
INTRODUCTION
Breast cancer (BC) consists in a heterogeneous group of malignancies deriving from the epithelial cells lining the milk ducts and still represents the leading cause of cancer-related mortality in women worldwide [1]. BC growth and metastasis are supported by the development of an intricate network of blood vessels which nourish cancer cells with oxygen and nutrients and remove their catabolic waste [2]. An increase in microvascular density positively correlates with the degree of metastasis, tumor recurrence and poorer outcome of invasive BC [3], which further hints at neovascularisation as a key determinant of BC malignancy. Vascular endothelial growth factor (VEGF) comprises a family of secreted polypeptides which include VEGF-A, VEGF-B, VEGF-C, VEGF-D and placental growth factor (PLGF) and act through three cognate receptor tyrosine kinases, designated as VEGFR1 (Flt1), VEGFR2 (Flk1/KDR), and VEGFR3 (Flt4) [4]. VEGF-A (hereafter termed as VEGF) is the prototypical member of the family and constitutes a master regulator of vascular development in health and disease [4–6]. VEGFR2 serves as the main transducer of VEGF signalling on endothelial cell differentiation, proliferation, migration and tubulogenesis [4]. Consistently, targeting VEGFR2 with either humanized monoclonal antibodies, such as bevacizumab and VEGF-Trap, or tyrosine kinase inhibitors (TKIs), such as sorafenib, sunitinib and pazopanib, halted neovessel formation and caused tumor shrinkage in immunodeficient mouse models of most malignancies [7, 8], including BC. Regrettably, these promising observations did not translate into an effective therapeutic benefit for cancer patients. Clinical trials have shown that anti-VEGF drugs, administered either as monotherapy or in combination with chemotherapy or radiation therapy, merely slowed down tumor progression, significantly prolonging only progression free survival (PFS), without improving overall survival (OS) in these individuals [7, 8]. A recent meta-analysis of randomised phase II and III clinical trials of bevacizumab (Avastin) revealed a lack of any substantial OS benefit either in the neoadjuvant or in the metastatic setting in BC patients [9]. Moreover, in 2010, the Food and Drug Administration (FDA) removed approval of Avastin for metastatic BC because of its toxicity and disease progression during long-term treatment [10]. The efficacy of anti-angiogenic therapy is severely limited by the development of refractoriness to anti-VEGF drugs, which causes tumor relapse after an initial period of tumor shrinkage or stasis. Also, a minority of cancer patients are intrinsically resistant to angiogenesis blockade, such that disease progression continues unabated and leads to patient death [11]. The failure of anti-VEGF drugs has been attributed to several mechanisms, such as the recruitment of VEGF-insensitive endothelial progenitor cells (EPCs), which are mobilized from either the bone marrow or the arterial wall and support angiogenesis in several types of cancer [12, 13], including BC. Among the several EPC subsets that have described in peripheral blood, endothelial colony forming cells (ECFCs) represent the only truly endothelial precursors [14], anastomose with the host vasculature and form patent vessels in vivo [14, 15], display an innate tumor tropism [13, 16–18] and may therefore drive the angiogenic switch by supplying endothelial cells to growing neovessels in BC and many other types of tumors, including BC [19–21]. A recent study revealed that ECFC frequency is remarkably increased in peripheral blood of naïve, i.e. not treated, BC patients [22]. Interestingly, VEGF fails to stimulate proliferation and in vitro tubulogenesis in ECFCs isolated from subjects suffering from solid tumors [23], such as renal cell carcinoma (RCC) [24] and infantile hemangioma (IH) [25], as well as in primary myelofibrosis (PMF) [26]. The effect of VEGF on BC-associated ECFCs (BC-ECFCs) is, however, still unknown.
VEGF has recently been shown to stimulate ECFC proliferation by inducing repetitive oscillations in intracellular Ca2+ concentration ([Ca2+]i) [27–29], which in turn promote the nuclear translocation of the Ca2+-sensitive transcription factor, NF-κB. Upon binding to its agonist, VEGFR2 recruits phospholipase Cγ (PLCγ) to synthesize inositol-1,4,5-trisphosphate (InsP3), which triggers the rhythmical Ca2+ discharge from the endoplasmic reticulum (ER), the largest Ca2+ reservoir in ECFCs [30]. VEGF-induced Ca2+ oscillations are sustained over time by the so-called store-operated Ca2+ entry (SOCE) mechanism [28], which is initiated by the activation of the ER Ca2+ sensor Stim1 following InsP3-induced ER Ca2+ depletion [31]. Once activated, Stim1 translocates towards the most peripheral regions of ER, where it traps and gates the two ubiquitous store-operated Ca2+-permeable channels, Orai1 and Transient Receptor Channel Canonical 1 (TRPC1) [24, 31, 32]. The Ca2+ toolkit is severely dysregulated in tumor-associated ECFCs [23, 27, 33, 34]]. For instance, the ER Ca2+ content is significantly reduced in RCC- and IH-derived ECFCs (RCC-ECFCs and IH-ECFCs, respectively) [25, 35], which might prevent VEGF from eliciting the periodical Ca2+ release [23]. Conversely, SOCE is up-regulated and controls proliferation in both RCC-ECFCs [24] and IH-ECFCs [25], thereby standing out as an alternative, promising target for highly angiogenic tumors [33, 36]. Of note, preliminary results indicated that VEGF-induced pro-angiogenic Ca2+ oscillations could be attenuated also in BC-ECFCs [22].
The present investigation was endeavoured to assess whether and how VEGF stimulates pro-angiogenic Ca2+ oscillations in BC-ECFCs. We exploited a multi-disciplinary approach, comprising electron microscopy (EM), Ca2+ imaging, real-time polymerase chain reaction (qRT-PCR), Western blotting, and functional assays to demonstrate that VEGF fails to promote proliferation and in vitro tubulogenesis in BC-ECFCs due to the down-regulation of the underlying repetitive Ca2+ spikes. The suppression of the Ca2+-dependent response to VEGF involves the decrease in ER Ca2+ levels. Conversely, SOCE is still functional in these cells and can be targeted to inhibit BC-ECFC proliferation. Our data contribute to shed light at cellular and molecular level on the failure of anti-VEGF therapies and hint at SOCE as an alternative target to halt vascularization in this disease.
RESULTS
Ultrastructural analysis reveals that BC-ECFCs are morphologically different as compared to normal cells
A recent microarray analysis unveiled that the genomic profile of BC-ECFCs was dramatically altered as compared to normal cells (N-ECFCs) as indicated by the identification of 342 differentially expressed genes (DEGs; 192 up-regulated, 150 down-regulated) in the former [22]. In order to assess whether this remarkable difference in the gene signature was associated to a significant ultrastructural rearrangement, as recently shown for RCC-ECFCs [35], we carried out a throughout analysis at electron microscope levels. This investigation revealed clear ultrastructural differences between N- and BC-ECFCs. Figure 1A and Figure 1D show that smooth ER (sER) vesicles were more abundant in BC-ECFCs. Likewise, rough ER (rER) cisternae occupied a much larger area and were more closely packed in BC-ECFCs as compared to normal cells (Figure 1B and Figure 1E). Finally, mitochondria were also more numerous and enlarged in BC-ECFCs (Figure 1C and Figure 1F). Therefore, not only the gene signature profile [22], but also the cellular microarchitecture was dramatically remodeled in BC-ECFCs.
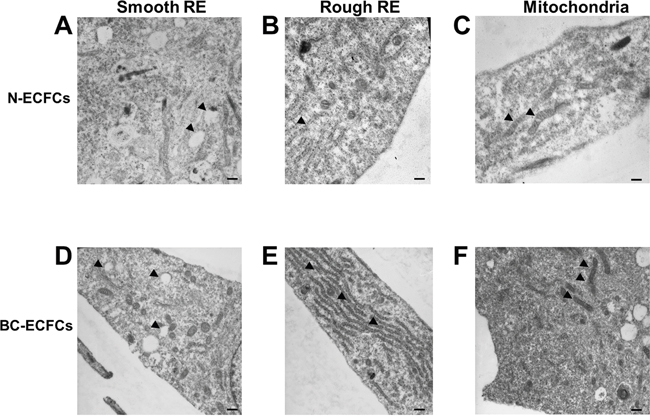
Figure 1: Ultrastructural remodelling in breast cancer-derived endothelial colony forming cells. Ultrastructural analysis of endothelial colony forming cell morphology. Representative images of two N-ECFCs and two BC-ECFCs samples analyzed. The bars in (A-F) correspond to 500 nm.
VEGF does not stimulate proliferation and in vitro tubulogenesis in BC-ECFCs
We have widely shown that 10 ng/mL is the most effective concentration to promote VEGF-dependent proliferation and in vitro tubulogenesis in ECFCs [26, 28, 29, 37]]. Both N- and BC-ECFCs were plated in the presence of EBM-2 supplemented with 2% foetal calf serum and 10 ng/mL VEGF, as described elsewhere [26]. Under such conditions, N-ECFCs reached confluence after 5 days in culture, while BC-ECFCs did not undergo any significant (p<0.05) increase in the rate of cell growth (Figure 2A). Neither N-ECFCs nor BC-ECFCs replicated in the absence of VEGF, while serum starvation did not affect the proliferative response to VEGF in N-ECFCs (data not shown). We further investigated the physiological outcome of VEGF by carrying out a tube formation assay in Matrigel, which reconstitutes the basement membrane extracellular matrix. This assay recapitulates many steps of the angiogenic process, including adhesion, migration, protease activity, and tubule formation, and is, therefore, extremely useful to evaluate whether VEGF differently affects N- and BC-ECFCs [22, 24, 26]. Ten ng/mL VEGF stimulated N-ECFCs to assemble into an extended bidimensional capillary-like network, while it was ineffective on BC-ECFCs (Figure 2B and Figure 2C). These data clearly show that VEGF stimulates proliferation and in vitro tubulogenesis in N-ECFCs, but not BC-ECFCs. In order to assess whether the distinct effect of VEGF was associated to the down-regulation of VEGFR-2 in tumor-associated cells, we analyzed its expression in both types of cells through flow cytometry, as shown in [24]. However, no significant difference was observed between N- and BC-ECFCs, as depicted in Figure 2D and in Supplementary Figure 1. Therefore, the inability of VEGF to stimulate proliferation in BC-ECFCs is likely to involve the downregulation of VEGFR2 signalling in tumor-associated ECFCs [13, 23].
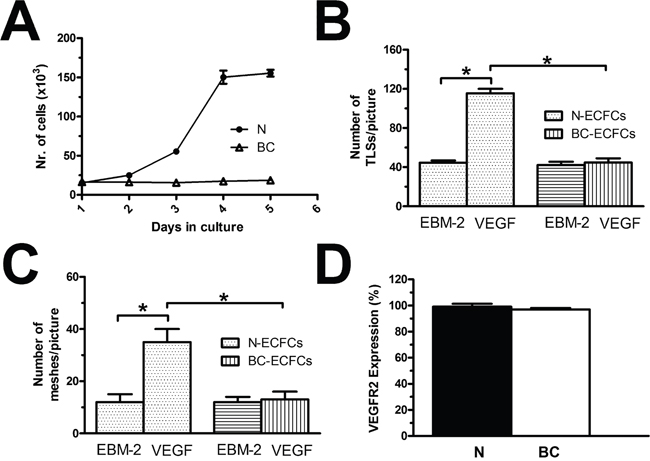
Figure 2: VEGF does not stimulate proliferation in breast cancer-derived endothelial colony forming cells. (A), mean±SE of ECFCs counted after five days in culture in the presence of EBM-2 + VEGF (10 ng/mL). The results are representative of three different experiments conducted on cells harvested from three different donors. VEGF stimulated proliferation in N-ECFCs, but not BC-ECFCs. EBM-2 alone, which was used as control, did not induce proliferation either in N- or BC-ECFCs. (B-C), statistical analysis of the dimensional (total TLSs/picture) and topologic (total number of meshes/picture) parameters of the capillary-like network established by N- and BC-ECFCs, respectively, plated in Matrigel scaffolds. In vitro angiogenesis was stimulated by plating the cells in the presence of the EBM-2 medium (which is devoid of growth factors) supplemented with VEGF (10 ng/mL), while EBM-2 alone was used as a control. The results are representative of three different experiments conducted on ECFCs deriving from three different donors. The asterisk indicates p<0.05. (D), mean±SE of the percentage of ECFCs expressing VEGFR-2, as assessed by flow cytometry. There was no statistically relevant difference between N- and BC-ECFCs.
