
INTRODUCTION
Colorectal cancer (CRC) is one of the most common and fatal forms of solid tumors in both men and women [1, 2]. Most CRC cases are sporadic, with about 15–25% having a family history [3, 4] and 5% diagnosed with inherited CRC syndrome [5]. Many genetic factors have been reported for their associations with the CRC syndrome [6]. For example, instability of the chromosomes and changes in the CpG island methylator phenotype may cause defects in the pathways, leading to CRC [6–8]. To date, a large number of somatic mutations have been identified in numerous genes for their involvement in colorectal cancer, such as KRAS [9, 10], PIK3CA [11], BRAF [12], MYH11 [13], and PARK2 [14]. Additionally, multiple mutations concurrently appearing in a gene may result in marked predisposition to CRC, such as those in the adenomatous polyposis coli (APC) gene, which is associated with familial adenomatous polyposis disease (FAP) [15]. Other examples of connections between CRC and genomic variations include mutations in genes such as MSH2 [16], MLH1 [17], PMS1 [18], TGFBR2 [19] etc, which are associated with hereditary nonpolyposis colorectal cancer (HNPCC), and those in the MUTYH gene, which are associated with autosomal recessive adenomatous polyposis [20]. However, the exact functions of the variations or mutations in these genes remain largely unknown.
APC is a key regulator in the Wnt/β-catenin signaling pathway, modulating the quantity ofβ-catenin in the cells. It plays key roles in several fundamental life processes, such as cell division, signal transduction, and tumor suppression [21, 22]. Several mutations and deletions in the APC gene and methylation in its promoter [23] have been described in FAP, and more than two thirds of CRC and adenomas patients have APC gene mutations [23–25]. Additionally, APC gene mutations have also been reported in tumors of other tissues and organs, such as the liver [4], stomach [5, 8, 26], lung [27], breast [28], and brain [29], further calling for the elucidation of the roles of these genetic changes in carcinogenesis [21, 30, 31].
In a previous study, we identified a novel mutation (p.1125Val>Ala) in the APC gene, which is associated with FAP and sporadic cases of CRC [32]. The polyposis syndrome is one of the most common syndromes associated with CRC [29, 30]. However, as many CRC cases do not present polyposis, and the associations between gene changes and the disease are still not established even though recent technical advances have demonstrated an apparent inheritance pattern of CRC [31, 33], contributions of specific changes in the APC gene to CRC have not been fully documented.
In this work, we profiled nucleotide changes of the APC gene and found that the SNP rs11954856 was associated with CRC. This SNP increased expression levels of genes in the Wnt/β-catenin signaling pathway in the CRC cancer patients, including APC, β-catenin, and transcription factor genes TCF7L1, TCF7L2, LEF1 in the pathway. Of particular significance, the p.1125Val>Ala mutation of the APC gene in the CRC cancer patients increased expression levels of not only genes downstream of APC in the Wnt/β-catenin signaling pathway but also a gene upstream of APC, GSK-3β, for feedback, providing further evidence indicating the involvement of the APC gene in the pathogenesis of CRC.
RESULTS
Clinical data
The clinical diagnosis was confirmed by three specialists in CRC in the Second Affiliated Hospital of Harbin Medical University, Harbin, China. There was no history of other systemic abnormalities of these CRC patients or previous tumor or familial history of tumor. All the CRC patients (n = 300, male 183, female 117, the min and max age were 16 and 87 respectively, and the average age was 58.59 years) and unrelated controls (n = 411, male 256, female 155, the min and max age were 50 and 70 respectively, and the average age was 59.39 years) were recruited specifically for this study, and there were no statistical differences in gender or age composition between the two groups (Supplementary Table 1).
SNP gene analyses
Using standard protocols, we extracted the genomic DNA from the peripheral blood leukocytes [34] and sequenced the APC gene to detect SNPs and test the hypothesis that germline common genetic variants in the gene may be associated with the susceptibility to CRC. We analyzed the SNPs distributed on the APC gene from the NCBI database (http://www.ncbi.nlm.nih.gov/) and eventually chose six of them to focus on, including rs11241185, rs11954856, rs2019720, rs2229992, rs2431238 and rs2289484 (Supplementary Figure 1A), based on the finding that the genetic heterozygosity of these six SNPs was high (Supplementary Figure 1B).
Polymorphism-disease association analyses
To test the hypothesized associations between APC variations and CRC, we conducted SNP analyses and found that the variant rs11954856 in the APC gene was associated with the risk of CRC in the Chinese Han population (Tables 1 and 2). We further analyzed the genotype frequencies in the CRC and control groups by three genetic models (allelic, dominant and recessive) and found that the variant rs11954856 was associated with the risk of CRC in allelic and dominant models (Table 3). On the other hand, we did not find statistical significance in the other analyzed APC gene SNPs rs11241185, rs2019720, rs2229992, rs2431238 and rs2289484 between the CRC and control groups (data not shown). We conducted the Hardy-Weinberg equilibrium test for the CRC and controls and the result was in line with equilibrium (Table 4). We also compared the genotype frequency of the rs11954856 in the CRC, control groups and the data from the HapMap HCB population, and the frequency in the control group was more consistent with the data from the HapMap HCB population (Table 5).
