
INTRODUCTION
FBXW7 is a tumor suppressor gene that is frequently inactivated in different types of cancer, including breast cancer, colon cancer and leukemia [1]. FBXW7 protein is a member of the F-box family of proteins, components of Skp1, Cul1, and F-box protein (SCF) ubiquitin ligase complexes. F-box proteins are responsible for recruiting specific substrates for ubiquitination and degradation [2]. FBXW7 targets several oncoproteins for proteolysis, such as cyclin E, c-Jun, c-Myc, Mcl-1 or Notch [3]. Mammalian cells contain three FBXW7 isoforms, FBXW7α, FBXW7β and FBXW7γ, that are produced by alternative splicing and localize to the nucleoplasm, cytoplasm and nucleolus, respectively [4, 5]. FBXW7α is the most highly expressed and stable FBXW7 isoform and expression levels of this protein do not vary significantly during the cell cycle [4, 6]. The FBXW7α transcript is ubiquitously expressed in all human tissues and is also induced by the p53 tumor suppressor in response to DNA damage [7, 8].
The FBXW7α protein contains several protein-protein interaction domains, including a dimerization domain, an F-box domain that recruits the SCF core complex, and eight WD40 repeats that form a β-propeller binding pocket [9-11]. Notably, it has been shown that WD40 β-propellers function as ubiquitin-binding domains and that ubiquitin interaction by FBXW7 promotes its auto-ubiquitination and turnover [12]. However, the importance of FBXW7α dimerization is still not entirely clear, but it has been proposed to increase the ubiquitination efficiency of low affinity substrates [11]. More recently, it has been reported that Pin1, a prolyl isomerase, interacts with FBXW7α in a phosphorylation-dependent manner and promotes FBXW7α auto-ubiquitination and protein degradation by disrupting FBXW7α dimerization, suggesting that inhibition of Pin1 could upregulate the expression of FBXW7α to retard the growth of human tumor cells [13].
FBXW7 binds to substrates via its WD40 domain located in the carboxy-terminus of the protein, which interacts with a phosphothreonine-containing motif, known as CPD (Cdc4 phosphodegron), in the substrates [14, 15]. SCFFBXW7 activity is regulated by various factors, among which are an active neddylation system [16], Pin1 and/or PP2A [17], and the deubiquitinating enzyme USP28 [18]. Interestingly, USP28 dissociates from FBXW7α in response to UV irradiation, providing a mechanism for how FBXW7α-mediated degradation of c-Myc is enhanced upon DNA damage [19]. Finally, FBXW7α-dependent substrate ubiquitination is also dependent on upstream signaling pathways, including the PI3K/Akt/GSK3β pathway [20], the ATM/ATR pathway upon induction of DNA damage [21], and the Ras signaling pathway [22].
Polo-like kinase 1 (PLK1) is a highly conserved serine/threonine kinase that plays a key role in eukaryotic cell division [23]. Expression of PLK1 increases in S phase and peaks during mitosis. PLK1 mediates many mitotic events, including entry into mitosis, centrosome maturation, assembly of the bipolar spindle, sister chromatid splitting, activation of the Anaphase-Promoting Complex/Cyclosome (APC/C), and exit from mitosis with the initiation of cytokinesis [24]. In addition, PLK1 has a plethora of roles being implicated in microtubule dynamics, DNA replication, chromosome dynamics, p53 regulation and recovery from the G2 DNA damage checkpoint [25]. Furthermore, PLK1 is degraded by the APC/CCDH1 from late anaphase, for the proper control of mitotic exit and cytokinesis, to the entry of cells into the G1 phase [26], and also after DNA-damage in G2 [27].
The transfer of genetic information with high fidelity from parent to daughter cells is one of the most important tasks of the cell cycle. Besides mitosis, where the replicated chromosomes are segregated, DNA replication during S phase is an essential stage for the maintenance of genome integrity. For the initiation of DNA replication, a series of proteins are assembled on each replication origin. Origin recognition complex (ORC) 1-6 subunits, which bind to the replication origins, and mini-chromosome maintenance (MCM) complex 2-7 subunits, which are loaded onto the origins, depending on Cdt1 and Cdc6, are involved in the formation of the pre-replicative complexes (pre-RCs). The MCM complex is the DNA helicase that plays a central role in the progression of replication forks. Later, other proteins are loaded onto pre-RCs to form pre-initiation complexes (pre-ICs), and two classes of kinases, DDK (Dbf4-dependent kinase) and CDK (cyclin-dependent kinase), play important roles in the assembly of pre-ICs and ensure that only a single round of DNA replication takes place in each cell cycle. Under unperturbed conditions, replication starts from a subset of these origins. Two replication forks move in opposite directions from each origin behind the CMG complex, formed by Cdc45, MCM and GINS, which unwinds the DNA, providing single-stranded DNA templates for the polymerases to duplicate. When progression of DNA replication forks is hindered, activation of checkpoint pathways temporarily halts the cell cycle progression, giving the cells time to solve replication problems before entering into mitosis (for review, see [28]).
Several reports associate PLK1 with DNA replication. In this regard, PLK1 depletion in the long term slows proliferation apparently due to attenuated progression through the S phase [29]. In addition, PLK1 interacts with MCM2, MCM3, MCM7 and ORC2, and phosphorylates Hbo1, regulating pre-RC loading of MCM2 and MCM6 [30-32]. Moreover, PLK has also been implicated in replication under stressful conditions. It has been shown that the Xenopus PLK homolog, Plx1, phosphorylates Claspin, one of the proteins that mediates the interaction between the MCM helicase and DNA polymerases, causing its dissociation from chromatin and resulting in the inactivation of the replication checkpoint kinase Chk1 after a prolonged checkpoint arrest [33]. The PLK homolog in budding yeast, Cdc5, is also required for the adaptation, that is the resumption of the cell cycle in the presence of a single unrepaired DSB after a prolonged arrest [34]. Furthermore, a novel role has been suggested for PLK1 in maintenance of genomic integrity by promoting DNA replication under conditions of stress [35]. Together, these findings indicate that PLK has an inhibitory effect on the checkpoint response.
In this study, we identify PLK1 as a novel SCFFBXW7α substrate and show how it is degraded in G1- and S-phase-arrested cells, and in response to DNA damage. Additionally, we demonstrate that PLK1 degradation impedes the formation of pre-RCs and, following DNA damage, avoids cell proliferation.
RESULTS
Identification of FBXW7α-interacting proteins
To identify new SCFFBXW7α substrates, we first searched for FBXW7α binding proteins by FBXW7α immunoprecipitation and tandem mass spectrometry assays (MS/MS). We transfected several cell lines with Flag-FBXW7α and confirmed by Western blot and immunofluorescence experiments its nuclear localization, as described for endogenous FBXW7α (supplementary Figs S1A and S1B) [6]. Duplicate MS/MS analysis of Flag-FBXW7α immunoprecipitation from Cos-7 nuclear extracts resulted in the identification of PLK1 as a novel FBXW7α-interacting protein. To further validate the authenticity of these results, we confirmed the presence of PLK1 within the FBXW7α immunocomplex using Western blot analysis (Fig 1A). In addition, we performed reciprocal immunoprecipitations using HCT116 transfected cells and monoclonal antibodies to PLK1. Immunoprecipitated proteins were resolved using SDS-PAGE and the band correlating to PLK1 was identified by Western blot (Fig 1B). Consistent with the results from the interaction assays, we localized by immunofluorescence both endogenous FBXW7α and PLK1 in the nuclei of U2OS cells (Fig 1C). Taken together, our findings show that FBXW7α and PLK1 associate in intact cells.