VEGF-induced intracellular Ca2+ oscillations are down-regulated in BC-ECFCs
As mentioned earlier, the mitogenic effect of VEGF impinges on the onset of intracellular Ca2+ oscillations, which develop with some delay after addition of the growth factor [26, 28, 29]. Consistent with our preliminary results [22], 10 ng/mL VEGF triggered a detectable increase in [Ca2+]i also in BC-ECFCs (Figure 3), while there was no spontaneous Ca2+ activity in absence of the agonist (not shown). The percentage of responding cells was 36.9±10.2% (n=477), which was significantly lower (p=0.00339) as compared to that measured in N-ECFCs (99.3±0.7%, n=380). The Ca2+ response recorded in BC-ECFCs consisted in either transient Ca2+ peaks or short, asynchronous Ca2+ oscillations, which did not arise in phase among the neighbouring cells from a given microscopic field (Figure 3). This feature represents the hallmark of VEGF-induced Ca2+ oscillations in both ECFCs [27] and mature endothelial cells [38]. Each baseline Ca2+ transient was preceded by a slow increase in [Ca2+]i (see inset in Figure 3), which is known as pacemaker Ca2+ rise and is indicative of InsP3-dependent Ca2+ release during the spiking response to VEGF [28]. The oscillating response to VEGF was reversibly abolished by removing the agonist from the perfusate (Figure 4A). When compared to N-ECFCs (Figure 4B), a careful statistical analysis revealed that the percentage of oscillating cells (Figure 4C) and the amplitude (Figure 4E) of the first Ca2+ transient were significantly (p<0.05) smaller in BC-ECFCs as compared to healthy cells. Conversely, the latency of the Ca2+ response was significantly (p<0.05) longer in BC-ECFCs (Figure 4D). We then exploited a recently described home-made software based on wavelet analysis to extract information encoded within the complex spatio-temporal pattern of Ca2+ spikes and obtain a straightforward quantitative evaluation of the differences between VEGF-induced Ca2+ oscillations in N- and BC-ECFCs [26, 39, 40]. This analysis confirmed that the spiking response to VEGF was significantly (p>0.05) reduced in BC-ECFCs (Figure 4F). Taken together, these data suggest that the down-regulation of intracellular Ca2+ oscillations underpins the little, if any, pro-angiogenic effect of VEGF in BC-ECFCs.
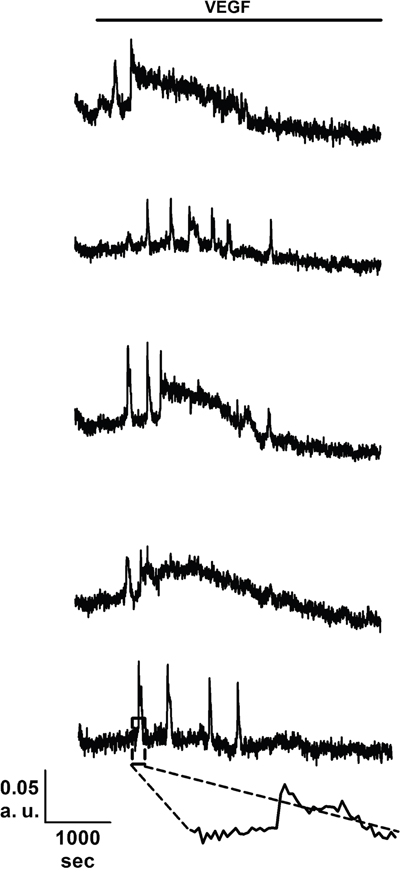
Figure 3: VEGF induces intracellular Ca2+ oscillations in breast cancer-derived endothelial colony forming cells. VEGF (10 ng/mL) elicits heterogeneous repetitive Ca2+ transients in five BC-ECFCs recorded from the same microscopic field. The dashed box in the lowermost trace marks the pacemaker increase in [Ca2+]i that features InsP3-dependent Ca2+ spikes. In these and the other figures, VEGF has been administrated during the time period indicated by the black bars placed above the Ca2+ traces.
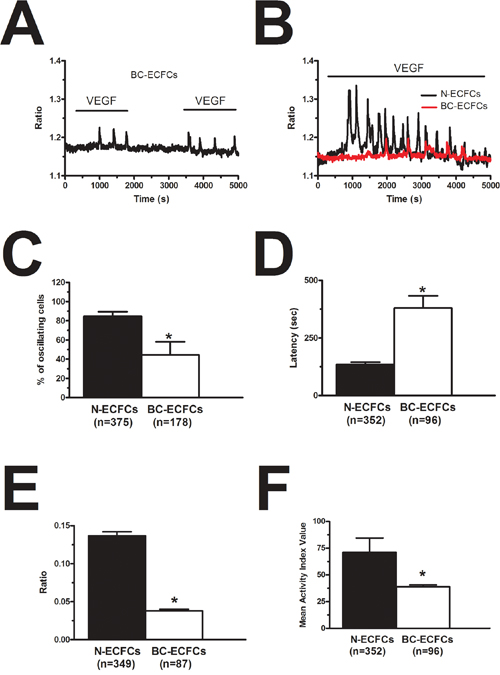
Figure 4: VEGF-induced intracellular Ca2+ oscillations are weaker in breast cancer-derived endothelial colony forming cells. (A), VEGF (10 ng/mL) removal from the bath reversibly inhibited the Ca2+ response to VEGF in BC-ECFCs. (B), VEGF-induced intracellular Ca2+ oscillations in N- and BC-ECFC. VEGF was applied at 10 ng/mL to both cell types. In the following panels, bar histograms have been used to compare the fraction of oscillating cells (C), the latency to the first spike (D), the magnitude of the initial Ca2+ transient (E) and the oscillatory index (F) between N- and BC-ECFCs. The asterisk indicates p<0.05.
VEGF-induced intracellular Ca2+ oscillations require InsP3-dependent Ca2+ release and SOCE in BC-ECFCs
The spiking response to VEGF is shaped by the concerted interplay between InsP3-dependent Ca2+ release and SOCE in N-ECFCs [28], however, this mechanism is subtly remodelled in PMF-derived cells [26]. In order to decipher the molecular underpinnings of VEGF-induced Ca2+ oscillations in BC-ECFCs, we first challenged the cells with the growth factor upon removal of Ca2+ from the perfusate (0Ca2+). Unlike cells bathed in the presence of extracellular Ca2+ (Figure 5A), VEGF still induced 1-2 Ca2+ transients in the absence of extracellular Ca2+, but the Ca2+ activity rapidly subsided despite for the prolonged exposure to the agonist (Figure 5B). The latency of the first Ca2+ transient was significantly (p<0.05) longer under 0Ca2+ conditions (Figure 5C), while there was no statistically relevant difference in its amplitude (Figure 5D). Ca2+ oscillations readily resumed upon Ca2+ restitution to the bath, thereby showing that the Ca2+ signal was initiated by Ca2+ mobilization from the intracellular stores and sustained by Ca2+ entry across the plasma membrane (Figure 5B). In agreement with this notion, removal of external Ca2+ reversibly suppressed ongoing VEGF-induced Ca2+ oscillations (Figure 5E). Therefore, we then focussed on the PLCγ/InsP3 signalling pathway, which underlies periodic ER Ca2+ release in N-ECFCs [28]. VEGF failed to ignite the Ca2+ train in the presence of U73122 (10 μM, 10 min) (1-[6-[[(17b)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione) (Figure 6A), an aminosteroid which selectivity inhibits PLC activity in ECFCs [26, 28, 29], while its inactive structural analogue U73343 (10 μM, 10 min) failed to block the Ca2+ response (Figure 6A). Likewise, 2-aminoethoxydiphenyl borate (2-APB; 50 μM, 20 min), a widely employed blocker of InsP3 receptors (InsP3Rs), suppressed VEGF-induced Ca2+ oscillations (Figure 6B). As 2-APB may also interfere with Stim1, Orai and TRP Vanilloid (TRPV) channels [36], these experiments were performed under 0Ca2+ conditions to prevent any contaminating effect from Ca2+ influx [28]. Finally, VEGF was applied after depletion of the ER Ca2+ pool with cyclopiazonic acid (CPA), which selectively inhibits the activity of Sarco-Endoplasmic Reticulum Ca2+-ATPase (SERCA), thereby preventing Ca2+ sequestration and emptying the endogenous Ca2+ stores [28]. As shown in Figure 6C, VEGF failed to increase intracellular Ca2+ levels following 30 min preincubation with CPA in the absence of extracellular Ca2+ (0Ca2+). The statistical analysis of these data has been synthesized in Figure 6D. SOCE represents the most important pathway for Ca2+ entry in N-ECFCs [27, 37]. Therefore, we assessed its contribution to the maintenance of VEGF-induced Ca2+ oscillations by pre-incubating the cells with BTP2 (N-(4-[3,5-bis(trifluoromethyl)-1H-pyrazo l-1-yl]phenyl)-4-methyl-1,2,3-thiadiazol e-5-carboxamide) (20 μM, 30 min), a pyrazole derivative which selectively inhibits SOCE in both ECFCs [31, 37] and a growing number of cell types [41, 42]. This treatment did not prevent the onset of the Ca2+ response to VEGF, but curtailed its duration to 1-2 Ca2+ spikes (Figure 5F), thereby mimicking the effect of 0Ca2+. Unlike 0Ca2+ conditions, however, BTP2 did not affect the latency of the first Ca2+ transient (Figure 5C), as well as it did not reduce its amplitude (Figure 5D). Overall, these observations clearly show that VEGF-induced Ca2+ oscillations required the InsP3-dependent rhythmical ER Ca2+ discharge and were sustained by SOCE also in BC-ECFCs. Therefore, the downregulation of the Ca2+-dependent pro-angiogenic response to VEGF in these cells must involve the remodeling of one or more components of their Ca2+ toolkit.
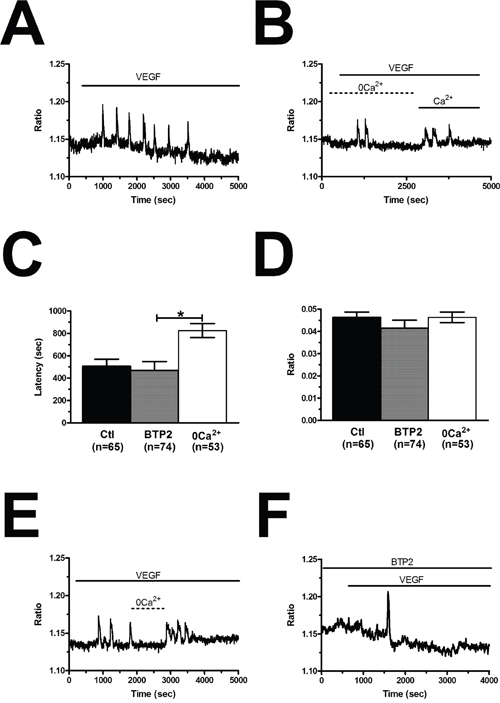
Figure 5: VEGF-induced intracellular Ca2+ oscillations are triggered by endogenous Ca2+ release and maintained by store-operated Ca2+ entry. (A), intracellular Ca2+ oscillations evoked by VEGF (10 ng/mL) in the presence of extracellular Ca2+. (B), VEGF induced only 2 Ca2+ spikes in the absence of extracellular Ca2+ (0Ca2+), whereas Ca2+ oscillations resumed upon Ca2+ re-addition to the extracellular solution. In the following panels, bar histograms have been used to compare the latency to the first spike (C) and the magnitude of the initial Ca2+ transient (D) under 0Ca2+ conditions or preincubation with BTP2 (20 μM, 30 min), which selectively inhibits SOCE in ECFCs. The asterisk indicates p<0.05. (E), removal of extracellular Ca2+ (0Ca2+) caused a reversible inhibition of ongoing VEGF-induced Ca2+ oscillations. VEGF was administered at 10 ng/mL. (F), BTP2 (20 μM, 30 min), a selective SOCE inhibitor, did not prevent the onset of VEGF-induced intracellular Ca2+ oscillations, but curtailed their duration to 1-2 transients.
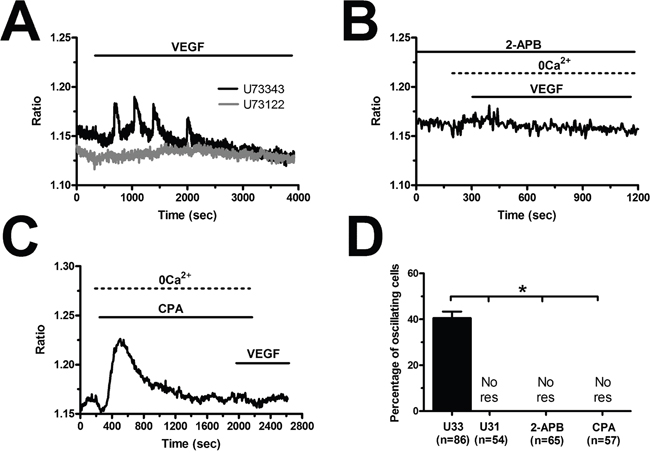
Figure 6: VEGF-induced intracellular Ca2+ oscillations are initiated by the PLCγ/InsP3 pathway in breast cancer-derived endothelial colony forming cells. (A), U73122 (10 μM, 10 min), a selective PLC blocker, prevented the Ca2+ response to VEGF, while U73343 (10 μM, 10 min), an inactive analogue of U7322, was without effect. (B), 2-APB (50 μM, 20 min), an InsP3R blocker, suppressed VEGF-induced Ca2+ oscillations under 0Ca2+ conditions to prevent any contaminating effect on plasmalemmal channels. (C), the depletion of the InsP3-sensitive ER Ca2+ store with CPA (10 μM) in the absence of external Ca2+ (0Ca2+) prevented the following Ca2+ response to VEGF (10 ng/mL). Note the transient increase in [Ca2+]i caused by CPA due to the depletion of the ER Ca2+ pool. (D), mean±SE of the percentage of BC-ECFCs responding to VEGF (10 ng/mL) under the designated treatments. The asterisk indicates p<0.05. NoR: No response.