Table 1: The genotype and allele frequency of rs11954856 variations in 300 Chinese Han sporadic colorectal cancer patients and 411 non-CRC controls
Group |
|
Genotype frequency (%) |
Allele frequency (%) |
|||
|---|---|---|---|---|---|---|
Genotype |
|
G/G |
G/T |
T/T |
G |
T |
CRC |
300 |
179(59.7) |
105(35.0) |
16(5.3) |
463(77.2) |
137(22.8) |
Controls |
411 |
283(68.9) |
116(28.2) |
12(2.9) |
682(83.0) |
140(17.0) |
Table 2: rs11954856 variation within APC gene associated with risk of sporadic colorectal cancer in Chinese populations
Variations |
Type |
|
Pearson Chi-square |
|
Risk |
|||
|---|---|---|---|---|---|---|---|---|
|
|
Value |
Min counta |
df |
Asymp. Sig. |
OR |
95%CI-Up |
95%CI-Low |
rs11954856 |
Genotype |
7.381a |
11.81 |
2 |
0.025 |
-- |
-- |
-- |
|
Allele |
7.443a |
116.88 |
1 |
0.006 |
0.694 |
0.533 |
0.903 |
a: The minimum expected count; b: Not assuming the null hypothesis; c: Using the asymptotic standard error assuming the null hypothesis; d: Based on normal approximation.
Table 3: SNP rs11954856 variation within APC gene associated with risk of sporadic colorectal cancer in allelic and dominant model
Value |
Allelic model |
Dominant model |
Recessive model |
|---|---|---|---|
ChisQ |
7.443 |
6.436 |
2.671 |
P |
0.0064 |
0.0112 |
0.1022 |
Table 4: The CRC and controls groups were in line with Hardy-Weinberg equilibrium
Group |
|
Genotype frequency (%) |
H-W equilibrium testing |
||||
|---|---|---|---|---|---|---|---|
Genotype |
|
G/G |
G/T |
T/T |
0 (HET) |
E (HET) |
P |
CRC |
300 |
179(59.7) |
105(35.0) |
16(5.3) |
0.3500 |
0.3524 |
0.8708 |
Controls |
411 |
283(68.9) |
116(28.2) |
12(2.9) |
0.2822 |
0.2826 |
1.0000 |
Table 5: The frequency in control group was more consistent with the data from the HapMap HCB population
Group |
Genotype frequency (%) |
||
|---|---|---|---|
Genotype |
G/G |
G/T |
T/T |
CRC |
0.597 |
0.350 |
0.160 |
Controls |
0.689 |
0.282 |
0.029 |
HCB data |
0.682 |
0.295 |
0.023 |
Gene expression analysis
We used Western blotting analysis to measure the expression levels of APC gene and related genes in the Wnt/β-catenin signaling pathway, including β-catenin, TCF7L1, TCF7L2, LEF1, MMP7, C-myc, C-jun, CYCLIND1 and GSK-3β in both cancer and normal tissues, for the patients who may have the wild or mutation types of the genes, the latter being either heterozygous or homozygous. We used the p.1125Val>Ala mutation in the APC gene, which is associated with the FAP syndrome [32], for the positive control. We found that the expression levels of the genes in the Wnt/β-catenin signaling pathway, including APC, β-catenin, TCF7L1, TCF7L2, LEF1, MMP7, C-myc, C-jun, CYCLIND1 and GSK-3β, were remarkably higher in cancer than non-cancer tissues in the p.1125Val>Ala mutant FAP family members (Figure 1A, 1B). Of significant importance, homozygous variation of the APC gene SNP rs11954856 was associated with higher expression levels of the APC andβ-catenin genes in the cancer tissue (Figure 2A, 2B, 2C). Notably, in patients with wild type or heterozygous variation types, the expression levels of the two genes in the normal tissue were higher than those in the cancer tissue (Figure 2A, 2B, 2C). The expression levels of cell cycle proteins MMP7, C-myc, C-jun, CYCLIND1 and β-catenin degradation protein GSK-3β in the Wnt/β-catenin signaling pathway were higher in cancer than in non-cancer tissues in all the wild type and the homozygous and heterozygous variations of the patients (Figure 2A, 2G and Figure 3A, 3B, 3C, 3D, 3E). Conversely, the expression levels of transcription factors TCF7L1, TCF7L2 and LEF1 in the Wnt/β-catenin signaling pathway were higher in cancer tissue in both homozygous and heterozygous variation types of the patients (Figure 2A, 2D, 2E, 2F).
