3). The significant differences are shown with superscript letters and p values are: a = 0.009; b = 0.008 and c = 0.021.
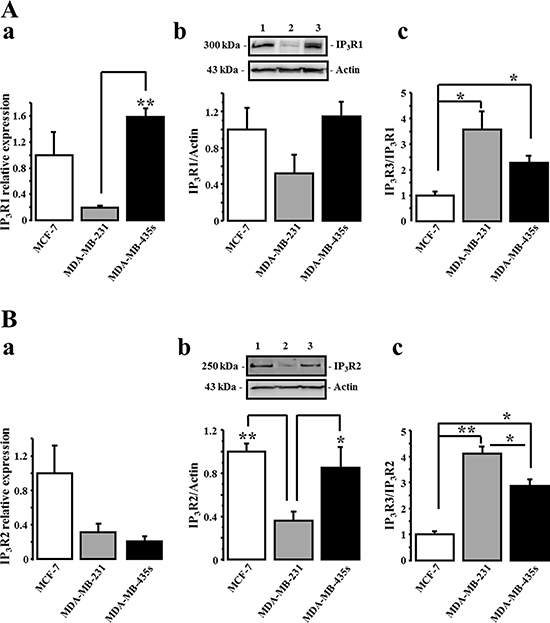
Figure 2: The expression level of IP3R1 and IP3R2 is independent of breast cancer migration capacity. IP3R1 (A) and IP3R2 (B) transcripts (a) and protein levels (b) were quantified in MCF-7 (1), MDA-MB-231 (2) and MDA-MB-435S (3) cells. IP3R3 protein expression level was rationalized to IP3R1 (Ac) and to IP3R2 (Bc) expression levels. Actin protein was used as loading control and, quantitative analyses of Western-blots are the average of three independent experiments. Values are reported as mean ± SEM normalized to the MCF-7 cells (N = 3). *p < 0.05, **p < 0.01.
IP3R3 silencing drastically decreases migration of breast cancer cell lines
In order to confirm that IP3R3 is implicated in cell migration, we realized cell migration assays in cells transfected with two siRNA targeting IP3R3 (siR3) compared to cells transfected with a control siRNA (siC). For each cell line, the migration was measured at different time duration considering the different migration rate of each cell line. IP3R3 being involved in breast cancer cell proliferation as we already described [17, 23], cell migration was assessed during a time frame which does not exceed 24 h where the cell viability, assessed by MTT, is not yet altered. This allows us to throw off the effect of IP3R3 silencing on cell proliferation and to ensure that the observed effects are due solely to the change in migration capacities of breast cancer cells. Cell migration and viability were thus tested 24 h after seeding, in the transwell chamber, for MCF-7 cells and 16 h after seeding for both MDA-MB-231 and MDA-MB-435S cells. In both cases, this time corresponds to 72 h post-transfection with respective siRNA. IP3R3 gene silencing reduced cell migration of the three cell lines, with a greater effect in the highly migrating MDA-MB-231 and MDA-MB-435S cells (Figure 3Aa, 3Ba, 3Ca, Table 2 and Supplementary Figure 1). In all conditions, the cell viability was not affected by siR3 (Figure 3Ab, 3Bb, 3Cb, Table 2 and Supplementary Figure 1). Figure 3Ac, 3Bc and 3Cc show the efficiency of siR3 transfection in MCF-7, MDA-MB-231 and MDA-MB-435S cell lines, respectively. IP3R2 gene silencing also reduces cell migration of the three cell lines (Figure 4Aa, 4Ba, 4Ca and Table 3), whereas IP3R1 silencing inhibits migration of the highly migrating MDA-MB-231 (Figure 4Ba) and MDA-MB-435S cells (Figure 4Ca) but not of MCF-7 (Figure 4Aa, Table 3). The efficiency and specificity of IP3R1, IP3R2 and IP3R3 gene silencing following the transfection by specific siRNAs were confirmed by quantitative real-time PCR (Supplementary Figure 2 and Table 4A) and Western-blot (Supplementary Figure 3 and Table 4B) on the three breast cancer cell lines. Nevertheless, IP3R1 or IP3R2 silencing remains less invalidating on cell migration compared to IP3R3 silencing (Figure 3). Like for siR3 conditions, IP3R1 or IP3R2 silencing has no effect on cell viability (Figure 4Ab, 4Bb, 4Cb and Table 3).
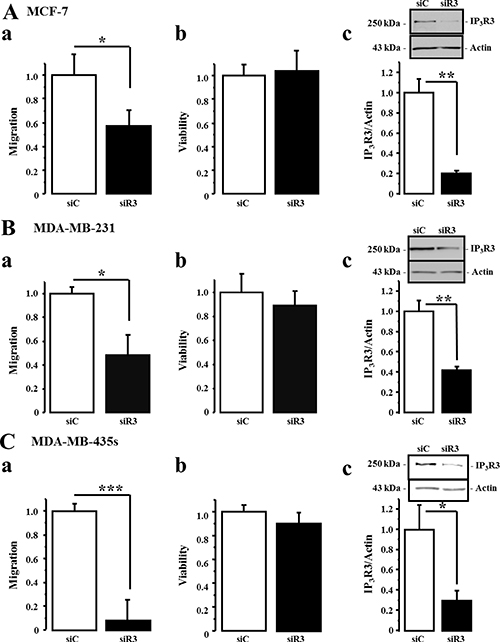
Figure 3: IP3R3 gene silencing decreases breast cancer cell migration. MCF-7, MDA-MB-231 and MDA-MB-435S breast cancer cell lines were transfected either by a small interfering RNA (siRNA) targeting IP3R3 (siR3) or a control siRNA (siC). The migration was then measured at 24 h for MCF-7 and 16 h for MDA-MB-231 and MDA-MB-435S cells, according to their different migration capacities and velocities. Migration tests were performed using Boyden chamber as described above. In all cases, measurements were carried out 72 h after the transfection with respective siRNAs (MCF-7, (Aa) MDA-MB-231, (Ba) and MDA-MB-435S cells, (Ca) compared to control conditions (siC-transfected cells). In the three cell lines, the cell viability was not affected by siR3 (Ab, Bb and Cb) for MCF-7, MDA-MB-231 and MDA-MB-435S cells, respectively). Silencing efficiency on IP3R3 protein expression was confirmed by Western-blot experiments on MCF-7, (Ac) MDA-MB-231, (Bc) and MDA-MB-435S cells, (Cc) Values are reported as mean ± SEM normalized to the corresponding cells transfected with control siRNA (siC) (N = 3). *p < 0.05, **p < 0.01.
Table 2: IP3R3 silencing decreases migration capacities of breast cancer cell lines
Cell type |
Migration assays |
Viability assays |
||
|---|---|---|---|---|
siC |
siR3 |
siC |
siR3 |
|
MCF-7 |
1 ± 0.24 |
0.57 ± 0.13a |
1 ± 0.09 |
1.04 ± 0.17 |
MDA-MB-231 |
1 ± 0.05 |
0.53 ± 0.15b |
1 ± 0.05 |
0.95 ± 0.14 |
MDA-MB-435S |
1 ± 0.06 |
0.08 ± 0.17c |
1 ± 0.05 |
0.91 ± 0.08 |
Results presented are mean of triplicate assays (N = 3). Significant differences are noted with superscript letters and correspond to p values listed below: a = 0.04; b = 0.03 and c = 0.001.
