
INTRODUCTION
Acute myeloid leukemia (AML) is a hematological neoplasia characterized by an abnormal growth and differentiation of immature myeloid cells that accumulate in bone marrow and interfere with the production of normal blood cells [1]. In spite of some improvement in its therapy, a minority of adults with AML are eventually cured, a fact stressing the need for new therapeutic approaches.
AML is thought to be organized as a hierarchy of functionally heterogeneous cells that is ultimately sustained by a small fraction of leukemic stem cells (LSCs) with enhanced self-renewal capacity, impaired differentiation ability and increased drug resistance [2]. Among all AML blasts constituting the bulk tumor, only LSCs initiate and sustain the disease. As the balance between self-renewal and differentiation is tightly regulated and favours the former in LSCs, induction of terminal differentiation of LSCs would exhaust the LSC pool [3, 4]. Since differentiating therapy could induce higher chemotherapy sensitivity and limited life span of more differentiated AML blasts, combinatorial strategies of differentiating and cytotoxic agents with the capability to eliminate LSCs are a promising therapeutic approach, which could result in a significant improved outcome of AML patients [5].
Although global mechanisms that block differentiation and increase self-renewal of AML cells are poorly understood, biological changes induced by agents that differentiate AML cells to diverse lineages are already described. Bioinformatics studies provided us the gene expression profile associated to granulocyte differentiation of AML cells treated with all-trans-retinoic acid (ATRA). This drug-induced gene expression pattern constituted the therapeutic goal in the search of agents with AML differentiating capacity. Following this approach and using Connectivity Maps [6], we identified the XIAP inhibitor Dequalinium chloride (DQA). XIAP inhibition induced cytotoxicity and differentiation in AML cells. Moreover DQA treatment preferentially affected leukemic stem cells, suggesting that targeting XIAP may contribute to eradicate the LSC population thought to be responsible for refractoriness and relapse episodes in AML and supporting further validation in pre-clinical and clinical studies.
RESULTS
In order to identify drugs that could induce myeloid differentiation in AML cells, a gene signature associated with ATRA-induced granulocytic differentiation of HL-60 AML cells was defined using public available gene expression profiles (GSE982). Differentially expressed probe sets were identified (Supplementary Table 1) and the given gene signature (Supplementary Table 2) was used to query and compare against the reference catalogue of gene expression profiles obtained after drug or other perturbagen treatment of cell lines using the Connectivity Map (http://www.broadinstitute.org/cmap/) [6]. The results were filtered at a P value <0.05; connectivity score > 0.75 in HL-60, and > 0.5 in PC3 and MCF7; and a concentration < 10 µM in all cell lines (Supplementary Table 3). Dequalinium chloride (DQA, CAS no. 522-51-0) was the only compound that met all the above-described criteria. DQA is an amphiphilic quinolinium derivative (Supplementary Figure 1).
We next studied the cytotoxic potential of DQA on well-characterized AML cell lines (HL-60, KG-1, MonoMac-1 and Kasumi-1). DQA induced cell death in all AML cell lines tested in a dose-response fashion after a 3-day treatment, achieving more than 50% of cell death at the highest concentration (5 µM) (Figure 1A). Since accumulating evidence shows that the bone marrow stromal environment might protect leukemia cells from chemotherapy-induced apoptosis, the cytotoxic potential of DQA was also tested on AML cell lines co-cultured with the HS-5 human bone marrow stroma cell line. Cellular viability of DQA-treated AML cells was impaired regardless the presence of HS-5 stroma cells (Figure 1A). Moreover, the proportion of cell death induced by DQA was similar regardless the presence of stroma cells, suggesting that the HS-5 stroma cells, that conferred chemoresistance to commonly used drugs such as ara-C and idarubicin [7], are unable to protect AML cell to the cytotoxic effect of DQA treatment. DQA was identified as a potential differentiation-inducer agent in our in silico screen. CD15 is up regulated in AML cells when differentiation is restored [8]. In all AML cell lines tested, DQA induced the upregulation of the CD15 surface marker (Figure 1B). These findings validated our in silico prediction of DQA as a differentiation-inducing drug of AML cells.