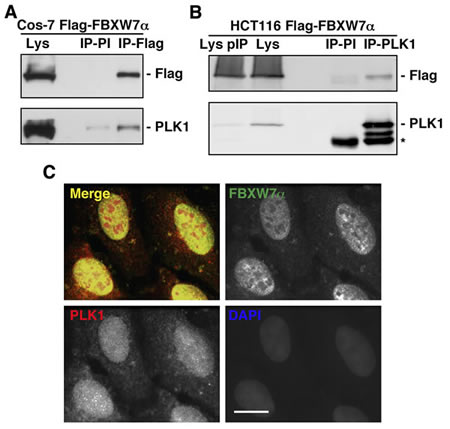
Figure 1: FBXW7α and PLK1 interact in the nuclei of mammalian cells. (A) Cos-7 cells were transiently transfected with pCDNA3.1-Flag-FBXW7α and nuclear extracts immunoprecipitated with anti-Flag monoclonal antibody or normal mouse serum (PI). Immunoprecipitates materials were analyzed by Western blotting. Lys: nuclear extracts from Cos-7 transfected cells. (B) Whole cell extracts from HCT116 transfected cells were used to immunoprecipitate PLK1, and complexes were analyzed by immunoblotting. IP-PI: immunoprecipitation with normal mouse serum. Lys and Lys pIP: whole cell extracts from HCT116 transfected cells before (Lys) and after (Lys pIP) PLK1 immunoprecipitation. Asterisk indicates IgG heavy chains. (C) U2OS cells were stained for FBXW7α, PLK1 and DNA. In the merge, FBXW7α staining is shown in green, PLK1 in red, and DAPI in blue. Bar, 10µm.
SCFFBXW7α targets PLK1 for ubiquitination
To investigate whether SCFFBXW7α promotes PLK1 ubiquitination, we performed both in vitro and in vivo ubiquitination assays. Figure 2A shows an increment of the in vitro-transcribed/translated PLK1 ubiquitination in the presence of FBXW7α. Other F-box proteins, βTrCP or SKP2, or FBXW7α∆F, a dominant-negative variant of FBXW7α lacking the F-box domain [36] which couples F-box proteins to the SCF complexes, were unable to trigger a significant ubiquitination of PLK1. Similar results were obtained when we used a recombinant SCFFBXW7α complex expressed in Sf21 insect cells (Fig 2B). In agreement with our findings in vitro, the in vivo results showed that FBXW7α promotes the ubiquitination of PLK1 in HCT116 transfected cells. As shown in Figure 2C, Flag-PLK1 immunoprecipitations presented an increment in the levels of poly-ubiquitinated forms when cells were co-transfected with FBXW7α and Myc-Ubiquitin, but not with FBXW7α∆F or an ubiquitin mutant that blocks the formation of the poly-ubiquitin chains (Myc-Ub (K48R)). These results show that SCFFBXW7α targets PLK1 for ubiquitination.
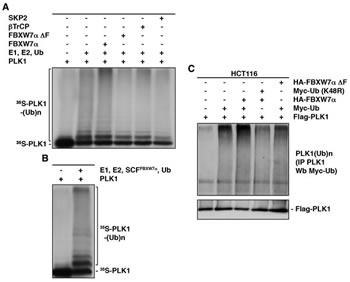
Figure 2: SCFFBXW7α ubiquitinates PLK1. (A) In vitro ubiquitin ligation assay of 35S labeled in vitro-transcribed/translated PLK1 was conducted in the presence or absence of the following products: cold in vitro-transcribed/translated SKP2, βTrCP, FBXW7α∆F or FBXW7α, E1 (His6-ubiquitin activating enzyme), E2 (His6-UbcH3 and UbcH5a) and Ub (ubiquitin). Samples were incubated at 30°C for 1h. The bracket on the left side marks a ladder of bands corresponding to poly-ubiquitinated PLK1. (B) The experiment was performed as in (A) except that the unlabeled F-box protein was substituted by a recombinant SCFFBXW7α complex expressed in Sf21 insect cells. (C) HCT116 cells were transfected with plasmids encoding the indicated proteins, and treated with LLnL for 4h before harvesting. Extracts were prepared as indicated in the Materials and Methods and poly-ubiquitinated PLK1 visualized after Western blots of the PLK1 immunoprecipitations.
SCFFBXW7α promotes proteasomal turnover of PLK1 in the G1 and S phases of the cell cycle
To examine whether SCFFBXW7α was responsible for PLK1 degradation, we first overexpressed FBXW7α in HeLa cells and found that increasing amounts of FBXW7α correlated with decreasing amounts of endogenous PLK1 (Fig 3A). We then down-regulated FBXW7α expression using an established siRNA [37] to test whether FBXW7α depletion stabilizes PLK1. Knock-down of FBXW7α was confirmed by expressing exogenous protein (supplementary Fig S2A), due to the inefficient detection of endogenous FBXW7α using commercial antibodies. Cytosolic (S100 fraction) and nuclear extracts interfered with siRNA-FBXW7α were analyzed for the presence of PLK1 in U2OS cells, and an important increase of PLK1 was observed in nuclear extracts (Fig 3B). Consistent with previous reports, cyclin E, a known target of FBXW7α [38, 39], was also stabilized by depletion of FBXW7α, but not Mre11, Nbs1 or α-Tubulin that acted as controls. To avoid possible indirect effects on transcriptional regulation of PLK1, we analyzed the effect of the overexpression of FBXW7α on ectopically expressed PLK1 using a constitutive heterologous promoter. As compared with empty vector, ectopic FBXW7α also reduced the levels of PLK1, including after lambda phosphatase treatment, to discount the possibility that PLK1 was phosphorylated (Fig 3C).
To demonstrate that the loss of PLK1 in the presence of FBXW7α was due to proteolysis, we carried out PLK1 half-life determinations after inhibition of protein synthesis by treatment with cycloheximide. HeLa cells interfered with siRNA-FBXW7α showed a longer PLK1 half-life than when cells were interfered with siRNA-EGFP (Figs 3D and 3E). As an internal control we analyzed the cyclin E half-life, obtaining similar results. In the same way, when we performed the reverse experiment using exogenous PLK1 and FBXW7α, we reduced the PLK1 half-life (supplementary Figs S2B and S2C). The FBXW7α-mediated degradation of PLK1 was alleviated by the inclusion of a proteasome inhibitor in the experiment, strongly suggesting that the ubiquitinated PLK1 is degraded by the 26S proteasome (Fig 3F).