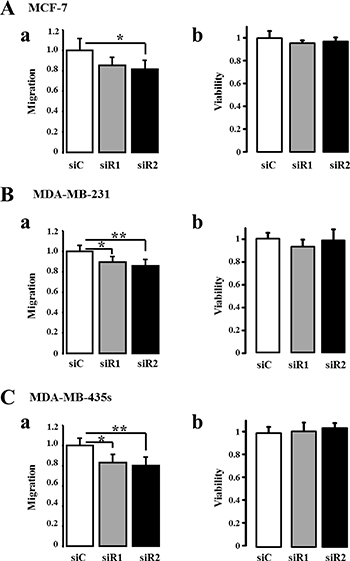
Figure 4: Silencing IP3R1 or IP3R2 reduce breast cancer cell migration to a lesser extent than IP3R3. The three cell lines were transfected either by a small interfering RNA (siRNA) targeting IP3R1 (siR1), IP3R2 (siR2) or a control siRNA (siC). The migration was then measured at 24 h for MCF-7 (Aa) and 18 h for MDA-MB-231 (Ba) and MDA-MB-435S (Ca) cells using Boyden chamber migration assay. In all cases, the measurement was carried out 48 h after the transfection with respective siRNAs. In all three-cell lines, the cell viability was neither affected by siR1 nor by siR2 (Ab, Bb and Cb) for MCF-7, MDA-MB-231 and MDA-MB-435S cells, respectively). Values are reported as mean ± SEM normalized to the corresponding cells transfected with control siRNA (siC) (N = 3). *p < 0.05, **p < 0.01.
Table 3: IP3R1 or IP3R2 silencing modulates breast cancer cell migration
Cell type |
Migration assays |
Viability assays |
||||
|---|---|---|---|---|---|---|
siC |
siR1 |
siR2 |
siC |
siR1 |
siR2 |
|
MCF-7 |
1 ± 0.11 |
0.85 ± 0.08 |
0.81 ± 0.08a |
1 ± 0.11 |
0.95 ± 0.02 |
0.97 ± 0.04 |
MDA-MB-231 |
1 ± 0.05 |
0.9 ± 0.05b |
0.86 ± 0.06c |
1 ± 0.05 |
0.93 ± 0.06 |
0.98 ± 0.01 |
MDA-MB-435S |
1 ± 0.07 |
0.83 ± 0.07d |
0.8 ± 0.08e |
1 ± 0.05 |
1.01 ± 0.08 |
1.05 ± 0.04 |
Results presented are mean of triplicate assays (N = 3). Significant differences are noted with superscript letters and correspond to p values listed below: a = 0.02; b = 0.04; c = 0.01; d = 0.02 and e = 0.008.
Table 4: Silencing of IP3R1, IP3R2 or IP3R3 specifically reduces gene (A) and protein (B) expression of IP3 corresponding receptor in breast cancer cell lines
A
Cell type |
IP3R1 |
IP3R2 |
IP3R3 |
|
|---|---|---|---|---|
MCF-7 |
siC |
1 ± 0.04 |
1 ± 0.03 |
1 ± 0.05 |
siR1 |
0.315 ± 0.06a |
1.13 ± 0.13 |
1.94 ± 0.94 |
|
siR2 |
0.86 ± 0.27 |
0.397 ± 0.09b |
1.58 ± 0.37 |
|
siR3 |
0.76 ± 0.33 |
1.24 ± 0.12 |
0.45 ± 0.03c |
|
MDA-MB-231 |
siC |
1 ± 0.03 |
1 ± 0.05 |
1 ± 0.06 |
siR1 |
0.307 ± 0.04d |
1.113 ± 0.22 |
1.113 ± 0.15 |
|
siR2 |
1.047 ± 0.18 |
0.32 ± 0.11e |
1.407 ± 0.28 |
|
siR3 |
0.876 ± 0.09 |
1.18 ± 0.1 |
0.228 ± 0.05f |
|
MDA-MB-435S |
siC |
1 ± 0.05 |
1 ± 0.06 |
1 ± 0.04 |
siR1 |
0.383 ± 0.06g |
1.38 ± 0.15 |
1.287 ± 0.16 |
|
siR2 |
1.14 ± 0.22 |
0.45 ± 0.02h |
1.463 ± 0.38 |
|
siR3 |
0.663 ± 0.17 |
0.91 ± 0.19 |
0.167 ± 0.02i |
|
B
Cell type |
IP3R1 |
IP3R2 |
IP3R3 |
|
|---|---|---|---|---|
MCF-7 |
siC |
1 ± 0.161 |
1 ± 0.122 |
1 ± 0.042 |
siR1 |
0.494 ± 0.066j |
0.927 ± 0.106 |
0.927 ± 0.057 |
|
siR2 |
0.983 ± 0.111 |
0.405 ± 0.077k |
0.945 ± 0.064 |
|
siR3 |
0.896 ± 0.211 |
1.096 ± 0.083 |
0.205 ± 0.038l |
|
MDA-MB-231 |
siC |
1 ± 0.117 |
1 ± 0.118 |
1 ± 0.0037 |
siR1 |
0.411 ± 0.092m |
0.927 ± 0.097 |
0.947 ± 0.081 |
|
siR2 |
0.939 ± 0.048 |
0.436 ± 0.058n |
1.049 ± 0.072 |
|
siR3 |
0.915 ± 0.095 |
0.964 ± 0.086 |
0.407 ± 0.029o |
|
MDA-MB-435S |
siC |
1 ± 0.103 |
1 ± 0.098 |
1 ± 0.041 |
siR1 |
0.551 ± 0.084p |
1.017 ± 0.12 |
0.939 ± 0.084 |
|
siR2 |
0.969 ± 0.065 |
0.436 ± 0.053q |
0.987 ± 0.086 |
|
siR3 |
0.985 ± 0.079 |
1.03 ± 0.092 |
0.289 ± 0.032r |
|
Results presented are mean of triplicate assays (N = 3). Significant differences are noted with superscript letters and correspond to p values listed below: a = 0.0008; b = 0.0026; c = 0.0001; d = 0.0001; e = 0.003; f = 0.0001; g = 0.0002; h = 0.0013; i = 0.0001; j = 0.01; k = 0.0009; l = 0.001; m = 0.007; n = 0.008; o = 0.01; p = 0.01; q = 0.009 and r = 0.0008.
ATP-induced maintained Ca2+ mobilization is an IP3R3-dependent signaling pathway
Migration processes implying Ca2+ flux [24–26], we investigated whether siR3-induced migration inhibition was associated with a modification of the Ca2+ homeostasis. Breast cancer cell lines transfected with siC or siR3 were loaded with Fura-2/AM (2 μM) for 45 min and ATP (5 μM) was applied, in a Ca2+-free solution, to prevent ionotropic/purinergic receptors activation [23, 27]. Statistical analyses revealed that IP3R3 silencing do not modify neither basal intracellular Ca2+ values (Figure 5A) nor the percentage of responding cells to ATP (Figure 5B) in all cell lines (Table 5). We have previously reported that the decrease of IP3R3 expression level changes the Ca2+ signal profile from a plateau-type to a sinusoidal oscillatory-shaped signal, which is in favor of a diminution of MCF-7 cell proliferation [17]. In this study, we investigated if such signaling pattern could also be related in highly migrating cells transfected with siR3. Statistical analyses revealed that at 72 h post-transfection, the number of oscillating cells in response to ATP is much higher in siR3-transfected cells compared to siC-transfected cells (Figure 5C) in all cell lines (Table 5). Figure 5D represents typical Ca2+ signals measured at 72 h post-transfection after perfusion with 5 μM ATP in a Ca2+-free medium. In order to determine if this IP3R3 signaling profile modification is associated with a change of ATP-induced Ca2+ mobilization, we measured the “area under curve” (AUC) for each trace in each cellular condition. Thus, the mean AUC values for Ca2+ signals elicited in siC-transfected versus siR3-transfected cells are statistically different (Figure 5E) in all cell lines (Table 5). The AUC values were decreased in MCF-7 and MDA-MB-231 siR3-transfected cells, whereas it increased in MDA-MB-435S siR3 transfected cells. Similar data were obtained with the ΔR/R ratio (Supplementary Figure 4A) except for MCF-7 cells. This discrepancy of AUC values between MCF-7, MDA-MB-231 vs MDA-MB-435S cells could not be the consequence of siR3 induced modifications of extracellular (Figure 6A) or intracellular (Figure 6B) ATP basal values as no significant differences could be detected for each condition. Moreover ATP induced-Ca2+ oscillations are specific to IP3R3 silencing since it is not observed in IP3R1- or IP3R2-silenced cells (Figure 7D). Indeed, IP3R1 silencing significantly decreased the resting ratio, whereas IP3R2 silencing increased it compared to control conditions (Figure 7A; 1.34 ± 0.005 vs. 1.26 ± 0.007 (p = 1.10–17) and 1.40 ± 0.01 (p = 3.10–6) for siC, siR1 and siR2 conditions respectively, N = 160 and 129 cells respectively). Silencing IP3R1 or IP3R2 does not affect the percentage of responding cells to ATP (5 μM) perfusion (Figure 7B). Interestingly, the global amount of Ca2+ released into the cell remained unchanged in siR2 conditions (Figure 7C), but is drastically increased in siR1 conditions (21.16 ± 0.81 vs. 38.06 ± 1.21, n =160 (p = 4.10–28) and 21.03 ± 0.86, n = 129, for siC, siR1 and siR2 conditions respectively).