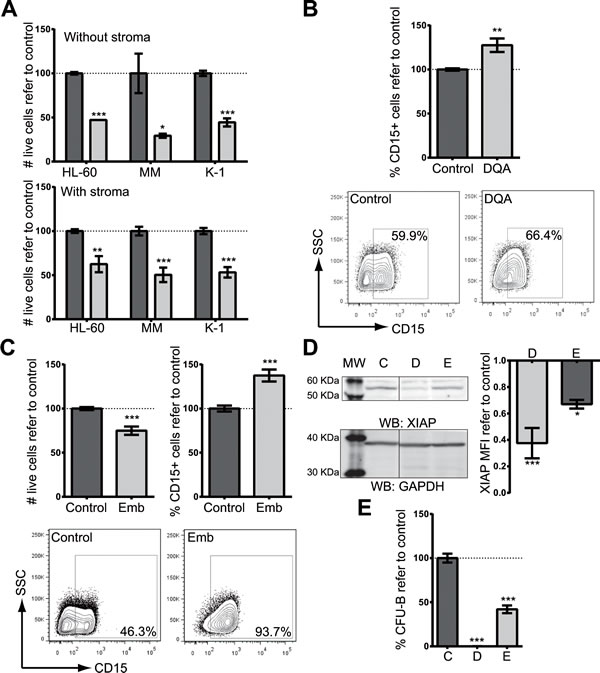
Figure 1: XIAP inhibitor treatment induces cytotoxicity and differentiation on AML cell lines. A. Cytotoxicity in HL-60, MonoMac-1 (MM) and Kasumi-1 (K-1) AML cell lines resulting from treatment with 5 µM DQA for 48 h in the absence (upper panel) or presence of HS-5 stroma cells (lower panel). Y-axis: relative number of live cells as assessed by flow cytometry (7-AAD-). B. Up-regulation of CD15 surface expression, measured by flow cytometry in AML cell lines (HL-60, KG-1, MonoMac-1 and Kasumi-1) treated with 5 µM DQA. Data from all AML cell lines are presented combined. Frequency of CD15-positive population normalized against control-treated samples is represented. CD15 surface expression representative plot of HL-60 untreated (left) or treated with 5 µM DQA (right). C. HL-60, Kasumi-1, MonoMac-1 and KG-1 AML cells were treated with different concentrations of Embelin for 48 h. Cell viability (upper left panel) and CD15 surface expression (upper right panel) were measured by flow cytometry. Representative flow cytometry plot of HL-60 untreated (left) or treated with 10 µM Embelin (right). D. XIAP protein was detected by Western blot upon DQA (D) and Emb (E) treatment of HL-60 cells. GAPDH was used as loading control. MFI refer to GAPDH and vehicle-treated control is represented. E. HL-60 cells were treated for 18 h with 5 µM DQA (left) and 10 µM embelin (right). Colonies were counted at day 7. * p<0.05; ** p<0.005; *** p<0.0005. Error bars correspond to SEM.
DQA has been identified as a XIAP inhibitor by its direct binding [9]. In order to confirm that XIAP inhibition was responsible for the cytotoxic and differentiation effects observed upon DQA treatment, a well-described XIAP inhibitor embelin was chosen[10]. As shown with DQA, embelin induced cytotoxicity and upregulation of CD15 surface expression (Figure 1C). In fact, both inhibitors reduced the amount of XIAP upon treatment (Figure 1D). Moreover, DQA and embelin treatment decreased the clonogenic capacity of AML cells (Figure 1E). These results suggest that XIAP inhibition overcomes the block in differentiation displayed by AML cells and reduces cell viability.
A way to promote differentiation is achieved through prevention of S-phase entry. This mechanism of action has been described for ATRA [11]. Similarly to ATRA, DQA treatment induced cell-cycle arrest in the G0/G1 phase whereas a reduction in G2/M phase was detected upon treatment of AML cell lines (Figure 2A and 2B).
