
INTRODUCTION
In 2015, breast cancer represented 26% of all cancer cases among Canadian women and was the second leading cause of cancer death constituting 14% of overall death due to cancer [1]. Like every common cancer, breast cancer shows some degree of familial clustering [2]. High-risk families having multiple cases of breast or ovarian cancer are associated with a higher risk of developing breast cancer during their lifetime than other families [3]. It is thought that approximately 10-15% of breast cancer cases are hereditary and associated with mutations in BRCA1 or BRCA2 genes and some other genes having high to moderate penetrance such as TP53, PTEN, ATM, CHEK2, PALB2 and BRIP1 and ATR, which account for approximately 5% of the risk [4-10]. Common variants have also been identified in additional susceptibility loci and would explain a further ~16% of the 2-fold familial risk of breast cancer [11]. Among our French Canadian cohort, 24% of high-risk breast cancer families were found to be carriers of a deleterious BRCA1 or BRCA2 mutation [12].
Therefore, susceptibility alleles for more than half of the high-risk families remain unknown. A portion of these remnant breast cancer families could be explained by modulation of gene expression, which is mainly regulated through methylation or alternative splicing (AS) mechanisms. Indeed, more than 90% of human genes undergo alternative splicing and it is now becoming clear that AS plays an important role in human cancer development [13-14]. RNA sequencing allows a genome-wide expression study of the transcriptome and can likely detect and quantify all coding and non-coding transcripts [15]. The use of RNA sequencing greatly enhanced our understanding of gene expression in cells [16].
Human immortalized lymphoblastoid cell lines (LCLs) provide information on gene expression without having to consider tissue specific expression [17]. LCLs used for the establishment of gene expression or splicing signatures are recognized as a reliable biological material to study a given disease [18-28], and some studies recently showed the heritability of splicing, as some exons were spliced in an allele-specific manner [18, 29, 30]. Moreover, it has been demonstrated that LCLs can be used to study life-course environmental epigenetics [31].
Previous studies attempted to discriminate BRCA1/2, non-BRCA1/2 (BRCAX) and sporadic breast cancers were based on gene expression levels and histological tests performed on breast tumor tissue [32-37]. In another study, although several genes or spliced transcripts were identified as differentially expressed in familial cases, they did not allow clusterization of BRCA1, BRCA2 and BRCAX tumor tissues [38].
In this study, we performed RNA sequencing on LCLs isolated from BRCA1/2 and BRCAX affected and unaffected individuals coming from high-risk breast cancer families in an attempt to distinguish breast cancer subgroups based on their transcriptome profile. This study revealed several transcripts involved in regulation of translation, apoptosis, cell cycle as well as cell growth and proliferation, which could discriminate BRCAX individuals from BRCA1/2 subgroups.
RESULTS
Our French Canadian cohort comprised three major familial breast cancer subgroups namely BRCA1 and BRCA2 carriers as well as BRCAX individuals, i.e. non BRCA1/2 (affected and unaffected). The BRCA1 cases included 25 individuals (ind) affected with breast cancer and 11 unaffected women, who were carriers of BRCA1 mutations namely R1443X (22 ind), 3705insA (2 ind), 2244insA (7 ind), 2953del3+C (2 ind) and three individuals carrying E352X, 4160delAG or 1723del9ins13 mutation, respectively. The BRCA2 subgroup was composed of 31 affected and 18 unaffected individuals carrying 8765delAG (44 ind), E3002K (2 ind), 6503delTT (1 ind), R3128X (1 ind) or 3036del4 mutation (1 ind). The BRCAX subgroup included 16 affected and 16 unaffected individuals, which represented 16 pairs of sisters (1 affected and 1 unaffected per family). It should be noted that the oldest unaffected sister available was purposely selected in BRCAX families. This subgroup of unaffected sisters was used as controls for comparison purpose in the analyses described below. The mutational profile and relationship status of BRCA1, BRCA2 and BRCAX individuals are highlighted in Supplementary Table 1.
RNA-Seq analyses generated an average of 68 million reads per sample, and more than 85% of the reads were aligned to the hg19 human reference genome using TopHat (data not shown). As displayed in Supplementary Table 2, out of a total of 173 259 transcripts detected, 95 transcripts (0.05 % of all transcripts) were found to be significantly (p<0.01) and differentially expressed based on the Bonferroni-corrected ANOVA analysis, when considering all four breast cancer subgroups (BRCA1, BRCA2, unaffected and affected BRCAX individuals). All these significant transcripts were encoded by 85 different genes. In addition to the main isoforms (one per gene), 10 mRNA isoforms were considered as alternatively spliced isoforms (11.8%). These significant transcripts included 54 gene isoforms showing a highly significant Bonferroni-corrected p-value (p < 0.001).
Principal component analysis (PCA) of these 95 transcripts identified as differentially expressed among BRCA1-carriers (n=36), BRCA2-carriers (n=49), unaffected BRCAX (n=16) and affected BRCAX (n=16) individuals is presented in Figure 1. The first three principal components of transcriptional variation accounted for 59.6 % of the total variance. PCA on the full dataset showed that the PC1 component accounted for 46 % of the variance, which is highly informative, while PC2 was also informative compared to the variance explained in the randomized data set.
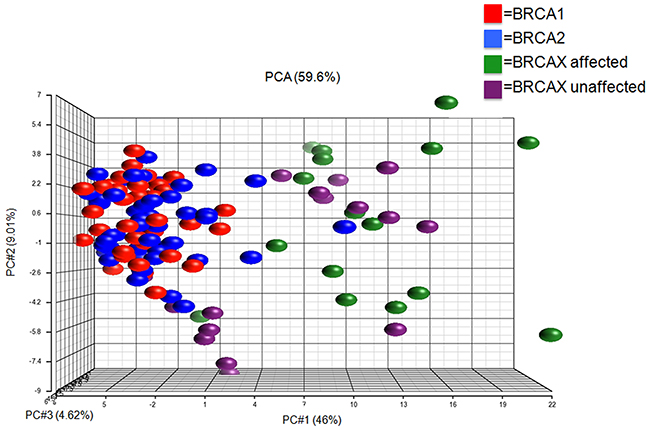
Figure 1: Principal component analysis (PCA) on lymphoblastoid cell lines. Unsupervised classification of the groups using a combination of PC1, PC2 and PC3. Distance between dots is a dimensional measure for the similarity of the expression profiles of the samples (red: BRCA1, blue: BRCA2, green: BRCAX unaffected and purple: BRCAX affected).
Unsupervised hierarchical clustering of all BRCA1, BRCA2 and BRCAX individuals was then performed using the 95 significant transcripts. As illustrated in Figure 2, gene expression levels of these significant transcripts allowed to discriminate distinctly BRCA1/2 from BRCAX (unaffected and affected) individuals. However, it was not possible to segregate BRCA1 from BRCA2 individuals as well as affected from unaffected BRCAX individuals. In addition, when considering BRCA1 and BRCA2 individuals, no specific clustering could be observed based on gene mutation or the status of the disease. Intra-group variance analysis using gene expression data was performed by Principle component analysis (PCA) for patients with the BRCA1 R1443X mutation (22 patients) and BRCA2 8765delAG mutation (44 patients). We did not find significance of BRCA1 R1443X and BRCA2 8765delAG mutation from their respective BRCA1 and BRCA2 subgroup. Thus, grouping of different mutations in BRCA1 or BRCA2 subgroup is justified (data not shown).
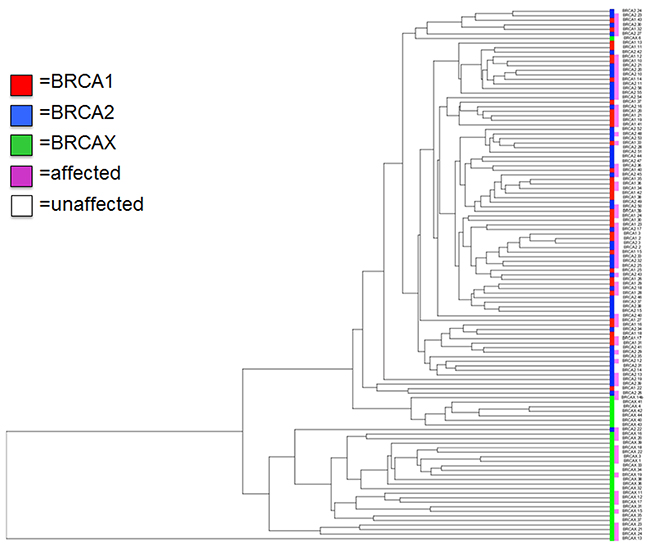
Figure 2: Hierarchical clustering of the 95 transcripts differentially expressed. Unsupervised LCLs classification based on the significantly and differentially expressed transcripts measured by RNA-sequencing using bonferroni corrected p-value <0.01. Color bar represents each of our groups (red: BRCA1, blue: BRCA2, Green: BRCAX) and status of disease (pink: affected and white: unaffected).
The ANOVA analysis followed by conservative post hoc Scheffé test, which is appropriate for comparing groups with unequal sample sizes, allowed to potentially identify transcripts discriminating BRCA1, BRCA2 and affected BRCAX individuals from unaffected BRCAX individuals, which were used as controls in this analysis. This analysis revealed 69, 71 and 28 gene isoforms differentially expressed from BRCAX unaffected for BRCA1, BRCA2 and affected BRCAX individuals, respectively (See Supplementary Table 2). It should be noted that the large majority of transcripts identified in BRCA1-carriers were also found in BRCA2-carriers.
As presented in Figure 3, these transcripts were then illustrated in Venn diagrams, which showed that 3 common transcripts (3.2%) were differentially expressed in all three subgroups, when compared to unaffected BRCAX individuals. In addition, a large portion of transcripts (65: 68.4%) was commonly identified in BRCA1 and BRCA2 subgroups, while only 1 transcript was specifically and exclusively associated with BRCA1, and another one different transcript with BRCA2. This illustrated the similarity between BRCA1 and BRCA2-carrier individuals regarding their gene expression profile. On the other hand, 23 gene isoforms were exclusively associated with affected BRCAX individuals and are not different in BRCA1 and BRCA2 subgroups. The name of the transcripts is presented in Supplementary Table 3.
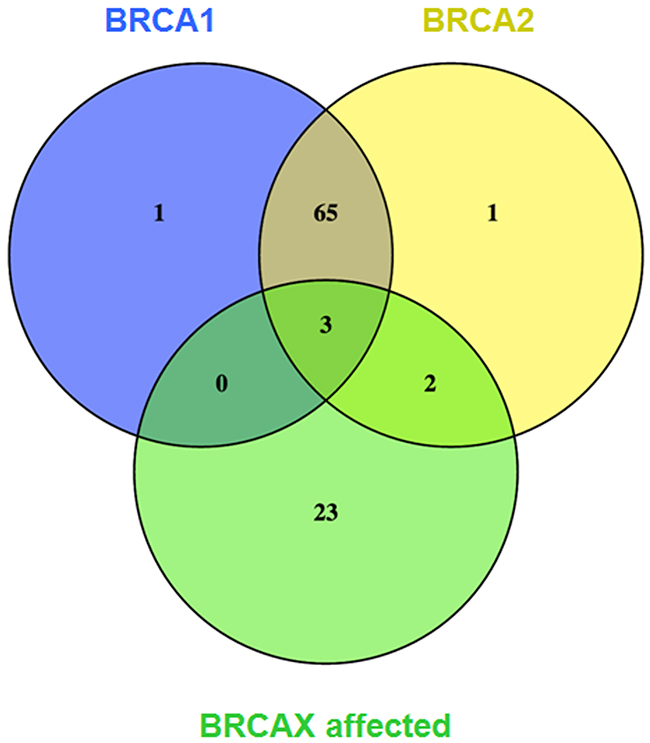
Figure 3: Venn diagram of significantly and differentially expressed transcripts compared with BRCAX unaffected individuals. An intersectional analysis of differentially expressed transcripts compared with BRCAX unaffected was performed. The cut-off value was Bonferroni corrected p-value ≤ 0.01.