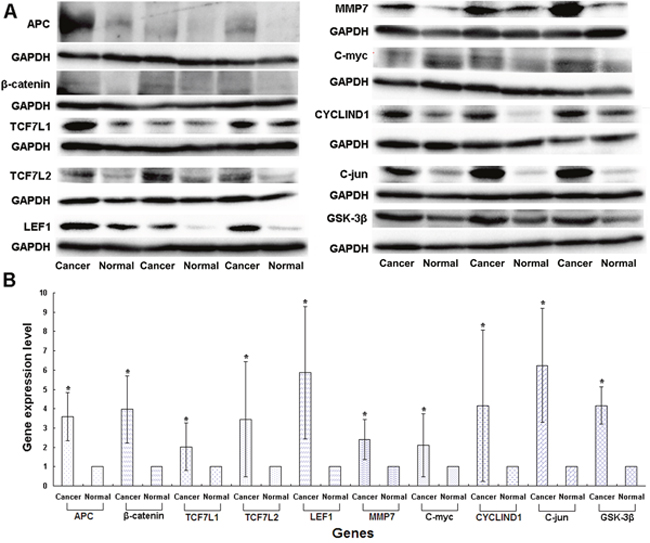
Figure 1: Expression levels of genes in the Wnt/β-catenin signaling pathway in CRC patients detected in the p.1125Val>Ala mutant FAP family patients by Western blotting. (A) Original experimental results; (B) Numerical experimental results with the band values in the original experimental pictures read by the image J software. The protein expression levels were normalized to GADPH.
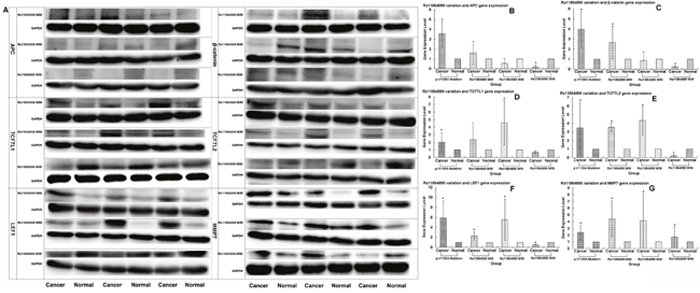
Figure 2: Expression levels of APC, β-catenin, TCF7L1, TCF7L2, LEF1 and MMP7 genes detected in patients with rs11954856 wild type or heterozygous or homozygous variations by Western blotting. (A) Original experimental results; (B, C, D, E, F, G) Numerical experimental results with the band values in the original experimental pictures read by the image J software. The protein expression levels were normalized to GADPH.
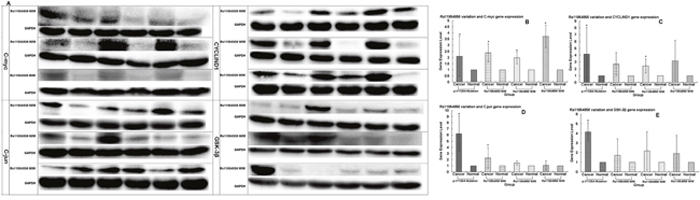
Figure 3: Expression levels of C-myc, C-jun, CYCLIND1 and GSK-3β genes detected in patients with wild type or heterozygous or homozygous variations by Western blotting. (A) Original experimental results; (B, C, D, E) Numerical experimental results with the band value in the original experimental pictures read by the image J software. The protein expression levels were normalized to GADPH.
Comparative analysis of clinical features
We also compared the clinical characteristicsbetween the wild type, heterozygous variant and homozygous variant groups of the CRC patients. We found statistically significant differences between the three groups in stool occult blood and CA199 but not in other clinical characteristics, such as gender composition, age, white blood cell counts, CEA, TNM stage, tumor sites, or pathological types and grades etc. (Table 6). Multiple testing assays on the three groups demonstrated statistically significant differences between the wild type, heterozygous, and homozygous variant groups of the CRC patients in stool occult blood (Table 6). For CA199, however, we found statistically significant differences only between the wild type and heterozygous variant groups (Table 6).