Table 1: Characteristics of patient samples. M, male; F, female. # x 109/L
AML sample |
Gender |
Age (yr) |
WHO subtype |
WBC count# |
% Blasts in PB |
% Blasts in BM |
Karyotype |
Additional molecular features |
#1 |
M |
28 |
AML without maturation |
143 |
98 |
90 |
46, XY |
FLT3-ITD |
#2 |
M |
40 |
AML without maturation |
52 |
66 |
80 |
46,XY |
Biallelic CEBPA mutation |
#3 |
F |
34 |
AML with myelodysplasia-related changes |
32 |
16 |
44 |
45,XX, -7 |
FLT3wt NPMwt |
#4 |
M |
45 |
AML with t(6,9) |
40 |
58 |
43 |
46,XY,t(6;9)(p23;q34) |
FLT3-ITD |
#5 |
M |
48 |
AML with t(8;21)(q22;q22); RUNX1-RUNX1T1 |
54 |
5 |
24 |
46,XY,t(8;21)(q22;q22) |
NPMwt / FLT3 N/A |
#6 |
M |
61 |
AML with t(8;21)(q22;q22); RUNX1-RUNX1T1 |
21.4 |
51 |
89 |
FLT3wt NPMwt |
|
#7 |
F |
58 |
AML with myelodysplasia-related changes |
100.7 |
45 |
80 |
46,XX,del(5)(q23q33), t(8;9)(p11;q34)[20] |
FLT3wt NPMwt |
#8 |
M |
24 |
AML with myelodysplasia-related changes |
7.1 |
83 |
30 |
46,XY[20] |
FLT3wt NPMwt |
#9 |
M |
49 |
AML with myelodysplasia-related changes |
76.4 |
42 |
26 |
46-47,XY,del(5)(q22q34),del(6)(q22q25),del(7)(q22q23),-8,-9,add(11)(q23),+i(11)(q11),-16,+mar1,+mar2,+mar3[cp8]. |
FLT3wt NPMwt |
#10 |
M |
22 |
AML with t(8;21)(q22;q22); RUNX1-RUNX1T1 |
20.4 |
83 |
69 |
FLT3 ITD |
|
#11 |
F |
60 |
AML with myelodysplasia-related changes |
218.1 |
68 |
36 |
48,XX,+8,+21[13] |
FLT3wt NPMwt |
Several signaling pathways are misregulated in AML. Activation of Erk and Akt pathways [12, 13] have been considered as critical for the survival and/or proliferation of AML cells. In this context, DQA treatment was observed to reduce the amount of activated signaling molecules in all AML cell lines tested after intracellular staining of P-Akt and P-Erk (Figure 2C and 2D). These results correlate with the observed cytotoxic effect of DQA, which might at least in part be due to Akt and Erk downregulation.
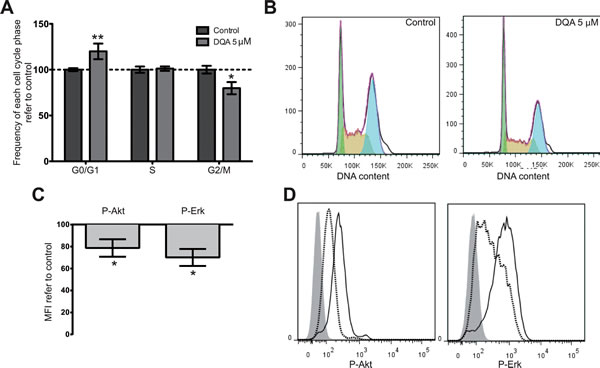
Figure 2: DQA treatment induces cell cycle arrest and downregulation of P-Akt, P-Erk and P-Stat3. HL-60, KG-1, MonoMac-1 and Kasumi-1 were treated with 5 µM DQA and cell cycle was analyzed by flow cytometry 48 h after treatment. A. Relative frequency of G0/G1, S and G2/M phases in control- vs. DQA-treated AML cells. Bars represent the mean value of all AML cell lines and error bars represent SEM. B. Representative DNA content flow profile of control- (left) and DQA-treated (right) HL-60 (green represents G0/G1 phase; yellow, S phase; blue, G2/M phase). P-Akt and P-Erk expression levels by flow cytometry in AML cell lines after treatment with 5 µM DQA for 24 h. C. Mean fluorescence intensity of each staining was normalized against vehicle-control treated sample and data from all AML cell lines tested is represented. D. Representative flow histograms of P-Akt (left), P-Erk (centre) and P-Stat3 (right) intracellular staining of DQA-treated HL-60 AML cells. Shadow, negative control; solid line, control treated sample; dashed line, DQA-treated sample. * p<0.05; ** p<0.005.
Next, the cytotoxicity of DQA and embelin treatment was evaluated in samples of patients with AML. The presence of these inhibitors reduced cell viability 24 and 72 h after treatment in the bulk AML population in a dose-dependent fashion (Figure 3A). Since the majority of the LSC population expresses the immature surface marker CD34 in the absence of CD38 [14], this marker combination (CD34+CD38-) was used to analyse the preferential effect of the drug on the LSC-enriched primitive population. Within the CD34+CD38- population, the reduction in cell viability was higher in the presence of DQA or embelin compared to the remaining leukemic blast population (Figure 3A), suggesting that XIAP inhibitor cytotoxic effect is preferentially displayed on the stem-cell like or primitive population. Interestingly, no effect was detected when healthy myeloid blood cells were incubated with DQA or embelin (Figure 3B). Taking into account that XIAP expression has been described as a prognostic marker [15, 16], we analysed the cytotoxic effect of DQA treatment on the most primitive AML blast cell fraction within each prognostic group [17]. In concordance with protein expression data [15], intermediate and unfavourable risk groups were more sensitive to DQA treatment (Figure 3C).