In an attempt to further discriminate unaffected and affected BRCAX individuals, hierarchical clustering was then performed using the 28 gene isoforms discriminating both BRCAX subgroups (Figure 4). Although a much better clustering could be observed between both subgroups, these genes could not differentiate distinctly unaffected from affected BRCAX individuals, with 4 affected individuals being located among the unaffected individuals.
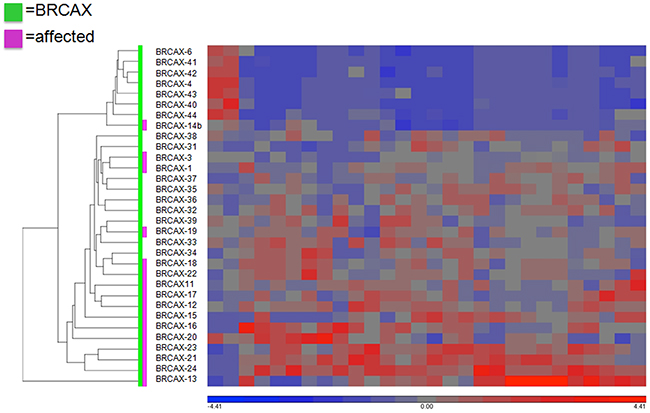
Figure 4: Hierarchical clustering of the 28 transcripts differentially expressed between BRCAX affected and BRCAX unaffected individuals. Heat map of the TPM (Transcripts Per Million) for 32 women using Euclidean distance with average linkage.
Further, we performed Scheffé analysis on all the 4 subgroups (BRCA1, BRCA2, unaffected and affected BRCAX) combined, this allowed us to identify specific transcripts, which are exclusively associated with each subgroup. As listed in Table 1, although no specific transcripts were specifically associated with BRCA1 or BRCA2 individuals, we could identify 67 transcripts specifically associated with BRCA1/BRCA2 following combination of both subgroups and compared to BRCAX individuals. In addition, 3 and 28 transcripts showed exclusive association with unaffected and affected BRCAX subgroups, respectively.
Table 1: Transcripts specifically associated with each group and their corrected p-value
Ensembl transcript ID |
HGNC symbol |
Bonferroni corrected p-value |
Relevant biological process |
|---|---|---|---|
Transcript specifically associated with BRCA1/2 carriers when compared to BRCAX unaffected and affected individuals |
|||
Ensembl transcript ID |
HGNC symbol |
Bonferroni corrected p-value |
Relevant biological process |
ENST00000598296 |
NOSIP |
1.16 E-06 |
negative regulation of nitric-oxide synthase activity |
ENST00000580799 |
GGA3 |
4.03 E-04 |
positive regulation of protein catabolic process |
ENST00000430762 |
PPP3CB |
1.11 E-03 |
positive regulation of transcription from RNA polymerase II promoter |
ENST00000486593 |
LAMP2 |
1.05 E-03 |
regulation of protein stability |
ENST00000366726 |
GUK1 |
1.18 E-03 |
ATP metabolic process |
ENST00000438462 |
RTN4 |
1.62 E-03 |
Regulation of apoptotic process, cell-cell adhesion, negative regulation of cell growth |
ENST00000588730 |
C18orf25 |
2.76 E-03 |
protein ubiquitination involved in ubiquitin-dependent protein catabolic process |
ENST00000471658 |
PSPC1 |
4.22 E-03 |
mRNA splicing, negative regulation of transcription |
ENST00000490523 |
EIF2AK1 |
3.26 E-07 |
negative regulation of cell proliferation, regulation of translational initiation by eIF2 alpha phosphorylation |
ENST00000586868 |
TBCB |
4.69 E-07 |
cell differentiation |
ENST00000572932 |
NOMO3 |
1.54 E-06 |
carbohydrate binding |
ENST00000596417 |
EEF2 |
1.09 E-06 |
positive regulation of translation, cell-cell adhesion, response to estradiol |
ENST00000485280 |
RAB7A |
1.99 E-06 |
positive regulation of protein catabolic process, regulation of autophagosome assembly |
ENST00000587393 |
AES |
3.25 E-06 |
negative regulation of canonical Wnt signaling pathway, negative regulation of transcription and protein binding |
ENST00000593582 |
TRIM28 |
4.78 E-06 |
positive regulation of DNA repair, positive regulation of transcription, epithelial to mesenchymal transition, protein sumoylation and ubiquitination |
ENST00000463243 |
HLA-DPA1 |
1.04 E-05 |
Immune process, positive regulation of interferon-gamma production |
ENST00000476642 |
HLA-DPA1 |
1.04 E-05 |
Immune process, positive regulation of interferon-gamma production |
ENST00000480481 |
HLA-DPA1 |
1.04 E-05 |
Immune process, positive regulation of interferon-gamma production |
ENST00000483480 |
HLA-DPA1 |
1.04 E-05 |
Immune process, positive regulation of interferon-gamma production |
ENST00000486449 |
HLA-DPA1 |
1.04 E-05 |
Immune process, positive regulation of interferon-gamma production |
ENST00000493893 |
COMT |
1.05 E-05 |
estrogen metabolic process, dopamine catabolic process |
ENST00000495074 |
HLA-DPA1 |
1.04 E-05 |
Immune process, positive regulation of interferon-gamma production |
ENST00000514979 |
HLA-DPA1 |
1.04 E-05 |
Immune process, positive regulation of interferon-gamma production |
ENST00000524786 |
DEAF1 |
7.14 E-06 |
regulation of mammary gland epithelial cell proliferation, regulation of transcription |
ENST00000368439 |
CKS1B |
1.40 E-05 |
regulation of mitotic cell cycle |
ENST00000524815 |
PACS1 |
1.83 E-05 |
positive regulation of protein binding, protein targeting to Golgi |
ENST00000515540 |
BAX |
3.35 E-05 |
DNA damage response, signal transduction by p53 class mediator resulting in cell cycle arrest, apoptotic process |
ENST00000548861 |
RP11-603J24.9 |
4.07 E-05 |
Unknown |
ENST00000529698 |
DGKZ |
5.99 E-05 |
protein kinase C-activating G-protein coupled receptor signaling pathway, cell migration |
ENST00000372077 |
VEGFA |
9.13 E-05 |
growth factor activity, cytokine activity |
ENST00000435720 |
PSMF1 |
1.51 E-04 |
MAPK cascade, regulation of Wnt signaling pathway and ubiquitin-protein ligase activity involved in mitotic cell cycle |
ENST00000461760 |
STK25 |
1.53 E-04 |
positive regulation of stress-activated MAPK cascade, signal transduction by protein phosphorylation |
ENST00000492277 |
RPL29 |
1.44 E-04 |
cell-cell adhesion, involved in nonsense-mediated decay |
ENST00000236957 |
EEF1B2 |
1.68 E-04 |
Involved in translational elongation |
ENST00000308774 |
TRMT112 |
2.13 E-04 |
Involved in RNA methylation and translational termination |
ENST00000494862 |
HDLBP |
2.89 E-04 |
cell-cell adhesion and cholesterol metabolic process |
ENST00000473991 |
PSMD2 |
3.14 E-04 |
MAPK cascade, regulation of Wnt signaling pathway and ubiquitin-protein ligase activity involved in mitotic cell cycle, tumor necrosis factor-mediated signaling pathway |
ENST00000394729 |
PRKCD |
4.02 E-04 |
apoptotic process, cell cycle, negative regulation of inflammatory response |
ENST00000563039 |
SPN |
3.96 E-04 |
apoptotic signaling pathway, immune response, signal transduction |
ENST00000406984 |
FTH1P15 |
6.76 E-04 |
Unknown |
ENST00000585935 |
RAVER1 |
0.000677355 |
mRNA splicing via spliceosome |
ENST00000528296 |
RPL8 |
0.000747246 |
cytoplasmic translation |
ENST00000456311 |
CAD |
0.000787707 |
cellular response to drug and epidermal growth factor stimulus |
ENST00000595355 |
GINS2 |
0.000895068 |
double-strand break repair, mitotic DNA replication initiation |
ENST00000620429 |
VPS11 |
0.000910435 |
positive regulation of cellular protein catabolic process, endosome to lysosome transport |
ENST00000630977 |
VPS11 |
0.000910435 |
positive regulation of cellular protein catabolic process, endosome to lysosome transport |
ENST00000352980 |
KAT5 |
0.000987787 |
double-strand break repair, regulation of growth and transcription |
ENST00000456818 |
TUBA4A |
0.00110052 |
G2/M transition of mitotic cell cycle, cytoskeleton organization |
ENST00000517577 |
FTH1P11 |
0.001117274 |
Unknown |
ENST00000591301 |
GNA11 |
0.00106912 |
G-protein coupled acetylcholine receptor signaling pathway, signal transduction |
ENST00000523037 |
MRPL22 |
0.001163245 |
mitochondrial translational elongation and termination |
ENST00000606722 |
NDUFA13 |
0.001321049 |
negative regulation of intrinsic apoptotic signaling pathway, of cell growth and transcription |
ENST00000381348 |
LINC00634 |
0.001422158 |
Unknown |
ENST00000594493 |
RPS11 |
0.001903649 |
Involved in nonsense-mediated decay and translation processes |
ENST00000568265 |
TAF1C |
0.002079705 |
positive regulation of transcription, epigenetic |
ENST00000597681 |
MAP1S |
0.002292852 |
apoptotic process, microtubule bundle formation |
ENST00000368436 |
CKS1B |
0.002744271 |
regulation of mitotic cell cycle and transcription |
ENST00000537533 |
PTPN6 |
0.004221708 |
Regulation of apoptotic process as well as cell differentiation and proliferation |
ENST00000569760 |
FUS |
0.004169852 |
mRNA splicing via spliceosome and regulation of nucleic acid-templated transcription |
ENST00000533397 |
RPL8 |
0.00427335 |
cytoplasmic translation |
ENST00000443451 |
NCOR2 |
0.005527765 |
negative regulation of transcription |
ENST00000487513 |
EHMT2 |
0.006799918 |
DNA methylation, regulation of transcription and DNA replication |
ENST00000552600 |
ESPL1 |
0.008498519 |
apoptotic process, regulation of mitotic metaphase/anaphase transition and mitotic sister chromatid segregation |
ENST00000543608 |
SPPL3 |
0.009823731 |
T cell receptor signaling pathway, positive regulation of protein dephosphorylation |
ENST00000405878 |
XRCC6 |
1.10805E-07 |
double-strand break repair via classical nonhomologous end joining, regulation of transcription |
ENST00000427834 |
SGSM3 |
1.38595E-06 |
Activates GTPase and binds to Rab GTPase |
ENST00000537739 |
HDGF |
4.52676E-06 |
Binds to the DNA and helps in cell proliferation and differentiation |
Transcript specifically associated with unaffected BRCAX when compared to BRCA1, BRCA2 and BRCAX affected individuals |
|||
ENST00000405878 |
XRCC6 |
1.10805E-07 |
double-strand break repair via classical nonhomologous end joining, regulation of transcription |
ENST00000427834 |
SGSM3 |
1.38595E-06 |
cell cycle arrest, regulation of Rab protein signal transduction |
ENST00000537739 |
HDGF |
4.52676E-06 |
cell proliferation, regulation of transcription and signal transduction |
Transcript specifically associated with affected BRCAX when compared to BRCA1, BRCA2 and BRCAX unaffected individuals |
|||
ENST00000419477 |
YWHAZ |
0.001164657 |
regulation of apoptotic process, cell-cell adhesion and establishment of Golgi localization |
ENST00000539269 |
CARS2 |
0.002107185 |
cysteinyl-tRNA aminoacylation |
ENST00000436614 |
ZNF687 |
5.67433E-07 |
regulation of transcription |
ENST00000237837 |
FGF23 |
6.98876E-06 |
MAPK cascade, fibroblast growth factor receptor signaling pathway, regulation of transcription |
ENST00000452722 |
CADM1 |
6.01183E-05 |
apoptotic process, regulation of cytokine secretion |
ENST00000459748 |
RP11-466H18.1 |
0.000157558 |
Unknown |
ENST00000460469 |
NMD3 |
0.000153464 |
protein transport |
ENST00000562465 |
CDAN1 |
0.000146598 |
chromatin assembly, negative regulation of DNA replication |
ENST00000495645 |
CHPF2 |
0.000410748 |