Table 6: Comparative analysis of clinical features between wild type, heterozygous variation and homozygous variation groups
Clinical Index |
Wild Type |
heterozygous variation |
homozygous variation |
Chi-Square test |
|---|---|---|---|---|
Gender (Male/Female) |
105/74 |
65/40 |
13/3 |
P=0.201 |
Blood in stool (Yes/No) |
119/60 |
56/49 |
5/11 |
P=0.005 |
BSMT |
WT-HE P=0.028 |
HE-HO P=0.100 |
WT-HO P=0.005 |
—— |
Smoking (Yes/No) |
64/115 |
35/70 |
6/10 |
P=0.897 |
Drinking (Yes/No) |
59/120 |
45/60 |
8/8 |
P=0.140 |
Type (Rectum/Colon) |
97/82 |
53/52 |
11/5 |
P=0.384 |
Age |
59.64±12.09 |
59.24±12.79 |
55.09±15.35 |
P=0.512 |
White Blood Cells |
6.69±2.35 |
6.71±2.27 |
7.83±2.72 |
P=0.299 |
NGP |
61.54±10.94 |
60.73±10.04 |
64.96±14.33 |
P=0.474 |
Hemoglobin |
126.81±26.31 |
127.69±23.53 |
130.64±16.61 |
P=0.876 |
Platelets |
251.02±86.04 |
242.45±72.04 |
244.64±74.08 |
P=0.760 |
ALT |
16.24±10.23 |
17.19±11.32 |
14.36±5.95 |
P=0.652 |
AST |
18.77±7.78 |
18.49±8.07 |
16.00±4.86 |
P=0.524 |
Prealbumin (SPA) |
180.68±75.07 |
171.99±64.97 |
192.30±70.56 |
P=0.569 |
Albumin |
40.51±6.42 |
44.85±41.60 |
40.13±4.97 |
P=0.471 |
Creatinine |
75.77±19.03 |
76.71±17.87 |
80.57±33.00 |
P=0.720 |
CEA |
17.92±87.83 |
21.04±71.96 |
3.58±3.06 |
P=0.769 |
CA199 |
36.95±133.43 |
107.30±279.63 |
14.05±12.97 |
P=0.034 |
CA199MT |
WT-HE P=0.013 |
HE-HO P=0.138 |
WT-HO P=0.707 |
—— |
TNM Stage (I/II/III/IV) |
25/85/58/11 |
6/56/38/5 |
0/12/4/0 |
P=0.115 |
Tumor Sites (left/right) |
133/46 |
73/32 |
15/1 |
P=0.117 |
Pathological Types (PU/PP) |
97/82 |
65/40 |
10/6 |
P=0.408 |
Pathological Grades (H/M/L/MA) |
21/148/4/6 |
9/88/4/4 |
4/12/0/0 |
P=0.522 |
NGP: neutrophilic granulocyte percentage; BSMT: blood in stool-multiple testing; CA199MT: CA199-multiple testing; PU: pathological ulcerative type; PP: pathological protuberant type; H: pathological high grades; M: pathological moderately grades; L: pathological low grades; MA: mucinous adenocarcinoma.
DISCUSSION
In this study, we found that the APC gene SNP rs11954856 was associated with colorectal cancer and increased the expression levels of genes in the Wnt/β-catenin signaling pathway in the CRC patients (Figure 4). Of remarkable significance, the p.1125Val>Ala mutation in the APC gene, previously reported for its association with the FAP syndrome [32], also increased the expression levels of all the genes downstream of APC gene in the Wnt/β-catenin signaling pathway in the CRC patients, including APC, β-catenin, TCF7L1, TCF7L2, LEF1, MMP7, C-myc, C-jun, and CYCLIND1, and GSK-3β for feedback located upstream of the APC gene (Figure 4).
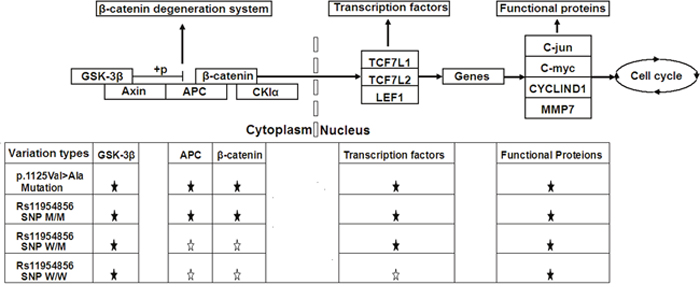
Figure 4: Schematic diagrams of Wnt/β-catenin signaling pathway. Shown here are influences of rs11954856 and the p.1125Val>Ala mutation in APC gene on the expression levels of genes in the pathway in CRC patients. ★ denotes statistically significant and ☆ denotes no statistical differences.
As an important integral part of the Wnt/β-catenin signaling pathway, the protein encoded by the APC gene plays key roles in tumor suppression by antagonizing the pathway [23]. Loss of APC functions can lead to inappropriate activation of the Wnt/β-catenin signaling pathway and thus may facilitate carcinogenesis [35]. Additionally, APC also has important roles in cell functions, such as migration, adhesion, chromosomal segregation, spindle assembly, apoptosis and differentiation [23]. The sequence about 40 kb upstream of the initiator codon is the promoter of the APC gene [36] and its hypermethylation is associated with dysfunction of the Wnt/β-catenin signaling pathway [37]. Transcripts bearing the untranslated exon of APC are spliced to either exon1 or exon2, which can be detected in mouse embryonic stem cells and all mouse tissues, demonstrating the housekeeping feature of these sequences [36]. We found in this study that the SNP rs11954856, located before the exon1 and within the 3,261 site upstream of the initiating methionine of the APC gene, was associated with colorectal cancer.