The ER Ca2+ content is decreased, while SOCE is unaffected, in BC-ECFCs
In order to assess whether and how the intracellular Ca2+ handling is altered in BC-ECFCs, we exploited the “Ca2+ add-back” protocol, which consists in first depleting the ER Ca2+ pool with CPA (10 μM) in the absence of extracellular Ca2+ (0Ca2+) and then restoring extracellular Ca2+ to monitor the following SOCE [24, 25]. This protocol has been largely used to assess both the ER Ca2+ content and the extent of SOCE activation in a myriad of cancer cells [43, 44], including tumor-associated ECFCs [26, 39, 40]. We found that CPA-induced ER Ca2+ release was significantly (p<0.05) reduced as compared to N-ECFCs, while SOCE amplitude was unaffected (Figure 7A and Figure 7B). To further corroborate these data, we repeated the “Ca2+ add-back” protocol in the presence of the physiological autacoid, ATP (100 μM), which binds to metabotropic P2Y receptors to stimulate InsP3 synthesis and promote InsP3-dependent ER Ca2+ release [26, 39, 40]. Again, ATP-induced InsP3-dependent ER Ca2+ release was significantly (p<0.05) lower in BC-ECFCs, while ATP-induced SOCE was unaltered as respect to N-ECFCs (Figure 7C and Figure 7D). As previously described [24], ATP was removed from the extracellular solution 100 sec before Ca2+ re-addition to prevent Ca2+ entry across store-independent pathways (Figure 7C). The reduction in ATP-induced intracellular Ca2+ release was not due to the down-regulation of InsP3Rs, as qRT-PCR analysis carried out by using the specific primers described in Supplementary Table 1 showed that there was no statistically relevant difference in the expression of all InsP3R isoforms between N- and BC-ECFCs (Supplementary Figure 2). Collectively, these data concur with the preliminary data we recently reported [22] and strongly suggest that the Ca2+ signalling toolkit is partially remodelled in BC-ECFCs. This dysregulation consists in a dramatic drop in ER Ca2+ levels, which might prevent the InsP3-dependent ER Ca2+ cycling that underlies the pro-angiogenic response to VEGF.
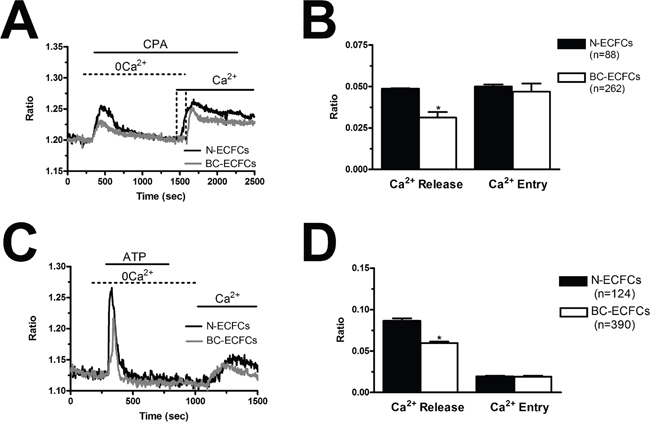
Figure 7: Remodelling of the Ca2+ toolkit in breast cancer-derived endothelial colony forming cells. (A), the intracellular Ca2+ pool was depleted by stimulating the cells with CPA (10 μM) in the absence of external Ca2+ (0Ca2+), and Ca2+ influx through store-operated channels was then assessed on Ca2+ replenishment to the bathing solution. (B), mean±SE of the amplitude of CPA-induced Ca2+ release and CPA-induced SOCE in N- and BC-ECFCs. The asterisk indicates p<0.05. (C), ATP (100 μM) evoked a transient increase in [Ca2+]i in N- and BC-ECFCs bathed in the absence of external Ca2+ (0Ca2+). (D) ATP was then removed from the bath, while Ca2+ was readded to the perfusate in order to measure SOCE. F, mean±SE of the amplitude of ATP-induced Ca2+ release and ATP-induced SOCE recorded from both N- and BC-ECFCs. The asterisk indicates p<0.05.
The pharmacological profile and molecular composition of SOCE is similar to that described in N-ECFCs
In order to confirm that BTP2 selectively inhibits agonist-induced SOCE in BC-ECFCs, we carried out the “Ca2+ add-back” protocol in the presence of this drug. BTP2 (20 μM, 30 min) selectively blocked both CPA- and ATP-induced SOCE, while it did not affect the initial phase of intracellular Ca2+ mobilization (Figure 8A–8D). The same effect was achieved by the trivalent cation, La3+ (10 μM, 30 min) (Figure 9A–9D). As discussed elsewhere [36, 37, 45], BTP2 and low micromolar doses of lanthanides specifically target store-operated channels (SOCs) whose pore-forming subunits are provided by Orai1 and/or TRPC1. These data further corroborate the notion that both passive CPA-facilitated and active InsP3-mediated ER Ca2+ store depletion led to the activation of the same plasmalemmal Ca2+-permeable pathway in ECFCs [23, 27]. Recent studies showed that Orai3 may replace Orai1 as pore-forming of store-operated channels [36, 43, 46]. To assess this issue in BC-ECFCs, we took advantage from the biphasic dependence of Orai1 on 2-APB. 2-APB activates Orai1 at 5 μM, while inhibits it at concentrations higher than 30 μM [24, 36]. Therefore, we fully activated SOCE by challenging BC-ECFCs with thapsigargin (2 μM), another SERCA inhibitor structurally unrelated to CPA, in the presence of extracellular Ca2+. As shown in RCC-ECFCs [24], this treatment caused a sustained increase in [Ca2+]i which was due to both passive ER Ca2+ release and SOCE. The following addition of 5 μM 2-APB caused a further increase in [Ca2+]i, which was in turn suppressed by the subsequent application of 50 μM 2-APB in 61 cells (Figure 10). This data further corroborates the role of Orai1 in SOCE activation in BC-ECFCs.
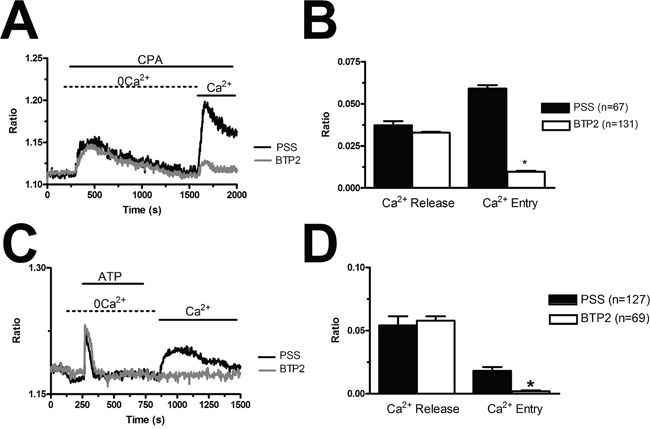
Figure 8: BTP2 inhibits store-dependent Ca2+ entry in breast cancer-derived endothelial colony forming cells. (A), CPA-elicited SOCE in the absence and presence of BTP2 (20 μM). The cells were pre-incubated with the drug for 30 min before the beginning of the experimental protocol. CPA was administered at 10 μM. (B), mean±SE of the amplitude of CPA-induced intracellular Ca2+ release and CPA-induced SOCE in the absence and presence of BTP2. The asterisk indicates p<0.05. (C), ATP-evoked intracellular Ca2+ release and SOCE in the presence and absence of BTP2 (20 μM). The cells were pre-incubated with the drug for 30 min before the beginning of the experimental protocol. ATP was applied at 100 μM. (D), mean±SE of the amplitude of CPA-induced Ca2+ release and CPA-induced SOCE in the absence and presence of BTP2. The asterisk indicates p<0.05.
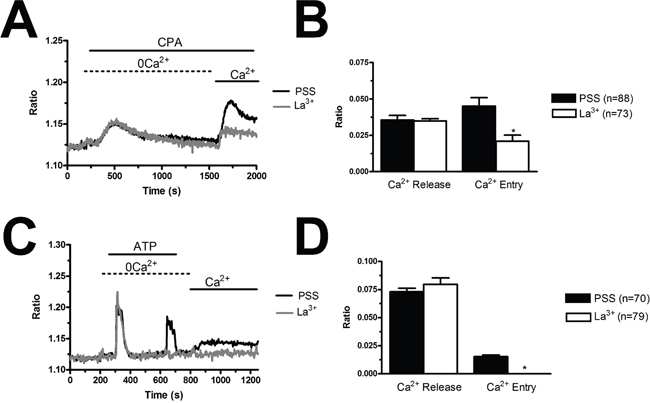
Figure 9: La3+ inhibits store-dependent Ca2+ entry in breast cancer-derived endothelial colony forming cells. (A), CPA-elicited SOCE in the absence and presence of La3+ (10 μM). The cells were pre-incubated with the drug for 20 min before the beginning of the experimental protocol. CPA was administered at 10 μM. (B), mean±SE of the amplitude of CPA-induced intracellular Ca2+ release and CPA-induced SOCE in the absence and presence of La3+. The asterisk indicates p<0.05. (C), ATP-evoked intracellular Ca2+ release and SOCE in the presence and absence of La3+ (10 μM). The cells were pre-incubated with the drug for 30 min before the beginning of the experimental protocol. ATP was applied at 100 μM. (D), mean±SE of the amplitude of CPA-induced Ca2+ release and CPA-induced SOCE in the absence and presence of BTP-2. The asterisk indicates p<0.05.
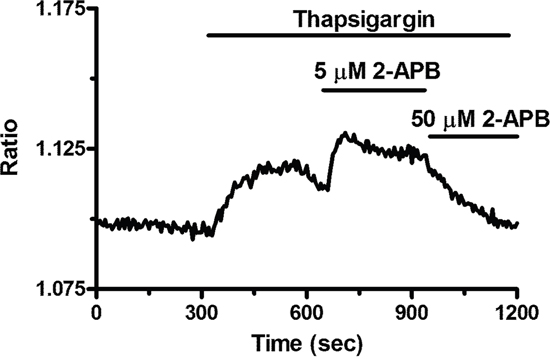
Figure 10: 2-APB exerts a dose-dependent effect on store-operated Ca2+ entry in breast cancer-derived endothelial colony forming cells. 5 μM 2-APB enhanced SOCE induced by thapsigargin (2 μM), whereas 50 μM inhibited it. This is a pharmacological property of Orai1 containing store-operated channels.
To extend this information at molecular level, we investigated the expression of the molecular components of SOCE through both qRT-PCR and immunoblotting. The expression of Stim1-2, Orai1-3, TRPC1-7 transcripts was assessed by qRT-PCR analysis of mRNA extracts from N- and BC-ECFCs, as previously shown [24, 25, 47]. We utilized the specific primers described in Supplementary Table 2, whereas negative controls were conducted by omitting the reverse transcriptase (not shown). The housekeeping gene β-actin served as reference gene for data normalization. We found that only Stim1 (Figure 11A) was significantly (p<0.05) up-regulated in BC-ECFCs as compared to N-ECFCs, while there was no remarkable difference in the pattern of expression of Stim2 (Figure 11B), Orai1 (Figure 11C), Orai2 (Figure 11D) and Orai3 (Figure 11E). Also, TRPC1 mRNA levels did not differ between N- and BC-ECFCs, while TRPC4 was significantly (p<0.05) over-expressed in tumor-associated cells (Figure 11F and Figure 11G). Similar to N-ECFCs, BC-ECFCs lacked TRPC2, TRPC3, TRPC5, TRPC6 and TRPC7 (data not shown). These data were confirmed at protein levels by a Western blot analysis performed by using affinity-purified antibodies directed against Stim1, Orai1, TRPC1 and TRPC4 (Figure 12). Immunoblots revealed a major band of 33 kDa for Orai1 and of 110 kDa for TRPC1 in both cell types, whereas the anti-Stim1 antibody detected two bands of 100 kDa and 77 kDa only in BC-ECFCs. Stim1 was detected as a double also in RCC-ECFCs [24], several human BC and RCC cell lines [43, 48], and primary cultures of human mestatic RCC cell lines [49]. Densitometric analysis of the gels showed that Stim1 protein was up-regulated in BC-ECFCs, while Orai1 and TRPC1 proteins were equally expressed in both cell types. Similar to Stim1, immunoblotting revealed a single band of 110 kDa for TRPC4 (Figure 12D), which was significantly (p<0.05) up-regulated compared to N-ECFCs. Taken together, these findings demonstrate that: 1) the pharmacological profile and molecular composition of SOCE in BC-ECFCs is similar to that described in N-ECFCs; and 2) the attenuation of VEGF-induced Ca2+ oscillations is unlikely to involve the recruitment of an alternative SOC pathway in tumor-associated cells. These data also confirm that BTP2 selectively targets SOCE as TRPC3 and TRPC5, that could be inhibited by this drug in heterologous expression systems [36], are absent in BC-ECFCs. Likewise, BC-ECFCs lack diacylglycerol-gated Ca2+-permeable channels, such as TRPC3, TRPC6 and TRPC7, which consists with our previous findings in N- and RCC-ECFCs [24].