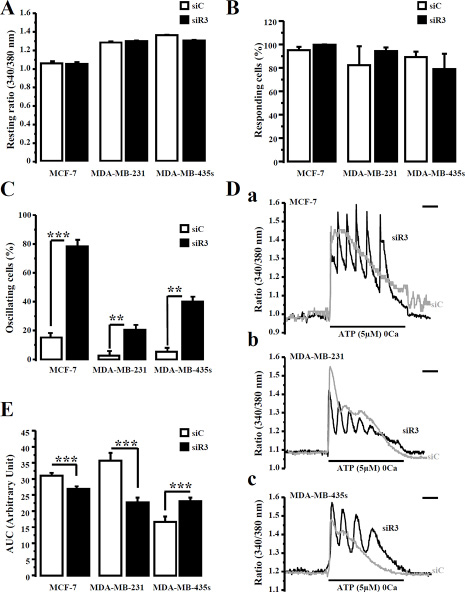
Figure 5: IP3R3 silencing reveals an oscillating calcium signal in breast cancer cell lines. (A) Fluorescence resting ratio (340/380 nm) was measured in Fura-2-loaded breast cancer cells after IP3R3 silencing (siR3, black columns) and compared to control cells (siC, white columns). (B) Cells number responding to application of ATP (5 μM) in absence of extracellular calcium was compared in the three breast cancer cell lines after IP3R3 silencing (black columns) vs control transfected cells (white columns). (C) In each cell lines, the number of ATP-induced oscillating responses was determined following IP3R3 silencing (black columns) vs control (white columns) transfected cells. (D) Representative ATP-induced Ca2+ responses in MCF-7 (Da), MDA-MB-231 (Db) and MDA-MB-435S cells (Dc) illustrate oscillating signal revealed by IP3R3 silencing (black traces) compare to control (grey traces) conditions. (E) The magnitude of the ATP-induced Ca2+ signal was quantified in each cell line after IP3R3 silencing (black columns) compared to control transfected cells siC (white columns). Scale bar = 120 s. Values are reported as mean ± SEM (n = 73 to 447 cells). **p < 0.01, ***p < 0.001.
Table 5: IP3R3 silencing modifies ATP-induced calcium signal on breast cancer cell lines
Cell type |
Resting ratio |
Responding cells (%) |
Oscillating cells (%) |
AUC |
Number of tested cells |
|
|---|---|---|---|---|---|---|
MCF-7 |
siC |
1.044 ± 0.02 |
94.97 ± 2.93 |
8.4 ± 3.5 |
34.5 ± 1.01 |
351 |
siR3 |
1.037 ± 0.02 |
99.65 ± 0.35 |
74.2 ± 8.6a |
30.1 ± 0.82d |
352 |
|
MDA-MB-231 |
siC |
1.267 ± 0.01 |
82.27 ± 16.16 |
6.5 ± 2.7 |
39.8 ± 2.63 |
73 |
siR3 |
1.280 ± 0.009 |
94.22 ± 3.22 |
21.6 ± 5.4b |
25.4 ± 1.63e |
85 |
|
MDA-MB-435S |
siC |
1.345 ± 0.005 |
89.22 ± 4.54 |
7.3 ± 2.5 |
18.6 ± 1.83 |
447 |
siR3 |
1.288 ± 0.007 |
78.95 ± 12.88 |
17.2 ± 3.7c |
25.7 ± 1.3f |
132 |
|
Results presented are mean of numerous assays (N = 6–12). Significant differences are noted with superscript letters and p values a = 4.10–4; b = 0.01; c = 0.009; d = 5.10–4; e = 3.10–6 and f = 0.001.
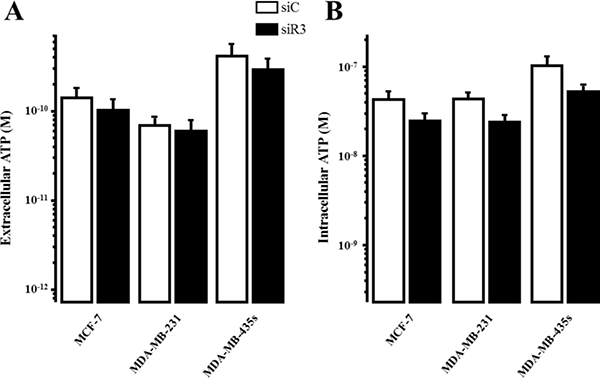
Figure 6: IP3R3 silencing has no impact on extracellular- or intracellular-ATP concentrations in breast cancer cell lines. (A) Extracellular ATP concentration (M) was measured in supernatant, incubated with luciferin/luciferase, of breast cancer cells after IP3R3 silencing (siR3, black columns) and compared to control cells (siC, white columns). (B) Intracellular ATP released of breast cancer cells transfected with siR3 (black columns) was measured, after incubation of cells with ATP releasing reagent and luciferin/luciferase, and compared to control cells (siC, white columns). Values are reported as mean ± SEM (N = 2).
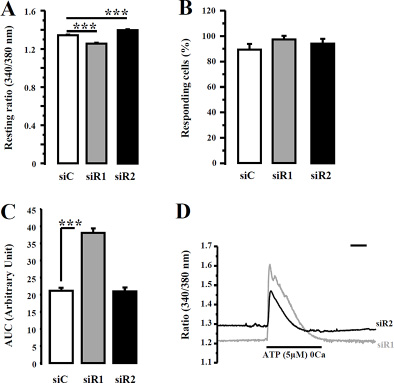
Figure 7: IP3R1 or IP3R2 silencing has no impact on ATP-induced Ca2+ profile in MDA-MB-435S cells. (A) IP3R1 (grey columns) or IP3R2 (black columns) expression was reduced using specific siRNAs and the fluorescence resting ratio (340/380 nm) was measured. (B) The number of the oscillating cells determined following IP3R1 and IP3R2 silencing (black columns) vs control transfected MDA-MB-435S cells (white columns). (C) The magnitude of ATP-induced Ca2+ response was evaluated by measuring the AUC after silencing of IP3R1 (grey column) or IP3R2 (black column). Typical ATP-induced Ca2+ response in siR1 (grey trace) or siR2 (black trace) MDA-MB-435S cells both present a transient profile without oscillating component (D). Scale bar = 120 s. Values are reported as mean ± SEM (n = 129 to 447 cells). ***p < 0.001.