Compared to the p.1125Val>Ala mutation in the APC gene, which increases the expression of not only the genes downstream of the APC gene in the signaling pathway but also the GSK-3β gene upstream of the APC gene, rs11954856 increased the expression of only APC and β-catenin in the CRC cancer patients, with lesser influences on those of the transcription factors TCF7L1, TCF7L2 and LEF1. The amino acids from 1265 to 2035 in the APC protein form the domain for bindingβ-catenin, which is important for β-catenin degradation [38, 39]. Since the variations in the binding domain may increase APC binding withβ-catenin, this domain has been believed to have key roles in the pathogenesis of CRC especially FAP [40]. The results of the present work provided new support for the involvement of APC in the pathogenesis of CRC.
The distinct influences between the SNP rs11954856 and mutation p.1125Val>Ala on the expression levels of genes in the Wnt/β-catenin signaling pathway in the CRC cancer patients may be associated with their different locations in the APC gene or different features of particular SNPs/mutations or both. Nucleotide changes may inactivate APC gene and in the meantime activate or stabilize β-catenin, resulting in the permanent activation of the Wnt/β-catenin signaling pathway, and the silenced β-catenin may lead to decreased colonosphere formation, which is an important feature in the tumorigenesis [41]. When TCF/LEF and C-myc are hyperactivated, the formation of colonospheres is increased [42]. Such events in stem cells may result in increased tumorigenesis and cause CRC [43]. Our findings in the present work are consistent with the previous reports [41–43], such as the APC gene p.1125Val>Ala mutation increasing the expression levels of all the genes in the Wnt/β-catenin signaling pathway in the CRC cancer patients (Figure 4).
The main features of the gastrointestinal epithelium cells include rapid cell proliferation and differentiation [43], so mutations may accumulate quickly, with some contributing to carcinogenesis. Additionally, other factors, such as advanced age [44], smoking [45], unhealthy lifestyle and polluted environment also play important roles in the pathogenesis of CRC syndrome [6]. The genes in the Wnt/β-catenin signaling pathway are affected by multiple genetic factors. For example, the expression of C-myc can be suppressed by hyperactivation of cell checkpoint genes such as CDKN1A, CDKN1B and CDKN2B [46, 47]. Therefore, the increased expression levels of cell cycle functional proteins MMP7, C-myc, C-jun and CYCLIND1 in the Wnt/β-catenin signaling pathway that we observed here in the cancer tissue (Figure 4) may have contributions also from other genes or factors.
In conclusion, we found that the APC gene SNP rs11954856 was associated with CRC by increasing expression levels of genes in the Wnt/β-catenin signaling pathway in the CRC cancer patients, compared to the p.1125Val>Ala mutation in APC, which increased expression levels of not only all the genes downstream of APC in the Wnt/β-catenin signaling pathway in CRC but also GSK-3β upstream of APC. Recently, some researchers have found that the SNP rs11954856 is associated with the risk of ovarian and gallbladder cancers [48, 49]. All such findings indicate the importance of the SNP rs11954856 in pathogenesis of cancer and may help develop new strategies for the treatment of cancers or new biomarkers for evaluation or diagnosis of colorectal cancer.
MATERIALS AND METHODS
Study population
We included 300 sporadic CRC cases and 411 normal controls (Supplementary Table 1) for this study, which were assembled at the Department of Colorectal Surgery and Medical Examination Center of the Second Affiliated Hospital of Harbin Medical University, Harbin, China. We obtained a written informed consent from each participant or guardian, and this work has been reviewed and approved by the Ethics Committee of Harbin Medical University. We also confirmed that all experiments were performed in accordance with relevant guidelines and regulations, consistent with the 1975 Declaration of Helsinki. Medical histories were recorded in detail for all the enrolled participants, and all participants received physical and enteroscopic examinations.
DNA analysis
We extracted genomic DNA from peripheral blood leukocytes of each participant using standard protocols [50]. The APC gene was amplified by polymerase chain reaction (PCR) with the primers (Supplementary Table 2) for analysis of SNPs. PCR products were sequenced using standard protocols [51] for genotype analysis.
APC SNP genotyping and statistical analysis
The variations of rs11241185, rs11954856, rs2019720, rs2229992, rs2431238 and rs2289484 within the APC gene (Supplementary Figure 1A) were determined for 300 sporadic colorectal cancer cases and 411 normal controls. We amplified the APC gene and sequenced the PCR products to determine the genotypes (Supplementary Figure 1B).
The statistical analyses were conducted using the SPSS software (version 19.0) and PLINK v1.07 software (http://pngu.mgh.harvard.edu/Bpurcell/plink/) by the methods as previous reported [52, 53]. P values less than 0.05 were considered statistically significant. The Hardy-Weinberg equilibrium test of the CRC and control populations was conducted with the online software OEGE [54].