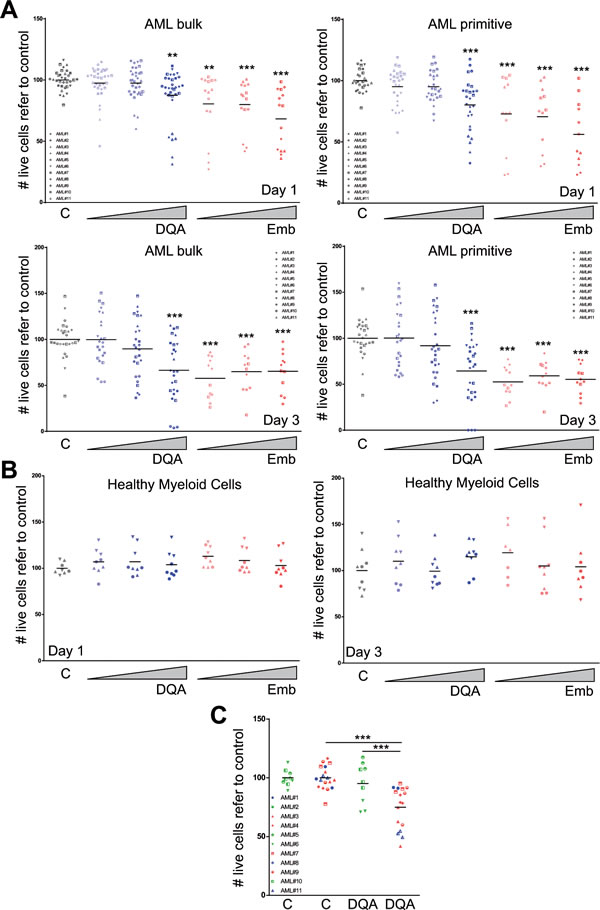
Figure 3: DQA and embelin treatment induces cell death in AML primary blasts by preferentially affecting LSC population and reduces clonogenic capacity. AML primary blasts A. or healthy myeloid blood cells B. were treated with different concentration of DQA (0.05, 0.5, 5 µM) and embelin (0.1, 1, 10 µM). Cell viability was analysed at day 1 (upper panels) and 3 (lower panels) after treatment. Each symbol corresponds to a single AML patient sample, specified in the graph legend. Bulk population corresponds to AML blast cells and the primitive fraction corresponds to a CD34+CD38- blast population. C. Primary AML patient samples were treated for 24 h with 5 mM DQA. Cell viability was measured by flow cytometry (volumetric counts on live 7-AAD- cells). Each symbol corresponds to an AML patient sample. Green, favourable risk group; blue, intermediate risk group; red, unfavourable risk group. * p<0.05; ** p<0.005; *** p<0.0005.
Moreover, clonogenic capacity, which constitutes a direct measure of stem cell function [18], was reduced after DQA and embelin treatment of AML primary cells (Figure 4A). Treatment of lineage-depleted umbilical cord blood cells with DQA or embelin had little effect on the clonogenic capacity, as measured by the total number of colonies or the frequency of each subtype (Figure 4B). Taken together, these results suggest that XIAP inhibition preferentially impairs LSC functionality in AML.
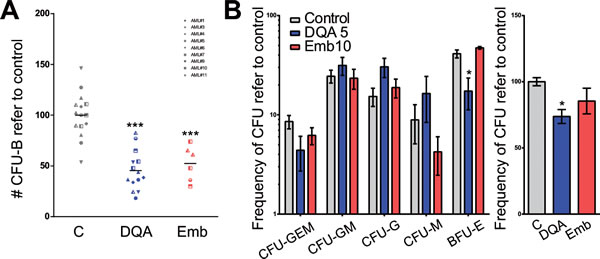
Figure 4: XIAP inhibitor treatment reduced the clonogenic capacity of AML cells with little effect on primitive healthy blood cells. D. AML primary cells or E. lineage-depleted umbilical cord blood cells were treated with 5 µM DQA or 10 µM embelin for 18 h. Colonies were screened at day 14 based on morphological criteria. Each symbol represents primary sample. * p<0.05; ** p<0.005; *** p<0.0005.