chondroitin sulfate biosynthetic process |
ENST00000377861 |
PCDH9 |
0.000632184 |
homophilic cell adhesion via plasma membrane adhesion molecules |
ENST00000415265 |
WDR6 |
0.000658126 |
cell cycle arrest, negative regulation of cell proliferation |
ENST00000552588 |
RPL18 |
0.00085376 |
Involved in nonsense-mediated decay and translational initiation |
ENST00000374752 |
ACAD8 |
0.001441532 |
lipid metabolic process, regulation of transcription |
ENST00000449683 |
ATP5J2 |
0.001764478 |
ATP biosynthetic process |
ENST00000513391 |
OCIAD1 |
0.001957078 |
protein binding |
ENST00000547276 |
HNRNPA1 |
0.002375206 |
fibroblast growth factor receptor signaling pathway, gene expression, mRNA splicing |
ENST00000525085 |
NDUFC2 |
0.003100189 |
mitochondrial electron transport, NADH to ubiquinone |
ENST00000500813 |
DCTD |
0.003245736 |
nucleotide biosynthetic process |
ENST00000612832 |
ARHGAP21 |
0.004173962 |
organelle transport along microtubule, Golgi organization |
ENST00000535413 |
MLEC |
0.004822683 |
carbohydrate metabolic process, protein folding |
ENST00000498022 |
NAGK |
0.004945162 |
UDP-N-acetylglucosamine biosynthetic process |
ENST00000444034 |
MED12 |
0.005486952 |
canonical Wnt signaling pathway, intracellular steroid hormone receptor signaling pathway, regulation of transcription |
ENST00000522754 |
NCALD |
0.006089983 |
calcium-mediated signaling |
ENST00000552819 |
PCBP2 |
0.002187131 |
mRNA metabolic process, mRNA splicing, defense response to virus |
ENST00000528413 |
IRF7 |
0.002697978 |
cellular response to DNA damage stimulus, interferon-gamma-mediated signaling pathway, regulation of transcription and immune response |
ENST00000405878 |
XRCC6 |
1.10805E-07 |
double-strand break repair via classical nonhomologous end joining, regulation of transcription |
ENST00000427834 |
SGSM3 |
1.38595E-06 |
cell cycle arrest, regulation of Rab protein signal transduction |
ENST00000537739 |
HDGF |
4.52676E-06 |
IRE1-mediated unfolded protein response, cell proliferation, regulation of transcription and signal transduction |
Of interest, out of 67 transcripts specifically associated with BRCA1/BRCA2 subgroups combined, 19 transcripts (28%) were involved in DNA repair, cell proliferation, apoptosis and cell cycle, 32 (48%) different transcripts exert an action in transcription, translation as well as mRNA and protein metabolic processes, while 10 transcripts (15%) were involved in immune processes. Regarding the 28 transcripts exclusively associated with BRCAX affected individuals, more than 28% were involved in DNA repair/proliferation/apoptosis/cell cycle mechanisms, and approximately 43% (12 transcripts) were implicated in transcription and translation-related processes. It should be noted that XRCC6, SGSM3 and HDGF transcripts were associated with BRCA1/BRCA2, BRCAX unaffected and BRCAX affected individuals given that their expression was differentially expressed between all three subgroups. The expression of SGSM3 and HDGF was validated by qPCR in BRCAX subgroup to differentiate affected from non-affected patients (Supplementary Figure 1). The expression level of these genes was also checked in different mammalian breast cancer cell lines. Further, ANOVA analysis highlighted that H3F3B was differentially expressed in BRCA1 and BRCA2 subgroups, which was also evaluated by qPCR (Supplementary Figure 1).
The 85 genes associated with the 95 significant transcripts identified as differentially expressed between BRCA1, BRCA2 and BRCAX (unaffected or affected) individuals were then submitted for pathway and molecular function analyses. Using Ingenuity Pathway analysis, enrichment of several canonical pathways and functions were identified. It should be noted that mapped genes can be classified in more than one biological process or metabolic process.
Moreover, as described in Table 2, cell death and survival (top p-value: 2.46 × 10−5 with 34 molecules), cellular function and maintenance (top p-value: 6.77 × 10−5 with 25 molecules), cell cycle (top p-value: 7.41 × 10−5 with 15 molecules), post-translational modification (top p-value: 2.03 × 10−4 with 11 molecules) as well as cell morphology (top p-value: 2.07 × 10−4 with 17 molecules) represent the top overrepresented functions associated with these 85 genes. In addition, organismal injury, cell signaling, cell cycle and cell death represent the top networks associated with the whole gene set. Of great interest, these genes were also associated with Cancer and Organismal Injury and Abnormalities (Table 2).
Table 2: Overrepresented functions, network and diseases for significantly and differentially expressed transcripts
Molecular and cellular functions |
Top p-value* |
|---|---|
Cell Death and Suvival [34] |
2.46E-05 |
Cellular Function and Maintenance [25] |
6.77E-05 |
Cell Cycle [15] |
7.41E-05 |
Post-Translational Modification [11] |
2.03E-04 |
Cell Morphology [17] |
2.07E-04 |
Top Networks |
Score |
Organismal Injury and Abnormalities, Renal and Urological Disease, Embryonic Development |
61 |
Psychological Disorders, Cancer, Cell Cycle |
29 |
Cell Signaling, Cell-To-Cell Signaling and Interaction, Cell Death and Survival |
25 |
Cell Cycle, Cancer, Organismal Injury and Abnormalities |
22 |
Cell Death and Survival, Cancer, Gastrointestinal Disease |
15 |
Diseases and disorders |
Top p-value* |
Cancer [71] |
1.45E-05 |
Organismal Injury and Abnormalities [73] |
1.45E-05 |
Renal and Urological Disease [27] |
1.45E-05 |
Neurological Disease [27] |
4.17E-05 |
Hematological Disease [34] |
1.86E-04 |
*Top P-value of the p-value range.
The IPA analysis was also performed using the 28 genes discriminating unaffected from affected BRCAX individuals. As presented in Table 3, BRCAX-related genes were particularly associated with Telomere Extension by Telomerase, DNA Double-Strand Break Repair by Non-Homologous End Joining as well as specific molecule degradation and biosynthesis (p-value ranging from 1.66 × 10−4 to 0.05). The top system development and functions represented by these genes were tissue development (top p-value: 1.28 × 10−3 with 5 molecules), embryonic development (top p-value: 1.28 × 10−3) and Immune Cell Trafficking (top p-value: 1.28 × 10−3) (data not shown).
Table 3: The most significant canonical pathways enriched for significantly and differentially expressed transcripts between BRCAX affected and BRCAX unaffected
IPA canonical enriched pathways |
Number of gene in pathways |
p-value |
|---|---|---|
Telomere Extension by Telomerase |
2 |
1.66E-04 |
N-acetylglucosamine Degradation II |
1 |
5.13E-03 |
CMP-N-acetylneuraminate Biosynthesis I (Eukaryotes) |
1 |
6.46E-03 |
Chondroitin and Dermatan Biosynthesis |
1 |
7.76E-03 |
DNA Double-Strand Break Repair by Non-Homologous End Joining |
1 |
1.78E-02 |
Isoleucine Degradation I |
1 |
1.78E-02 |
Valine Degradation I |
1 |
2.29E-02 |
tRNA Charging |
1 |
4.90E-02 |
Analyses were performed using genes associated with significantly and differentially expressed transcripts between BRCAX affected and BRCAX unaffected using bonferroni correct p-value <0.01.
In addition, cell-to-cell signaling and interaction, cellular development, cellular growth and proliferation, cellular movement and lipid metabolism (top p-value at 1.28 × 10−3) represented the enriched functions (Table 4). As also described in this table, IPA analysis revealed that these genes were involved in networks such as “Cell death and survival” as well as “Connective tissue disorders and metabolic diseases” with 38 and 26 molecules, respectively.
Table 4: Overrepresented functions, network and diseases for significantly and differentially expressed transcripts between BRCAX affected and BRCAX unaffected
Molecular and cellular functions |
Top p-value* |
|---|---|
Cell-To-Cell Signaling and Interaction [4] |
1.28E-03 |
Cellular Development [7] |
1.28E-03 |
Cellular Growth and Proliferation [4] |
1.28E-03 |
Cellular Movement [2] |
1.28E-03 |
Lipid Metabolism [1] |
1.28E-03 |
Top networks |
Score |
Cell Death and Survival, Cell Morphology, Hair and Skin Development and Function |
38 |
Connective Tissue Disorders, Metabolic Disease, Nutritional Disease |
26 |
Cellular Assembly and Organization, DNA Replication, Recombination, and Repair, Nucleic Acid Metabolism |
2 |
Diseases and disorders |
Top p-value* |
Connective Tissue Disorders [3] |
2.37E-05 |
Metabolic Disease [3] |
2.37E-05 |
Nutritional Disease [2] |
2.37E-05 |
Skeletal and Muscular Disorders [4] |
2.37E-05 |
Cancer [26] |
5.46E-04 |
* Top P-value of the p-value range.
The number of genes involved in the process is given in parentheses.
Analyses were performed using genes associated with significantly and differentially expressed transcripts between BRCAX affected and BRCAX unaffected using bonferroni correct p-value <0.01.
Moreover, “cancer” (top p-value: 5.46 × 10−4 with 26 molecules) represented the disease associated with the highest number of molecules. Altogether, these analyses revealed enrichment of several pathways and functions involved in key mechanisms required for carcinogenesis development.
DISCUSSION
In this study, we analyzed the genome-wide transcription profile observed in LCLs immortalized from high-risk breast cancer families. To our knowledge, this is the first study describing clustering of BRCA1, BRCA2 and unaffected/affected BRCAX individuals based on their whole gene expression profile observed in corresponding LCLs.
The reliability of using LCLs from affected individuals for a given disease with regard to expression studies or splicing signatures has already been established [18-28]. A particular study conducted by Hussain and colleague concluded that LCLs were a good reflection of isolated lymphocytes given their close resemblance at the genetic and phenotypic levels to parent lymphocytes and were a valuable resource for studies regarding genotype-phenotype interactions [39] and inter-individual variations associated with various diseases and disorders such as cancer or infectious disease [40-42]. In addition, peripheral blood mononuclear cells (PBMCs) have also been used to investigate the links between DNA damage response, immunity and cancer [43] and to study the early stages of breast cancer development on gene expression patterns [44].