As protein degradation constitutes the major post-translational control of cell cycle regulators, we examined whether PLK1 degradation occurs in a cell cycle phase-specific manner. HeLa cells were interfered with siRNA-FBXW7α and arrested in the different phases of the cell cycle (supplementary Fig S2D), and we found that the amount of PLK1 augmented both in G1 and S, but not in G2 or M phases (Fig 3G). Complementary results were found when we overexpressed FBXW7α, namely, FBXW7α caused the PLK1 diminution in both butyrate (G1 phase) and hydroxyurea (S phase) treated cells (supplementary Fig S2E). Taken together, our results clearly show that SCFFBXW7α promotes proteasomal degradation of PLK1 in the G1 and S phases of a normal cell cycle.

Figure 3: SCFFBXW7α mediates PLK1 proteasomal degradation in the G1 and S phases. (A) HeLa cells were transfected with increasing amount of pCMVHA-FBXW7α and, 18h later, cytosolic (S100) and nuclear extracts (NE) were subjected to Western blot. (B) U2OS cells interfered with siRNA-FBXW7α or siRNA-EGFP as a control, were used to prepare cytosolic (S100) and nuclear extracts (NE), and fractions were analyzed for the presence of different proteins as indicated. (C) Whole cell extracts from U2OS cells transfected with plasmids encoding the indicated proteins were treated or not with lambda phosphatase (λ-PP), migrated, electroblotted and probed with different antibodies. (D) HeLa cells were interfered with EGFP- or FBXW7α-siRNA and, after 48h, cycloheximide (CHX) was added to the medium and cells were collected at the indicated times. Extracts were analyzed by Western blot. (E) Quantification of PLK1 and cyclin E protein levels presented in (D) using the ImageJ software. Error bars represent the S.D. (n=3). (F) U2OS cells were transiently transfected with plasmids encoding the indicated proteins and, treated or not with LLnL for 4h before harvesting. Lysates were analyzed by Western blotting. (G) HeLa cells were interfered with the indicated siRNA and arrested in the different phases of the cell cycle. Extracts were blotted with different antibodies. Grb2 expression was used as a loading control. The quantitative fold change in PLK1 was determined relative to the loading control.
SCFFBXW7α regulates PLK1 levels in response to UV irradiation
It has been published that PLK1 is degraded by the APC/CCDH1/proteasome in response to DNA damage in G2, avoiding entry into mitosis and instead initiating DNA repair [27]. On the other hand, it is also known that PLK1 has an important role during S phase [40, 41]. In this study, we have demonstrated that SCFFBXW7α mediates PLK1 proteolytic degradation in S. However, little is known about the potential effects of DNA damage on the role of PLK1 in S phase. Therefore, we decided to investigate whether PLK1 might be degraded after DNA damage in S phase and whether SCFFBXW7α would be involved in this degradation. To this end, we analyzed the PLK1 protein level in several cell lines synchronized in S phase (by double thymidine block followed by a 4h release, or by treatment with hydroxyurea or aphidicolin), before and after UV irradiation. In Figure 4A (and supplementary Figs S3A-C) a significant decrease of PLK1 was observed following UV treatment, and this decrease could be reversed using a proteasome inhibitor. In addition, FBXW7α∆F was able to avoid PLK1 degradation after UV treatment (Fig 4B), indicating that SCFFBXW7α is also responsible for the regulation of PLK1 levels in response to DNA damage. In fact, silencing of FBXW7α reduced the PLK1 turnover after UV irradiation in S phase compared with mock treated cells (Fig 4C).
The ATR and ATM protein kinases are recruited to defective replication forks or to sites of DNA damage, and their homologs are thought to initiate the DNA damage response in all eukaryotes [42]. To determine whether these kinases are involved in PLK1 degradation in S phase after UV irradiation, we carried out down-regulation experiments in this phase and detected PLK1 levels by Western blot before and after UV treatment. As shown in Figure 4D, both ATM- and ATR-depleted cells prevented the degradation of PLK1 after UV irradiation in a similar way to the knock-down of FBXW7α (Fig 4C). Comparable results were obtained after treatment with caffeine, an inhibitor of ATM and ATR kinases, or UCN-01, which inhibits the ATR-Chk1 pathway after UV treatment (supplementary Fig S3D). Overall, these results show that SCFFBXW7α mediates PLK1 degradation induced by UV irradiation and suggest the involvement of ATM/ATR in this response.
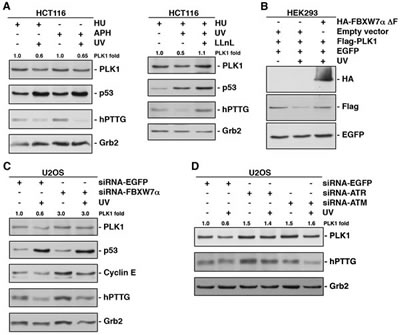
Figure 4: UV irradiation accelerates PLK1 degradation in S phase of the cell cycle via SCFFBXW7α/proteasome. (A) HCT116 cells arrested in S phase using hydroxyurea (HU) or aphidicolin (APH) were irradiated (30J/m2) or not, and extracts analyzed by Western blot. In the right panel, cells were treated with LLnL 30 min before irradiation. p53 and hPTTG were used as controls whose degradation is avoided or induced by UV. Grb2 expression was used as a loading control. (B) HEK293 cells were transfected and subjected or not to UV irradiation before harvesting. Extracts were analyzed by Western blot. (C) U2OS cells were interfered with EGFP- or FBXW7α-siRNA, arrested in S phase with HU and irradiated where indicated. Extracts were analyzed by Western blot. Cyclin E and hPTTG were used as a positive and a negative control, respectively, of FBXW7 depletion. (D) U2OS cells interfered with the indicated siRNA and arrested in S phase (HU) were irradiated or not 4h before harvesting, and proteins immunoblotted with different antibodies. The quantitative fold change in PLK1 was determined relative to the loading control.
Identification of a Cdc4 phosphodegron in PLK1
Several reports have described that the CPD recognized by FBXW7α contains the motif L/I-L/I/P-pT-P-X-X-X-X (where X refers to any residue other than K or R) [14, 36, 43]. However, other authors have established a slightly different motif, (L)-X-pT/pS-P-(P)-X-pS/pT/E/D (where X is any amino acid) (revised in [15]). With these data in mind, we analyzed the PLK1 protein sequence and found a single non-canonical CPD motif between residues 212 and 219, C-G-T-P-N-Y-I-A. This CPD-like sequence is almost completely conserved in other human PLK family members, as well as, in PLK1 orthologs from other species (Fig 5A). To ascertain whether this sequence is a functional CPD, we mutated threonine 214 to glycine. We proved that this mutant was unable to be ubiquitinated by FBXW7α in vitro (Fig 5B) and degraded in transfected cells (Fig 5C). Furthermore, when we overexpressed FBXW7α the half-life of PLK1-T214G was longer than the half-life of wild-type (Figs 5D and 5E), indicating that threonine 214 is involved in the regulation of PLK1 stability. Given that threonine 214 is found within the PLK1 kinase domain, we performed an in vitro kinase assay using dephosphorylated α-casein as a substrate. This assay confirmed that the PLK1-T214G mutant still retained its kinase activity (Fig 5F), suggesting that the overall structure of this mutant protein remains largely intact. Finally, we analyzed the effect of UV irradiation on the degradation of the PLK1-T214G mutant. We found that point mutation of threonine 214 clearly prevented the PLK1 degradation induced by UV, while other point mutant (PLK1-KD) was degraded (Fig 5G). Therefore, our findings show that PLK1 contains a CPD motif that promotes PLK1 degradation following UV irradiation and that this motif is highly conserved from yeast to humans.