DISCUSSION
Although the notion that induction of differentiation by chemical agents in leukemic cells could reprogram the cells toward proliferation arrest and/or programmed cell death was established four decades ago [19, 20], only retinoic acid for the treatment of acute promyelocytic leukemia has revealed as a successful clinical application of this hypothesis. We performed an in silico screening seeking for FDA-approved drugs that produced a gene expression regulation similar to ATRA in HL-60 AML cells. A XIAP inhibitor, DQA, was identified and validated both in different AML cells lines and primary patient samples. XIAP inhibition also induced myeloid differentiation and cell death in AML cells. Interestingly, the clonogenic capacity was severely reduced upon treatment. Additionally, the most primitive fraction of AML blasts appeared to be more cytotoxic-sensitive to XIAP inhibition than more mature blasts. Interestingly, little effect was detected in healthy blood cells and primitive lineage-negative cord blood cells.
DQA is a classic broad bacteriostatic with anti-fungicidal and anti-protozoal activity [21, 22]. At a molecular level, DQA interrupts the interaction XIAP/Caspase-3 [9]. XIAP protein and mRNA levels have been correlated with chemoresistance [23] and poor clinical outcome in AML patients [15, 16], suggesting that XIAP may constitute an interesting therapeutic target for AML treatment. In fact, only most aggressive AML patient samples seemed to respond to DQA treatment. Phase I/II clinical trials evaluating the effect of a XIAP antisense oligonucleotide showed that this strategy seems to be effective when combined with chemotherapy in patients with AML refractory to a single induction regimen[24]. Interestingly, DQA-mediated cytotoxic effect seems to be stroma-independent. Microenvironment has been linked with primary LSC resistance to therapy where the niche signaling could regulate cell cycle and chemoresistance [25]. Here, XIAP inhibitors are described as AML differentiation inducer and, consequently, a reduction in clonogenic capacity and cell cycle arrest is produced. Similarly, other cell cycle arrest-inducing and pro-apoptotic drugs are effective in pre-clinical studies [26]. XIAP inhibitors already approved for clinical use, such as DQA, may be of great interest to explore their anti-leukemia effect and their potential therapeutic use in combination with conventional chemotherapy.
AML is hierarchically organized, with quiescent “stem-cell”-like cells (LSCs) at the apex, that have the ability to perpetuate themselves through self-renewal and generate more mature progeny through differentiation [2]. According with this model, the inability to eradicate LSCs represents the cause of relapse and therapeutic failure. Thus, effective tumor therapy will require eradication of these cells. Designing agents that can differentiate leukemia cells into non-dividing/non-malignant growth-arrested cells constitutes a new experimental approach for non-APL AML treatment. Here, XIAP inhibitors are described as AML differentiation inducer and, consequently, a reduction in clonogenic capacity and cell cycle arrest is produced. Therefore, there is a cessation of self-renewal capacity and XIAP may constitute a therapeutic target. Similarly, Bcl2L10 (another anti-apoptotic molecule) has also been described as a therapeutic target for AML and MDS [27]. Interestingly, an inhibitor against both Bcl-2 and Bcl-X, ABT-737, displayed anti-leukemia effect on AML [28]. Moreover, Pten/mTOR and the PI3K/Akt signalling pathway have been shown to be implicated in AML development and leukemia stem cell population maintenance [13, 29]. Here, XIAP inhibition reduced P-Akt levels in concordance with previous work and may explain the differential effect on the LSC population. XIAP inhibitors already approved for clinical use, such as DQA, may be of great interest to explore their anti-leukemia effect and their potential therapeutic use in combination with conventional chemotherapy.
In summary, our in silico screen for differentiation-inducing agents lead to the identification of DQA, an agent with a known capacity of XIAP inhibition. Interestingly, XIAP inhibitors proved to result in a significant cytotoxic and differentiating activity both in AML cell lines and primary samples, with a preferential effect on the immature leukemic stem-cell fraction and sparing healthy blood cells. These findings warrant further investigation in pre-clinical and clinical setting, in combination with currently used chemotherapy.
MATERIALS AND METHODS
Identification of genes related to myeloid differentiation with Connectivity Maps
Gene signature associated with ATRA-induced differentiation in HL-60 cells was obtained from GSE982. Raw files (.cel) were normalized and probe sets with a differential expression of at least 2-fold of change and p value <0.005 were chosen (Supplementary Table 1) using GenePattern software (Broad Institute Cancer Program; http://www.broadinstitute.org/cancer/software/genepattern/). The 49 top-ranking downregulated and 269 top-ranking upregulated probes during ATRA treatment were selected for in silico signature-based screening (Supplementary Table 2) (Connectivity Maps; http://www.broadinstitute.org/cmap/).