As described in other studies [45-47], we used ANOVA analysis of variance with the Scheffé multiple post-hoc test to identify transcripts specifically regulated in BRCA1, BRCA2 as well as in unaffected and affected BRCAX individuals. Using gene expression data of the 95 significant gene isoforms, our clustering results allowed discrimination of BRCA subgroups, particularly BRCA1/2 from BRCAX individuals.
This is in agreement with other studies in which gene expression in LCLs was successfully used for clustering analysis in several diseases including autism spectrum disorders and spinocerebellar ataxia (SCA28) [48, 49]. Moreover, LCLs served as a model system to assess genotype–phenotype relationships in human cells, including studies for quantitative trait loci influencing levels of individual mRNAs and responses to drugs and radiation [50-53], as well as regarding the haploinsufficiency effects of various BRCA1 mutation [54].
Over the last decade, several investigations clustered breast tumors based on single gene expression levels as well as their gene/splicing expression profile or molecular and clinical characteristics. The first correlation between the tumor phenotypic diversity (histopathological and clinical characteristics) and gene expression patterns was demonstrated in 2000 by Perou and colleague [55-58]. Van’t Veer et al. conducted clustering analyses of breast tumors based on their gene expression profile and determined a predictive signature of metastases development (poor prognosis) in patients without tumoral cells in local lymph node at diagnosis and established a specific signature of BRCA1 tumors [56]. Clustering analyses of breast tumors based on whole gene expression revealed familial aggregation of BRCA-related tumors and of specific molecular subtypes including Basal, HER2-enriched, Luminal A and B as well as Normal-like and sporadic tumors [38, 55-57, 59-70]. In addition, alternative splicing expression profile was also successfully used in several studies aiming to discriminate subtypes of breast tumors [71-73], and specific gene expression profiles have also been identified for BRCA1, BRCA2 and CHEK2-associated breast tumors [62, 74].
To our knowledge, the only clustering analysis involving LCLs in breast cancer classification distinguished BRCA1 carrier from non-carrier individuals [75], in which 133 genes were found to be differentially expressed between BRCA1-mutated and non-carriers. However, hierarchical clustering of these genes did not result in an accurate discrimination between both subgroups. Of these 133 genes identified by Vuillaume et al. [75], the RPL29 and PSMF1 genes have also been identified in our comparison between BRCA1/2 and BRCAX individuals. RPL29 is a ribosomal protein, involved in RNA interaction and protein synthesis, while PSMF1 gene encodes a proteasome inhibitor protein involved in protein folding and degradation [76, 77].
We then compared our results with GTEx Portal database, which contains normalized expression data from RNA sequencing for each gene and transcripts for different types of tissues. The normalization of expression was done by similar method for all databases. The expression values for genes from EBV transformed lymphocytes are available, and the normalized expression values in this database are similar with the ones we obtained [78]. In addition, we performed correlation between TPM value and FPKM value by doing a regression analysis using the values for each sample in the analysis and we found significant correlation between both of them.
We also compared our gene lists with the lists for up and down regulated genes associated with breast cancer using BioXpress, a curated gene expression and disease association database using RNA sequencing from TCGA database [79]. A certain number of our significant genes were common. Indeed, there were 5 genes that were present in our list and in the up regulated list for breast cancer (RAP2C, SPN, GINS2, ESPL1 and IRF7). Regarding the list for down regulated genes associated with breast cancer, 3 genes were also present in our list (HLA-DPA1, PCDH9 and NCALD).
Among the highest significant genes associated with BRCA1/2 subgroups, some of them (p-values ranging from 1.1 × 10−7 to 4.5 × 10−6) namely NOSIP, EIF2AK1, TBCB, XRCC6, SGSM3 and HDGF are involved in key mechanisms implicated in carcinogenesis susceptibility.
The expression of the NOSIP gene was upregulated and EIF2AK1 and TBCB were downregulated in BRCA1/2 individuals when compared to BRCAX subgroups. The eNOS Interacting Protein NOSIP was identified as an interacting protein of the endothelial isoform of nitric oxide synthase (eNOS) to enhance its translocation to intracellular membrane [80].
The EIF2AK1 gene is involved in the modulation of the basal hepatic endoplasmic reticulum stress tone [81]. Although no information links this gene to breast cancer, it has been demonstrated in a mouse xenograft model of human breast cancer that an activator of EIF2AK1 protein was associated with tumor growth inhibition compared with vehicle [82]. Thus, its downregulation in BRCA1/2 individuals could promote tumor development.
Regarding TBCB, this protein is involved in regulation of axonal growth and microtubule functional diversity and dynamics [83]. This protein was shown to be overexpressed and phosphorylated in breast tumors [84]. Therefore the effect of its decreased expression in BRCA1/2 individuals remains to be investigated.
Of great interest, specific expression levels of XRCC6, SGSM3 and HDGF genes were also associated with BRCAX individuals (unaffected and affected). These genes are involved in DNA repair as well as in the regulation of cell cycle, cell proliferation and transcription, and were differentially expressed between BRCA1/2, unaffected BRCAX and affected BRCAX individual. These genes showed the highest expression in affected BRCAX and the lowest expression in BRCA1 and BRCA2 subgroups. Indeed, a similar and progressive pattern of expression values for all three genes was observed between subgroups (BRCAX affected > BRCAX unaffected > BRCA1/2 individuals) as presented in Supplementary Table 2. Futhermore, the study highlighted few genes which could discriminate BRCA1 from BRCA2 subgroup, amongst them the highest difference was depicted by H3F3B.
XRCC6 gene encodes the Ku70 protein, which is a component of the non-homologous end joining (NHEJ) DNA repair pathway. This pathway is an alternative mechanism to homologous recombination (HR) repair pathway involved in double-strand break (DSB) repair in mammalian cells [85]. Defect or variation of expression of NHEJ genes such as XRCC6, might escape cell cycle checkpoint surveillance and could lead to suboptimal DNA repair and subsequently to accumulation of DNA damage and carcinogenesis initiation [86-88]. Given the key roles of BRCA1/2 in HR repair pathway [89], defective activity of BRCA1/2 proteins found in some individuals combined with low expression of NHEJ-associated genes could likely increase the accumulation of DNA damage in BRCA1/2 individuals. Indeed, this low expression of Ku70 was previously observed in BRCA1-deficient cell lines [90]. On the other hand, the high expression of XRCC6 in BRCAX individuals affected with breast cancer remains to be elucidated. Moreover, polymorphisms in XRCC6 gene have been shown to increase breast cancer susceptibility as well as other types of cancer [91-96].
SGSM3 is a member of the small G protein signaling modulators, which is associated with small G protein coupled receptor signal transduction pathway [97]. The human SGSM3 proteins were demonstrated to coprecipitate with RAP and RAB subfamily members of the small G protein superfamily. Therefore it has been suggested that the SGSM family members exert a role as modulators of the small G protein RAP and RAB-mediated neuronal signal transduction and vesicular transportation pathways [97]. The only information in the literature associating this protein with breast cancer described a decrease of SGSM3 mRNA in breast cancer tissue compared to normal tissue [98], which is in contrast with the significant increase of expression in LCLs of BRCAX affected individuals observed in our study. However, in the study performed by Nourashrafeddin et al. using basic RT-PCR method and visualization on agarose gel [98], SGSM3 was not detected in normal and cancerous tissues, illustrating the very low expression of this gene in breast tissues. It should be noted that a polymorphism (rs17001868) found in the SGSM3 gene has been associated with mammographic dense areas of the breast [99], which represents a factor of breast cancer risk [100-102]. SGSM3 has also been associated with hepatocellular carcinoma [103].
The Hepatoma-derived growth factor (HDGF) is now recognized as a breast cancer-associated gene and promotes the epithelial-mesenchymal transition (EMT) [104]. EMT is a hallmark of many cancers characterized by an increased cell invasion, which enhances the initial phase of metastatic progression [105, 106]. HDGF is overexpressed in several types of cancers including breast cancer cell lines and tissues and correlates with poor prognosis [104, 107-112]. Blockade of HDGF using a specific antibody results in the inhibition of malignant features and EMT of breast cancer cells [104]. Thus, its overexpression in breast cancer tissues is in concordance with our results demonstrating the overexpression of HDGF in LCLs of BRCAX individuals affected with breast cancer. Hence, this protein could be considered as a prognostic factor for tumor metastasis and recurrence.
H3 Histone Family Member 3B (H3F3B) is part of core histone molecule and has a role in gene regulation, DNA repair, DNA replication and chromosomal stability. Mutations in H3F3B gene have been associated with several cancers including brain cancer, giant cell tumor of bone and colorectal cancer [113-115]. Overexpression of this gene is also associated with colorectal cancer. In breast cancer, the copy number of the chromosome carrying this gene is significantly high [116], which is further confirmed by the data from the human protein atlas data.
In addition to XRCC6, SGSM3 and HDGF genes as described above, ZNF687 and FGF23 genes were also associated with and upregulated in BRCAX affected individuals when compared to unaffected individuals.
ZNF687 encodes a zinc finger protein and represents an important regulator of skeletal development and maintenance [117]. Overexpression of ZNF687 has been observed in tumor tissue of individual giant cell tumor of bone associated with Paget disease of bone, and this high expression is also observed in the peripheral blood of patients affected with Paget disease [118]. The role of ZNF687 in breast cancer is unknown.
As to fibroblast growth factor 23 (FGF23), it is a binding partner of Klotho proteins for endocrine signaling through the action of FGFRs. These FGFR receptors are involved in several mechanisms such as regulation of cell survival, proliferation, differentiation and motility during embryogenesis as well as tissue homeostasis and carcinogenesis [119-122]. Indeed, FGF23 signaling promotes proliferation in myeloma cells [123], while increase of FGF23 levels in serum were observed in cancer patients, and were also elevated in patients with non-cancerous diseases, such as hypophosphatemic rickets and chronic kidney diseases [124].
Considering all significant genes identified following ANOVA analysis of gene expression data observed in four BRCA subgroups, several interesting pathways seem to be affected by the regulation of specific genes. Among these pathways, EIF2 signaling, 14-3-3-mediated pathway and mTOR signaling are particularly significant and relevant to breast cancer. Moreover, both the EIF2 (through the activation of the PI3K pathway) and 14-3-3-mediated signaling cascades regulate the mTOR pathway [125, 126], which is involved in the response to hormones and growth factor stimulation and is well known to exert a significant role in tumor cell growth and proliferation as well as in breast cancer development [127] and references therein.
On the other hand, cell death and survival, cellular function and maintenance as well as cell cycle represent the highest enriched functions, while cancer remains among the diseases/disorders having the highest p-value, which is associated with a high number of genes involved in cancer-related pathways.
Taken together, in the present study, we compared gene expression profiles in lymphoblastoid cell lines in BRCA1- and BRCA2- carriers as well as BRCAX affected and unaffected individuals from high-risk breast cancer families in order to determine specific markers which could be of great relevance for further studies. Indeed, several transcripts have been identified as potential valuable markers of interest for breast cancer, and deserve further analysis.
MATERIALS AND METHODS
Ascertainment of high-risk families
Recruitment of high-risk French Canadian breast and ovarian cancer families started in 1996 as a large interdisciplinary research program designated INHERIT BRCAs [12]. The High risk group is defined as families with a history of breast cancer with at least 3 cases in 1st degree relative or 4 cases in 2nd degree relative, the full selection criteria have been published previously [12]. Patients were screened for deleterious mutations in BRCA1 and BRCA2 genes. The BRCA testing was done by complete sequencing of the BRCA1/2 gene by using primers in both directions (forward and reverse). Confirmation was done by Myriad Laboratories. A subset of 96 high-risk families with no deleterious mutation in BRCA1 or BRCA2 were recruited (BRCAX families) as described elsewhere [128, 129]. For the purpose of this study, carriers of a BRCA1 or BRCA2 mutation were selected. As for the BRCAX families, the youngest available breast cancer case in the family was selected, along with the oldest non-affected sister. All unaffected women were post-menopausal. All individuals provided their written informed consent in order for their genetic material to be part of a biobank (Dr J. Simard, director). The age range of affected BRCA1, BRCA2 and BRCAX were 23-65, 29-72 and 35-70 respectively. For unaffected BRCA1, BRCA2 and BRCAX the age range was 35-66, 37-77 and 41-86 respectively.