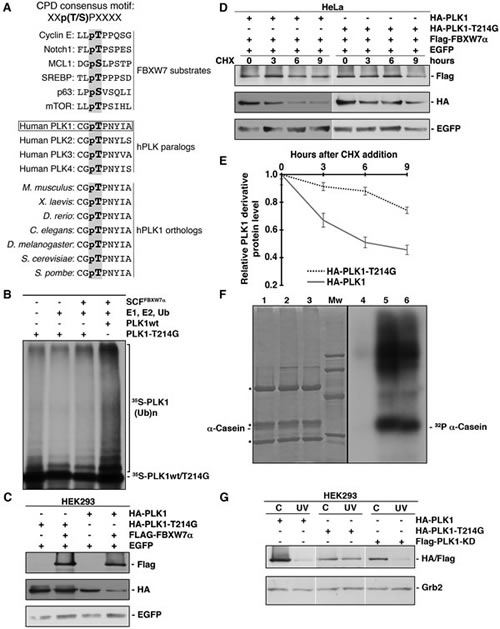
Figure 5: PLK1 possesses a functional CPD motif. (A) Alignment of some CPDs from different FBXW7 substrates and comparison of the CPD-like motif identified in human PLK1 (residues 212 to 219) with other human PLKs and PLK1 orthologs. X refers to any residue other than K or R. pT in bold in human PLK1 (residue 214) is the putative phosphothreonine in CPDs. (B) In vitro ubiquitin ligation assay of 35S labeled in vitro-transcribed/translated PLK1-T214G was conducted in the presence or absence of the following products: recombinant SCFFBXW7α complex expressed in Sf21 insect cells, E1 (His6-ubiquitin activating enzyme), E2 (His6-UbcH3 and UbcH5a) and Ub (ubiquitin). 35S-PLK1wt was used as a positive control. Samples were incubated at 30°C for 1h. The bracket on the right side marks a ladder of bands corresponding to poly-ubiquitinated PLK1. (C) HEK293 cells were transiently transfected with plasmids encoding the indicated proteins, and extracts analyzed by Western blot. (D) HeLa cells were transfected with plasmids encoding the indicated proteins and, after 48h, CHX was added to the medium and cells were collected at the indicated times. Whole cell extracts were analyzed by Western blot. (E) Protein levels from (D) were quantified using the ImageJ software. Error bars represent the S.D. (n=3). (F) Anti-HA immunoprecipitates from lysates of HCT116 cells transfected with pCMVHA (lanes 1 and 4), pCMVHA-PLK1 (lanes 2 and 5) and pCMVHA-PLK1-T214G (lanes 3 and 6) were incubated with α-casein dephosphorylated, (γ-32P) ATP and a kinase buffer as described in Materials and Methods. Reactions were analyzed by SDS-PAGE, stained with Coomassie Blue (lanes 1-3), and autoradiographed (lanes 4-6). Mw: molecular mass markers (95, 66, 45 and 31 kDa). Asterisks indicate IgG heavy and light chains. (G) PLK1 (and derivatives) transfected cells were irradiated (30J/m2) or not and lysates subjected to Western blotting. Grb2 expression was used as a loading control.
PLK1 degradation avoids the cell proliferation after DNA damage in S-phase
In this study, we have shown that PLK1 is ubiquitinated by SCFFBXW7α and degraded in the G1 and S phases of the cell cycle and in response to DNA damage. To elucidate the role of this ubiquitination and degradation, we analyzed the involvement of PLK1 in the intra-S-phase checkpoint. Firstly, we tested the possibility that FBXW7α overexpression could reduce the amount of MCM proteins loaded onto chromatin, as mediated by PLK1. Figure 6A shows that the chromatin loading of MCM2 and MCM7 in FBXW7α-transfected HEK293 cells is less than that found in non-transfected control cells. Similar results were obtained from cells treated with BI2536, a potent and selective PLK1 inhibitor. Moreover, we observed an important diminution of MCM proteins in the chromatin fraction of UV irradiated HEK293 cells (data not shown). Next, we examined the distribution of MCM proteins after UV irradiation of HEK293 cells transfected with PLK1 or PLK1-T214G. Cells transfected with the non-degradable PLK1 mutant conserved more MCM proteins on chromatin than cells transfected with wild-type PLK1, indicating that the PLK1 degradation mediated by SCFFBXW7α impedes the formation of pre-RCs (Fig 6B). To determine whether PLK1-T214G altered cell proliferation following UV treatment, we transfected HeLa cells with PLK1, PLK1-T214G or empty vector, and then arrested cells in S-phase by the addition of hydroxyurea. After release, cells were irradiated or not and cell proliferation was quantified. As shown in Figure 6C (and supplementary Fig S4A), the three transfected cell lines had a similar proliferation pattern. However, after UV irradiation, the presence of PLK1-T214G mutant in HeLa cells accelerated cell proliferation (Fig 6D and supplementary Fig S4B). Similar results were obtained in U2OS transfected cells (data not shown). Therefore, we can conclude that PLK1 degradation by SCFFBXW7α avoids cell proliferation after DNA damage in the S-phase of the cell cycle.
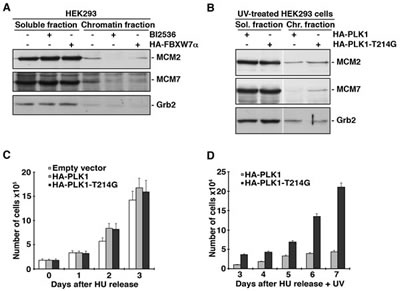
Figure 6: PLK1 degradation by SCFFBXW7α reduces pre-RC formation and cell proliferation. (A) HEK293 cells were transfected with pCMVHA-FBXW7α for 18h or treated with BI2536 for 8h, and subjected to chromatin fractionation as described in Materials and Methods. Fractions were treated with λ-PP and blotted with the indicated antibodies. (B) HEK293 cells were transfected with pCMVHA-PLK1 and pCMVHA-PLK1-T214G, irradiated and subjected to chromatin fractionation as described. Extracts were treated with λ-PP and analyzed by Western blot. (C) HeLa cells were transfected with pCMVHA (empty vector), pCMVHA-PLK1 and pCMVHA-PLK1-T214G for 18h, arrested in S phase with HU for 24h and, after releasing, trypan blue-excluding cells were counted using a hemocytometer at different time points. The analysis began with equal numbers of the differently transfected cells. Error bars represent the S.D. (n=3). (D) Transfected cells were treated as in (C) but just after HU release, cells were UV irradiated. Viable cells were counted, starting three days after release, for 5 days.