AML cell lines and cell cultures
AML cell lines HL-60 (ACC-3)[30], KG-1 (ACC-14)[31], MonoMac-1 (ACC-252)[32] and Kasumi-1 (ACC-220)[33] were obtained from DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen) and the human stroma cell line HS-5 was purchased from ATCC (American Type Culture Collection). Experiments were performed within 6 months after receipt or resuscitation. AML cell lines were cultured in complete RPMI medium (PAA laboratories) supplemented with fetal bovine serum (Lonza), sodium pyruvate (Lonza) and/or non-essential amino acids (Lonza) according to manufacturers’ recommendations. HS-5 cell line was cultured in complete DMEM medium (PAA laboratories) supplemented with 10% fetal bovine serum (Lonza). Primary AML blasts were cultured in IMDM (PAA laboratories) supplemented with 3% heat-inactivated fetal bovine serum (Lonza), 1x BIT (StemCell Technologies), 5 ng/ml IL3 (Peprotech), sodium pyruvate (Lonza) and β-mercaptoethanol (Sigma).
Primary samples
Primary AML samples were obtained from patients diagnosed with AML at Hospital Clínic of Barcelona (Spain). AML diagnosis and classification was based on accepted WHO criteria. Main AML patient’s characteristics are summarized in Table 1. Samples were obtained from bone marrow and mononuclear cells (MNCs) were isolated by Ficoll density gradient centrifugation (GE). All patients provided written informed consent in accordance with the Declaration of Helsinki, and the study was approved by the Ethics Committee of Hospital Clínic of Barcelona. Blood mature MNCs were isolated from healthy-donor buffy coats provided by Banc de Sang i Teixits (Barcelona, Spain). Umbilical cord blood MNCs were obtained after Ficoll density gradient centrifugation and were depleted for lineage marker-positive cells (Milteny).
Drugs
Dequalinium chloride hydrate was obtained from Sigma-Aldrich, resuspended in ultrapure H2O (Sigma) and stored at -20ºC at 5mM. Embelin was purchased from Enzo Life Science, resuspended in DMSO and stored at -20ºC at 10 mM.
Citotoxicity assay
Five-hundred thousand cells per ml were cultured in 96-well plates in complete medium. DQA or embelin were added at different concentrations. Co-culture experiments were performed seeding 30 x 103 HS-5 cells with 1 x 105 AML cells per 200 µl of complete medium. Cell viability was measured by 7-AAD (eBioscience) exclusion by flow cytometry and cell count was obtained by volume in a FACSVerse cytometer (BD). In co-culture experiments, AML cells were discriminated based on CD45 expression. Statistical analysis and IC50 determination were calculated in GraphPad (Prism software). FlowJo software (TriStar) was used for flow cytometry analysis.
Myeloid differentiation
Cells were treated as indicated for cytotoxicity assays. Forty-eight hours after treatment, cells were stained with anti-human CD15-APC (BD) and surface expression of the antigen was analyzed by flow cytometry (FACSVerse, BD).
Western Blot
HL-60 cells (10 x 106 cells) were incubated with 5 µM DQA or 10 µM embelin for 48 h. Cells were harvested and lysed in RIPA buffer. The protein extract was subjected to SDS-polyacrylamide gel (BioRad) electrophoresis and transferred to PVDF membrane (BioRad). Mouse anti-XIAP and goat anti-GAPDH antibodies were purchased from BD and Abcam respectively.
Cell cycle analysis
Cells were cultured and treated with DQA as described above for experiments of differentiation induction. Twenty-four hours after treatment, cells were harvested, washed with PBS (Sigma), and fixed and permeabilized in 70% ethanol (Sigma) at 4ºC. DNA content was stained with 7-AAD (eBioscience) and measured by flow cytometry (FACSVerse, Becton Dickinson). Data were analyzed using Watson cell cycle model in FlowJo 7.6.5.
Intracellular staining
Cells were fixed with 2% formaldehyde (Applichem Lifescience), permabilized with 100% methanol (VWR BDH Prolabo), and stained with anti-Phospho-Akt (Thr308) clone C31E5E and anti-Phospho-Erk1/2 (Thr202/Tyr204) clone E10 from Cell Signaling Technologies following manufacturers’ recommendations. RPE-coupled goat anti-mouse and anti-rabbit secondary antibodies (Life Technologies) were used and samples were acquired in a FACSVerse cytometer (BD). FCS files were analyzed in FlowJo software (TriStar).
Clonogenic assay
Primary AML (50 x 103) or HL-60 (1 x 103) cells were treated for 18 h with the compound indicated and mixed with 1 mL of MethoCult H4034 Optimum (StemCell Technologies). Colonies were screened based on morphology and cellularity at day 5 (AML cell lines) or day 14 (primary AML cells).