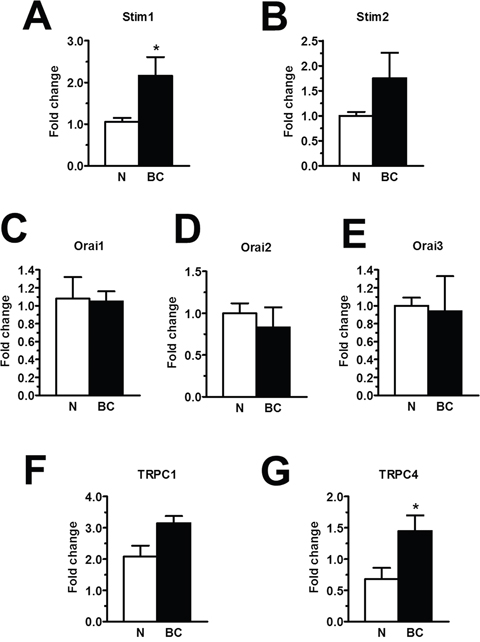
Figure 11. The expression of Stim1-2, Orai1-3, TRPC1 and TRPC4 transcripts in breast cancer-derived endothelial colony forming cells. qRT-PCR showing increased expression of Stim1 (A) mRNA in BC-ECFCs compared to N-ECFCs. Conversely, Stim2 (B), Orai1 (C), Orai2 (D), Orai3 (E), TRPC1 (F), were not differently expressed in BC-ECFCs. Also, the expression of TRPC4 mRNA was enhanced in BC-ECFCs (G). Bars represent mean±SE of at least 4 different experiments each from different RNA extracts. The asterisk indicates p<0.05 vs. N-ECFCs (ANOVA followed by Newman-Keuls’ Q test). The PCR products were of the expected size (not shown): Orai1, 257 bp; Orai2, 334 bp; Orai3, 159 bp; Stim1, 347 bp; Stim2, 186 bp; TRPC1, 307 bp and TRPC4, 300 bp.
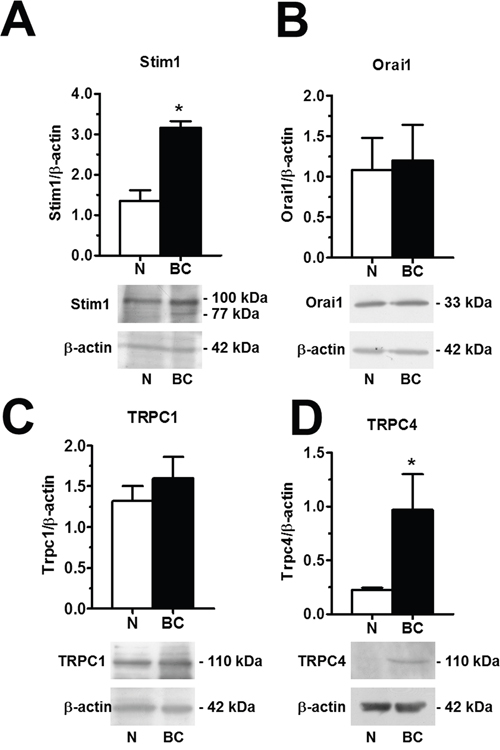
Figure 12: Orai1, Stim1, TRPC1 and TRPC4 proteins are up-regulated in breast cancer-associated endothelial colony forming cells. Western blot and densitometry depicting the significant elevation in Orai1 (A), Stim1 (B), TRPC1 (C), and TRPC4 (D) proteins in BC-ECFCs as compared to N-ECFCs. Blots for Orai1, Stim1, TRPC1 and TRPC4 representative of 4 different experiments are shown in the lower panel. Lanes were loaded with 20 μg of proteins. Major bands of the expected molecular weight were observed in both cell types. One additional band of 77 kDa was detected by anti-Stim1 in RCC-EPCs. When both Stim1 bands (77 and 100 kDa) were compared to the single band detected at 100 kDa in N-ECFCs, the expression of Stim1 protein became significantly higher in BC-ECFCs. Each bar in the upper panel represents the mean±SE of the densitometric analysis of four different experiments. The asterisk indicates p<0.01 (Student's t-test).
The pharmacological blockade of SOCE inhibits BC-ECFC proliferation and in vitro tubulogenesis
The observation that VEGF does not stimulate proliferation and tube formation in BC-ECFC leads to the quest for alternative targets to halt BC vascularization. Our previous work provided the evidence that SOCE represents a druggable signalling pathway to inhibit the angiogenic activity of tumor-associated ECFCs [24, 25, 47]. Our recent study showed that there was no difference in either growth kinetics or tubulogenic rate between N- and BC-ECFCs cultured in EGM-2 [22]. Therefore, we first ascertained whether BC-ECFC proliferation was inhibited in BC-ECFCs cultured in EGM-2 supplemented with either of the following drugs: BAPTA (30 μM, 1 hour), BTP2 (20 μM, 30 min), and La3+ (10 μM, 30 min). As observed earlier for N-, RCC-, and IH-ECFCs, all of those treatments also prevented BC-ECFCs from reaching confluence at 5 days from plating (Figure 13A). Finally, we probed the effect of carboxyamidotriazole (CAI), a synthetic small molecule non-specific inhibitor of various types of Ca2+-permeable channels which hastens proliferation in BC endothelial cells [50] and RCC-ECFCs [24]. We first confirmed that CAI (10 μM, 20 min) fully abolished both phases of the Ca2+ response to CPA (Figure 14A and Figure 14B) and ATP (Figure 14C and Figure 14D). Then we found that CAI prevented VEGF-induced Ca2+ oscillations (Supplementary Figure 3) and blocked proliferation in BC-ECFCs (Figure 13A). Furthermore, we found that BAPTA (30 μM, 1 hour), BTP2 (20 μM, 30 min), and CAI (10 μM, 20 min) also blocked in vitro tubulogenesis when BC-ECFCs were plated in Matrigel in the presence of the EGM-2 medium. Again, we evaluated both dimensional (total number of TLSs per picture) (Figure 13B) and topological (number of meshes per picture) (Figure 13C) parameters of the capillary-like network generated by BC-ECFCs plated in a Matrigel scaffold in the presence and the absence of the aforementioned drugs. Therefore, Ca2+ signalling in general and, particularly, SOCE could serve as a suitable target to dampen vascularization also in BC.
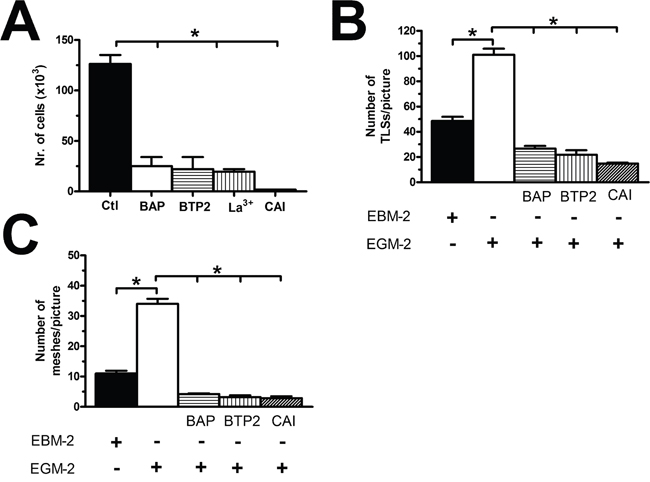
Figure 13: The pharmacological inhibition of store-operated Ca2+ entry blocks proliferation in breast cancer-derived endothelial colony forming cells. (A), BAPTA (30 μM, 2 hours), a membrane-permeable buffer of intracellular Ca2+ levels, BTP2 (20 μM, 30 min), La3+ (10 μM, 30 min) and CAI (10 μM, 30 min) blocked proliferation in BC-ECFCs cultured in the presence of EGM-2. The asterisk indicates p<0.05. (B-C), statistical analysis of the dimensional (total TLSs per picture) and topological (total number of junctions between adjacent TLS and total number of meshes per picture) parameters of the capillary-like networks established by BC-ECFCs plated in Matrigel scaffolds in the presence and absence of BAPTA (30 μM, 2 hours), BTP2 (20 μM, 30 min), and CAI (10 μM, 30 min). In vitro angiogenesis was stimulated by plating the cells in the presence of EGM-2, while the EBM-2 medium (which is devoid of growth factors) was used as a control. The results are representative of three different experiments conducted on cells derived from three different donors. The asterisk indicates p<0.05.
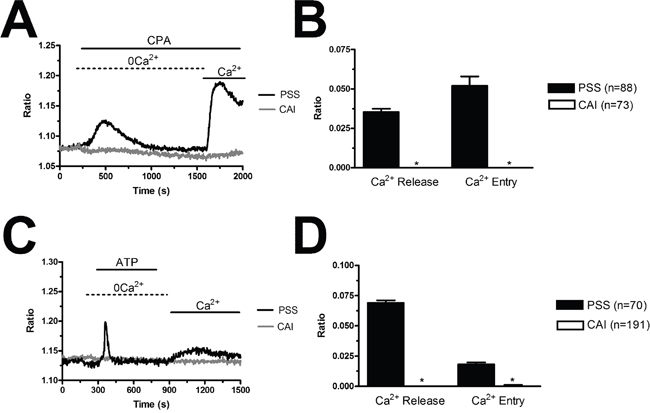
Figure 14: Carboxyamidotriazole suppresses intracellular Ca2+ signalling in endothelial progenitor cells. (A), CAI (10 μM, 20 min) abolishes the Ca2+ response to CPA (10 μM) in BC-ECFCs. (B), mean±SE of the amplitude of CPA-induced intracellular Ca2+ release and SOCE in BC-ECFCs. (C), CAI (10 μM, 20 min) abolishes the Ca2+ response to ATP (100 μM) in BC-ECFCs. B, mean±SE of the amplitude of ATP-induced intracellular Ca2+ release and SOCE in BC-ECFCs. The asterisk indicates p<0.05.
DISCUSSION
VEGFR2 has long been regarded as the most suitable target to interfere with tumor vascularization and increase OS in patients affected by highly angiogenic tumors, such as RCC and BC. The majority of in vitro studies which led to this assertion were, however, carried out on normal endothelial cell lines, such as human umbilical vein endothelial cells (HUVECs) and human dermal microvascular endothelial cell (HMVECs-D) [24]. Herein, we took advantage from the availability of patients-derived cells to assess whether VEGF stimulates proliferation in ECFCs, which are likely to support the angiogenic switch during BC development [19–21]. Our results provide the first evidence that ECFCs, which contribute to the angiogenic switch in BC [19, 21], are insensitive to VEGF due to the attenuation of pro-angiogenic intracellular Ca2+ oscillations that arise upon VEGFR-2 activation in normal cells. These results may help to understand the cellular and molecular underpinnings of primary and secondary refractoriness of BC patients to anti-VEGF therapies.
In stringent disagreement with the traditional assumption [6, 51], VEGF was unable to stimulate proliferation and tube formation in BC-ECFCs. This feature was not due to the downregulation of VEGFR2, the receptor isoform that mediates its pro-angiogenic effect also in ECFCs [52], and suggested that the signalling pathways downstream VEGFR2 could be silenced/suppressed in tumor-associated ECFCs. Intracellular Ca2+ oscillations represent the main mode whereby growth factors stimulate angiogenesis in both mature endothelial cells [38, 39] and their more immature precursors [28, 29]. An increase in [Ca2+]i represents a central hub in the sophisticated network of interconnected signalling pathways that are recruited following VEGFR2 stimulation. For instance, intracellular Ca2+ oscillations deliver the most effective signal to engage the Ca2+-dependent transcription factors, NF-κB and nuclear factor of activated T cells (NFAT), which play a key role in VEGF-induced ECFC proliferation [28, 53], extracellular signal-regulated kinase (ERK), and phosphatidyl inositol 3-kinase (PI3K)/Akt/protein kinase B [54], following VEGF stimulation. Our Ca2+ imaging recordings revealed that VEGF-induced intracellular Ca2+ oscillations were dramatically down-regulated in BC-ECFCs as compared to healthy cells. This observation is fully consistent with the results obtained from other types of tumor-associated ECFCs. Accordingly, VEGF failed to induce detectable Ca2+ spikes in RCC- and IH-ECFCs [24, 25], although VEGFR2 was normally expressed in these cells. Similarly, VEGF-induced Ca2+ oscillations were rather weak in ECFCs isolated from individuals affected from PMF [26], a chronic myeloproliferative neoplasm that is characterized by the development of a robust vascular network in both the bone marrow and spleen. Interestingly, VEGF failed to induce proliferation and tube formation also in these cells, a finding that has been invoked to explain the failure of anti-VEGF in this disease [13, 26, 34]. We, therefore, suggest that the weaker Ca2+ burst induced by VEGF in BC-ECFCs and PMF-ECFCs as compared to N-ECFCs does not reach the threshold of activation of endothelial Ca2+-dependent pro-angiogenic transcription factors, such as NF-κB and NFAT.