IP3R3 overexpression enhances the migration capacity of MCF-7 cell line
Based on our results showing that siR3 decreased cancer cell migration, we investigated the effect of a stable overexpression of IP3R3 in the low migrating MCF7 cell line. The transfection of MCF-7 cells with a pcDNA(3.1) plasmid encoding IP3R3 [28] led to a 2.59 fold increase in IP3R3 expression level (2.59 ± 0.6 in pcDNA(3.1)/IP3R3-transfected cells vs. 1 ± 0.4 in empty pcDNA(3.1)-transfected cells; N = 3, p = 0.03) (Figure 8A). In order to confirm that the effects recorded were correlated to IP3R3 level increase and not to a modification of IP3R1 and/or IP3R2 expression levels, RT-qPCR analysis of all IP3R isoform mRNAs was carried out. Our results show no significant change in the expression of IP3R1 and IP3R2 at the mRNA level in MCF-7 cells overexpressing IP3R3 (Figure 8B). The relative expression level is 0.77 ± 0.17 (p = 0.15) of control (empty pcDNA(3.1)-transfected cells) for IP3R1 and 0.78 ± 0.51 (p = 0.32) of control for IP3R2, whereas it is significantly elevated to 11.96 ± 1.81 (p = 0.03) of control for IP3R3. We then compared migration ability of MCF-7 overexpressing IP3R3 vs empty pcDNA(3.1)-transfected MCF-7 cells (Figure 8C). The relative migration of IP3R3 overexpressing MCF-7 cells is enhanced by more than a 2 fold factor (2.26 ± 0.37 vs. 1 ± 0.33; N = 3; p = 0.008). Furthermore, the migration of MCF-7 stably overexpressing IP3R3 is reduced to control values by siR3 (0.82 ± 0.19; N = 3; p = 0.01; IP3R3+siR3). This effect on cell migration is independent of any effect on cell viability (Figure 8D). We also record intracellular calcium variations in these cells. IP3R3 overexpression does not modify Ca2+ homeostasis in these cells (1.04 ± 0.001 vs. 1.04 ± 0.001 in empty and IP3R3 transfected cells respectively, Figure 9A). The perfusion of ATP (5 μM) induced a plateau-type signal that appears more sustained in IP3R3 overexpressing cells (Figure 9B) without modify the amount of Ca2+mobilized (Figure 9C; 5.02 ± 0.37 vs. 4.72 ± 0.39 in empty and IP3R3 transfected cells respectively). Indeed, the measure of ΔR/R ratio to Thapsigargin (TG)- or ATP-induced Ca2+ response shows no significant differences (Supplementary Figure 4B and 4C). This result indicates that IP3R3 overexpression modify rather intracellular Ca2+ availability than intracellular Ca2+ homeostasis. This Ca2+ mobilization maintained during ATP application stimulates migration capacities of MCF-7 cells (Figure 10A). Indeed, migration of MCF-7 cells transfected with the pcDNA(3.1)-empty plasmid is increased following ATP stimulation (2 μM) (8.41 ± 1.2 in ATP treated cells vs. 1 ± 0.24 in Ctrl; N = 3, p = 0.0001). This increase seems to occur independently of IP3R3 since IP3R3 silencing failed to decrease the ATP-stimulated migration. However, in MCF-7 stably overexpressing IP3R3, and was not correlated to a proliferative effect (Figure 10B). ATP strongly increases cell migration (10 ± 1.24 in ATP treated cells vs. 1 ± 0.24 in Ctrl; N = 3, p = 0.03) that is abolished following IP3R3 silencing.
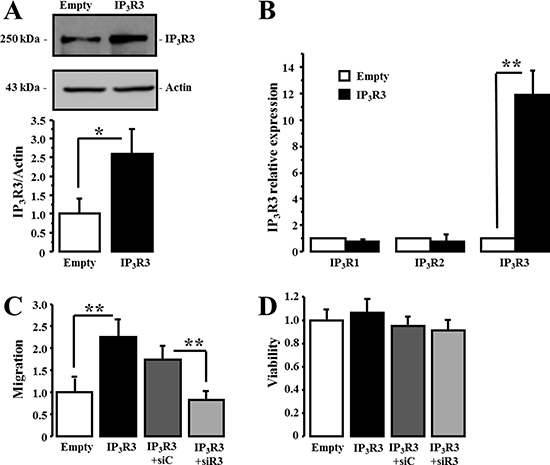
Figure 8: IP3R3 overexpression enhances the migration of MCF-7 cells. (A) MCF-7 cells stably overexpressing IP3R3 were generated following transfection with pcDNA(3.1)-IP3R3 plasmid. Protein expression level of IP3R3 was evaluated by Western-blot (black column) and compared to control conditions (pcDNA(3.1)-empty plasmid) (white column). (B) RT-qPCR was carried out to investigate the impact of stable IP3R3 overexpression on IP3R1 and IP3R2 expression levels. (C) The effect of stable IP3R3 overexpression on the migration capacities of MCF-7 cells was evaluated by Boyden chamber migration assay and the specificity of IP3R3-overexpression effect was controlled by an siRNA targeting IP3R3 in the pcDNA(3.1)-IP3R3 transfected cells. In all experiments, the migration was always measured 72 h after cell transfection with the respective siRNAs and at a time at which there was no effect on cell viability (D). Values are reported as mean ± SEM normalized to MCF-7 cells transfected with empty plasmid (N = 3). *p < 0.05, **p < 0.01.
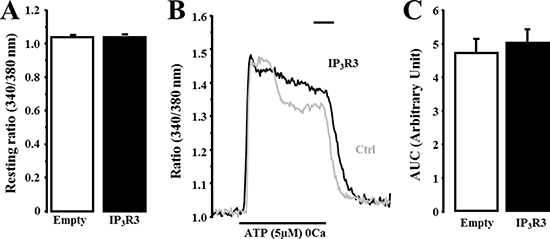
Figure 9: IP3R3 overexpression modifies ATP-induced Ca2+ transient profile into a sustained Ca2+ signal. (A) Fluorescence resting ratio (340/380 nm) was measured in MCF-7 overexpressing IP3R3. (B) ATP-induced Ca2+ signal was investigated in MCF-7 cells transfected with IP3R3 plasmid (B, black trace) compared to the empty plasmid condition (grey trace). (C) Magnitude of ATP-induced Ca2+ response was evaluated by measuring the area under the curve (AUC) in both IP3R3- and empty-plasmid transfected MCF-7 cells. Scale bar = 120 seconds. Values are reported as mean ± SEM (n = 32 to 285 cells).
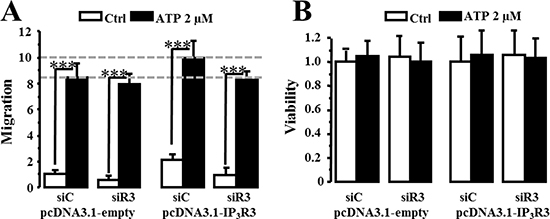
Figure 10: ATP-induced migration is modulated by IP3R3 expression. (A) Migration capacities of MCF-7 stably overexpressing IP3R3 were evaluated after incubation with ATP (2 μm) during 24 h (black columns) and compared to non-stimulated cells (white columns). The specificity of IP3R3-overexpression effect was controlled by an siRNA targeting IP3R3 in the pcDNA(3.1)-IP3R3 and pcDNA(3.1)-empty plasmid-transfected cells. (B) IP3R3 overexpression and ATP (2 μM) stimulation impact on MCF-7 cells viability is controlled by MTT assay. Values are reported as mean ± SEM normalized to the MCF-7 cells transfected with empty plasmid and control siRNA (siC pcDNA3.1-empty) (N = 3). ***p < 0.001.