ACKNOWLEDGEMENTS
We would like to thank Robin Hallett for his help with bioinformatics analysis and Josep Maria Cornet-Masana for critical reading of the manuscript. RMR is supported by Ramón y Cajal program/Ministry of Economy (RYC-2011-07998), MN is supported by Premi Fi de Residència Emili Letang of Hospital Clínic, and MP holds a “Rio Hortega” fellowship (ISCIII). This work was in part supported by the Plan Nacional SAF2012-34352 (RMR), AECC de Barcelona (RMR), Fondo de Investigaciones Sanitarias PI040135 (EM) and PI041357 (AUI), and RD06/0020/0004 (JE). The support of the Fundació Internacional Josep Carreras and Obra Social La Caixa is also acknowledged.
AUTHORS CONTRIBUTIONS
DM-M, MN, MCLC, NT and AE, acquisition of data, analysis and interpretation of data, and revision of the manuscript; MP, MD-B, MR, DC, EM, AUI and JE, revision of the manuscript and material support; RMR, conception and design, acquisition of data, analysis and interpretation of data and writing the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL, Le Beau MM, Hellstrom-Lindberg E, Tefferi A and Bloomfield CD. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009; 114(5):937-951.
2. Bonnet D and Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997; 3(7):730-737.
3. Sachlos E, Risueno RM, Laronde S, Shapovalova Z, Lee JH, Russell J, Malig M, McNicol JD, Fiebig-Comyn A, Graham M, Levadoux-Martin M, Lee JB, Giacomelli AO, Hassell JA, Fischer-Russell D, Trus MR, et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell. 2012; 149(6):1284-1297.
4. Zheng R, Friedman AD and Small D. Targeted inhibition of FLT3 overcomes the block to myeloid differentiation in 32Dcl3 cells caused by expression of FLT3/ITD mutations. Blood. 2002; 100(12):4154-4161.
5. Petrie K, Zelent A and Waxman S. Differentiation therapy of acute myeloid leukemia: past, present and future. Curr Opin Hematol. 2009; 16(2):84-91.
6. Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006; 313(5795):1929-1935.
7. Garrido SM, Appelbaum FR, Willman CL and Banker DE. Acute myeloid leukemia cells are protected from spontaneous and drug-induced apoptosis by direct contact with a human bone marrow stromal cell line (HS-5). Exp Hematol. 2001; 29(4):448-457.
8. Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML, Dayaram T, Owens BM, Shigematsu H, Levantini E, Huettner CS, Lekstrom-Himes JA, Akashi K and Tenen DG. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity. 2004; 21(6):853-863.
9. Orzaez M, Gortat A, Sancho M, Carbajo RJ, Pineda-Lucena A, Palacios-Rodriguez Y and Perez-Paya E. Characterization of dequalinium as a XIAP antagonist that targets the BIR2 domain. Apoptosis. 2011; 16(5):460-467.
10. Nikolovska-Coleska Z, Xu L, Hu Z, Tomita Y, Li P, Roller PP, Wang R, Fang X, Guo R, Zhang M, Lippman ME, Yang D and Wang S. Discovery of embelin as a cell-permeable, small-molecular weight inhibitor of XIAP through structure-based computational screening of a traditional herbal medicine three-dimensional structure database. J Med Chem. 2004; 47(10):2430-2440.
11. Fang Y, Zhou X, Lin M, Jing H, Zhong L, Ying M, Luo P, Yang B and He Q. The ubiquitin-proteasome pathway plays essential roles in ATRA-induced leukemia cells G0/G1 phase arrest and transition into granulocytic differentiation. Cancer Biol Ther. 2010; 10(11):1157-1167.
12. Kornblau SM, Womble M, Qiu YH, Jackson CE, Chen W, Konopleva M, Estey EH and Andreeff M. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood. 2006; 108(7):2358-2365.
13. Peng C, Chen Y, Li D and Li S. Role of Pten in leukemia stem cells. Oncotarget. 2010; 1(2):156-160.
14. Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, Canty AJ, Danska JS, Bohlander SK, Buske C, Minden MD, Golub TR, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011; 17(9):1086-1093.
15. Tamm I, Richter S, Oltersdorf D, Creutzig U, Harbott J, Scholz F, Karawajew L, Ludwig WD and Wuchter C. High expression levels of x-linked inhibitor of apoptosis protein and survivin correlate with poor overall survival in childhood de novo acute myeloid leukemia. Clin Cancer Res. 2004; 10(11):3737-3744.