The down-regulation of VEGF-induced intracellular Ca2+ oscillations could depend on the recruitment of signalling components other than those at work in N-ECFCs [26] or on the remodelling of the Ca2+ toolkit [24, 25, 35]. However, the following pieces of evidence confirmed that the PLCγ/InsP3/SOCE signalling pathway was engaged by VEGF also in BC-ECFCs. First, the Ca2+ signal arose in the absence of extracellular Ca2+, which indicated that the Ca2+ response was driven by intracellular Ca2+ mobilization rather than Ca2+ entry, as described in PMF-ECFCs [26]. Second, the pharmacological blockade of PLCγ with U73122 or of InsP3Rs with 2-APB abrogated the onset of the Ca2+ spikes. Third, the pharmacological blockade of SOCE with BTP2 mimicked the effect of 0Ca2+ by curtailing the duration of the Ca2+ train without preventing its onset. Unlike 0Ca2+ conditions, however, BTP2 did not delay the onset of the 1st Ca2+ spike. This apparent discrepancy could be explained by anticipating that BTP does not fully abrogate SOCE in BC-ECFCs (see Figure 7). We hypothesize that SOCE represents the source of Ca2+ necessary to sensitize InsP3Rs to PLCγ-derived InsP3, by acting either on the luminal or the cytosolic side [55, 56], thereby regulating the latency of the 1st Ca2+ spike. If BTP2 does not fully abrogate SOCE, then some extremely localized Ca2+ influx is predict to occur in proximity of InsP3Rs and maintain the latency of the signal unaltered. Obviously, no Ca2+ entry occurs in the absence of external Ca2+, which could lead to a significant delay in the onset of the oscillations.
Based on the evidences illustrated above, the most likely interpretation to account for the attenuation of the pro-angiogenic Ca2+ oscillations was the remodelling of the Ca2+ toolkit in BC-ECFCs. This phenomenon has recently been proposed to underlie the resistance to chemotherapy and radiation therapy in both tumor cells [57, 58] and tumor-associated ECFCs [23, 34]. For instance, the reduction in ER Ca2+ concentration ([Ca2+]ER) and the hypo-expression of InsP3Rs prevent VEGF from triggering robust Ca2+ spikes in RCC-ECFCs [24, 35]. Our Ca2+ imaging recordings revealed that the ER Ca2+ pool was reduced also in BC-ECFCs. Accordingly, CPA-induced intracellular Ca2+ release was significantly dampened as compared to healthy cells. CPA, as well as its structurally unrelated analogue, thapsigargin, unmasks the physiological Ca2+ leakage through ER membrane by inhibiting SERCA-mediated Ca2+ sequestration, thereby leading to the rapid depletion of the ER Ca2+ pool. Previously, we exploited this strategy to show that ER Ca2+ levels were decreased in RCC-, IH-, and PMF-ECFCs [24, 25, 47]. Notably, the chronic underfilling of ER in tumor-associated ECFCs was confirmed by directly measuring [Ca2+]ER through recombinant ER-targeted aequorin [35, 59]. This observation was further supported by the finding that InsP3-dependent Ca2+ release, which was monitored by challenging the cells with the InsP3-synthesizing autacoid ATP [24, 44], was also smaller in BC-ECFCs, while InsP3Rs were normally expressed. On the other hand, there was no difference in the amplitude and molecular composition of SOCE between N- and BC-ECFCs. Consistently, there was no difference in the expression profile of Orai1 and TRPC1, which provide the Ca2+-permeable routes on the plasma membrane gated following ER depletion, between N- and BC-ECFCs. The overexpression of Stim1, which functions as the sensor of [Ca2+]ER and activates SOCs, was not sufficient to enhance SOCE in the latter, as previously shown in human salivary gland cells [60]. This feature confirms that Orai1 and TRPC1 represent the two limiting structural components of the SOCE machinery and that a correct stoichiometric expression of Stim1, Orai1 and TRPC1 is necessary for full SOCE activation in ECFCs [37, 61]. Moreover, SOCE was inhibited by BTP2 and 10 μM La3+, which block SOCs contributed by Orai1 and TRPC1 in a growing number of cell types [36, 62–65], including tumor-associated ECFCs [24, 25]. Unlike IH-ECFCs [25], pre-incubating the cells with either BTP2 or La3+ did not cause the depletion of the InsP3-sensitive ER Ca2+ pool. This result suggests that SOCE is not, or just minimally, activated under resting conditions and requires agonist-induced ER depletion to arise. Altogether, these observations strongly suggest that the down-regulation of VEGF-induced Ca2+ oscillations is due to the reduction in [Ca2+]ER, which prevents InsP3 from triggering the dynamic interaction between InsP3-dependent Ca2+ release and SOCE which reliably engages the Ca2+-dependent pro-angiogenic genetic program in N-ECFCs. The drop in ER Ca2+ levels could be due to the up-regulation of TMTC1 recently reported in BC-ECFCs [22]. TMTC1 is a novel ER-resident tetratricopeptide repeat-containing adapter protein that binds to SERCA2B to curb its activity [66]. It has been shown that over-expression of TMTC1 in HEK293T caused a strong reduction in acetylcholine- and ionomycin-induced intracellular Ca2+ mobilization [66]. Although this hypothesis remains to be experimentally probed, we speculate that the increase in TMTC1 levels leads to the chronic Ca2+ underfilling of ER in BC-ECFCs. Likewise, future work will have to address whether lysosomal Ca2+ signalling contributes to VEGF-induced intracellular Ca2+ oscillations and is dysregulated in BC-ECFCs. Accordingly, nicotinic acid adenine dinucleotide phosphate (NAADP) has been recently shown to underpin VEGF-induced endothelial Ca2+ signals and neo-angiogenesis in melanoma [67]. An NAADP-sensitive lysosomal Ca2+ store is also present in N-ECFCs [30, 68], although it is seemingly down-regulated in BC-ECFCs (unpublished observations from our group).
As widely discussed elsewhere [13, 23], ECFC insensitivity to VEGF could contribute to the resistance to anti-VEGF therapies observed in cancer patients. Accordingly, ECFCs resident within the vascular “stem cell niches” provide the building blocks for neovessel formation in growing tumors. Additionally, ECFCs paracrinally may boost angiogenesis by releasing a myriad of growth factors and cytokines that stimulate endothelial cells to undergo angiogenesis [13, 16–21, 69, 70]. Limited evidence has been provided to show that human TECs require VEGF for proliferation, survival and migration [20, 71–73], while only one study revealed VEGF-induced Ca2+ signals in B-TECs [72]. In the clinical practice, anti-VEGF inhibitors are administered as adjuvant for standard chemotherapy or radiation therapy when tumor vasculature has already been established. At this stage, ECFCs have already been diluted/replaced by endothelial cells sprouting from neighbouring capillaries and B-TEC mainly derive from VEGF-sensitive cancer stem cells or adjoining sprouting capillaries [12, 74–76]. It turns out that tumor blood vessels, which are mainly lined by VEGF-sensitive B-TECs, regress in the presence of anti-angiogenic inhibitors. We hypothesize that the consequent dismantling of tumor vasculature exacerbates the hypoxic conditions of tumor microenvironment, thereby boosting the activation of hypoxia-inducible factors (HIFs) and inducing a second wave of ECFC mobilization [23]. Consequently, circulating ECFCs will be again recruited to the tumor site, in which they will be able to proliferate and re-establish the vascular network in spite of the presence of anti-VEGF drugs as they are not sensitive to VEGF [13, 23]. Although this scenario remains speculative and does not rule out the contribution of other mechanisms to the development of acquired refractoriness, including VEGFR2 downregulation in B-TECs [77], it could explain the limited increase in OS and PFS observed in BC patients treated with anti-angiogenic inhibitors. Unfortunately, no study has hitherto assessed the impact of anti-VEGF drugs on ECFC frequency either in BC or in any other tumor type. Of note, earlier studies showed that the systemic administration of bevacizumab caused an increase in the frequency of CD45dim, CD133+, VEGFR2+ EPCs in BC patients not responding to the therapy, while a reduction could not always be observed in those who did not show any change in disease progression [78]. Likewise, there was no significant relationship between the frequency of CD45–, CD133+/CD34+_EPCs and the therapeutic outcome of bevacizumab in BC patients enrolled in another study [79].
If VEGF does not stimulate BC-ECFC proliferation and tube formation, VEGFR2 cannot serve as a suitable target to prevent or interfere with BC vascularization. Nevertheless, the finding that the pharmacological blockade of SOCE with either BTP2 or 10 μM La3+ suppresses BC-ECFC growth and in vitro tubulogenesis provides further hints at SOCE as a promising candidate to develop alternative treatments to treat BC [36, 80]. Several studies showed that SOCE drives proliferation and migration also in several BC cell lines [43, 81, 82]. Therefore, SOCE stands out as a very attractive target to simultaneously halt BC cell growth and prevent ECFC activation at the tumor site [23, 33]. It would be interesting to assess whether SOCE controls proliferation (or migration) also in B-TECs, in which only store-independent Ca2+ pathways have hitherto been described [72, 83]. The rationale to include SOCE among the most promising molecular anti-cancer targets has been provided by pre-clinical and clinical studies conducted on CAI. CAI is a non-specific synthetic blocker of various types of Ca2+ channels, including InsP3Rs, TRP channels, SOCs, and the mitochondrial Ca2+ uniporter [33, 84]. CAI suppresses a multitude of Ca2+-dependent signalling pathways and has been validated as an effective anti-tumor agent due to its ability to inhibit angiogenesis as well as tumor development, migration and metastasis [33]. Preliminary studies revealed that CAI also blocked proliferation and/or migration in several human BC cell lines and in B-TECs [85]. Previous work from our group confirmed that CAI prevented N- and RCC-ECFC proliferation and in vitro tubulogenesis by interfering with both ER-dependent Ca2+ release and SOCE activation [24]. The present results extend these observations by showing that CAI interferes with intracellular Ca2+ signalling and inhibits both growth and tube formation also in BC-ECFCs. Of note, CAI represents the sole Ca2+ antagonist currently under investigation as an orally administered tumoristatic and anti-angiogenic agent in clinical phase I-III trials of several solid cancers (http://clinicaltrials.gov/). Future work will have to assess whether CAI is capable of halting BC vascularization and causing tumor shrinkage also in preclinical models.
In conclusion, we provided the first clear-cut evidence that VEGF does not stimulate angiogenesis in ECFCs isolated from peripheral blood of metastatic BC patients. Several authors proposed that the acquired resistance to anti-VEGF therapy could result from the recruitment of vascular progenitor cells and myeloid cells from their bone marrow/vascular niches, which may obviate the necessity of VEGF signalling [7, 8, 11, 23, 86]. Therefore, our data further corroborate the notion that tumor-associated ECFCs are insensitive to VEGF and lend the first strong cellular and molecular support to the notion that VEGFR2 is unlikely to be the most suitable target to halt vascularization and prevent tumor relapse in BC patients. VEGF fails to stimulate proliferation in BC-ECFCs due to the down-regulation of the intracellular Ca2+ spikes arising in normal cells following VEGFR-2 activation. The dramatic reduction in ER Ca2+ content is a key determinant to prevent VEGF from triggering the periodic InsP3-depedent Ca2+ release that underlies the Ca2+ train. This feature further suggests that the remodelling of the Ca2+ toolkit stands out as a crucial player in the development of the refractoriness to anti-cancer treatments [23, 57, 58]. Targeting the intracellular Ca2+ machinery could represent an alternative, yet promising strategy to overcome the resistance to anti-angiogenic therapies and prevent tumor vascularization. In this view, SOCE is emerging as an attractive candidate to design innovative approaches to treat BC patients as it controls proliferation in both BC cells and BC-ECFCs, as shown by the present investigation.
MATERIALS AND METHODS
Isolation and cultivation of ECFCs
Blood samples (40 mL) were obtained from patients who were out of ongoing cytoreductive therapy at the moment of blood sampling for ECFC isolation and from healthy donors. The ECFC samples used in the present investigation belong to the stocks recently described and characterized in [22]. To isolate ECFCs, mononuclear cells (MNCs) were separated from peripheral blood by density gradient centrifugation on lymphocyte separation medium for 30 min at 400g and washed twice in endothelial basal medium-2 (EBM-2) with 2% foetal bovine serum (FBS). A median of 36 x 106 MNCs (range 18-66) were plated on collagen-coated culture dishes (BD Biosciences) in the presence of the endothelial cell growth medium EGM-2 MV Bullet Kit (Lonza) containing EBM-2, 5% FBS, recombinant human (rh) EGF, rhVEGF, rhFGF-B, rhIGF-1, ascorbic acid and heparin, and maintained at 37°C in 5% CO2 and humidified atmosphere. Discard of non-adherent cells was performed after 2 days; thereafter medium was changed three times a week. The outgrowth of endothelial colonies from adherent MNCs was characterized by the formation of a cluster of cobblestone-appearing cells, resembling endothelial cells. That ECFCs-derived colonies belonged to endothelial lineage was confirmed as described in [22, 87]. In more detail, ECFCs-derived colonies were stained with anti-CD31, anti-CD105, anti-CD144, anti-CD146, anti-von Willebrand factor (vWF), anti-CD45, and anti-CD14 monoclonal antibodies and by assessment of capillary-like network formation in an in vitro Matrigel assay. For our experiments, we have mainly used endothelial cells obtained from early passage ECFCs (P1-3, which roughly encompasses a 15-18 day period) with the purpose to avoid (or maximally reduce) any potential bias due to possible cell differentiation. As already show in [24], the immunophenotype of both normal and tumor ECFCs does not change at different passages in culture. We also tested whether functional differences occurred when early (P2) and late (P6) passage-ECFCs were used by testing the in vitro capacity of capillary network formation in a Matrigel assay and found no differences between early and late passage ECFC-derived cells (data not shown).