DISCUSSION
It is well known that Ca2+ ions, together with many other Ca2+-dependent proteins and signaling pathways, play a crucial role in regulating cell migration [25, 26, 29, 30]. For example, capacitive Ca2+ entry has been shown to be involved in the migration of breast cancer cells [31], hepatocarcinoma cells [32] and in vascular smooth muscle cells [33]. Conversely, few studies have shown the role of the secretory pathway of Ca2+ in cancer cell migration. Thereby RyRs have been found to control astrocyte cell migration [34] whereas the SPCA1 Ca2+-ATPase has been shown to be involved in cell migration during Caenorhabditis elegans embryonic development [35]. In this context, and based on previous works showing a role of Ca2+ and IP3R Ca2+ release channels in cell motility, we have studied the potential implication of IP3R3 in cell migration process in three different breast cancer cell lines showing distinct migration capacities. Our study demonstrates, for the first time, that IP3R3 by modifying the calcium signal profile can regulate different breast cancer cell migration capacities. Indeed we establish that (i) MCF-7, MDA-MB-231 and MDA-MB-435S breast cancer cell lines express different levels of IP3R3 protein, (ii) cells expressing IP3R3 in a larger amount migrate more extensively than the others, (iii) silencing of IP3R3 changes a sustained ATP-induced Ca2+ increase to an oscillatory one, (iv) overexpression of IP3R3 increases the migration capacities of the non-invasive MCF-7 cell line and switch the ATP-induced transient Ca2+ response to a sustained phase.
Interestingly, we were able to discriminate a modulation of ATP-induced Ca2+ response specifically correlated to IP3R3 expression. Indeed, down-regulation of IP3R3 in high migrating cell lines reveals an oscillatory ATP-induced Ca2+ signal, whereas overexpression of IP3R3 in low migrating breast cancer cell line modify the Ca2+ signal in a sustained one. These results are in agreement with previous studies showing that IP3R3 forms an anti-oscillating unit, its down-regulation revealing a Ca2+ oscillating profile specific to IP3R2 activation [15]. Interestingly, Ca2+ oscillations associated to IP3R3 silencing were associated to a reduction of the intracellular Ca2+ concentrations in MCF-7 and MDA-MB-231, and an increase in MDA-MB435s cells. Thus, even if IP3R3 silencing systematically induces an oscillating Ca2+ signature in breast cancer cells, the Ca2+ amount involved in this response differ between the various cell lines. This discrepancy could be explained by differences in the expression levels of the purinergic receptors P2 [36–39] or activation of P1 adenosine receptors due to the ATP hydrolysis by ectonucleotidases such as CD39 or CD73 [40]. Moreover a variation of intracellular Ca2+ availability through the Mitochondrial Calcium Uniport (MCU) activity [41] could also convey such difference between MCF-7 and MDA-MB-231 vs MDA-435s cells.
One of the major issues of the present work lies in the identification, for the first time, that IP3R3, by regulating the Ca2+ homeostasis in an anti-oscillating profile, impact the migratory capacities of breast cancer cells. Our results are thus supported by other studies showing that IP3R3 is overexpressed in various cancer tissues. Indeed, IP3R3 expression is found to be increased in gastric carcinoma [21] and glioblastoma [20] where it seems to be involved respectively in peritoneal dissemination and in brain invasion. Kang et al. [20] elegantly demonstrate that caffeine, at concentration that inhibits IP3R3 preferentially to the two other IP3R subtypes, is able to inhibit migration and invasion of glioblastoma cells in vitro. Moreover, IP3Rs inhibition markedly reduces the migration of pancreatic adenocarcinoma cells by modulating the Ca2+ signaling complexes [42]. We establish a link between an oscillating IP3- dependent Ca2+ signal and the migration capacity of breast cancer cells. Indeed, if Ca2+ oscillating signature has been described to modulate dendritic [43] or astrocytoma [44, 45] cell migration; such correlation has never been characterized on cancer cells. These intracellular Ca2+ fluctuations, by modulating the availability of free Ca2+, may regulate migration processes. A variety of intracellular signaling molecules and their associated biochemical pathways have been identified in the regulation of cell migration such as actin and myosin complexes [46, 47], and Rho subfamily of small G proteins (Rho, Rac, and Cdc42) [48, 49]. This Ca2+ signaling has also been shown to activate myosin light chain kinase (MLCK) and control the local disassembly of focal adhesions [24, 50–54]. IP3R3-dependent Ca2+ oscillations may be the consequences of binding partners like IRBIT [55], Ca2+ binding proteins [56], or a close interaction between IP3Rs and the Mitochondrial Calcium Uniport (MCU) [57, 58]. Further studies are needed to determine the implication of these actors in IP3Rs dependent oscillations genesis.
Altogether, our results clearly indicate that IP3R3 is involved in the migration of human breast cancer cells through a specific calcium signature. Overexpression of IP3R3 increases cell migration capacities by inducing a sustained Ca2+ signature, whereas down-regulation of IP3R3 expression reveals an oscillating Ca2+ signature along with a slowing down of cell migration. Thus, this work led us to put forward the hypothesis that IP3R3, by remodeling the Ca2+ signal, is a key player in the migration of human breast cancer cells.
MATERIALS AND METHODS
Cell culture
Human breast epithelial cancer cell lines MCF-7, MDA-MB-231 and MDA-MB-435S were obtained from the ATCC (American Type Culture Collection). Cell lines were cultured in the Minimum Essential Medium (MEM, Gibco, LifeSciences, Cergy Pontoise, France) complemented with 0.45% of sodium bicarbonate, 0.06% Hepes, 0.1% of non-essential amino acids, 2 mM L-glutamine, and 5% fetal calf serum (FCS, Lonza, Aubergenville, France). Cell lines were maintained at 37°C and 5% CO2 in a humidified atmosphere. Cells were used up to 20 passages after ATCC vial defrosting.
Cell migration
Migration tests were performed in 8 μm pore sized membrane Boyden chamber (BD FALCONTM Cell Culture Inserts, BD Biosciences, Le Pont de Claix, France) according to the manufacturer’s protocol. The upper compartment was seeded with 4.104 cells 48 h after transfection (siC and siR3 or pcDNA(3.1) empty-plasmid and pcDNA(3.1) IP3R3-plasmid) in MEM medium supplemented with 5% FCS. The lower compartment was filled with MEM medium supplemented with 5% FCS. After 16 h of further incubation at 37°C for MDA-MB-231 and MDA-MB-435S cells and 24 h for the low migrating MCF-7 cells (as previously described [59, 60]), migrated cells which have passed to the lower side were washed by PBS, fixed in ice-cold methanol and stained by hematoxylin. The remaining cells were removed from the upper side of the membrane by scrubbing. Migrated cells were counted under an inverted microscope (Nikon eclipse TS100 microscope, Champigny-sur-Marne, France) in duplicate (20 contiguous areas at 400 × magnification for each insert). For each experiment, the number of migrating cells per area for each condition (siC and siR3 or pcDNA(3.1) empty-plasmid and pcDNA(3.1) IP3R3-plasmid) was normalized to control (siC and pcDNA(3.1) empty-plasmid). For each experiment, a cell viability test was carried out in the same condition than the migration assays.
Total RNA isolation and quantitative RT-qPCR
After transfection, 5.105 cells were seeded in 100 mm petri dishes in MEM medium 5% FCS. Total RNA was harvested from the cells with the standard trizol-phenol-choloroform protocol. Complementary DNA was synthesized from 2 μg of RNA with random hexamers and MultiScribe™ Reverse Transcriptase (Applied Biosystems, Carlsbad, CA). Relative abundance of mRNA was quantified based on Ct difference on a LightCycler real-time PCR machine (Roche, Basel, Switzerland) using a mix containing SYBR green, Taq polymerase and specific primers (Table 6). Results were expressed as gene expression normalized to β-actin expression.