16. Tamm I, Richter S, Scholz F, Schmelz K, Oltersdorf D, Karawajew L, Schoch C, Haferlach T, Ludwig WD and Wuchter C. XIAP expression correlates with monocytic differentiation in adult de novo AML: impact on prognosis. Hematol J. 2004; 5(6):489-495.
17. Mrozek K, Marcucci G, Nicolet D, Maharry KS, Becker H, Whitman SP, Metzeler KH, Schwind S, Wu YZ, Kohlschmidt J, Pettenati MJ, Heerema NA, Block AW, Patil SR, Baer MR, Kolitz JE, et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol. 2012; 30(36):4515-4523.
18. Sutherland HJ, Blair A and Zapf RW. Characterization of a hierarchy in human acute myeloid leukemia progenitor cells. Blood. 1996; 87(11):4754-4761.
19. Friend C, Scher W, Holland JG and Sato T. Hemoglobin synthesis in murine virus-induced leukemic cells in vitro: stimulation of erythroid differentiation by dimethyl sulfoxide. Proc Natl Acad Sci U S A. 1971; 68(2):378-382.
20. Sartorelli AC. The 1985 Walter Hubert lecture. Malignant cell differentiation as a potential therapeutic approach. Br J Cancer. 1985; 52(3):293-302.
21. D’Auria FD, Simonetti G and Strippoli V. [Antimicrobial characteristics of a tincture of dequalinium chloride]. Ann Ig. 1989; 1(5):1227-1241.
22. Della Casa V, Noll H, Gonser S, Grob P, Graf F and Pohlig G. Antimicrobial activity of dequalinium chloride against leading germs of vaginal infections. Arzneimittelforschung. 2002; 52(9):699-705.
23. Wilkinson JC, Cepero E, Boise LH and Duckett CS. Upstream regulatory role for XIAP in receptor-mediated apoptosis. Mol Cell Biol. 2004; 24(16):7003-7014.
24. Schimmer AD, Estey EH, Borthakur G, Carter BZ, Schiller GJ, Tallman MS, Altman JK, Karp JE, Kassis J, Hedley DW, Brandwein J, Xu W, Mak DH, LaCasse E, Jacob C, Morris SJ, et al. Phase I/II trial of AEG35156 X-linked inhibitor of apoptosis protein antisense oligonucleotide combined with idarubicin and cytarabine in patients with relapsed or primary refractory acute myeloid leukemia. J Clin Oncol. 2009; 27(28):4741-4746.
25. Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, Fukata M, Miyamoto T, Lyons B, Ohshima K, Uchida N, Taniguchi S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007; 25(11):1315-1321.
26. Tan BX, Khoo KH, Lim TM and Lane DP. High Mdm4 levels suppress p53 activity and enhance its half-life in acute myeloid leukaemia. Oncotarget. 2014; 5(4):933-943.
27. Cluzeau T, Robert G, Mounier N, Karsenti JM, Dufies M, Puissant A, Jacquel A, Renneville A, Preudhomme C, Cassuto JP, Raynaud S, Luciano F and Auberger P. BCL2L10 is a predictive factor for resistance to azacitidine in MDS and AML patients. Oncotarget. 2012; 3(4):490-501.
28. Bose P, Rahmani M and Grant S. Coordinate PI3K pathway and Bcl-2 family disruption in AML. Oncotarget. 2012; 3(12):1499-1500.
29. Jacque N and D B. Targeting glutamine uptake in AML. Oncoscience. 2014; 1(1):1-2.
30. Gallagher R, Collins S, Trujillo J, McCredie K, Ahearn M, Tsai S, Metzgar R, Aulakh G, Ting R, Ruscetti F and Gallo R. Characterization of the continuous, differentiating myeloid cell line (HL-60) from a patient with acute promyelocytic leukemia. Blood. 1979; 54(3):713-733.
31. Koeffler HP and Golde DW. Acute myelogenous leukemia: a human cell line responsive to colony-stimulating activity. Science. 1978; 200(4346):1153-1154.
32. Steube KG, Teepe D, Meyer C, Zaborski M and Drexler HG. A model system in haematology and immunology: the human monocytic cell line MONO-MAC-1. Leuk Res. 1997; 21(4):327-335.
33. Asou H, Tashiro S, Hamamoto K, Otsuji A, Kita K and Kamada N. Establishment of a human acute myeloid leukemia cell line (Kasumi-1) with 8;21 chromosome translocation. Blood. 1991; 77(9):2031-2036.