Western blotting analysis
Proteins of the tumor and normal tissues near the tumor were extracted using standard protocols, and the contents were determined by the BCA protein assay kit (from BOSTER) and ELISA. The proteins were separated by 8% SDS-PAGE and transferred to PVDF membrane. The membranes were then incubated with the primary antibodies against the proteins, including APC (No.ab58, Abcam, Cambridge, USA), β-catenin (No.ab32572, Abcam, Cambridge, USA), TCF7L1 (No.ab133360, Abcam, Cambridge, USA), TCF7L2 (No.ab76151, Abcam, Cambridge, USA), LEF1 (No.ab137872, Abcam, Cambridge, USA), C-myc (No.sc40, Santa, California, USA), C-jun (No.ab32137, Abcam, cambridge, USA), CYCLIND1(No.ab134175, Abcam, cambridge, USA), MMP7 (No.ab205525, Abcam, cambridge, USA), GSK-3β (No.sc53931, Santa, California USA) and GAPDH (No.ta08, ZSGB-BIO, Beijing China) in 5% non-fat milk in TBST at room temperature for two hours. After washing for three times using TBST, the membranes were incubated with secondary antibodies (No.zdr5306 and 5307, ZSGB-BIO, Beijing China) at room temperature for two hours. Then the membranes were developed using the enhanced chemiluminescence plus reagent and imaged using the Bio-Rad gel imaging system [55]. Finally, the band values were read using the image J software.
Author contributions
Conceptualization: FF L, SL L; methodology: FF L, SL L, CH S, XS W, GY W; software: FF L, SL L, Q Z; formal analysis: FF L, SL L, Q Z; investigation: ZU Z, P Y, S W, Z L, XN Z; resources: ZU Z, P Y, S W, Z L, XS W, GY W; data curation: FF L, SL L, CH S, XS W, GY W; writing: FF L, SL L; funding acquisition: FF L, SL L, XS W, GY W.
ACKNOWLEDGMENTS
The authors thank the patients and their families for their cooperation and participation in this study.
CONFLICTS OF INTEREST
All the authors have declared that no competing interest exist.
FINANCIAL SUPPORT
There are no financial disclosures from any authors.
FUNDING
This work was supported by grant from Chinese Postdoctoral Science Foundation (2016M601460), and grants of National Natural Science Foundation of China (NSFC81271786, 81030029, 81671980), Health and Family Planning Commission of Heilongjiang province Foundation (2016-189), Heilongjiang Innovation Research Foundation for College Students (201610226096), Heilongjiang province postdoctoral Science Foundation (LBH-Z14175). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ethics approval
Ethics Committee of Harbin Medical University.
Informed consent
Informed consent was obtained from all individual participants included in the study.
REFERENCES
1. Knopperts AP, Nielsen M, Niessen RC, Tops CM, Jorritsma B, Varkevisser J, Wijnen J, Siezen CL, Heine-Broring RC, van Kranen HJ, Vos YJ, Westers H, Kampman E, et al. Contribution of bi-allelic germline MUTYH mutations to early-onset and familial colorectal cancer and to low number of adenomatous polyps: case-series and literature review. Fam Cancer. 2013; 12:43-50.
2. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64:9-29.
3. Lovett E. Family studies in cancer of the colon and rectum. Br J Surg. 1976; 63:13-18.
4. Stephenson BM, Finan PJ, Gascoyne J, Garbett F, Murday VA, Bishop DT. Frequency of familial colorectal cancer. Br J Surg. 1991; 78:1162-1166.
5. Tops CM, Wijnen JT, Hes FJ. Introduction to molecular and clinical genetics of colorectal cancer syndromes. Best Pract Res Clin Gastroenterol. 2009; 23:127-146.
6. Schweiger MR, Hussong M, Rohr C, Lehrach H. Genomics and epigenomics of colorectal cancer. Wiley Interdiscip Rev Syst Biol Med. 2013; 5:205-219.
7. Kok-Sin T, Mokhtar NM, Ali Hassan NZ, Sagap I, Mohamed Rose I, Harun R, Jamal R. Identification of diagnostic markers in colorectal cancer via integrative epigenomics and genomics data. Oncol Rep. 2015; 34:22-32.
8. Jarvinen HJ. Hereditary cancer: guidelines in clinical practice. Colorectal cancer genetics. Ann Oncol. 2004; 15:iv127-131.
9. Kranenburg O. The KRAS oncogene: past, present, and future. Biochim Biophys Acta. 2005; 1756:81-82.
10. Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, Settleman J, Giovannini M, Jacks T. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008; 40:600-608.