Cell line immortalization and RNA extraction
Lymphocytes (LCLs) were isolated and immortalized from 7 to 9 mL of blood samples from breast cancer individuals using Epstein-Barr virus in 15% RPMI medium as previously described [128, 130, 131]. Total RNA was extracted from LCLs using TRI Reagent (Molecular Reasearch Center Inc, Cincinnati, OH, USA) according to the manufacturer’s instructions as described previously [128]. The viral strain, number of passage and conditions for cell lines were kept identical to avoid bias in gene expression [133].
RNA-seq experiments
The quality of RNA samples was evaluated with an Agilent Bioanalyzer 2100 to determine the RIN (RNA Integrity) score using the Agilent RNA 6000 Nano chip and reagents. Samples with a RIN score >7 were retained and converted to cDNA with the Illumina RNA seq kit for sequence library preparation based on the Illumina TruSeq RNA Sample Preparation protocol. The final libraries were pooled in triplicate and then sequenced on an Illumina HiSeq 2000 at the McGill University and Génome Québec Innovation Centre.
Raw reads were trimmed for length (n>=32), quality (phred33 score >= 30) and adaptor sequence using fastxv0.0.13.1. Trimmed paired-end reads (read length: 100 bp) were aligned to the hg19 human reference genome using Tophat version v1.4.0 [132]. The resulting alignment file was indexed using samtools v0.1.18. Raw, trimmed and aligned read numbers were retrieved after alignment to determine the quality of the sequence data. GATK (v1.0.5777) [134] was then used to compute the coding sequence coverage for each sample. Raw read counts and normalized read counts (in transcript per million, TPM) were obtained using the Kallisto v0.43.0 quant command [135] with the default parameter on the GRCh38.rel79 version of the human transcriptome, while Pearson correlation values were obtained pairwise for each sample using R v2.12.0. Differential gene expression was determined using edgeRv2.2.6 [136] on R v2.12.0 and DESeqv1.6.1 [137] on R v2.14.0. Transcript differential expression was performed using cuffdiff v1.3.0. A gene ontology analysis was then launched on gene and transcript differential expression results using goseqv1.2.1 [138] on R v2.12.0. Finally, UCSC compatible wiggle tracks were generated using FindPeaks v4.0.16.
Statistical analysis
Statistical analyses were carried out using the R Package v3.3. In regard to mRNA levels, One-factor analysis of variance (ANOVA) was performed to compare the breast cancer subgroups. First a model was fitted using the lm function from the stats package then the ANOVA analysis was performed with the Anova command from the car package [139, 140]. Bonferroni correction was performed with the p.adjust function from the stats package using “BH”, “BY” and “bonferroni” methods and statistically significant differences were considered at p < 0.01 for the “bonferroni” method [140]. The Scheffé test was carried out with the scheffé test function from the agricolae R package for post-hoc analysis for comparisons between two of the multiple groups [141]. We performed intra-group variance analysis using gene expression data of patients with the BRCA1 R1443X mutation and BRCA2 8765delAG mutation by Principle component analysis (PCA).
Pathways, network and clustering analyses
Partek Genomics Suites® software package (copyright © 2009 Partek Incorporated. St. Louis, MO) was used for hierarchical clustering using the default setting (Euclidean dissimilarity and average linkage method) as well as for Principal component analysis (PCA).
Identification of overrepresented pathways, functions and gene-associated diseases were performed using QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity) software. Default settings in IPA for expression dataset analyses were used for gene list functional analysis. Gene lists were uploaded using NCBI Entrez gene IDs or gene symbols and submitted for IPA Core Analysis. IPA calculates p-values that reflect the statistical significance of association between the genes and the networks by the Fisher’s exact test. P-value ≤ 0.05 were considered significant.
Author contributions
FD designed the research protocol. MCP, CK and GO conducted experiments. JS and AD contributed samples and analytic tools. MCP, CK, CJB and YL performed data analysis. MCP, CK, CJB, YL, FD drafted manuscript. Critical revision of the manuscript was done by all the authors.
ACKNOWLEDGMENTS
The authors are indebted to the participants and their families for their generosity and providing DNA samples.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
GRANT SUPPORT
This study was supported by research grant from the Canadian Cancer Society and Quebec Breast Cancer Foundation. MCP holds a studentship from Laval University, Cancer Research Center at Laval University, Fondation CHU de Quebec and Fondation René Bussières. CK holds a Bourse de recrutement au doctorat- Pierre J. Durand. CJB holds a Fond de Recherche du Québec - Santé (FRQS) Doctoral award. AD is chairholder of the L’Oreal Research Chair in Digital Biology. JS is chairholder of the Canada Research Chair in Oncogenetics.
REFERENCES
1. Canadian Cancer Society’s Advisory Committee on Cancer Statistics. Canadian Cancer Statistics 2014. Toronto, ON; 2014. 1 p.
2. Peto J, Houlston RS. Genetics and the common cancers. Eur J Cancer. 2001; 37: S88-96. https://doi.org/10.1016/S0959-8049(01)00255-6.
3. Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormonal contraceptives: collaborative reanalysis of individual data on 53297 women with breast cancer and 100239 women without breast cancer from 54 epidemiological studies. Lancet. 1996; 347: 1713-27. https://doi.org/10.1016/S0140-6736(96)90806-5.
4. Aloraifi F, McCartan D, McDevitt T, Green AJ, Bracken A, Geraghty J. Protein-truncating variants in moderate-risk breast cancer susceptibility genes: a meta-analysis of high-risk case-control screening studies. Cancer Genet. 2015; 208: 455-63. https://doi.org/10.1016/j.cancergen.2015.06.001.
5. Easton DF. How many more breast cancer predisposition genes are there? Breast Cancer Res. 1999; 1: 14-7. https://doi.org/10.116:bcr6.
6. StadlerZK, Thom P, Robson ME, Weitzel JN, Kauff ND, Hurley KE, Devlin V, Gold B, Klein RJ, Offit K. Genome-wide association studies of cancer. J Clin Oncol. 2010; 28: 4255-67. https://doi.org/10.1200/JCO.2009.25.7816.
7. Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, Hollestelle A, Houben M, Crepin E, van Veghel-Plandsoen M, Elstrodt F, van Duijn C, Bartels C, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in non-carriers of BRCA1 or BRCA2 mutations. Nat Genet. 2002; 31: 55-9. https://doi.org/10.1038/ng879.
8. Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, McGuffog L, Evans DG, Eccles D, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006; 38: 873-5. https://doi.org/10.1038/ng1837.
9. Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007; 39: 165-7. https://doi.org/10.1038/ng1959.
10. Malkin D, Li FP, Strong LC, Fraumeni JF, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990; 250: 1233-8.
11. Michailidou K, Beesley J, Lindstrom S, Canisius S, Dennis J, Lush MJ, Maranian MJ, Bolla MK, Wang Q, Shah M, Perkins BJ, Czenz K, Eriksson M, et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet. 2015; 47: 373-80. https://doi.org/10.1038/ng.3242.
12. Simard J, Dumont M, Moisan AM, Gaborieau V, Vézina H, Durocher F, Chiquette J, Plante M, Avard D, Bessette P, Brousseau C, Dorval M, Godard B, et al. Evaluation of BRCA1 and BRCA2 mutation prevalence, risk prediction models and a multistep testing approach in French-Canadian families with high risk of breast and ovarian cancer. J Med Genet. 2007; 44: 107-21. https://doi.org/10.1136/jmg.2006.044388.
13. Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge BC. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008; 456: 470-6. https://doi.org/10.1038/nature07509.
14. Venables JP. Aberrant and alternative splicing in cancer. Cancer Res. 2004; 64: 7647-54. https://doi.org/10.1158:0008-5472.CAN-04-1910.
15. Hoen PA, Friedländer MR, Almlöf J, Sammeth M, Pulyakhina I, Anvar SY, Laros JF, Buermans HP, Karlberg O, Brännvall M, Dunnen den JT, van Ommen GJ, Guigo R, et al. Reproducibility of high-throughput mRNA ans small RNA sequencing across laboratories. Nat Biotechnol. 2013; 31: 1015-22. https://doi.org/10.1038/nbt.2702.
16. Ching T, Huang S, Garmire LX. Power analysis and sample size estimation for RNA-Seq differential expression. RNA. 2014; 20: 1684-96. https://doi.org/10.1261/rna.046011.114.
17. Cheung VG, Conlin LK, Weber TM, Arcaro M, Jen KY, Morley M, Spielman RS. Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Genet. 2003; 33: 422-5. https://doi.org/10.1038/ng1094.
18. Hull J, Campino S, Rowlands K, Chan MS, Copley RR, Taylor MS, Rockett K, Elvidge G, Keating B, Knight J, Kwiatkowski D. Identification of common genetic variation that modulates alternative splicing. PLoS Genet. 2007; 3: e99-9. https://doi.org/10.1371/journal.pgen.0030099.
19. Cheung HC, Baggerly KA, Tsavachidis S, Bachinski LL, Neubauer VL, Nixon TJ, Aldape KD, Cote GJ, Krahe R. Global analysis of aberrant pre-mRNA splicing in glioblastoma using exon expression arrays. BMC Genomics. 2008; 9: 216-6. https://doi.org/10.1186:1471-2164-9-216.
20. Gardina PJ, Clark TA, Shimada B, Staples MK, Yang Q, Veitch J, Schweitzer A, Awad T, Sugnet C, Dee S, Davies C, Williams A, Turpaz Y. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genomics. 2006; 7: 325-5. https://doi.org/10.1186/1471-2164-7-325.
21. Tuthill A, Semple RK, Day R, Soos MA, Sweeney E, Seymour PJ, Didi M, O’Rahilly S. Functional characterization of a novel insulin receptor mutation contributing to Rabson-Mendenhall syndrome. Clin Endocrinol (Oxf). 2007; 66: 21-6. https://doi.org/10.1111/j.1365-2265.2006.02678.x.
22. Rivolta C, McGee TL, Rio Frio T, Jensen RV, Berson EL, Dryja TP. Variation in retinitis pigmentosa-11 (PRPF31 or RP11) gene expression between symptomatic and asymptomatic patients with domain RP11 mutations. Hum Mutat. 2006; 27: 644-53. https://doi.org/10.1002/humu.20325.
23. Philibert RA, Crowe R, Ryu GY, Yoon JG, Secrest D, Sandhu H, Madan A. Transcriptional profiling of lymphoblast cell lines from subjects with panic disorder. Am J Med Genet B Neuropsychiatr Genet. 2007; 144B: 674-82. https://doi.org/10.1002/ajmg.b.30502.
24. Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, Spielman RS, Cheung VG. Genetic analysis of genome-wide variation in human gene expression. Nature. 2004; 430:743-7. https://doi.org/10.1038/nature02797.
25. Morello F, de Bruin TW, Rotter JI, Pratt RE, van der Kallen CJ, Hladik GA, Dzau VJ, Liew CC, Chen YD. Differential gene expression of blood-derived cell lines in familial combined hyperlipidemia. Arterioscler Throm Vasc Biol. 2004; 24: 2149-54. https://doi.org/10.1161/01.ATV.0000145978.70872.63.
26. Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, Wong KC, Taylor J, Burnett E, Gut I, Farall M, Lathrop GM, Abecasis GR, Cookson WO. A genome-wide association study of global gene expression. Nat Genet. 2007; 39: 1202-7. https://doi.org/10.1038/ng2109.
27. Kwan T, Benovoy D, Dias C, Gurd S, Provencher C, Beaulieu P, Hudson TJ, Sladek R, Majewski J. Genome-wide analysis of transcript isoform variation in humans. Nat Genet. 2008; 40: 225-31. https://doi.org/10.1038/ng.2007.57.
28. Thorsen K, Sørensen KD, Brems-Eskildsen AS, Modin C, Gaustadnes M, Hein AM, Kruhøffer M, Laurberg S, Borre M, Wang K, Brunak S, Krainer AR, Tørring N, et al. Alternative splicing in colon, bladder, and prostate cancer identified by exon array analysis. Mol Cell Proteomics. 2008; 7: 1214-24. https://doi.org/10.1074/mcp.M700590-MCP200.
29. Kwan T, Benovoy D, Dias C, Gurd S, Serre D, Zuzan H, Clark TA, Schweitzer A, Staples MK, Wang H, Blume JE, Husdon TJ, Sladek R, et al. Heritability of alternative splicing in the human genome. Genome Res. 2007; 17: 1210-8. https://doi.org/10.1101/gr.6281007.
30. Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, Ingle CE, Dunning M, Flicek P, Koller D, Montgomery S, Tavaré S, Deloukas P, et al. Population genomics of human gene expression. Nat Genet. 2007; 39:1217-24. https://doi.org/10.1038/ng2142.
31. Suderman M, Pappas JP, Borghol N, Buxton JL, McArdle WL, Ring SM, Hertzman C, Power C, Szyf M, Pembrey M. Lymbhoblastoid cell lines reveal associations of adult DNA methylation with childhood and current adversity that are distinct from whole blood associations. Int J Epidemiol. 2015; 44: 1331-40. https://doi.org/10.1093/ije/dyv168.
32. Aloraifi F, Alshehhi M, McDevitt T, Cody N, Meany M, O’Doherty A, Quinn CM, Green AJ, Bracken A, Geraghty JG. Phenotypic analysis of familial breast cancer: comparison of BRCAx tumors with BRCA1-, BRCA2-carriers and non-familial breast cancer. Eur J Surg Oncol. 2015; 41: 641-6. https://doi.org/10.1016/j.ejso.2015.01.021.
33. Hedenfalk I, Duggan D, Chen Y, Radmacher M, Bittner M, Simon R, Meltzer P, Gusterson B, Esteller M, Kallioniemi OP, Wilfond B, Borg A, Trent J, et al. Gene-expression profiles in hereditary breast cancer. N Engl J Med. 2001; 344: 539-48. https://doi.org/10.1056/NEJM200102223440801.
34. Honrado E, Osorio A, Milne RL, Paz MF, Melchor L, Cascón A, Urioste M, Cazorla A, Díez O, Lerma E, Esteller M, Palacios J, Benítez J. Immunohistochemical classification of non-BRCA1/2 tumors identifies different groups that demonstrate the heterogeneity of BRCAX families. Modern Pathol. 2007; 20: 1298-306. https://doi.org/10.1038/modpathol.3800969.
35. Hedenfalk I, Ringner M, Ben-Dor A, Yakhini Z, Chen Y, Chebil G, Ach R, Loman N, Olsson H, Meltzer P, Borg A, Trent J. Molecular classification of familial non-BRCA1/BRCA2 breast cancer. Proc Natl Acad Sci U S A. 2003; 100: 2532-7.https://doi.org/10.1073/pnas.0533805100.
36. Lakhani SR, Gusterson BA, Jacquemier J, Sloane JP, Anderson TJ, van de Vijver MJ, Venter D, Freeman A, Antoniou A, McGuffog L, Smyth E, Steel CM, Haites N, et al. The pathology of familial breast cancer: histological features of cancers in families not attributable to mutations in BRCA1 or BRCA2. Clin Cancer Res. 2000; 6: 782-9.
37. Fernández-Ramires R, Gómez G, Muñoz-Repeto I, de Cecco L, Llort G, Cazorla A, Blanco I, Gariboldi M, Pierotti MA, Benítez J, Osorio A. Transcriptional characteristics of familial non-BRCA1/BRCA2 breast tumors. Int J Cancer. 2011; 128: 2635-44. https://doi.org/10.1002/ijc.25603.
38. Larsen MJ, Thomassen M, Tan Q, Lænkholm AV, Bak M, Sørensen KP, Andersen MK, Kruse TA, Gerdes AM. RNA profiling reveals familial aggregation of molecular subtypes in non-BRCA1/2 breast cancer families. BMC Med Genomics. 2014; 7: 9. https://doi.org/10.1186/1755-8794-7-9.
39. Hussain T, Kotnis A, Sarin R, Mulherkar R. Establishment & characterization of lymphoblastoid cell lines from patients with multiple primary neoplasms in the upper aero-digestive tract & healthy individuals. Indian J Med Res. 2012; 135: 820-9.
40. Chaussabel D, Allman W, Mejias A, Chung W, Bennett L, Ramilo O, Pascual V, Palucka AK, Banchereau J. Analysis of significance patterns identifies ubiquitous and disease-specific gene-expression signatures in patient peripheral blood leukocytes. Ann N Y Acad Sci. 2005; 1062: 146-54. https://doi.org/10.1196/annals.1358.017.
41. Radich JP, Mao M, Stepaniants S, Biery M, Castle J, Ward T, Schimmack G, Kobayashi S, Carleton M, Lampe J, Linsley PS. Individual-specific variation of gene expression in peripheral blood leukocytes. Genomics. 2004; 83: 980-8. https://doi.org/10.1016/j.ygeno.2003.12.013.
42. Whitney AR, Diehn M, Popper SJ, Alizadeh AA, Boldrick JC, Relman DA, Brown PO. Individuality and variation in gene expression patterns in human blood. Proc Natl Acad Sci U S A. 2003; 100: 1896-901. https://doi.org/10.1073/pnas.252784499.
43. Gasser S, Raulet D. The DNA damage response, immunity and cancer. Semin Cancer Biol. 2006; 16: 344-7. https://doi.org/10.1016/j.semcancer.2006.07.004.
44. Sharma P, Sahni NS, Tibshirani R, Skaane P, Urdal P, Berghagen H, Jensen M, Kristiansen L, Moen C, Sharma P, Zaka A, Arnes J, Sauer T, et al. Early detection of breast cancer based on gene-expression patterns in peripheral blood cells. Breast Cancer Res. 2005; 7: R634-44. https://doi.org/10.1186/bcr1203.
45. Honma N, Takubo K, Sawabe M, Arai T, Akiyama F, Sakamoto G, Utsumi T, Yoshimura N, Harada N. Alternative use of multiple exons 1 of aromatase gene in cancerous and normal breast tissues from women over the age of 80 years. Breast Cancer Res. 2009; 11: R48-8. https://doi.org/10.1186/bcr2335.
46. Mougeot JL, Bahrani-Mostafavi Z, Vachris JC, McKinney KQ, Gurlov S, Zhang J, Naumann RW, Higgins RV, Hall JB. Gene expression profiling of ovarian tissues for determination of molecular pathways reflective of tumorigenesis. J Mol Biol. 2006; 358: 310-29. https://doi.org/10.1016/j.jmb.2006.01.092.
47. Shin JC, Lee JH, Yang DE, Moon HB, Rha JG, Kim SP. Expression of insulin-like growth factor-II and insulin-like growth factor binding protein-1 in the placental basal plate from pre-eclamptic pregnancies. Int J Gynaecol Obstet. 2003; 81: 273-80. https://doi.org/10.1016/S0020-7292(02)00444-7.
48. Hu VW, Lai Y. Developing a predictive gene classifier for autism spectrum disorders based upon differential gene expression profiles of phenotypic subgroups. N Am J Med Sci (Boston). 2013; 6. https://doi.org/10.7156/najms.2013.0603107.
49. Mancini C, Roncaglia P, Brussino A, Stevanin G, Buono Lo N, Krmac H, Maltecca F, Gazzano E, Stella AB, Calvaruso MA, Iommarini L, Cagnoli C, Forlani S, et al. Genome-wide expression profiling and functional characterization of SCA28 lymphoblastoid cell lines reveal impairment in cell growth and activation of apoptotic pathways. BMC Med Genomics. 2013; 6: 22-2. https://doi.org/10.1186/1755-8794-6-22.
50. Choy E, Yelensky R, Bonakdar S, Plenge RM, Saxena R, De Jager PL, Shaw SY, Wolfish CS, Slavik JM, Cotsapas C, Rivas M, Dermitzakis ET, Cahir-McFarland E, et al. Genetic analysis of human traits in vitro: drug response and gene expression in lymphoblastoid cell lines. PLoS Genet. 2008; 4: e1000287-7. https://doi.org/10.1371/journal.pgen.1000287.
51. Duan S, Bleibel WK, Huang RS, Shukla SJ, Wu X, Badner JA, Dolan ME. Mapping genes that contribute to daunorubicin-induced cytotoxicity. Cancer Res. 2007; 67: 5425-33. https://doi.org/10.1158/0008-5472.CAN-06-4431.
52. Huang RS, Duan S, Bleibel WK, Kistner EO, Zhang W, Clark TA, Chen TX, Schweitzer AC, Blume JE, Cox NJ, Dolan ME. A genome-wide approach to identify genetic variants that contribute to etoposide-induced cytotoxicity. Proc Natl Acad Sci U S A. 2007; 104: 9758-63. https://doi.org/10.1073/pnas.0703736104.
53. Huang RS, Duan S, Shukla SJ, Kistner EO, Clark TA, Chen TX, Schweitzer AC, Blume JE, Dolan ME. Identification of genetic variants contributing to cisplatin-induced cytotoxicity by use of a genomewide approach. Am J Hum Genet. 2007; 81: 427-37. https://doi.org/10.1086/519850.
54. Vaclová T, Gómez-López G, Setién F, Bueno JM, Macías JA, Barroso A, Urioste M, Esteller M, Benítez J, Osorio A. DNA repair capacity is impaired in healthy BRCA1 heterozygous mutation carriers. Breast Cancer Res Treat. 2015; 152: 271-82. https://doi.org/10.1007/s10549-015-3459-3.
55. Perou CM, Sørlie T, Eisen MB, van de Rijn M. Molecular portraits of human breast tumours. Nature. 2000; 406: 747-52. https://doi.org/10.1038/35021093.
56. van ’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002; 415: 530-6. https://doi.org/10.1038/415530a.
57. Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001; 98: 10869-74. https://doi.org/10.1073/pnas.191367098.
58. Bertucci F, Finetti P, Cervera N, Charafe-Jauffret E, Mamessier E, Adélaïde J, Debono S, Houvenaeghel G, Maraninchi D, Viens P, Charpin C, Jacquemier J, Birnbaum D. Gene expression profiling shows medullary breast cancer is a subgroup of basal breast cancers. Cancer Res. 2006; 66: 4636-44. https://doi.org/10.1158/0008-5472.CAN-06-0031.
59. Sørlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, Demeter J, Perou CM, Lønning PE, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003; 100: 8418-23. https://doi.org/10.1073/pnas.0932692100.