Solutions
Physiological salt solution (PSS) had the following composition (in mM): 150 NaCl, 6 KCl, 1.5 CaCl2, 1 MgCl2, 10 Glucose, 10 Hepes. In Ca2+-free solution (0Ca2+), Ca2+ was substituted with 2 mM NaCl, and 0.5 mM EGTA was added. Solutions were titrated to pH 7.4 with NaOH. The osmolality of PSS as measured with an osmometer (Wescor 5500, Logan, UT) was 338 mmol/kg.
Electron microscopy
ECFCs were fixed with 2% paraformaldehyde and 2.5% glutaraldehyde (Sigma–Aldrich) in 0.1 M sodium phosphate buffer, pH 7.4, as described in [35]. After fixation, cells were postfixed in 2% osmium (OsO4) for 1 h at room temperature, dehydrated, and embedded in LR White (Sigma–Aldrich). Polymerization was performed for 24 h at 60°C. Ultrathin sections were cut with a Reichert (Depew, New York) OM-U3 ultramicrotome. The sections were stained with uranyl acetate and lead citrate (Sigma–Aldrich), examined and photographed at 3000×, 7.000×, or 20.000× magnifications on a Zeiss (Jena, Germany) EM900 (80 kV, objective diaphragm 30 μm) electron microscope.
Flow cytometry
Early passage ECFCs were isolated by trypsinization and resuspended in EBM2 medium (Lonza, Basel CH) supplemented with 10% FBS, as described in [88]. ECFC-derived cells (1x105 cells) were incubated with 5 μl of phycoerythrin (PE)-conjugated isotype control (IgG1) (BD Biosciences, San Josè, CA, USA) or 5 μl of PE-conjugated anti- VEGFR-2 antibody (R&D Systems, Inc Minneapolis, MN, USA) for 30 minutes at 4°C. ECFCs were than washed and resuspended in PBS with 1% FBS. Cells were aquired by a flow cytometer (NaviosTM; Beckman Coulter, Inc, Brea, CA) and analyzed by Kaluza® flow analysis software (Beckman Coulter). The number of dead/apoptotic cells was negligible, therefore the analysis was performed excluding cellular debris in a side scatter/forward scatter dot plot.
[Ca2+]i measurements and statistical analysis of Ca2+ signals
ECFCs were loaded with 4 μM fura-2 acetoxymethyl ester (fura-2/AM; 1 mM stock in dimethyl sulfoxide) in PSS for 1 hour at room temperature. After washing in PSS, the coverslip was fixed to the bottom of a Petri dish and the cells observed by an upright epifluorescence Axiolab microscope (Carl Zeiss, Oberkochen, Germany), usually equipped with a Zeiss ×40 Achroplan objective (water-immersion, 2.0 mm working distance, 0.9 numerical aperture). ECFCs were excited alternately at 340 and 380 nm, and the emitted light was detected at 510 nm. A first neutral density filter (1 or 0.3 optical density) reduced the overall intensity of the excitation light and a second neutral density filter (optical density=0.3) was coupled to the 380 nm filter to approach the intensity of the 340 nm light. A round diaphragm was used to increase the contrast. The excitation filters were mounted on a filter wheel (Lambda 10, Sutter Instrument, Novato, CA, USA). Custom software, working in the LINUX environment, was used to drive the camera (Extended-ISIS Camera, Photonic Science, Millham, UK) and the filter wheel, and to measure and plot on-line the fluorescence from 10 up to100 rectangular “regions of interest” (ROI). Each ROI was identified by a number. Since cell borders were not clearly identifiable, a ROI may not include the whole cell or may include part of an adjacent cell. Adjacent ROIs never superimposed. [Ca2+]i was monitored by measuring, for each ROI, the ratio of the mean fluorescence emitted at 510 nm when exciting alternatively at 340 and 380 nm (shortly termed “ratio”). An increase in [Ca2+]i causes an increase in the ratio [87]. Ratio measurements were performed and plotted on-line every 3 s. The experiments were performed at room temperature (22°C).
All the data have been collected ECFCs isolated from three different healthy donors. VEGF-induced intracellular Ca2+ oscillations were analysed by evaluating: 1) the latency before the onset of the pacemaker potential leading to the first regenerative Ca2+ spike; 2) the amplitude of the first Ca2+ spike (which was calculated as the difference as the ratio at the peak and the mean ratio of 1 min baseline before the slow pacemaker potential); 3) the number of Ca2+ spikes during 1 hour recording. The amplitude of the peak Ca2+ response to CPA and ATP was measured as the difference between the ratio at the peak (either of intracellular Ca2+ mobilization in 0Ca2+ or of Ca2+ entry occurring upon Ca2+ restoration to the bath) and the mean ratio of 1 min baseline before the peak. Pooled data are given as mean±SE and statistical significance (p<0.05) was evaluated by the Student’s t test for unpaired observations.
Wavelet analysis of Ca2+ signals
To give a quantitative evaluation of the differences in Ca2+ oscillatory activity between N- and BC-ECFCs after VEGF administration, an approach based on wavelet analysis was employed, using KYM 0.5 software (http://sourceforge.net/projects/kym/) as presented in [26, 39, 40]. Briefly, the wavelet transform W(a,b) = |a|-1/2 ∫ ψ*[(t – b)/a] f(t) dt was computed for each calcium trace using Morlet function ψ(t) ≈ π-1/4 exp(–0.5 t2) exp(ist) as mother wavelet, then, after having renamed b as t and having done the substitution ν = s/(2πa), the activity index J(t) ≈ ∫ |W(t,ν)|2 ν dν was calculated according to equations 3,4,5 and 9 of [40]. Notably, weighting wavelet transform modulus by the frequency, J keeps into account both the amplitudes and the values of the frequency components making up the oscillatory signal. Activity index has been evaluated for each calcium trace as a function of time, then its time-averaged value <J> has been calculated for each cell thus allowing to statistically compare the two <J> mean values representative of the two group of interest (N- and BC-ECFCs).
RNA isolation and qRT-PCR
Total RNA was extracted from N- and BC-ECFCs by using the QIAzol Lysis Reagent (QIAGEN, Italy). Single cDNA was synthesized from RNA (1 μg) using random hexamers and M-MLV Reverse Transcriptase (Invitrogen S.R.L., Italy). Reverse transcription was always performed in the presence or absence (negative control) of the reverse transcriptase enzyme. qRT-PCR was performed in triplicate using 1 μg cDNA and specific primers (intron-spanning primers) for Stim1-2, Orai1-3, TRPC1-7, and InsP3Rs1-3, as previously described [24, 47] (Supplementary Tables 1-2). Briefly, GoTaq qPCR Mastermix (Promega, Italy) was used according to the manufacturer instruction and qRT-PCR performed using Rotor Gene 6000 (Corbett, Concorde, NSW, Australia). The conditions were as follows: initial denaturation at 95°C for 5 min; 40 cycles of denaturation at 95°C for 30 sec; annealing at 58°C for 30 sec, and elongation at 72°C for 40 sec. The qRT-PCR reactions were normalized using β-actin as housekeeping gene. Melting curves were generated to detect the melting temperatures of specific products immediately after the PCR run. The triplicate threshold cycles (Ct) values for each sample were averaged resulting in mean Ct values for both the gene of interest and the housekeeping gene β-actin. The gene Ct values were then normalized to the housekeeping gene by taking the difference: ΔCt = Ct[gene] - Ct[β-actin], with high ΔCt values reflecting low mRNA expression levels. The sequences of the bands were checked by using the Big dye terminator cycle sequencing kit (Applied Biosystem, PE, USA). PCR products were also separated with agarose gel electrophoresis, stained with ethidium bromide, and acquired with the Image Master VDS (Amersham Biosciences Europe, Italy). The molecular weight of the PCR products was compared to the DNA molecular weight marker VIII (Roche Molecular Biochemicals, Italy).
Sample preparation and immunoblotting
ECFCs were homogenized by using a Dounce homogenizer in a solution containing: 250 mM Sucrose, 1 mM EDTA, 10 mM Tris-HCl, pH 7.6, 0.1 mg/mL PMSF, 100 mM β-mercaptoethanol and Protease Inhibitor Cocktail (P8340, Sigma, USA). The homogenates were solubilized in LaemmLi buffer (Dragoni et al., 2014) and 30 μg proteins were separated on 10% SDS-polyacrilamide gel electrophoresis and transferred to the Hybond-P PVDF Membrane (GE Healthcare, Italy) by electroelution. After 1 h blocking with Tris buffered saline (TBS) containing 3% BSA and 0.1% Tween (blocking solution), the membranes were incubated for 3 h at room temperature with the following affinity purified antibodies diluted 1:200 in the TBS and 0.1% Tween: anti-Stim1 (sc-166840), anti-Orai1 (sc-68895), anti-TRPC1 (sc-133076), anti-TRPC3/6/7 (sc-15056), and anti-IP3R-I/II/III (sc-377518) from Santa Cruz Biotechnology, anti-Orai3 (HPA015022), anti-Stim2 (PRS4123) from Sigma-Aldrich (Italy), and anti β-actin rabbit antibody as control (Rockland Immunochemicals for Research, U.S.A.; code, 600-401-886). The membranes were washed and incubated for 1 h with peroxidase-conjugated mouse, rabbit or goat IgG (1:120000 in blocking solution), from Dakocytomation (P0260), Chemicon (AP132P), and Santa Cruz (sc-2354), respectively. The bands were detected with the ECL™ Select western blotting detection system (GE Healthcare Europe GmbH, Italy). Prestained molecular weight markers (SDS7B2, Sigma, Italy) were used to estimate the molecular weight of the bands. Control experiments were performed by using the antibody preadsorbed with a 20-fold molar excess of the immunizing peptide or by incubating the blots with non immune serum.
Protein content
Protein contents of all the samples were determined by the Bradford’s method using bovine serum albumin (BSA) as standard [24, 47].
Chemicals
EBM and EGM-2 were purchased from Clonetics (Cell System, St. Katharinen, Germany). Fura-2/AM was obtained from Molecular Probes (Molecular Probes Europe BV, Leiden, The Netherlands). BTP2 was purchased from Calbiochem (La Jolla, CA, USA). All other chemicals were obtained from Sigma Chemical Co. (St. Louis, MO, USA).
Proliferation assays
As described elsewhere [24, 87], growth kinetics were evaluated by plating a total of 1 x 105 ECFCs-derived cells (first passage) in 30-mm collagen-treated dishes in the presence of EBM-2 and 5% FBS supplemented with 10 ng/mL VEGF and one of the either compounds: BAPTA (30 μM, 1 hour), BTP2 (20 μM, 30 min), La3+ (10 μM, 30 min), or CAI (10 μM, 20 min). Cultures were incubated at 37°C (in 5% CO2 and humidified atmosphere) and cell growth assessed every day until confluence was reached in control cultures. At this point, cells were recovered by trypsinization from all dishes and the cell number assessed by counting in a haemocytometer. Preliminary experiments showed no unspecific or toxic effect for each agent when used at these concentrations. Each assay was repeated in triplicate.
In vitro tube formation assay
The tube formation assay was carried out as previously shown [26]. To evaluate the effect of VEGF, early passage (P2-P3) BC-ECFCs were detached by trypsinization and resuspended in EBM-2 supplemented with 2% FBS. ECFC-derived cells were plated at 1.5 x 104 per well in Cultrex (Trevigen)-coated 96 well plates, in the presence of EBM-2 ± 10 ng/mL VEGF. Capillary network formation was assessed starting from 4 to 24 hours later. Three different sets of experiments, each performed in duplicate, were carried out. Experiments were repeated a minimum of three times and the vasculogenic response was measured by evaluating both dimensional and topological parameters. As illustrated in [26], we analyzed the length of endothelial tube-like structures (TLSs) and the number of the polygon structures established by TLSs, which are referred to as complex meshes and are indicative of endothelial cell migration. These analyses were performed by using the Angiogenesis Analyzer plugin of ImageJ (Gilles Carpentier, Faculte´ des Sciences et Technologie, Universite´ Paris Est, Creteil Val de Marne, France) [26]. To evaluate the effect of SOCE inhibitors, the same protocol was repeated by stimulating in vitro angiogenesis with the EGM-2 medium in the presence and absence of the following drugs: BAPTA (30 μM, 1 hour), BTP2 (20 μM, 30 min), or CAI (10 μM, 20 min).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Polyak K, Metzger Filho O. SnapShot: breast cancer. Cancer Cell. 2012; 22: 562-.e1. https://doi.org/10.1016/j.ccr.2012.06.021.
2. Le Bourhis X, Romon R, Hondermarck H. Role of endothelial progenitor cells in breast cancer angiogenesis: from fundamental research to clinical ramifications. Breast Cancer Res Treat. 2010; 120: 17-24. https://doi.org/10.1007/s10549-009-0686-5.