DISCUSSION
Cancer is the consequence of intra- and extracellular signaling network dysregulation that derives from the activation of oncogenes or inactivation of tumor suppressor genes. Cancer cells exhibit altered signaling pathways with adaptations that overcome cellular safeguards that prevent oncogenic transformation. Both PLK1 and FBXW7α are factors involved in tumorigenesis. PLK1 is considered a proto-oncogene, whose overexpression is often observed in tumor cells and FBXW7α is a tumor suppressor whose mutation occurs in multiple neoplasms. Overexpression of PLK1 has been identified in samples taken from patients with lung, breast, colon, pancreas, prostate and ovary tumors, and approximately 6% of all primary human tumors harbor mutations in FBXW7α, with the greatest mutation rates found in cholangio-carcinoma and T-cell acute lymphoblastic leukemia [1, 44]. The misregulated degradation of tumor suppressors or oncoproteins can also drive tumorigenesis. Accordingly, an overexpressed (or underexpressed) F-box protein can function as an oncoprotein or as a tumor suppressor depending on whether their substrates are tumor suppressors or oncoproteins, respectively. Here we show that PLK1 interacts with FBXW7α in vivo, is specifically ubiquitinated both in vitro and in vivo by SCFFBXW7α and is degraded via the proteasome. This degradation occurs in control conditions and after UV irradiation. These results led us to propose that, as for other SCFFBXW7α substrates, such c-Myc, c-Jun, cyclin E and Notch [3], FBXW7α is also acting as a tumor suppressor, avoiding excessive cell proliferation in unstressed conditions and after DNA damage via control of PLK1. Down-regulation of endogenous PLK1 in several human cell lines significantly decreases cell proliferation and migrating ability, and overexpression of PLK1 in NIH3T3 cells induces oncogenic transformation [45, 46]. Our proliferation experiments in S phase after UV irradiation using PLK1-transfected cells versus transfected cells with a non-degradable SCFFBXW7α PLK1 point mutant (PLK1-T214G) showed that PLK1 degradation is necessary for prolonging cell proliferation arrest after DNA damage.
During undisturbed DNA replication, PLK1 promotes pre-RC loading via phosphorylation of Hbo1. When DNA replication is under stress, checkpoint activation causes active replication forks to stall and PLK1 phosphorylates ORC2, promoting the maintenance of pre-RC on dormant replication origins [30-32]. Importantly, our findings show that the monitoring performed by PLK1 on DNA replication is regulated by PLK1 degradation via SCFFBXW7α. These results are supported by the fact that the PLK1-T214G mutant was still able to promote MCM loading onto chromatin after UV irradiation, and that, as mentioned above, PLK1-T214G transfected cells reduced the cell cycle arrest after DNA damage. Interestingly, the new CPD motif identified in PLK1 is phylogenetically conserved, suggesting that it has an important role in PLK activity regulation. Together, these data demonstrate that the PLK1 inhibitory effect on the intra-S-checkpoint response is determined by its degradation via SCFFBXW7α/proteasome.
During the progression of this work, it has been reported that FBXW7 governs CDH1 activity in a cyclin E-dependent manner, indicating that loss of FBXW7 increases expression of APC/CCDH1 substrates, PLK1 among them [47]. These results raise the possibility that the degradation of PLK1 via FBXW7α that we report may be indirect and that CDH1 is indeed responsible for this degradation. However, a number of arguments support that FBXW7α directly governs PLK1 levels. First, PLK1 is only an APC/CCDH1 substrate between late anaphase and G1, or in response to genotoxic stress in G2, since the APC/CCDH1 is only active during this period [26, 27]. Second, PLK1 and FBXW7α are both located in the nucleus (Fig 1C), whereas CDH1 is located in the nucleus during G1 but redistributes to the cytosol between S phase and the end of mitosis [48]. Third, APC/CCDH1 mediates proteasomal degradation of the ubiquitin-conjugating enzyme UbcH10, providing a negative feedback mechanism that inactivates APC/CCDH1 during G1/S transition [49]. Fourth, when the replication fork is stalled, APCCDH1 activation is prevented by ATR/Chk1 activity-promoted degradation of CDH1 (supplementary Fig S5 and [50]). Therefore, at least during the APC/CCDH1 inactivation period, FBXW7 cannot modulate CDH1 activity. Finally, our in vitro ubiquitination assays clearly demonstrate a direct ubiquitination of PLK1 by SCFFBXW7α. In fact, the PLK1 mutant in the FBXW7 phosphodegron was unable to be degraded by FBXW7 signaling pathway. Hence, we can conclude that PLK1 is a direct target of SCFFBXW7α for degradation via the proteasome.
Prior to our work, multiple SCFFBXW7α substrates had been identified. However, it remained largely unknown how these substrates contribute to the tumor suppression function of FBXW7α. We speculate that PLK1 may contribute to this function. For example, it is known that PLK1 is involved in checkpoint adaptation, a process originally described in Saccharomyces cerevisiae, whereby cells can override the checkpoint and resume cycling with damaged DNA if lesions are not repaired or are incompletely repaired. In human U2OS cells, adaptation was promoted by inhibiting Chk1 and delayed by depleting PLK1 [51]. The authors proposed that Chk1 and PLK1 might control the process of adaptation by independent signaling pathways. In human cells, checkpoint adaptation might potentially promote genomic instability and lead to cancer [52]. Based on our proliferation assays using the non-degradable PLK1 mutant, where transfected cells displayed accelerated proliferation following UV irradiation compared with wild-type PLK1, we could predict that human tumors lacking FBXW7α could have enhanced checkpoint adaptation, making this an interesting area for future research. Nevertheless, we cannot forget the essential role of PLK1 in checkpoint recovery by directly targeting multiple DNA damage checkpoint factors and allowing checkpoint-desactivation [53]. Perhaps, tumors lacking FBXW7α, where PLK1 is not degraded, do not block cell cycle reentry after DNA damage, a possibility that warrants further study.
METHODS
Plasmids, cloning, point mutation and sequencing
pCDNA3.1-Flag-FBXW7α, p3xFlag-myc-CMV-24-FBXW7α∆F, pCS2HA-βTrCP, pFlag-CMV2-PLK1, pFlag-CMV2-PLK1-KD, and empty vectors have been previously described [36, 54-56]. pINCY-SPK2, pEGFP-N1, and pCW7 and pCW8 were from Thermo Fisher Scientific, BD Biosciences and ATCC, respectively. pCMVHA-FBXW7α, pCMVHA-FBXW7α∆F and pCMVHA-PLK1 were obtained by cloning the corresponding PCR fragments in pCMVHA. HA-PLK1-T214G was constructed using the “QuickChange II XL Site-Directed Mutagenesis Kit” from Stratagene. Sequence of constructs and point mutation was verified on both strands with an automatic sequencer.