60. Waddell N, Arnold J, Cocciardi S, da Silva L, Marsh A, Riley J, Johnstone CN, Orloff M, Assie G, Eng C, Reid L, Keith P, Yan M, et al. Subtypes of familial breast tumours revealed by expression and copy number profiling. Breast Cancer Res Treat. 2010; 123: 661-77. https://doi.org/10.1007/s10549-009-0653-1.
61. Jönsson G, Staaf J, Vallon-Christersson J, Ringnér M, Holm K, Hegardt C, Gunnarsson H, Fagerholm R, Strand C, Agnarsson BA, Kilpivaara O, Luts L, Heikkilä P, et al. Genomic subtypes of breast cancer identified by array-comparative genomic hybridization display distinct molecular and clinical characteristics. Breast Cancer Res. 2010; 12: R42-2. https://doi.org/10.1186/bcr2596.
62. Nagel JH, Peeters JK, Smid M, Sieuwerts AM, Wasielewski M, de Weerd V, Trapman-Jansen AM, van den Ouweland A, Brüggenwirth H, van I Jcken WF, Klijn JG, van der Spek PJ, Foekens JA, et al. Gene expression profiling assigns CHEK2 1100delC breast cancers to the luminal intrinsic subtypes. Breast Cancer Res Treat. 2012; 132: 439-48. https://doi.org/10.1007/s10549-011-1588-x.
63. Hu Z, Fan C, Oh DS, Marron JS, He X, Qaqish BF, Livasy C, Carey LA, Reynolds E, Dressler L, Nobel A, Parker J, Ewend MG, et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics. 2006; 7: 96. https://doi.org/10.1186/1471-2164-7-96.
64. Perou CM, Jeffrey SS, van de Rijn M, Rees CA, Eisen MB, Ross DT, Pergamenschikov A, Williams CF, Zhu SX, Lee JC, Lashkari D, Shalon D, Brown PO, et al. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci U S A. 1999; 96: 9212-7.
65. Sørlie T. Molecular portraits of breast cancer: tumour subtypes as distinct disease entities. Eur J Cancer. 2004; 40: 2667-75. https://doi.org/10.1016/j.ejca.2004.08.021.
66. Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler L, Akslen LA, Ragaz J, Gown AM, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004; 10: 5367-74. https://doi.org/10.1158/1078-0432.CCR-04-0220.
67. Abd El-Rehim DM, Ball G, Pinder SE, Rakha E, Paish C, Robertson JF, Macmillan D, Blamey RW, Ellis IO. High-throughput protein expression analysis using tissue microarray technology of a large well-characterised series identifies biologically distinct classes of breast cancer confirming recent cDNA expression analyses. Int J Cancer. 2005; 116: 340-50. https://doi.org/10.1002/ijc.21004.
68. Jacquemier J, Ginestier C, Rougemont J, Bardou VJ, Charafe-Jauffret E, Geneix J, Adélaïde J, Koki A, Houvenaeghel G, Hassoun J, Maraninchi D, Viens P, Birnbaum D, et al. Protein expression profiling identifies subclasses of breast cancer and predicts prognosis. Cancer Res. 2005; 65: 767-79.
69. Sørlie T, Wang Y, Xiao C, Johnsen H, Naume B, Samaha RR, Børresen-Dale AL. Distinct molecular mechanisms underlying clinically relevant subtypes of breast cancer: gene expression analyses across three different platforms. BMC Genomics. 2006; 7: 127. https://doi.org/10.1186/1471-2164-7-127.
70. Sotiriou C, Neo SY, McShane LM, Korn EL, Long PM, Jazaeri A, Martiat P, Fox SB, Harris AL, Liu ET. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci U S A. 2003; 100: 10393-8. https://doi.org/10.1073/pnas.1732912100.
71. Venables JP, Klinck R, Bramard A, Inkel L, Dufresne-Martin G, Koh C, Gervais-Bird J, Lapointe E, Froehlich U, Durand M, Gendron D, Brosseau JP, Thibault P, et al. Identification of alternative splicing markers for breast cancer. Cancer Res. 2008; 68: 9525-31. https://doi.org/10.1158/0008-5472.CAN-08-1769.
72. Li C, Kato M, Shiue L, Shively JE, Ares M, Lin RJ. Cell type and culture condition-dependent alternative splicing in human breast cancer cells revealed by splicing-sensitive microarrays. Cancer Res. 2006; 66: 1990-9. https://doi.org/10.1158/0008-5472.CAN-05-2593.
73. André F, Michiels S, Dessen P, Scott V, Suciu V, Uzan C, Lazar V, Lacroix L, Vassal G, Spielmann M, Vielh P, Delaloge S. Exonic expression profiling of breast cancer and benign lesions: a retrospective analysis. Lancet Oncol. 2009; 10: 381-90. https://doi.org/10.1016/S1470-2045(09)70024-5.
74. Honrado E, Osorio A, Palacios J, Benitez J. Pathology and gene expression of hereditary breast tumors associated with BRCA1, BRCA2 and CHEK2 gene mutations. Oncogene. 2006; 25: 5837-45. https://doi.org/10.1038/sj.onc.1209875.
75. Vuillaume ML, Uhrhammer N, Vidal V, Vidal VS, Chabaud V, Jesson B, Kwiatkowski F, Bignon YJ. Use of gene expression profiles of peripheral blood lymphocytes to distinguish BRCA1 mutation carriers in high risk breast cancer families. Cancer Inform. 2009; 7: 41-56.
76. Li C, Ge M, Yin Y, Luo M, Chen D. Silencing expression of ribosomal protein L26 and L29 by RNA interfering inhibits proliferation of human pancreatic cancer PANC-1 cells. Mol Cell Biochem. 2012; 370: 127-39. https://doi.org/10.1007/s11010-012-1404-x.
77. McCutchen-Maloney SL, Matsuda K, Shimbara N, Binns DD, Tanaka K, Slaughter CA, DeMartino GN. cDNA cloning, expression, and functional characterization of PI31, a proline-rich inhibitor of the proteasome. J Biol Chem. 2000; 275: 18557-65. https://doi.org/10.1074/jbc.M001697200.
78. GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013; 45: 580-5. https://doi.org/10.1038/ng.2653.
79. Wan Q, Dingerdissen H, Fan Y, Gulzar N, Pan Y, Wu TJ, Yan C, Zhang H, Mazumder R. BioXpress: an integrated RNA-seq-derived gene expression database for pan-cancer analysis. Database (Oxford). 2015; 2015: bav019. https://doi.org/10.1093/database/bav019.
80. König P, Dedio J, Oess S, Papadakis T, Fischer A, Müller-Esterl W, Kummer W. NOSIP and its interacting protein, eNOS, in the rat trachea and lung. J Histochem Cytochem. 2005; 53: 155-64. https://doi.org/10.1369/jhc.4A6453.2005.
81. Acharya P, Chen JJ, Correia MA. Hepatic heme-regulated inhibitor (HRI) eukaryotic initiation factor 2alpha kinase: a protagonist of heme-mediated translational control of CYP2B enzymes and a modulator of basal endoplasmic reticulum stress tone. Mol Pharmacol. 2010; 77: 575-92. https://doi.org/10.1124/mol.109.061259.
82. Chen T, Ozel D, Qiao Y, Harbinski F, Chen L, Denoyelle S, He X, Zvereva N, Supko JG, Chorev M, Halperin JA, Aktas BH. Chemical genetics identify eIF2α kinase heme-regulated inhibitor as an anticancer target. Nat Chem Biol. 2011; 7: 610-6. https://doi.org/10.1038/nchembio.613.
83. Lopez-Fanarraga M, Carranza G, Bellido J, Kortazar D, Villegas JC, Zabala JC. Tubulin cofactor B plays a role in the neuronal growth cone. J Neurochem. 2007; 100: 1680-7. https://doi.org/10.1111/j.1471-4159.2006.04328.x.
84. Vadlamudi RK, Barnes CJ, Rayala S, Li F, Balasenthil S, Marcus S, Goodson HV, Sahin AA, Kumar R. p21-activated kinase 1 regulates microtubule dynamics by phosphorylating tubulin cofactor B. Mol Cell Biol. 2005; 25: 3726-36. https://doi.org/10.1128/MCB.25.9.3726-3736.2005.
85. Davis AJ, Chen DJ. DNA double strand break repair via non-homologous end-joining. Transl Cancer Res. 2013; 2: 130-43. https://doi.org/10.3978/j.issn.2218-676X.2013.04.02.
86. Tseng R-C, Hsieh F-J, Shih C-M, Hsu H-S, Chen C-Y, Wang Y-C. Lung cancer susceptibility and prognosis associated with polymorphisms in the nonhomologous end-joining pathway genes: a multiple genotype-phenotype study. Cancer. 2009; 115: 2939-48. https://doi.org/10.1002/cncr.24327.
87. Dombernowsky SL, Weischer M, Freiberg JJ, Bojesen SE, Tybjaerg-Hansen A, Nordestgaard BG. Missense polymorphisms in BRCA1 and BRCA2 and risk of breast and ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2009; 18: 2339-42. https://doi.org/10.1158/1055-9965.EPI-09-0447.
88. Pucci S, Mazzarelli P, Rabitti C, Giai M, Gallucci M, Flammia G, Alcini A, Altomare V, Fazio VM. Tumor specific modulation of KU70/80 DNA binding activity in breast and bladder human tumor biopsies. Oncogene. 2001; 20: 739-47. https://doi.org/10.1038/sj.onc.1204148.
89. Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997; 88: 265-75.
90. Alshareeda AT, Negm OH, Albarakati N, Green AR, Nolan C, Sultana R, Madhusudan S, Benhasouna A, Tighe P, Ellis IO, Rakha EA. Clinicopathological significance of KU70/KU80, a key DNA damage repair protein in breast cancer. Breast Cancer Res Treat. 2013; 139: 301-10. https://doi.org/10.1007/s10549-013-2542-x.
91. Jia J, Ren J, Yan D, Xiao L, Sun R. Association between the XRCC6 polymorphisms and cancer risks: a systematic review and meta-analysis. Medicine (Baltimore). 2015; 94: e283-3. https://doi.org/10.1097/MD.0000000000000283.
92. Rajaei M, Saadat I, Omidvari S, Saadat M. Association between polymorphisms at promoters of XRCC5 and XRCC6 genes and risk of breast cancer. Med Oncol. 2014; 31: 885-5. https://doi.org/10.1007/s12032-014-0885-8.
93. Henríquez-Hernández LA, Valenciano A, Foro-Arnalot P, Álvarez-Cubero MJ, Cozar JM, Suárez-Novo JF, Castells-Esteve M, Fernández-Gonzalo P, De-Paula-Carranza B, Ferrer M, Guedea F, Sancho-Pardo G, Craven-Bartle J, et al. Association between single-nucleotide polymorphisms in DNA double-strand break repair genes and prostate cancer aggressiveness in the Spanish population. Prostate Cancer Prostatic Dis. 2016; 19: 28-34. https://doi.org/10.1038/pcan.2015.63.
94. Dimberg J, Skarstedt M, Slind Olsen R, Andersson RE, Matussek A. Gene polymorphism in DNA repair genes XRCC1 and XRCC6 and association with colorectal cancer in Swedish patients. APMIS. 2016; 124: 736-40.. https://doi.org/10.1111/apm.12563.
95. Hsu CM, Yang MD, Chang WS, Jeng LB, Lee MH, Lu MC, Chang SC, Tsai CW, Tsai Y, Tsai FJ, Bau DT. The contribution of XRCC6/Ku70 to hepatocellular carcinoma in Taiwan. Anticancer Res. 2013; 33: 529-35.