Table 6: Primers sequences used for RT-PCR experiments
Primer |
Sequence |
|---|---|
IP3R1 forward |
5′-CAGGTTCAACTGCTGGTTAC-3′ |
IP3R1 reverse |
5′-CCTTCTCATAGGGAATCTGC -3′ |
IP3R2 forward |
5′-AAGCAGGTGCAATTACTGG-3′ |
IP3R2 reverse |
5′-CAAAGTGTTGTACAACTCTCTCG-3′ |
IP3R3 forward |
5′-CACAGCCATCACCATCAAG-3′ |
IP3R3 reverse |
5′-GCTGTAGAAGCCGAAGTAG-3′ |
β-actin forward |
5′-CAGAGCAAGAGAGGCATCCT-3′ |
β-actin reverse |
5′-ACGTACATGGCTGGGGTG-3′ |
Western-blot
After transfection, 5.105 cells were seeded in 100 mm petri dishes in MEM medium 5% FCS. Western-blots experiments were performed as previously described [23]. After protein transfer, the membranes were incubated with blocking solution (1-3% bovine serum albumin (BSA) in Tris-buffered saline solution containing 0.1% Tween 20) at room temperature for 1 h. Then, membranes were incubated overnight at 4°C with mouse monoclonal anti-IP3R1 (1/500, NeuroMab, Davis, CA, USA), goat monoclonal anti-IP3R2 (1/250, Santa Cruz, CA, USA) or mouse monoclonal anti-IP3R3 (1/500, BD Biosciences, Le Pont de Claix, France) and goat polyclonal anti-actin (1/1000, Santa Cruz, CA, USA) primary antibodies. Membranes were then incubated with respective secondary antibodies (1/2500–1/5000, Santa Cruz, CA, USA), developed using ECL substrate solution (ECL RevelBlot Intense, Cell signaling, Ozyme, Saint Quentin Yvelines, France), exposed with the MF-ChemiBIS (DNR bio-imaging systems, Neve Yamin, Israel) and analyzed using Quantity one software (Biorad, Marnes-la-Coquette, France). For quantification, actin was used as a loading control.
Cell transfection
Cell transfection was performed using the nucleofection technology according to the Amaxa Biosystems manufacturer’s instructions. For the IP3R silencing experiments cells were transfected with siRNAs targeting the three IP3R isoforms (siR1, 2, 3, ON-TARGET plus, Dharmacon, GE Healthcare Life Sciences, Velizy-Villacoublay, France) or with scrambled siRNA as a control (siC; siGENOME Non-Targeting siRNA, Dharmacon, GE Healthcare Life Sciences, Velizy-Villacoublay, France). MCF-7 cell line was also transfected with plasmid encoding IP3R3 (pcDNA(3.1)-IP3R3 plasmid, gift from Professor Jan B. Parys [28]) or with the corresponding empty plasmid (pcDNA(3.1)-empty plasmid) for the overexpression experiments. Briefly, 1.106 cells (MDA-MB-231 and -435 s) or 2.106 (MCF-7 cells) were transfected with 2 μg siRNA or pcDNA(3.1) plasmid. After the electroporation, 500 μL of prewarmed MEM medium supplemented with 5% FCS was added and cells were placed at 37°C for 15 min in a CO2 incubator. Immediately after transfection, cells were cultured 48 to 72 h for Western-blot, RT-qPCR, cell migration and calcium imaging experiments.
Cell viability
In parallel to the migration assay, cell viability was evaluated for the different conditions. Briefly, 2.104 cells were seeded in 6-well plates in EMEM medium 5% FCS. After 16 to 24 h (according to the migration time for each cell line), cells were washed with phosphate-buffered saline (PBS) and incubated with a medium containing 0.5 mg/ml of 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma, Saint-Quentin Fallavier, France) and incubated for 1 h at 37°C. Medium was removed and 800 μL of Dimethyl-sulfoxide (DMSO) was added to solubilize crystals. The optical density of each sample was read on the microplate reader Infinite F200 Pro (TECAN, Lyon, France) at 570 nm.
Immunofluorescence microscopy
Cells were cultured on coverslips for 72 h. After washing 3 times with PBS, cells were fixed in 3% paraformaldehyde for 10 min at RT. Cells were then washed twice in PBS, blocked with and permeabilized with PBS containing Triton (0.1%) and BSA (5%) for 30 min and incubated overnight with primary antibodies at 4°C. The following day, cells were washed four times in PBS and labeled with an Alexa Fluor 488-labeled goat anti-mouse and a DyLight549-conjugated goat anti-rabbit secondary antibodies for 1 h in the dark at room temperature. After labeling, cells were washed with PBS and incubated with 4′, 6-diamidino-2-phenylindole1% for 1 min. Cells were then washed two more times and mounted onto slides with ProLong® Gold antifade reagent (Life Technologies, Villebon-sur-Yvette, France). Images were acquired with a Zeiss LSM780 confocal microscope (Marly-le-Roi, France) and analyzed with ZEN 2012 software.
Calcium imaging
After transfection, 7.104 cells were seeded in MEM medium 5% FCS in 35 mm petri dishes on glass cover slips. After 3 days, cells were loaded for 45 min with Fura-2/AM (2 μM in medium solution) at 37°C in a CO2 incubator and subsequently washed with MEM medium. The cover slip was then transferred into a perfusion chamber of a fluorescence Zeiss inverted microscope (Marly-le-Roi, France). Fluorescence was excited at 340 and 380 nm alternately, using a monochromator (Polychrome IV; TILL Photonics, Planegg, Germany), and captured by a Cool SNAP HQ camera (Princeton Instruments, Evry, France) after filtration through a long-pass filter (510 nm). Background fluorescence was determined at 340 and 380 nm from an area of the cover slip free of cells. These values were routinely subtracted. Metafluor software (version 7.1.7.0, Molecular Devices, St. Grégoire, France) was used for acquisition and analysis. All recordings were carried out at room temperature. Cells were continuously perfused with the saline solution, and chemicals were added via the perfusion system. The flow rate of the whole-chamber perfusion system was set at 10 mL/min, and the chamber volume was 1 mL. Recording solution had the following composition (in mM): NaCl (145), KCl (5), CaCl2 (2), MgCl2 (1), glucose (5) and Hepes (10) at pH 7.4 (NaOH). In experiments where Ca2+-free solution was used, Ca2+ was omitted and EGTA (0.4 mM) was added to the solution.
ATP measurements
The luciferin⁄luciferase detection of ATP was performed with the microplate reader Infinite F200 Pro (TECAN, Lyon, France) with ATP bioluminescent somatic cell assay kit (FLASC, Sigma, Saint-Quentin Fallavier, France). Breast cancer cell lines MCF-7, MDA-MB-231 and MDA-MB-435S transfected with siRNA control (siC) or siRNA targeting IP3R3 (siR3) were seeded at 2000 cells/well in white 96-well Nunc dishes with clear bottoms 72 hours before ATP measurements. For extracellular ATP measurements, 100μl of supernatant were incubated with the luciferin⁄luciferase (FLAAM, Sigma) at a final concentration of 0.04%. For intracellular ATP measurements cells were incubated with 100 μl of ATP releasing reagent (FLSAR, Sigma), before incubation with the luciferin⁄luciferase (FLAAM, Sigma). To determine the amount of ATP released, a calibration curve was constructed using known concentrations of ATP in solution (1, 10, 100, 1000, 10 000, 100 000 pM). Control experiments were performed to eliminate any drug effect on luciferase activity.