Cell culture, cell synchronization, transient transfections, drugs and UV irradiation
Routinely, HeLa, HCT116, Cos-7, U2OS and HEK293 cells (from ATCC) were grown in Dulbecco’s modified Eagle’s medium (Lonza) as described [57]. Cells enriched in the different phases of the cell cycle were also obtained as previously described [58] and confirmed by flow cytometry. DNA constructs were transiently transfected by electroporation or using lipid transfection reagents (Lipofectamine (Invitrogen) or Xfect (Clontech)), and 18h or 48h post-transfection, respectively, cells were harvested and lysed. For some experiments, cells were pretreated with the proteasome and calpain inhibitor Ac-LLnL-CHO (LLnL 100µM, Sigma), cycloheximide (CHX 50µg/ml, Sigma), BI2536 (100nM, Selleck Chemicals), caffeine (10mM, Sigma) or UCN-01 (1µM, supplied by the Division of Cancer Treatment and Diagnosis, National Cancer Institute) and harvested at various times. Where indicated, cells were UV irradiated with 30J/m2 and harvested 4h later [57].
Subcellular fractionation and lysis
Subcellular fractionation was carried out as described [59]. Whole cell extracts were prepared at 4°C in 420mM NaCl, 10mM Tris-HCl (pH 7.5), 1% Nonidet P-40 (NP40), 10% glycerol, 1mM PMSF (phenylmethylsulfonyl fluoride), 1µg/ml aprotinin, 1µg/ml pepstatin, 1µg/ml leupeptin and 10µg/ml chymostatin for 20min. Extracts were centrifuged at 20,000 g for 20min and supernatants frozen in liquid nitrogen and stored at -80°C. Protein concentration was determined using the Bradford assay (Bio-Rad). When necessary, extracts were treated with lambda protein phosphatase (λ-PP) [60].
Small interfering RNA (siRNA) and half-life assays
Cells were interfered with FBXW7α-, CDH1-, ATM- or ATR-siRNA [37, 61, 62] using the Oligofectamine method (Invitrogen) to suppress the expression of endogenous genes. EGFP-siRNA [61] was used as a non-specific control. Cells were harvested 48h post-transfection and reduction of protein levels confirmed by Western blotting.
Half-life experiments were performed by interfering cells with FBXW7α-siRNA or EGFP-siRNA and, 48h later, adding cycloheximide to the medium. Cells were harvested at the indicated times.
Electrophoresis, Western blot analysis and antibodies
Proteins were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and gels were electroblotted onto nitrocellulose membranes and probed with the following antibodies: anti-HA-peroxidase monoclonal antibody (Roche); anti-Cdc25A, anti-cyclin E and anti-p53 monoclonal antibodies, and anti-MCM7 and anti-Grb2 polyclonal antibodies from Santa Cruz; anti-α-Tubulin, anti-Flag and anti-β-actin monoclonal antibodies from Sigma; anti-GFP polyclonal antibody (Immune Systems Ltd.); anti-MCM2 polyclonal antibody (Cell Signaling); anti-PLK1 and anti-CDH1 monoclonal antibodies from Millipore; anti-Nbs1 polyclonal antibody (Novus); anti-Myc monoclonal antibody (Clontech); and anti-Mre11 monoclonal antibody (Gene Tex). Peroxidase-coupled donkey anti-rabbit IgG and sheep anti-mouse IgG were obtained from GE Healthcare. Immunoreactive bands were visualized using the Enhanced Chemiluminescence Western blotting system (ECL, GE Healthcare).
In vitro kinase assays
Anti-HA immunoprecipitates from HCT116 cells transfected with pCMVHA-PLK1, pCMVHA-PLK1-T214G or pCMVHA were incubated with α-casein dephosphorylated (Sigma), (γ-32P) ATP and a kinase buffer (50mM Tris-HCl (pH 7.5), 10mM MgCl2, 0.1mM Na3VO4, 2mM DTT and 100µM unlabeled ATP) for 15 min at 30°C. Reactions were terminated by adding 4x SDS-sample buffer and proteins analyzed by SDS-PAGE and autoradiography.
Co-immunoprecipitation experiments
Nuclear extracts or whole cell extracts diluted to 150mM NaCl (1-2 mg) were incubated with normal mouse serum (Santa Cruz) for 30 minutes and subsequently with protein G-sepharose beads (GE Healthcare) for 1 hour at 4°C. After centrifugation, beads were discarded and supernatants incubated for 2 hours with anti-PLK1 (Millipore) or anti-Flag (Sigma) monoclonal antibodies or normal mouse serum, followed by protein G-sepharose beads for 1 hour. Beads were washed and bound proteins were solubilized by the addition of SDS-sample buffer heated at 95°C for 5 minutes.
For immunoprecipitation to in vitro kinase assays, an anti-HA monoclonal antibody from Covance was used.
Protein identification by liquid chromatography-tandem mass spectrometry (LC-MS/MS)
Flag-FBXW7α was transfected into Cos-7 cells and, after 18h, cells were treated for 4h with LLnL and FBXW7α immunoprecipitated from nuclear extracts with anti-Flag previously coupled to a resin from Pierce (AminoLink Plus Coupling Resin). Resins were washed three times in NP-40 lysis buffer and six times in ammonium bicarbonate 50mM. Samples were analyzed by LC-MS/MS using a LTQ mass spectrometer (Thermo Electron) as described previously [63].
Immunofluorescence microscopy
U2OS cells were grown on coverslips, fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. Immunostaining, using monoclonal anti-PLK1 (Millipore) and polyclonal anti-FBXW7α (Zymed) antibodies, and counterstaining with DAPI to visualize the nuclei, was carried out according to standard protocols. Epifluorescence microscopy was performed using a Leica microscope.
Chromatin fractionation
The soluble fraction of cells was prepared by lysis in CSK buffer (10mM PIPES pH 6.8, 100mM NaCl, 300mM sucrose, 3mM MgCl2, 1mM EGTA, 1mM DTT, 1mM PMSF, 50mM NaF, 0.1mM Na3VO4, 20mM sodium pyrophosphate, 0.5% Triton X-100, 1µg/ml aprotinin, 1µg/ml pepstatin, 1µg/ml leupeptin and 10µg/ml chymostatin) for 5 min on ice. Cell lysates were centrifuged at 7,500 rpm for 5 min at 4°C, and the supernatants collected and clarified by high-speed centrifugation (10 min at 14,000 rpm, 4°C). The chromatin pellet was washed again with CSK buffer and centrifuged at 7,500 rpm for 5 min at 4°C. For nuclease digestions, chromatin pellets were resuspended in lysis buffer containing 50 units of RNase-free DNase I (Roche) and 1mM CaCl2 and then incubated at 37°C for 30 min, followed by chilling on ice and centrifugation. The DNase pellet was washed once in 1ml of ice-cold CSK buffer (5 min, 14,000 rpm, 4°C) and incubated in ice-cold CSK buffer containing 500mM NaCl for 10 min at 4°C. This extract was clarified by centrifugation (10 min, 14,000 rpm, 4°C) and pooled with the DNase-treated fraction, constituting the chromatin-associated fraction. After the protein concentration was adjusted, proteins were resolved by SDS-PAGE and analyzed by Western blotting.