11. Gustin JP, Karakas B, Weiss MB, Abukhdeir AM, Lauring J, Garay JP, Cosgrove D, Tamaki A, Konishi H, Konishi Y, Mohseni M, Wang G, Rosen DM, et al. Knockin of mutant PIK3CA activates multiple oncogenic pathways. Proc Natl Acad Sci U S A. 2009; 106:2835-2840.
12. Millington GW. Mutations of the BRAF gene in human cancer, by Davies et al. (Nature 2002; 417: 949-54). Clin Exp Dermatol. 2013; 38:222-223.
13. Alhopuro P, Phichith D, Tuupanen S, Sammalkorpi H, Nybondas M, Saharinen J, Robinson JP, Yang Z, Chen LQ, Orntoft T, Mecklin JP, Jarvinen H, Eng C, et al. Unregulated smooth-muscle myosin in human intestinal neoplasia. Proc Natl Acad Sci U S A. 2008; 105:5513-5518.
14. Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W, Ladanyi M, Sander C, Heguy A, et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet. 2010; 42:77-82.
15. Homma MK, Li D, Krebs EG, Yuasa Y, Homma Y. Association and regulation of casein kinase 2 activity by adenomatous polyposis coli protein. Proc Natl Acad Sci U S A. 2002; 99:5959-5964.
16. Lamers MH, Perrakis A, Enzlin JH, Winterwerp HH, de Wind N, Sixma TK. The crystal structure of DNA mismatch repair protein MutS binding to a G x T mismatch. Nature. 2000; 407:711-717.
17. Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM, Adams MD, Venter JC, Hamilton SR, Petersen GM, et al. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994; 263:1625-1629.
18. Liu T, Yan H, Kuismanen S, Percesepe A, Bisgaard ML, Pedroni M, Benatti P, Kinzler KW, Vogelstein B, Ponz de Leon M, Peltomaki P, Lindblom A. The role of hPMS1 and hPMS2 in predisposing to colorectal cancer. Cancer Res. 2001; 61:7798-7802.
19. Tannergard P, Liu T, Weger A, Nordenskjold M, Lindblom A. Tumorigenesis in colorectal tumors from patients with hereditary non-polyposis colorectal cancer. Hum Genet. 1997; 101:51-55.
20. Sampson JR, Dolwani S, Jones S, Eccles D, Ellis A, Evans DG, Frayling I, Jordan S, Maher ER, Mak T, Maynard J, Pigatto F, Shaw J, Cheadle JP. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003; 362:39-41.
21. Bodmer WF, Bailey CJ, Bodmer J, Bussey HJ, Ellis A, Gorman P, Lucibello FC, Murday VA, Rider SH, Scambler P, Sheer∥ D, Solomon E, Spurret NK. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature. 1987; 328:614-616.
22. Schneikert J, Behrens J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut. 2007; 56:417-425.
23. Hanson CA, Miller JR. Non-traditional roles for the Adenomatous Polyposis Coli (APC) tumor suppressor protein. Gene. 2005; 361:1-12.
24. Nagase H, Nakamura Y. Mutations of the APC (adenomatous polyposis coli) gene. Hum Mutat. 1993; 2:425-434.
25. Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1992; 1:229-233.
26. Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Clendenning M, Sotamaa K, Prior T, Westman JA, Panescu J, Fix D, Lockman J, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008; 26:5783-5788.
27. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004; 96:261-268.
28. Lynch HT, Lynch JF. What the physician needs to know about Lynch syndrome: an update. Oncology (Williston Park). 2005; 19:455-463; discussion 463-454, 466, 469.
29. Farrington SM, Tenesa A, Barnetson R, Wiltshire A, Prendergast J, Porteous M, Campbell H, Dunlop MG. Germline susceptibility to colorectal cancer due to base-excision repair gene defects. Am J Hum Genet. 2005; 77:112-119.
30. Cleary SP, Cotterchio M, Jenkins MA, Kim H, Bristow R, Green R, Haile R, Hopper JL, LeMarchand L, Lindor N, Parfrey P, Potter J, Younghusband B, Gallinger S. Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology. 2009; 136:1251-1260.
31. Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomaki P, Chadwick RB, Kaariainen H, Eskelinen M, Jarvinen H, Mecklin JP, de la Chapelle A. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med. 1998; 338:1481-1487.
32. Li FF, Liu Z, Yan P, Shao X, Deng X, Sam C, Chen YG, Xu YP, Wang XS, Wang GY, Liu SL. Identification of a novel mutation associated with familial adenomatous polyposis and colorectal cancer. Int J Mol Med. 2015; 36:1049-1056.
33. Burt RW, Bishop DT, Cannon LA, Dowdle MA, Lee RG, Skolnick MH. Dominant inheritance of adenomatous colonic polyps and colorectal cancer. N Engl J Med. 1985; 312:1540-1544.