Reagents
All the products were from Sigma (Saint-Quentin Fallavier, France) unless otherwise stated. Final concentrations were obtained by appropriate dilution of stock solutions so that the solvent never exceeded 1/1,000.
Statistical analysis
All data are expressed as mean ± SEM of at least three independent experiments. N refers to the number of experiments repeated and n to the number of tested cells. The Student’s t-test and one-way analysis of variance (ANOVA) with Bonferroni post-hoc analysis were used to group comparison. Statistical significance is indicated in the figures (* P < 0.05; ** P < 0.01; *** P < 0.001).
Author contributions
A.M. & L.R.D.: design, execution, interpretation of data, statistical analysis and manuscript preparation. A.V., A.F., B.B & H.F.: execution and interpretation of data. J.B.P.: plasmid gift and manuscript preparation. H.O.A.: interpretation of data and manuscript preparation.
ACKNOWLEDGMENTS AND FUNDINGS
This article is dedicated to the memory of Pr Fabrice MATIFAT, a highly talented scientist who initiated this project.
We acknowledge Luciane ZABIJAK from Plateforme d’Ingénierie Cellulaire & Analyses des Protéines (ICAP) for confocal acquisitions.
This study was supported by Hauts-de-France (Picardie) Région, Fonds européen de développement économique régional (A.M. and A.V. were supported by grant from Picardie région, France), le Ministère de la Recherche et de l’Enseignement Supérieur and «Ligue Nationale Contre le Cancer (SEPTENTRION)».
CONFLICTS OF INTEREST
The authors declare that they have no interest of any kind affecting this study.
REFERENCES
1. Spano D, Heck C, De Antonellis P, Christofori G, Zollo M. Molecular networks that regulate cancer metastasis. Semin Cancer Biol. 2012; 22:234–249.
2. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. The Journal of clinical investigation. 2009; 119:1420–1428.
3. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009; 139:871–890.
4. Li W, Kang Y. Probing the Fifty Shades of EMT in Metastasis. Trends in cancer. 2016; 2:65–67.
5. Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer cell. 2012; 22:725–736.
6. Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nature reviews Cancer. 2011; 11:609–618.
7. Chen YF, Chen YT, Chiu WT, Shen MR. Remodeling of calcium signaling in tumor progression. J Biomed Sci. 2013; 20:23.
8. Gautier M, Dhennin-Duthille I, Ay AS, Rybarczyk P, Korichneva I, Ouadid-Ahidouch H. New insights into pharmacological tools to TR(i)P cancer up. British journal of pharmacology. 2014; 171:2582–2592.
9. Faouzi M, Kischel P, Hague F, Ahidouch A, Benzerdjeb N, Sevestre H, Penner R, Ouadid-Ahidouch H. ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells. Biochimica et biophysica acta. 2013; 1833:752–760.
10. Fiorio Pla A, Kondratska K, Prevarskaya N. STIM and ORAI proteins: crucial roles in hallmarks of cancer. Am J Physiol Cell Physiol. 2016; 310:C509–519.
11. Dai L, Zhuang L, Zhang B, Wang F, Chen X, Xia C. DAG/PKCdelta and IP3/Ca(2)(+)/CaMK IIbeta Operate in Parallel to Each Other in PLCgamma1-Driven Cell Proliferation and Migration of Human Gastric Adenocarcinoma Cells, through Akt/mTOR/S6 Pathway. Int J Mol Sci. 2015; 16:28510–28522.
12. Zhang L, Liu Y, Song F, Zheng H, Hu L, Lu H, Liu P Hao X, Zhang W, Chen K. Functional SNP in the microRNA-367 binding site in the 3′UTR of the calcium channel ryanodine receptor gene 3 (RYR3) affects breast cancer risk and calcification. Proc Natl Acad Sci USA. 2011; 108:13653–13658.
13. Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007; 87:593–658.
14. Miyakawa T, Maeda A, Yamazawa T, Hirose K, Kurosaki T, Iino M. Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 1999; 18:1303–1308.
15. Hattori M, Suzuki AZ, Higo T, Miyauchi H, Michikawa T, Nakamura T, Inoue T, Mikoshiba K. Distinct roles of inositol 1,4,5-trisphosphate receptor types 1 and 3 in Ca2+ signaling. J Biol Chem. 2004; 279:11967–11975.
16. de Mattia F, Gubser C, van Dommelen MM, Visch HJ, Distelmaier F, Postigo A, Luyten T, Parys JB, de Smedt H, Smith GL, Willems PH, van Kuppeveld FJ. Human Golgi antiapoptotic protein modulates intracellular calcium fluxes. Molecular biology of the cell. 2009; 20:3638–3645.
17. Szatkowski C, Parys JB, Ouadid-Ahidouch H, Matifat F. Inositol 1,4,5-trisphosphate-induced Ca2+ signalling is involved in estradiol-induced breast cancer epithelial cell growth. Molecular cancer. 2010; 9:156.
18. Tsunoda T, Koga H, Yokomizo A, Tatsugami K, Eto M, Inokuchi J, Hirata A, Masuda K, Okumura K, Naito S. Inositol 1,4,5-trisphosphate (IP3) receptor type1 (IP3R1) modulates the acquisition of cisplatin resistance in bladder cancer cell lines. Oncogene. 2005; 24:1396–1402.
19. Vervloessem T, Yule DI, Bultynck G, Parys JB. The type 2 inositol 1,4,5-trisphosphate receptor, emerging functions for an intriguing Ca(2)(+)-release channel. Biochim Biophys Acta. 2015; 1853:1992–2005.
20. Kang SS, Han KS, Ku BM, Lee YK, Hong J, Shin HY, Almonte AG, Woo DH, Brat DJ, Hwang EM, Yoo SH, Chung CK, Park SH, et al. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 2010; 70:1173–1183.
21. Sakakura C, Hagiwara A, Fukuda K, Shimomura K, Takagi T, Kin S, Nakase Y, Fujiyama J, Mikoshiba K, Okazaki Y, Yamagishi H. Possible involvement of inositol 1,4,5-trisphosphate receptor type 3 (IP3R3) in the peritoneal dissemination of gastric cancers. Anticancer Res. 2003; 23:3691–3697.
22. Shibao K, Fiedler MJ, Nagata J, Minagawa N, Hirata K, Nakayama Y, Iwakiri Y, Nathanson MH, Yamaguchi K. The type III inositol 1,4,5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium. 2010; 48:315–323.
23. Mound A, Rodat-Despoix L, Bougarn S, Ouadid-Ahidouch H, Matifat F. Molecular interaction and functional coupling between type 3 inositol 1,4,5-trisphosphate receptor and BK channel stimulate breast cancer cell proliferation. Eur J Cancer. 2013; 49:3738–51.
24. Schwab A, Finsterwalder F, Kersting U, Danker T, Oberleithner H. Intracellular Ca2+ distribution in migrating transformed epithelial cells. Pflugers Arch. 1997; 434:70–76.
25. Ouadid-Ahidouch H, Dhennin-Duthille I, Gautier M, Sevestre H, Ahidouch A. TRP channels: diagnostic markers and therapeutic targets for breast cancer? Trends Mol Med. 2013; 19:117–124.
26. Prevarskaya N, Ouadid-Ahidouch H, Skryma R, Shuba Y. Remodelling of Ca2+ transport in cancer: how it contributes to cancer hallmarks? Philos Trans R Soc Lond B Biol Sci. 2014; 369:20130097.
27. Bilbao PS, Santillan G, Boland R. ATP stimulates the proliferation of MCF-7 cells through the PI3K/Akt signaling pathway. Arch Biochem Biophys. 2010; 499:40–48.