96. Zhu B, Cheng D, Li S, Zhou S, Yang Q. High expression of XRCC6 promotes human osteosarcoma cell proliferation through the β-catenin/Wnt signaling pathway and is associated with poor prognosis. Int J Mol Sci. 2016; 1: 1188. https://doi.org/10.3390/ijms17071188.
97. Yang H, Sasaki T, Minoshima S, Shimizu N. Identification of three novel proteins (SGSM1, 2, 3) which modulate small G protein (RAP and RAB)-mediated signaling pathway. Genomics. 2007; 90: 249-60. https://doi.org/10.1016/j.ygeno.2007.03.013.
98. Nourashrafeddin S, Aarabi M, Modarressi MH, Rahmati M, Nouri M. The evaluation of WBP2NL-related genes expression in breast cancer. Pathol Oncol Res. 2015; 21: 293-300. https://doi.org/10.1007/s12253-014-9820-8.
99. Lindstrom S, Thompson DJ, `Paterson AD, Li J, Gierach GL, Scott C, Stone J, Douglas JA, Santos Silva Dos I, Fernandez-Navarro P, Verghase J, Smith P, Brown J, et al. Genome-wide association study identifies multiple loci associated with both mammographic density and breast cancer risk. Nat Commun. 2014; 5: 5303-3. https://doi.org/10.1038/ncomms6303.
100. Vachon CM, van Gils CH, Sellers TA, Ghosh K, Pruthi S, Brandt KR, Pankratz VS. Mammographic density, breast cancer risk and risk prediction. Breast Cancer Res. 2007; 9: 217-7. https://doi.org/10.1186/bcr1829.
101. Pettersson A, Graff RE, Ursin G, Santos Silva ID, McCormack V, Baglietto L, Vachon C, Bakker MF, Giles GG, Chia KS, Czene K, Eriksson L, Hall P, et al. Mammographic density phenotypes and risk of breast cancer: a meta-analysis. J Natl Cancer Inst. 2014; 106. https://doi.org/10.1093/jnci/dju078.
102. Pettersson A, Hankinson SE, Willett WC, Lagiou P, Trichopoulos D, Tamimi RM. Nondense mammographic area and risk of breast cancer. Breast Cancer Res. 2011; 13: R100. https://doi.org/10.1186/bcr3041.
103. Wang C, Zhao H, Zhao X, Wan J, Wang D, Bi W, Jiang X, Gao Y. Association between an insertion/deletion polymorphism within 3’UTR of SGSM3 and risk of hepatocellular carcinoma. Tumour Biol. 2014; 35: 295-301. https://doi.org/10.1007/s13277-013-1039-x.
104. Chen SC, Kung ML, Hu TH, Chen HY, Wu JC, Kuo HM, Tsai HE, Lin YW, Wen ZH, Liu JK, Yeh MH, Tai MH. Hepatoma-derived growth factor regulates breast cancer cell invasion by modulating epithelial--mesenchymal transition. J Pathol. 2012; 228: 158-69. https://doi.org/10.1002/path.3988.
105. Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW. Epithelial--mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J Cell Physiol. 2007; 213: 374-83. https://doi.org/10.1002/jcp.21223.
106. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004; 117: 927-39. https://doi.org/10.1016/j.cell.2004.06.006.
107. Chang KC, Tai MH, Lin JW, Wang CC, Huang CC, Hung CH, Chen CH, Lu SN, Lee CM, Changchien CS, Hu TH. Hepatoma-derived growth factor is a novel prognostic factor for gastrointestinal stromal tumors. Int J Cancer. 2007; 121: 1059-65. https://doi.org/10.1002/ijc.22803.
108. Hu TH, Huang CC, Liu LF, Lin PR, Liu SY, Chang HW, Changchien CS, Lee CM, Chuang JH, Tai MH. Expression of hepatoma-derived growth factor in hepatocellular carcinoma. Cancer. 2003; 98: 1444-56. https://doi.org/10.1002/cncr.11653.
109. Ren H, Tang X, Lee JJ, Feng L, Everett AD, Hong WK, Khuri FR, Mao L. Expression of hepatoma-derived growth factor is a strong prognostic predictor for patients with early-stage non-small-cell lung cancer. J Clin Oncol. 2004; 22: 3230-7. https://doi.org/10.1200/JCO.2004.02.080.
110. Hu TH, Lin JW, Chen HH, Liu LF, Chuah SK, Tai MH. The expression and prognostic role of hepatoma-derived growth factor in colorectal stromal tumors. Dis Colon Rectum. 2009; 52: 319-26. https://doi.org/10.1007/DCR.0b013e31819d1666.
111. Wu D, Niu X, Pan H, Zhang Z, Zhou Y, Qu P, Zhou J. MicroRNA-497 targets hepatoma-derived growth factor and suppresses human prostate cancer cell motility. Mol Med Rep. 2016; 13: 2287-92. https://doi.org/10.3892/mmr.2016.4756.
112. Yamamoto S, Tomita Y, Hoshida Y, Takiguchi S, Fujiwara Y, Yasuda T, Doki Y, Yoshida K, Aozasa K, Nakamura H, Monden M. Expression of hepatoma-derived growth factor is correlated with lymph node metastasis and prognosis of gastric carcinoma. Clin Cancer Res. 2006; 12: 117-22. https://doi.org/10.1158/1078-0432.CCR-05-1347.
113. Yuen BT, Knoepfler PS. Histone H3.3 mutations: a variant path to cancer. Cancer Cell. 2013; 24: 567-74. https://doi.org/10.1016/j.ccr.2013.09.015.
114. Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, Nik-Zainal S, Martin S, McLaren S, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013; 45: 1479-82. https://doi.org/10.1038/ng.2814.
115. Ayoubi HA, Mahjoubi F, Mirzaei R. Investigation of the human H3.3B (H3F3B) gene expression as a novel marker in patients with colorectal cancer. J Gastrointest Oncol. 2017; 8: 64-9. https://doi.org/10.21037/jgo.2016.12.12.
116. Shadeo A, Lam WL. Comprehensive copy number profiles of breast cancer cell model genomes. Breast Cancer Res. 2006; 8: R9. https://doi.org/10.1186/bcr1370.
117. Ganss B, Jheon A. Zinc finger transcription factors in skeletal development. Crit Rev Oral Biol Med. 2004; 15: 282-97. https://doi.org/10.1177/154411130401500504.
118. Divisato G, Formicola D, Esposito T, Merlotti D, Pazzaglia L, Del Fattore A, Siris E, Orcel P, Brown JP, Nuti R, Strazzullo P, Benassi MS, Cancela ML, et al. ZNF687 mutations in severe paget disease of bone associated with giant cell tumor. Am J Pathol. 2016; 98: 275-86. https://doi.org/10.1016/j.ajhg.2015.12.016.
119. Katoh M. FGFR2 abnormalities underlie a spectrum of bone, skin, and cancer pathologies. J Invest Dermatol. 2009; 129: 1861-7. https://doi.org/10.1038/jid.2009.97.
120. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010; 10: 116-29. https://doi.org/10.1038/nrc2780.
121. Kelleher FC, O’Sullivan H, Smyth E, McDermott R, Viterbo A. Fibroblast growth factor receptors, developmental corruption and malignant disease. Carcinogenesis. 2013; 34: 2198-205. https://doi.org/10.1093/carcin/bgt254.
122. Helsten T, Schwaederle M, Kurzrock R. Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: biologic and clinical implications. Cancer Metastasis Rev. 2015; 34: 479-96. https://doi.org/10.1007/s10555-015-9579-8.
123. Suvannasankha A, Tompkins DR, Edwards DF, Petyaykina KV, Crean CD, Fournier PG, Parker JM, Sandusky GE, Ichikawa S, Imel EA, Chirgwin JM. FGF23 is elevated in multiple myeloma and increases heparanase expression by tumor cells. Oncotarget. 2015; 6: 19647-60. https://doi.org/10.18632/oncotarget.3794.
124. Degirolamo C, Sabbà C, Moschetta A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat Rev Drug Discov. 2016; 15: 51-69. https://doi.org/10.1038/nrd.2015.9.
125. Kazemi S, Mounir Z, Baltzis D, Raven JF, Wang S, Krishnamoorthy JL, Pluquet O, Pelletier J, Koromilas AE. A novel function of eIF2alpha kinases as inducers of the phosphoinositide-3 kinase signaling pathway. Mol Biol Cell. 2007; 18: 3635-44. https://doi.org/10.1091/mbc.E07-01-0053.
126. Gardino AK, Yaffe MB. 14-3-3 proteins as signaling integration points for cell cycle control and apoptosis. Semin Cell Dev Biol. 2011; 22: 688-95. https://doi.org/10.1016/j.semcdb.2011.09.008.
127. Paplomata E, O’Regan R. The PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkers. Ther Adv Med Oncol. 2014; 6: 154-66. https://doi.org/10.1177/1758834014530023.
128. Durocher F, Labrie Y, Soucy P, Sinilnikova O, Labuda D, Bessette P, Chiquette J, Laframboise R, Lépine J, Lespérance B, Ouellette G, Pichette R, Plante M, et al. Mutation analysis and characterization of ATR sequence variants in breast cancer cases from high-risk French Canadian breast/ovarian cancer families. BMC Cancer. 2006; 6: 230. https://doi.org/10.1186/1471-2407-6-230.
129. Guénard F, Pedneault CS, Ouellette G, Labrie Y, Simard J, Durocher F. Evaluation of the contribution of the three breast cancer susceptibility genes CHEK2, STK11, and PALB2 in non-BRCA1/2 French Canadian families with high risk of breast cancer. Genet Test Mol Biomarkers. 2010; 14: 515-26. https://doi.org/10.1089/gtmb.2010.0027.
130. Litim N, Labrie Y, Desjardins S, Ouellette G, Plourde K, Belleau P, Durocher F. Polymorphic variations in the FANCA gene in high-risk non-BRCA1/2 breast cancer individuals from the French Canadian population. Mol Oncol. 2013; 7: 85-100. https://doi.org/10.1016/j.molonc.2012.08.002.
131. Desjardins S, Belleau P, Labrie Y, Ouellette G, Bessette P, Chiquette J, Laframboise R, Lépine J, Lespérance B, Pichette R, Plante M, Durocher F. Genetic variants and haplotype analyses of the ZBRK1/ZNF350 gene in high-risk non BRCA1/2 French Canadian breast and ovarian cancer families. Int J Cancer. 2008; 122: 108-16. https://doi.org/10.1002/ijc.23058.
132. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Endocrinol Metabol Syndrome. 2009; 25: 1105-11. https://doi.org/10.1093/bioinformatics/btp120.
133. Yuan Y, Tian L, Lu D, Xu S. Analysis of genome-wide RNA-sequencing data suggests age of the CEPH/Utah (CEU) lymphoblastoid cell lines systematically biases gene expression profiles. Sci Rep. 2015; 5: 7960. https://doi.org/10.1038/srep07960.
134. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011; 43: 491-8. https://doi.org/10.1038/ng.806.
135. Bray N, Pimentel H, Melsted P, Pachter L. Near-optimal RNA-Seq quantification. arXiv.org. 2015.
136. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26: 139-40. https://doi.org/10.1093/bioinformatics/btp616.
137. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010; 11: R106. https://doi.org/10.1186/gb-2010-11-10-r106.
138. Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010; 11: R14. https://doi.org/10.1186/gb-2010-11-2-r14.
139. Fox J, Weisberg S. An R Companion to Applied Regression. SAGE; 2010. 1 p.
140. R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria; 2016. Available from https://www.r-project.org/.
141. De Mendiburu F. Agricolae: Statistical procedures for agricultural research. R package version 12-3. 2015. Available from https://cran.r-project.org/web/packages/agricolae/index.html.