34. Tan ZX, Li FF, Qu YY, Liu J, Liu GR, Zhou J, Zhu YL, Liu SL. Identification of a known mutation in Notch 3 in familiar CADASIL in China. PLoS One. 2012; 7:e36590.
35. Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007; 21:2525-2538.
36. Karagianni N, Ly MC, Psarras S, Chlichlia K, Schirrmacher V, Gounari F, Khazaie K. Novel adenomatous polyposis coli gene promoter is located 40 kb upstream of the initiating methionine. Genomics. 2005; 85:231-237.
37. Wang ZK, Liu J, Liu C, Wang FY, Chen CY, Zhang XH. Hypermethylation of adenomatous polyposis coli gene promoter is associated with novel Wnt signaling pathway in gastric adenomas. J Gastroenterol Hepatol. 2012; 27:1629-1634.
38. Sturt NJ, Gallagher MC, Bassett P, Philp CR, Neale KF, Tomlinson IP, Silver AR, Phillips RK. Evidence for genetic predisposition to desmoid tumours in familial adenomatous polyposis independent of the germline APC mutation. Gut. 2004; 53:1832-1836.
39. Schiessling S, Kihm M, Ganschow P, Kadmon G, Buchler MW, Kadmon M. Desmoid tumour biology in patients with familial adenomatous polyposis coli. Br J Surg. 2013; 100:694-703.
40. Polakis P. The adenomatous polyposis coli (APC) tumor suppressor. Biochim Biophys Acta. 1997; 1332:F127-147.
41. Kanwar SS, Yu Y, Nautiyal J, Patel BB, Majumdar AP. The Wnt/beta-catenin pathway regulates growth and maintenance of colonospheres. Mol Cancer. 2010; 9:212.
42. van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, Tjon-Pon-Fong M, Moerer P, van den Born M, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002; 111:241-250.
43. Abetov D, Mustapova Z, Saliev T, Bulanin D. Biomarkers and signaling pathways of colorectal cancer stem cells. Tumour Biol. 2015; 36:1339-1353.
44. U.S. Preventive Services Task Force. Screening for colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2008; 149:627-637.
45. Morrison DS, Batty GD, Kivimaki M, Davey Smith G, Marmot M, Shipley M. Risk factors for colonic and rectal cancer mortality: evidence from 40 years’ follow-up in the Whitehall I study. J Epidemiol Community Health. 2011; 65:1053-1058.
46. Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000; 103:295-309.
47. Siegel PM, Shu W, Massague J. Mad upregulation and Id2 repression accompany transforming growth factor (TGF)-beta-mediated epithelial cell growth suppression. J Biol Chem. 2003; 278:35444-35450.
48. Mostowska A, Pawlik P, Sajdak S, Markowska J, Pawalowska M, Lianeri M, Jagodzinski PP. An analysis of polymorphisms within the Wnt signaling pathway in relation to ovarian cancer risk in a Polish population. Mol Diagn Ther. 2014; 18:85-91.
49. Yadav A, Gupta A, Yadav S, Rastogi N, Agrawal S, Kumar A, Kumar V, Misra S, Mittal B. Association of Wnt signaling pathway genetic variants in gallbladder cancer susceptibility and survival. Tumour Biol. 2016; 37:8083-8095.
50. Deng X, Zhou J, Li FF, Yan P, Zhao EY, Hao L, Yu KJ, Liu SL. Characterization of nodal/TGF-lefty signaling pathway gene variants for possible roles in congenital heart diseases. PLoS One. 2014; 9:e104535.
51. Li FF, Wang X, Zhu M, Xiao H, Yang Q, Shao X, Feng H, Lin G, Liu L. Identification of a location at chromosome 19p in a big Chinese family with charcot-marie-tooth disease. J Mol Biomark Diagn. 2013; 4: 144. https://doi.org/10.4172/2155-9929.1000144.
52. Li FF, Zhou J, Zhao DD, Yan P, Li X, Han Y, Li XS, Wang GY, Yu KJ, Liu SL. Characterization of SMAD3 gene variants for possible roles in ventricular septal defects and other congenital heart diseases. PLoS One. 2015; 10:e0131542.
53. Li FF, Han Y, Shi S, Li X, Zhu XD, Zhou J, Shao QL, Li XQ, Liu SL. Characterization of transcriptional repressor gene MSX1 variations for possible associations with congenital heart diseases. PLoS One. 2015; 10:e0142666.
54. Rodriguez S, Gaunt TR, Day IN. Hardy-Weinberg equilibrium testing of biological ascertainment for Mendelian randomization studies. Am J Epidemiol. 2009; 169:505-514.
55. Li FF, Yan P, Zhao ZX, Liu Z, Song DW, Zhao XW, Wang XS, Wang GY, Liu SL. Polymorphisms in the CHIT1 gene: associations with colorectal cancer. Oncotarget. 2016; 7:39572-39581. https://doi.org/10.18632/oncotarget.9138.