In vitro and in vivo ubiquitination assays
The in vitro ubiquitination of PLK1 was performed in a volume of 10µl containing 50mM Tris-HCl (pH 7.6), 5mM MgCl2, 0.6mM DTT, 2mM ATP, 2µl in vitro transcribed/translated unlabeled F-box protein, 1.5ng/µl E1 (His6-ubiquitin activating enzyme, Boston Biochem), 10ng/µl His6-UbcH3 (E2, Boston Biochem), 10ng/µl UbcH5a (E2, Boston Biochem), 2.5µg/µl ubiquitin (Sigma), 1µM ubiquitin aldehyde (Boston Biochem), and 1µl 35S-methionine-labelled in vitro transcribed/translated PLK1 as substrate. The reactions were incubated at 30ºC for 1h and analyzed by SDS-PAGE and autoradiography. For some experiments the unlabeled F-box protein was substituted by a recombinant SCFFBXW7α complex expressed in Sf21 insect cells.
The in vivo ubiquitination experiments were performed in Cos-7 cells transfected and treated with LLnL 4 hours before harvesting. Cells were washed in PBS, lysed at 95ºC for 15 minutes in NP40 buffer supplemented with 5% SDS and 10mM iodoacetamide, and diluted in NP40 buffer supplemented with 10mM iodoacetamide. PLK1 was immunoprecipitated and proteins separated by SDS-PAGE, electroblotted and probed with different antibodies.
Proliferation assays
Transfected HeLa cells were arrested in S phase with hydroxyurea (0.5mM) for 24h and, after releasing, cells were untreated or irradiated (30J/m2) and counted at different time points. Adherent cells were trypsinized, collected and viable cells (trypan blue-excluding) were counted using a hemocytometer. In parallel, as another method for measuring cell proliferation, the protein amount was quantified using the Bradford assay (Bio-Rad).
ACKNOWLEDGEMENTS
We are grateful to B.E. Clurman, F. Margottin, P. Wang and X. Ye for their generous contributions of reagents, E. Calvo for LC-MS/MS assistance and J.C. Meadows for critical reading of the manuscript.
This work was supported by Spanish grants from Ministerio de Economía y Competitividad, MINECO (SAF2011-30003) and Dirección General de Investigación, Tecnología y Empresa, Junta de Andalucía, DGITE (P08-CVI-03603). SG was supported by a fellowship from DGITE, and JH-R and MM-S by MINECO.
The authors declare that they have no conflict of interest.
REFERENCES
1. Lau AW, Fukushima H and Wei W. The Fbw7 and betaTRCP E3 ubiquitin ligases and their roles in tumorigenesis. Frontiers in bioscience : a journal and virtual library. 2012; 17:2197-2212.
2. Ho MS, Ou C, Chan YR, Chien CT and Pi H. The utility F-box for protein destruction. Cellular and molecular life sciences : CMLS. 2008; 65(13):1977-2000.
3. Wang Z, Inuzuka H, Zhong J, Wan L, Fukushima H, Sarkar FH and Wei W. Tumor suppressor functions of FBW7 in cancer development and progression. FEBS letters. 2012; 586(10):1409-1418.
4. Spruck CH, Strohmaier H, Sangfelt O, Muller HM, Hubalek M, Muller-Holzner E, Marth C, Widschwendter M and Reed SI. hCDC4 gene mutations in endometrial cancer. Cancer research. 2002; 62(16):4535-4539.
5. Welcker M, Orian A, Grim JE, Eisenman RN and Clurman BE. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Current biology : CB. 2004; 14(20):1852-1857.
6. Grim JE, Gustafson MP, Hirata RK, Hagar AC, Swanger J, Welcker M, Hwang HC, Ericsson J, Russell DW and Clurman BE. Isoform- and cell cycle-dependent substrate degradation by the Fbw7 ubiquitin ligase. The Journal of cell biology. 2008; 181(6):913-920.
7. Kimura T, Gotoh M, Nakamura Y and Arakawa H. hCDC4b, a regulator of cyclin E, as a direct transcriptional target of p53. Cancer science. 2003; 94(5):431-436.
8. Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S and Balmain A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004; 432(7018):775-779.
9. Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW and Elledge SJ. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996; 86(2):263-274.
10. Smith TF, Gaitatzes C, Saxena K and Neer EJ. The WD repeat: a common architecture for diverse functions. Trends in biochemical sciences. 1999; 24(5):181-185.
11. Welcker M and Clurman BE. Fbw7/hCDC4 dimerization regulates its substrate interactions. Cell division. 2007; 2:7.
12. Pashkova N, Gakhar L, Winistorfer SC, Yu L, Ramaswamy S and Piper RC. WD40 repeat propellers define a ubiquitin-binding domain that regulates turnover of F box proteins. Molecular cell. 2010; 40(3):433-443.
13. Min SH, Lau AW, Lee TH, Inuzuka H, Wei S, Huang P, Shaik S, Lee DY, Finn G, Balastik M, Chen CH, Luo M, Tron AE, Decaprio JA, Zhou XZ, Wei W, et al. Negative regulation of the stability and tumor suppressor function of Fbw7 by the Pin1 prolyl isomerase. Molecular cell. 2012; 46(6):771-783.
14. Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T and Tyers M. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001; 414(6863):514-521.
15. Skaar JR, Pagan JK and Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nature reviews Molecular cell biology. 2013.
16. Enchev RI, Scott DC, da Fonseca PC, Schreiber A, Monda JK, Schulman BA, Peter M and Morris EP. Structural basis for a reciprocal regulation between SCF and CSN. Cell reports. 2012; 2(3):616-627.
17. Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR and Sears R. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nature cell biology. 2004; 6(4):308-318.
18. Popov N, Wanzel M, Madiredjo M, Zhang D, Beijersbergen R, Bernards R, Moll R, Elledge SJ and Eilers M. The ubiquitin-specific protease USP28 is required for MYC stability. Nature cell biology. 2007; 9(7):765-774.
19. Popov N, Herold S, Llamazares M, Schulein C and Eilers M. Fbw7 and Usp28 regulate myc protein stability in response to DNA damage. Cell Cycle. 2007; 6(19):2327-2331.
20. Kitagawa K, Kotake Y, Hiramatsu Y, Liu N, Suzuki S, Nakamura S, Kikuchi A and Kitagawa M. GSK3 regulates the expressions of human and mouse c-Myb via different mechanisms. Cell division. 2010; 5:27.
21. Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP and Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007; 316(5828):1160-1166.
22. Minella AC, Welcker M and Clurman BE. Ras activity regulates cyclin E degradation by the Fbw7 pathway. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102(27):9649-9654.
23. Barr FA, Sillje HH and Nigg EA. Polo-like kinases and the orchestration of cell division. Nature reviews Molecular cell biology. 2004; 5(6):429-440.
24. Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nature reviews Drug discovery. 2010; 9(8):643-660.
25. Liu XS, Song B and Liu X. The substrates of Plk1, beyond the functions in mitosis. Protein & cell. 2010; 1(11):999-1010.
26. Lindon C and Pines J. Ordered proteolysis in anaphase inactivates Plk1 to contribute to proper mitotic exit in human cells. The Journal of cell biology. 2004; 164(2):233-241.
27. Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A and Pagano M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell. 2008; 134(2):256-267.
28. Leman AR and Noguchi E. The Replication Fork: Understanding the Eukaryotic Replication Machinery and the Challenges to Genome Duplication. Genes. 2013; 4(1):1-32.