3. Uzzan B, Nicolas P, Cucherat M, Perret GY. Microvessel density as a prognostic factor in women with breast cancer: a systematic review of the literature and meta-analysis. Cancer Res. 2004; 64: 2941-55.
4. Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med. 2012; 2: a006502. https://doi.org/10.1101/cshperspect.a006502.
5. Claesson-Welsh L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med. 2013; 273: 114-27. https://doi.org/10.1111/joim.12019.
6. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011; 146: 873-87. https://doi.org/10.1016/j.cell.2011.08.039.
7. Loges S, Schmidt T, Carmeliet P. Mechanisms of resistance to anti-angiogenic therapy and development of third-generation anti-angiogenic drug candidates. Genes Cancer. 2010; 1: 12-25. https://doi.org/10.1177/1947601909356574.
8. Ribatti D. Tumor refractoriness to anti-VEGF therapy. Oncotarget. 2016; 7: 46668-77. https://doi.org/10.18632/oncotarget.8694.
9. Kumler I, Christiansen OG, Nielsen DL. A systematic review of bevacizumab efficacy in breast cancer. Cancer Treat Rev. 2014; 40: 960-73. https://doi.org/10.1016/j.ctrv.2014.05.006.
10. Montero AJ, Escobar M, Lopes G, Gluck S, Vogel C. Bevacizumab in the treatment of metastatic breast cancer: friend or foe? Curr Oncol Rep. 2012; 14: 1-11. https://doi.org/10.1007/s11912-011-0202-z.
11. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008; 8: 592-603. https://doi.org/10.1038/nrc2442.
12. Gao DC, Nolan D, McDonnell K, Vahdat L, Benezra R, Altorki N, Mittal V. Bone marrow-derived endothelial progenitor cells contribute to the angiogenic switch in tumor growth and metastatic progression. Biochim Biophys Acta. 2009; 1796: 33-40. https://doi.org/10.1016/j.bbcan.2009.05.001.
13. Moccia F, Zuccolo E, Poletto V, Cinelli M, Bonetti E, Guerra G, Rosti V. Endothelial progenitor cells support tumour growth and metastatisation: implications for the resistance to anti-angiogenic therapy. Tumour Biol. 2015; 36: 6603-14. https://doi.org/10.1007/s13277-015-3823-2.
14. Ingram DA, Mead LE, Tanaka H, Meade V, Fenoglio A, Mortell K, Pollok K, Ferkowicz MJ, Gilley D, Yoder MC. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004; 104: 2752-60.
15. Tasev D, Koolwijk P, van Hinsbergh VW. Therapeutic potential of human-derived endothelial colony-forming cells in animal models. Tissue Eng Part B Rev. 2016; 22: 371-82. https://doi.org/10.1089/ten.TEB.2016.0050.
16. Bieback K, Vinci M, Elvers-Hornung S, Bartol A, Gloe T, Czabanka M, Klüter H, Augustin H, Vajkoczy P. Recruitment of human cord blood-derived endothelial colony-forming cells to sites of tumor angiogenesis. Cytotherapy. 2013; 15: 726-39. https://doi.org/10.1016/j.jcyt.2013.01.215.
17. Margheri G, Zoppi A, Olmi R, Trigari S, Traversi R, Severi M, Bani D, Bianchini F, Torre E, Margheri F, Chilla A, Biagioni A, Calorini L, et al. Tumor-tropic endothelial colony forming cells (ECFCs) loaded with near-infrared sensitive Au nanoparticles: A “cellular stove” approach to the photoablation of melanoma. Oncotarget. 2016; 7: 39846-60. https://doi.org/10.18632/oncotarget.9511.
18. Wei J, Jarmy G, Genuneit J, Debatin KM, Beltinger C. Human blood late outgrowth endothelial cells for gene therapy of cancer: determinants of efficacy. Gene Ther. 2007; 14: 344-56. https://doi.org/10.1038/sj.gt.3302860.
19. Chou CP, Jiang SS, Pan HB, Yen YC, Tseng HH, Hung YT, Wang SH, Chen YL, Chen YW. Endothelial cell colony forming units derived from malignant breast diseases are resistant to tumor necrosis factor-alpha-induced apoptosis. Sci Rep. 2016; 6: 37450. https://doi.org/10.1038/srep37450.
20. Naito H, Wakabayashi T, Kidoya H, Muramatsu F, Takara K, Eino D, Yamane K, Iba T, Takakura N. Endothelial side population cells contribute to tumor angiogenesis and antiangiogenic drug resistance. Cancer Res. 2016; 76: 3200-10. https://doi.org/10.1158/0008-5472.CAN-15-2998.
21. Smadja DM, d'Audigier C, Weiswald LB, Badoual C, Dangles-Marie V, Mauge L, Evrard S, Laurendeau I, Lallemand F, Germain S, Grelac F, Dizier B, Vidaud M, et al. The Wnt antagonist Dickkopf-1 increases endothelial progenitor cell angiogenic potential. Arterioscler Thromb Vasc Biol. 2010; 30: 2544-52. https://doi.org/10.1161/ATVBAHA.110.213751.
22. Moccia F, Fotia V, Tancredi R, Della Porta MG, Rosti V, Bonetti E, Poletto V, Marchini S, Beltrame L, Gallizzi G, Da Prada GA, Pedrazzoli P, Riccardi A, et al. Breast and renal cancer-Derived endothelial colony forming cells share a common gene signature. Eur J Cancer. 2017; 77: 155-164. https://doi.org/10.1016/j.ejca.2017.01.025.
23. Moccia F, Poletto V. May the remodeling of the Ca(2)(+) toolkit in endothelial progenitor cells derived from cancer patients suggest alternative targets for anti-angiogenic treatment? Biochim Biophys Acta. 2015; 1853: 1958-73. https://doi.org/10.1016/j.bbamcr.2014.10.024.
24. Lodola F, Laforenza U, Bonetti E, Lim D, Dragoni S, Bottino C, Ong HL, Guerra G, Ganini C, Massa M, Manzoni M, Ambudkar IS, Genazzani AA, et al. Store-operated Ca2+ entry is remodelled and controls
25. Zuccolo E, Bottino C, Diofano F, Poletto V, Codazzi AC, Mannarino S, Campanelli R, Fois G, Marseglia GL, Guerra G, Montagna D, Laforenza U, Rosti V, et al. Constitutive store-operated Ca(2+) entry leads to enhanced nitric oxide production and proliferation in infantile hemangioma-derived endothelial colony-forming cells. Stem Cells Dev. 2016; 25: 301-19. https://doi.org/10.1089/scd.2015.0240.
26. Dragoni S, Reforgiato M, Zuccolo E, Poletto V, Lodola F, Ruffinatti FA, Bonetti E, Guerra G, Barosi G, Rosti V, Moccia F. Dysregulation of VEGF-induced proangiogenic Ca2+ oscillations in primary myelofibrosis-derived endothelial colony-forming cells. Exp Hematol. 2015; 43: 1019-30 e3. https://doi.org/10.1016/j.exphem.2015.09.002.
27. Moccia F, Guerra G. Ca(2+) Signalling in endothelial progenitor cells: Friend or Foe? J Cell Physiol. 2016; 231: 314-27. https://doi.org/10.1002/jcp.25126.
28. Dragoni S, Laforenza U, Bonetti E, Lodola F, Bottino C, Berra-Romani R, Carlo Bongio G, Cinelli MP, Guerra G, Pedrazzoli P, Rosti V, Tanzi F, Moccia F. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells. 2011; 29: 1898-907. https://doi.org/10.1002/stem.734.
29. Dragoni S, Laforenza U, Bonetti E, Lodola F, Bottino C, Guerra G, Borghesi A, Stronati M, Rosti V, Tanzi F, Moccia F. Canonical transient receptor potential 3 channel triggers vascular endothelial growth factor-induced intracellular Ca2+ oscillations in endothelial progenitor cells isolated from umbilical cord blood. Stem Cells Dev. 2013; 22: 2561-80. https://doi.org/10.1089/scd.2013.0032.
30. Zuccolo E, Dragoni S, Poletto V, Catarsi P, Guido D, Rappa A, Reforgiato M, Lodola F, Lim D, Rosti V, Guerra G, Moccia F. Arachidonic acid-evoked Ca2+ signals promote nitric oxide release and proliferation in human endothelial colony forming cells. Vascul Pharmacol. 2016; 87: 159-71. https://doi.org/10.1016/j.vph.2016.09.005.
31. Moccia F, Lodola F, Dragoni S, Bonetti E, Bottino C, Guerra G, Laforenza U, Rosti V, Tanzi F. Ca2+ signalling in endothelial progenitor cells: a novel means to improve cell-based therapy and impair tumour vascularisation. Curr Vasc Pharmacol. 2014; 12: 87-105.
32. Li J, Cubbon RM, Wilson LA, Amer MS, McKeown L, Hou B, Majeed Y, Tumova S, Seymour VAL, Taylor H, Stacey M, O'Regan D, Foster R, et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ Res. 2011; 108: 1190-8. https://doi.org/10.1161/circresaha.111.243352.
33. Moccia F, Dragoni S, Poletto V, Rosti V, Tanzi F, Ganini C, Porta C. Orai1 and transient receptor potential channels as novel molecular targets to impair tumor neovascularisation in renal cell carcinoma and other malignancies. Anticancer Agents Med Chem. 2014; 14: 296-312.
34. Moccia F. Remodelling of the Ca2+ toolkit in tumor endothelium as a crucial responsible for the resistance to anticancer therapies. Curr Signal Transduct Ther. 2017; 12. https://doi.org/10.2174/1574362412666170207113636.
35. Poletto V, Dragoni S, Lim D, Biggiogera M, Aronica A, Cinelli M, De Luca A, Rosti V, Porta C, Guerra G, Moccia F. Endoplasmic reticulum Ca(2+) handling and apoptotic resistance in tumor-derived endothelial colony forming cells. J Cell Biochem. 2016; 117: 2260-71. https://doi.org/10.1002/jcb.25524.
36. Moccia F, Zuccolo E, Poletto V, Turin I, Guerra G, Pedrazzoli P, Rosti V, Porta C, Montagna D. Targeting stim and orai proteins as an alternative approach in anticancer therapy. Curr Med Chem. 2016; 23: 3450-80.
37. Moccia F, Dragoni S, Lodola F, Bonetti E, Bottino C, Guerra G, Laforenza U, Rosti V, Tanzi F. Store-dependent Ca(2+) entry in endothelial progenitor cells as a perspective tool to enhance cell-based therapy and adverse tumour vascularization. Curr Med Chem. 2012; 19: 5802-18.
38. Noren DP, Chou WH, Lee SH, Qutub AA, Warmflash A, Wagner DS, Popel AS, Levchenko A. Endothelial cells decode VEGF-mediated Ca2+ signaling patterns to produce distinct functional responses. Sci Signal. 2016; 9: ra20. https://doi.org/10.1126/scisignal.aad3188.
39. Moccia F, Ruffinatti FA, Zuccolo E. Intracellular Ca(2)(+) signals to reconstruct a broken heart: still a theoretical approach? Curr Drug Targets. 2015; 16: 793-815.
40. Ruffinatti FA, Lovisolo D, Distasi C, Ariano P, Erriquez J, Ferraro M. Calcium signals: analysis in time and frequency domains. J Neurosci Methods. 2011; 199: 310-20. https://doi.org/10.1016/j.jneumeth.2011.05.009.
41. Kar P, Mirams GR, Christian HC, Parekh AB. Control of NFAT isoform activation and NFAT-dependent gene expression through two coincident and spatially segregated intracellular Ca2+ signals. Mol Cell. 2016; 64: 746-59. https://doi.org/10.1016/j.molcel.2016.11.011.
42. Somasundaram A, Shum AK, McBride HJ, Kessler JA, Feske S, Miller RJ, Prakriya M. Store-operated CRAC channels regulate gene expression and proliferation in neural progenitor cells. J Neurosci. 2014; 34: 9107-23. https://doi.org/10.1523/JNEUROSCI.0263-14.2014.
43. Motiani RK, Abdullaev IF, Trebak M. A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J Biol Chem. 2010; 285: 19173-83. https://doi.org/10.1074/jbc.M110.102582.
44. Pierro C, Cook SJ, Foets TC, Bootman MD, Roderick HL. Oncogenic K-Ras suppresses IP(3)-dependent Ca(2)(+) release through remodelling of the isoform composition of IP(3)Rs and ER luminal Ca(2)(+) levels in colorectal cancer cell lines. J Cell Sci. 2014; 127: 1607-19. https://doi.org/10.1242/jcs.141408.
45. Putney JW. Pharmacology of store-operated calcium channels. Mol Interv. 2010; 10: 209-18. https://doi.org/10.1124/mi.10.4.4.
46. Ay AS, Benzerdjeb N, Sevestre H, Ahidouch A, Ouadid-Ahidouch H. Orai3 constitutes a native store-operated calcium entry that regulates non small cell lung adenocarcinoma cell proliferation. PLoS One. 2013; 8: e72889. https://doi.org/10.1371/journal.pone.0072889.