28. Van Acker K, Nadif Kasri N, De Smet P, Parys JB, De Smedt H, Missiaen L, Callewaert G. IP(3)-mediated Ca(2+) signals in human neuroblastoma SH-SY5Y cells with exogenous overexpression of type 3 IP(3) receptor. Cell Calcium. 2002; 32:71–81.
29. Vanderheyden V, Devogelaere B, Missiaen L, De Smedt H, Bultynck G, Parys JB. Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim Biophys Acta. 2009; 1793:959–970.
30. Saidak Z, Boudot C, Abdoune R, Petit L, Brazier M, Mentaverri R, Kamel S. Extracellular calcium promotes the migration of breast cancer cells through the activation of the calcium sensing receptor. Exp Cell Res. 2009; 315: 2072–2080.
31. Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009; 15:124–134.
32. Yang N, Tang Y, Wang F, Zhang H, Xu D, Shen Y, Sun S, Yang G. Blockade of store-operated Ca(2+) entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Lett. 2013; 330:163–169.
33. Potier M, Gonzalez JC, Motiani RK, Abdullaev IF, Bisaillon JM, Singer HA, Trebak M. Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. FASEB J. 2009; 23:2425–2437.
34. Matyash M, Matyash V, Nolte C, Sorrentino V, Kettenmann H. Requirement of functional ryanodine receptor type 3 for astrocyte migration. FASEB J. 2002; 16:84–86.
35. Praitis V, Simske J, Kniss S, Mandt R, Imlay L, Feddersen C, Miller MB, Mushi J, Liszewski W, Weinstein R, Chakravorty A, Ha DG, Schacht Farrell A, et al. The secretory pathway calcium ATPase PMR-1/SPCA1 has essential roles in cell migration during Caenorhabditis elegans embryonic development. PLoS Genet. 2013; 9:e1003506.
36. Azimi I, Beilby H, Davis FM, Marcial DL, Kenny PA, Thompson EW, Roberts-Thomson SJ, Monteith GR. Altered purinergic receptor-Ca(2)(+) signaling associated with hypoxia-induced epithelial-mesenchymal transition in breast cancer cells. Mol Oncol. 2016; 10:166–178.
37. Tan C, Han LI, Zou L, Luo C, Liu A, Sheng X, Xi D. Expression of P2X7R in breast cancer tissue and the induction of apoptosis by the gene-specific shRNA in MCF-7 cells. Exp Ther Med. 2015; 10:1472–1478.
38. Chadet S, Jelassi B, Wannous R, Angoulvant D, Chevalier S, Besson P, Roger S. The activation of P2Y2 receptors increases MCF-7 breast cancer cells migration through the MEK-ERK1/2 signalling pathway. Carcinogenesis. 2014; 35:1238–1247.
39. Eun SY, Ko YS, Park SW, Chang KC, Kim HJ. P2Y2 nucleotide receptor-mediated extracellular signal-regulated kinases and protein kinase C activation induces the invasion of highly metastatic breast cancer cells. Oncol Rep. 2015; 34:195–202.
40. Zhou JZ, Riquelme MA, Gao X, Ellies LG, Sun LZ, Jiang JX. Differential impact of adenosine nucleotides released by osteocytes on breast cancer growth and bone metastasis. Oncogene. 2015; 34:1831–1842.
41. Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, Duchen MR, Rosato A, Bogeski I, Szabadkai G, Rizzuto R, Mammucari C. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO Mol Med. 2016; 8:569–585.
42. Okeke E, Parker T, Dingsdale H, Concannon M, Awais M, Voronina S, Molgo J, Begg M, Metcalf D, Knight AE, Sutton R, Haynes L, Tepikin AV. Epithelial-mesenchymal transition, IP3 receptors and ER-PM junctions: translocation of Ca2+ signalling complexes and regulation of migration. Biochem J. 2016; 473:757–767.
43. Solanes P, Heuze ML, Maurin M, Bretou M, Lautenschlaeger F, Maiuri P, Terriac E, Thoulouze MI, Launay P, Piel M, Vargas P, Lennon-Dumenil AM. Space exploration by dendritic cells requires maintenance of myosin II activity by IP3 receptor 1. EMBO J. 2015; 34:798–810.
44. Ronde P, Giannone G, Gerasymova I, Stoeckel H, Takeda K, Haiech J. Mechanism of calcium oscillations in migrating human astrocytoma cells. Biochim Biophys Acta. 2000; 1498:273–280.
45. Hamadi A, Giannone G, Takeda K, Ronde P. Glutamate involvement in calcium-dependent migration of astrocytoma cells. Cancer Cell Int. 2014; 14:42.
46. Giannone G, Dubin-Thaler BJ, Dobereiner HG, Kieffer N, Bresnick AR, Sheetz MP. Periodic lamellipodial contractions correlate with rearward actin waves. Cell. 2004; 116:431–443.
47. Burnette DT, Manley S, Sengupta P, Sougrat R, Davidson MW, Kachar B, Lippincott-Schwartz J. A role for actin arcs in the leading-edge advance of migrating cells. Nat Cell Biol. 2011; 13:371–381.
48. Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009; 461:99–103.
49. Tkachenko E, Sabouri-Ghomi M, Pertz O, Kim C, Gutierrez E, Machacek M, Groisman A, Danuser G, Ginsberg MH. Protein kinase A governs a RhoA-RhoGDI protrusion-retraction pacemaker in migrating cells. Nat Cell Biol. 2011; 13:660–667.
50. Brundage RA, Fogarty KE, Tuft RA, Fay FS. Calcium gradients underlying polarization and chemotaxis of eosinophils. Science. 1991; 254:703–706.
51. Clark AJ, Petty HR. Observation of calcium microdomains at the uropod of living morphologically polarized human neutrophils using flash lamp-based fluorescence microscopy. Cytometry A. 2008; 73:673–678.
52. Wei C, Wang X, Chen M, Ouyang K, Song LS, Cheng H. Calcium flickers steer cell migration. Nature. 2009; 457:901–905.
53. Giannone G, Dubin-Thaler BJ, Rossier O, Cai Y, Chaga O, Jiang G, Beaver W, Dobereiner HG, Freund Y, Borisy G, Sheetz MP. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell. 2007; 128: 561–575.
54. Tsai FC, Meyer T. Ca2+ pulses control local cycles of lamellipodia retraction and adhesion along the front of migrating cells. Curr Biol. 2012; 22:837–842.
55. Mikoshiba K. Role of IP3 receptor signaling in cell functions and diseases. Adv Biol Regul. 2015; 57:217–227.
56. Kang D, Hur CG, Park JY, Han J, Hong SG. Acetylcholine increases Ca2+ influx by activation of CaMKII in mouse oocytes. Biochem Biophys Res Commun. 2007; 360:476–482.
57. Qi H, Li L, Shuai J. Optimal microdomain crosstalk between endoplasmic reticulum and mitochondria for Ca2+ oscillations. Sci Rep. 2015; 5:7984.
58. Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012; 13:566–578.
59. Hammadi M, Chopin V, Matifat F, Dhennin-Duthille I, Chasseraud M, Sevestre H, Ouadid-Ahidouch H. Human ether a-gogo K(+) channel 1 (hEag1) regulates MDA-MB-231 breast cancer cell migration through Orai1-dependent calcium entry. J Cell Physiol. 2012; 227: 3837–3846.
60. Darakhshan S, Bidmeshkipour A, Mansouri K, Saeid HM, Ghanbari A. The Effects of Tamoxifen in Combination with Tranilast on CXCL12-CXCR4 Axis and Invasion in Breast Cancer Cell Lines. Iran J Pharm Res. 2014; 13:683–693.