29. Lei M and Erikson RL. Plk1 depletion in nontransformed diploid cells activates the DNA-damage checkpoint. Oncogene. 2008; 27(28):3935-3943.
30. Stuermer A, Hoehn K, Faul T, Auth T, Brand N, Kneissl M, Putter V and Grummt F. Mouse pre-replicative complex proteins colocalise and interact with the centrosome. European journal of cell biology. 2007; 86(1):37-50.
31. Tsvetkov L and Stern DF. Interaction of chromatin-associated Plk1 and Mcm7. The Journal of biological chemistry. 2005; 280(12):11943-11947.
32. Wu ZQ and Liu X. Role for Plk1 phosphorylation of Hbo1 in regulation of replication licensing. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105(6):1919-1924.
33. Yoo HY, Kumagai A, Shevchenko A, Shevchenko A and Dunphy WG. Adaptation of a DNA replication checkpoint response depends upon inactivation of Claspin by the Polo-like kinase. Cell. 2004; 117(5):575-588.
34. Vidanes GM, Sweeney FD, Galicia S, Cheung S, Doyle JP, Durocher D and Toczyski DP. CDC5 inhibits the hyperphosphorylation of the checkpoint kinase Rad53, leading to checkpoint adaptation. PLoS biology. 2010; 8(1):e1000286.
35. Song B, Liu XS, Davis K and Liu X. Plk1 phosphorylation of Orc2 promotes DNA replication under conditions of stress. Molecular and cellular biology. 2011; 31(23):4844-4856.
36. Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN and Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101(24):9085-9090.
37. van Drogen F, Sangfelt O, Malyukova A, Matskova L, Yeh E, Means AR and Reed SI. Ubiquitylation of cyclin E requires the sequential function of SCF complexes containing distinct hCdc4 isoforms. Molecular cell. 2006; 23(1):37-48.
38. Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW and Elledge SJ. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001; 294(5540):173-177.
39. Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O and Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001; 413(6853):316-322.
40. Tsou MF, Wang WJ, George KA, Uryu K, Stearns T and Jallepalli PV. Polo kinase and separase regulate the mitotic licensing of centriole duplication in human cells. Developmental cell. 2009; 17(3):344-354.
41. Yim H and Erikson RL. Polo-like kinase 1 depletion induces DNA damage in early S prior to caspase activation. Molecular and cellular biology. 2009; 29(10):2609-2621.
42. Nyberg KA, Michelson RJ, Putnam CW and Weinert TA. Toward maintaining the genome: DNA damage and replication checkpoints. Annual review of genetics. 2002; 36:617-656.
43. Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K and Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. The EMBO journal. 2004; 23(10):2116-2125.
44. Weiss L and Efferth T. Polo-like kinase 1 as target for cancer therapy. Experimental hematology & oncology. 2012; 1(1):38.
45. Smith MR, Wilson ML, Hamanaka R, Chase D, Kung H, Longo DL and Ferris DK. Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochemical and biophysical research communications. 1997; 234(2):397-405.
46. Zhao C, Gong L, Li W and Chen L. Overexpression of Plk1 promotes malignant progress in human esophageal squamous cell carcinoma. Journal of cancer research and clinical oncology. 2010; 136(1):9-16.
47. Lau AW, Inuzuka H, Fukushima H, Wan L, Liu P, Gao D, Sun Y and Wei W. Regulation of APC E3 ligase activity by the Fbw7/cyclin E signaling axis contributes to the tumor suppressor function of Fbw7. Cell research. 2013.
48. Jaquenoud M, van Drogen F and Peter M. Cell cycle-dependent nuclear export of Cdh1p may contribute to the inactivation of APC/C(Cdh1). The EMBO journal. 2002; 21(23):6515-6526.
49. Rape M and Kirschner MW. Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature. 2004; 432(7017):588-595.
50. Yamada M, Watanabe K, Mistrik M, Vesela E, Protivankova I, Mailand N, Lee M, Masai H, Lukas J and Bartek J. ATR-Chk1-APC/CCdh1-dependent stabilization of Cdc7-ASK (Dbf4) kinase is required for DNA lesion bypass under replication stress. Genes & development. 2013; 27(22):2459-2472.
51. Syljuasen RG, Jensen S, Bartek J and Lukas J. Adaptation to the ionizing radiation-induced G2 checkpoint occurs in human cells and depends on checkpoint kinase 1 and Polo-like kinase 1 kinases. Cancer research. 2006; 66(21):10253-10257.
52. Lupardus PJ and Cimprich KA. Checkpoint adaptation; molecular mechanisms uncovered. Cell. 2004; 117(5):555-556.
53. van Vugt MA and Medema RH. Checkpoint adaptation and recovery: back with Polo after the break. Cell Cycle. 2004; 3(11):1383-1386.
54. Liu N, Li H, Li S, Shen M, Xiao N, Chen Y, Wang Y, Wang W, Wang R, Wang Q, Sun J and Wang P. The Fbw7/human CDC4 tumor suppressor targets proproliferative factor KLF5 for ubiquitination and degradation through multiple phosphodegron motifs. The Journal of biological chemistry. 2010; 285(24):18858-18867.
55. Liu T, Deng M, Li J, Tong X, Wei Q and Ye X. Phosphorylation of right open reading frame 2 (Rio2) protein kinase by polo-like kinase 1 regulates mitotic progression. The Journal of biological chemistry. 2011; 286(42):36352-36360.
56. Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K and Benarous R. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Molecular cell. 1998; 1(4):565-574.
57. Romero F, Gil-Bernabe AM, Saez C, Japon MA, Pintor-Toro JA and Tortolero M. Securin is a target of the UV response pathway in mammalian cells. Molecular and cellular biology. 2004; 24(7):2720-2733.
58. Jin DY, Spencer F and Jeang KT. Human T cell leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell. 1998; 93(1):81-91.
59. Romero F, Germani A, Puvion E, Camonis J, Varin-Blank N, Gisselbrecht S and Fischer S. Vav binding to heterogeneous nuclear ribonucleoprotein (hnRNP) C. Evidence for Vav-hnRNP interactions in an RNA-dependent manner. The Journal of biological chemistry. 1998; 273(10):5923-5931.
60. Gil-Bernabe AM, Romero F, Limon-Mortes MC and Tortolero M. Protein phosphatase 2A stabilizes human securin, whose phosphorylated forms are degraded via the SCF ubiquitin ligase. Molecular and cellular biology. 2006; 26(11):4017-4027.
61. Lee JY, Yu SJ, Park YG, Kim J and Sohn J. Glycogen synthase kinase 3beta phosphorylates p21WAF1/CIP1 for proteasomal degradation after UV irradiation. Molecular and cellular biology. 2007; 27(8):3187-3198.
62. Cotto-Rios XM, Jones MJ, Busino L, Pagano M and Huang TT. APC/CCdh1-dependent proteolysis of USP1 regulates the response to UV-mediated DNA damage. The Journal of cell biology. 2011; 194(2):177-186.
63. Barquilla A, Saldivia M, Diaz R, Bart JM, Vidal I, Calvo E, Hall MN and Navarro M. Third target of rapamycin complex negatively regulates development of quiescence in Trypanosoma brucei. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109(36):14399-14404.