47. Dragoni S, Laforenza U, Bonetti E, Reforgiato M, Poletto V, Lodola F, Bottino C, Guido D, Rappa A, Pareek S, Tomasello M, Guarrera MR, Cinelli MP, et al. Enhanced expression of Stim, Orai, and TRPC transcripts and proteins in endothelial progenitor cells isolated from patients with primary myelofibrosis. PLoS One. 2014; 9: e91099. https://doi.org/10.1371/journal.pone.0091099.
48. Kim JH, Lkhagvadorj S, Lee MR, Hwang KH, Chung HC, Jung JH, Cha SK, Eom M. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem Biophys Res Commun. 2014; 448: 76-82. https://doi.org/10.1016/j.bbrc.2014.04.064.
49. Dragoni S, Turin I, Laforenza U, Potenza DM, Bottino C, Glasnov TN, Prestia M, Ferulli F, Saitta A, Mosca A, Guerra G, Rosti V, Luinetti O, et al. Store-operated Ca2+ entry does not control proliferation in primary cultures of human metastatic renal cellular carcinoma. Biomed Res Int. 2014; 2014: 739494. https://doi.org/10.1155/2014/739494.
50. Fiorio Pla A, Grange C, Antoniotti S, Tomatis C, Merlino A, Bussolati B, Munaron L. Arachidonic acid-induced Ca2+ entry is involved in early steps of tumor angiogenesis. Mol Cancer Res. 2008; 6: 535-45. https://doi.org/10.1158/1541-7786.MCR-07-0271.
51. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011; 473: 298-307. https://doi.org/10.1038/nature10144.
52. Joo HJ, Song S, Seo HR, Shin JH, Choi SC, Park JH, Yu CW, Hong SJ, Lim DS. Human endothelial colony forming cells from adult peripheral blood have enhanced sprouting angiogenic potential through up-regulating VEGFR2 signaling. Int J Cardiol. 2015; 197: 33-43. https://doi.org/10.1016/j.ijcard.2015.06.013.
53. Yang L, Guan H, He J, Zeng L, Yuan Z, Xu M, Zhang W, Wu X, Guan J. VEGF increases the proliferative capacity and eNOS/NO levels of endothelial progenitor cells through the calcineurin/NFAT signalling pathway. Cell Biol Int. 2012; 36: 21-7. https://doi.org/10.1042/CBI20100670.
54. Lee SH, Lee JH, Asahara T, Kim YS, Jeong HC, Ahn Y, Jung JS, Kwon SM. Genistein promotes endothelial colony-forming cell (ECFC) bioactivities and cardiac regeneration in myocardial infarction. PLoS One. 2014; 9: e96155. https://doi.org/10.1371/journal.pone.0096155.
55. Berridge MJ. Inositol trisphosphate and calcium oscillations. Biochem Soc Symp. 2007: 1-7. https://doi.org/10.1042/BSS0740001.
56. Berridge MJ. Inositol trisphosphate and calcium signalling mechanisms. Biochim Biophys Acta. 2009; 1793: 933-40. https://doi.org/10.1016/j.bbamcr.2008.10.005.
57. Bittremieux M, Parys JB, Pinton P, Bultynck G. ER functions of oncogenes and tumor suppressors: modulators of intracellular Ca(2+) signaling. Biochim Biophys Acta. 2016; 1863: 1364-78. https://doi.org/10.1016/j.bbamcr.2016.01.002.
58. Marchi S, Pinton P. Alterations of calcium homeostasis in cancer cells. Curr Opin Pharmacol. 2016; 29: 1-6. https://doi.org/10.1016/j.coph.2016.03.002.
59. Lim D, Bertoli A, Sorgato MC, Moccia F. Generation and usage of aequorin lentiviral vectors for Ca(2+) measurement in sub-cellular compartments of hard-to-transfect cells. Cell Calcium. 2016; 59: 228-39. https://doi.org/10.1016/j.ceca.2016.03.001.
60. Cheng KT, Liu X, Ong HL, Ambudkar IS. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J Biol Chem. 2008; 283: 12935-40. https://doi.org/10.1074/jbc.C800008200.
61. Berna-Erro A, Galan C, Dionisio N, Gomez LJ, Salido GM, Rosado JA. Capacitative and non-capacitative signaling complexes in human platelets. Biochim Biophys Acta. 2012; 1823: 1242-51. https://doi.org/10.1016/j.bbamcr.2012.05.023.
62. Liu XR, Zhang MF, Yang N, Liu Q, Wang RX, Cao YN, Yang XR, Sham JS, Lin MJ. Enhanced store-operated Ca(2)+ entry and TRPC channel expression in pulmonary arteries of monocrotaline-induced pulmonary hypertensive rats. Am J Physiol Cell Physiol. 2012; 302: C77-87. https://doi.org/10.1152/ajpcell.00247.2011.
63. Olah T, Fodor J, Ruzsnavszky O, Vincze J, Berbey C, Allard B, Csernoch L. Overexpression of transient receptor potential canonical type 1 (TRPC1) alters both store operated calcium entry and depolarization-evoked calcium signals in C2C12 cells. Cell Calcium. 2011; 49: 415-25. https://doi.org/10.1016/j.ceca.2011.03.012.
64. Sabourin J, Bartoli F, Antigny F, Gomez AM, Benitah JP. Transient receptor potential canonical (TRPC)/Orai1-dependent store-operated Ca2+ channels: new targets of aldosterone in cardiomycytes. J Biol Chem. 2016; 291: 13394-409. https://doi.org/10.1074/jbc.M115.693911.
65. Pani B, Cornatzer E, Cornatzer W, Shin DM, Pittelkow MR, Hovnanian A, Ambudkar IS, Singh BB. Up-regulation of transient receptor potential canonical 1 (TRPC1) following sarco(endo)plasmic reticulum Ca2+ ATPase 2 gene silencing promotes cell survival: a potential role for TRPC1 in Darier's disease. Mol Biol Cell. 2006; 17: 4446-58. https://doi.org/10.1091/mbc.E06-03-0251.
66. Sunryd JC, Cheon B, Graham JB, Giorda KM, Fissore RA, Hebert DN. TMTC1 and TMTC2 are novel endoplasmic reticulum tetratricopeptide repeat-containing adapter proteins involved in calcium homeostasis. J Biol Chem. 2014; 289: 16085-99. https://doi.org/10.1074/jbc.M114.554071.
67. Favia A, Pafumi I, Desideri M, Padula F, Montesano C, Passeri D, Nicoletti C, Orlandi A, Del Bufalo D, Sergi M, Ziparo E, Palombi F, Filippini A. NAADP-dependent Ca(2+) signaling controls melanoma progression, metastatic dissemination and neoangiogenesis. Sci Rep. 2016; 6: 18925. https://doi.org/10.1038/srep18925.
68. Di Nezza F, Zuccolo E, Poletto V, Rosti V, De Luca A, Moccia F, Guerra G, Ambrosone L. Liposomes as a putative tool to investigate NAADP signaling in vasculogenesis. J Cell Biochem. 2017. https://doi.org/10.1002/jcb.26019.
69. Yoder MC, Ingram DA. The definition of EPCs and other bone marrow cells contributing to neoangiogenesis and tumor growth: is there common ground for understanding the roles of numerous marrow-derived cells in the neoangiogenic process? Biochim Biophys Acta. 2009; 1796: 50-4. https://doi.org/10.1016/j.bbcan.2009.04.002.
70. Laurenzana A, Margheri F, Chilla A, Biagioni A, Margheri G, Calorini L, Fibbi G, Del Rosso M. Endothelial progenitor cells as shuttle of anticancer agents. Hum Gene Ther. 2016. https://doi.org/10.1089/hum.2016.066.
71. Bussolati B, Deambrosis I, Russo S, Deregibus MC, Camussi G. Altered angiogenesis and survival in human tumor-derived endothelial cells. FASEB J. 2003; 17: 1159-61. https://doi.org/10.1096/fj.02-0557fje.
72. Pupo E, Pla AF, Avanzato D, Moccia F, Cruz JE, Tanzi F, Merlino A, Mancardi D, Munaron L. Hydrogen sulfide promotes calcium signals and migration in tumor-derived endothelial cells. Free Radic Biol Med. 2011; 51: 1765-73. https://doi.org/10.1016/j.freeradbiomed.2011.08.007.
73. Hernandez-Garcia R, Iruela-Arispe ML, Reyes-Cruz G, Vazquez-Prado J. Endothelial RhoGEFs: a systematic analysis of their expression profiles in VEGF-stimulated and tumor endothelial cells. Vascul Pharmacol. 2015; 74: 60-72. https://doi.org/10.1016/j.vph.2015.10.003.
74. Gao DC, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008; 319: 195-8. https://doi.org/10.1126/science.1150224.
75. Nolan DJ, Ciarrocchi A, Mellick AS, Jaggi JS, Bambino K, Gupta S, Heikamp E, McDevitt MR, Scheinberg DA, Benezra R, Mittal V. Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev. 2007; 21: 1546-58. https://doi.org/10.1101/gad.436307.
76. Brossa A, Grange C, Mancuso L, Annaratone L, Satolli MA, Mazzone M, Camussi G, Bussolati B. Sunitinib but not VEGF blockade inhibits cancer stem cell endothelial differentiation. Oncotarget. 2015; 6: 11295-309. https://doi.org/10.18632/oncotarget.3123.
77. Li JL, Sainson RC, Oon CE, Turley H, Leek R, Sheldon H, Bridges E, Shi W, Snell C, Bowden ET, Wu H, Chowdhury PS, Russell AJ, et al. DLL4-Notch signaling mediates tumor resistance to anti-VEGF therapy
78. Jain S, Ward MM, O'Loughlin J, Boeck M, Wiener N, Chuang E, Cigler T, Moore A, Donovan D, Lam C, Cobham MV, Schneider S, Christos P, et al. Incremental increase in VEGFR1(+) hematopoietic progenitor cells and VEGFR2(+) endothelial progenitor cells predicts relapse and lack of tumor response in breast cancer patients. Breast Cancer Res Treat. 2012; 132: 235-42. https://doi.org/10.1007/s10549-011-1906-3.
79. Dellapasqua S, Bertolini F, Bagnardi V, Campagnoli E, Scarano E, Torrisi R, Shaked Y, Mancuso P, Goldhirsch A, Rocca A, Pietri E, Colleoni M. Metronomic cyclophosphamide and capecitabine combined with bevacizumab in advanced breast cancer. J Clin Oncol. 2008; 26: 4899-905. https://doi.org/10.1200/JCO.2008.17.4789.
80. Vashisht A, Trebak M, Motiani RK. STIM and Orai proteins as novel targets for cancer therapy. A review in the theme: cell and molecular processes in cancer metastasis. Am J Physiol Cell Physiol. 2015; 309: C457-69. https://doi.org/10.1152/ajpcell.00064.2015.
81. Davis FM, Peters AA, Grice DM, Cabot PJ, Parat MO, Roberts-Thomson SJ, Monteith GR. Non-stimulated, agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on calcium entry. PLoS One. 2012; 7: e36923. https://doi.org/10.1371/journal.pone.0036923.
82. Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009; 15: 124-34. https://doi.org/10.1016/j.ccr.2008.12.019.
83. Fiorio Pla A, Avanzato D, Munaron L, Ambudkar IS. Ion channels and transporters in cancer. 6. Vascularizing the tumor: TRP channels as molecular targets. Am J Physiol Cell Physiol. 2012; 302: C9-15. https://doi.org/10.1152/ajpcell.00280.2011.
84. Mignen O, Brink C, Enfissi A, Nadkarni A, Shuttleworth TJ, Giovannucci DR, Capiod T. Carboxyamidotriazole-induced inhibition of mitochondrial calcium import blocks capacitative calcium entry and cell proliferation in HEK-293 cells. J Cell Sci. 2005; 118: 5615-23. https://doi.org/10.1242/jcs.02663.
85. Guo L, Li ZS, Wang HL, Ye CY, Zhang DC. Carboxyamido-triazole inhibits proliferation of human breast cancer cells via G(2)/M cell cycle arrest and apoptosis. Eur J Pharmacol. 2006; 538: 15-22. https://doi.org/10.1016/j.ejphar.2006.03.036.
86. Abdullah SE, Perez-Soler R. Mechanisms of resistance to vascular endothelial growth factor blockade. Cancer. 2012; 118: 3455-67. https://doi.org/10.1002/cncr.26540.
87. Sanchez-Hernandez Y, Laforenza U, Bonetti E, Fontana J, Dragoni S, Russo M, Avelino-Cruz JE, Schinelli S, Testa D, Guerra G, Rosti V, Tanzi F, Moccia F. Store-operated Ca(2+) entry is expressed in human endothelial progenitor cells. Stem Cells Dev. 2010; 19: 1967-81. https://doi.org/10.1089/scd.2010.0047.
88. Rosti V, Bonetti E, Bergamaschi G, Campanelli R, Guglielmelli P, Maestri M, Magrini U, Massa M, Tinelli C, Viarengo G, Villani L, Primignani M, Vannucchi AM, et al. High frequency of endothelial colony forming cells marks a non-active myeloproliferative neoplasm with high risk of splanchnic vein thrombosis. PLoS One. 2010; 5: e15277. https://doi.org/10.1371/journal.pone.0015277.