
Sex hormone signaling in the endometrium
The inner layer of the human uterus, the endometrium is a dynamic tissue that undergoes hundreds of cycles of proliferation, differentiation and shedding during a woman’s reproductive years [1]. The endometrium can be divided into two layers, the functionalis and the basalis. Whereas the functionalis layer comprises the upper two-thirds of the endometrium and is shedded during menstruation, the basalis includes the lower one-third which remains intact and is responsible for producing a new functionalis during each subsequent menstrual cycle [1] [2].
It is the fine balance between the activities of the two female sex hormones, estradiol and progesterone, which determines lineage faith in the endometrium. During the first two weeks of the menstrual cycle the thecal cells of the ovary produce large amounts of estradiol. Simultaneously, endometrial estrogen receptor (ER) levels (mainly ERα) are increased [3]. Upon ligand binding, ER dimers will translocate to the nucleus where they activate transcription of downstream target genes (e.g. insulin like growth factor; IGF-1), that stimulate endometrial proliferation [4]. Later, during the third and fourth week of the menstrual cycle, the corpus luteum starts producing progesterone thus inhibiting estradiol-induced proliferation of endometrial cells and stimulating cellular differentiation [5] [6]. If no fertilized oocytes are implanted in the uterus, the corpus luteum cannot be maintained due to lack of human chorion gonadotropine (HCG), and both estrogen and progesterone levels will decline. Withdrawal of progesterone leads to endometrial cell apoptosis and tissue breakdown of the functionalis of the endometrium, resulting in its shedding from the uterus [7].
The inhibition of estrogens’ mitotic activity exerted by progesterone is of clinical relevance as unopposed or increased estrogen action in women is a well-established risk factor for endometrial cancer [8] [9].
Unbalanced sex hormone signaling can induce endometrial cancer
Endometrial cancer is one of the most common cancers of the female genital tract [10]. It accounts each year for approximately 142.000 new cases diagnosed worldwide, and for 42.000 deaths [11] [12]. Endometrial cancer is the seventh most common malignant disorder and its incidence is expected to increase in the near future due to the increase in life span expectancy and obesity [13].
Based on epidemiology, conventional histopathology, and clinical behavior, endometrial carcinoma can be divided into two subtypes. Type I endometrial cancer, comprising approximately 85% of the total endometrial carcinoma burden among western societies, resembles normal endometrial hyperplasia in morphology and is associated with increased or unopposed estrogen signaling [12]. Type I endometrial cancer often shows mutations in the PTEN and in DNA mismatch repair genes (MLH1, MSH2, MSH6). Also, oncogenic mutations in KRAS and/or CTNNB1 (β-catenin) are recognized major alterations [14] [15] [16]. Type II endometrial cancer occurs predominantly in older post-menopausal women, is not correlated to increased estrogen exposure, and is generally associated with a poorer prognosis. Type II endometrial cancers often show mutations in TP53 and ERBB2 (Her-2/neu) [12] [15] [16].
In western, industrialized countries, two large groups of women are at increased risk of developing endometrial cancer: (i) women with significant overweight [13], and (ii) those receiving tamoxifen for breast cancer treatment [17]. Tamoxifen is a selective estrogen receptor modulator (SERM) acting as an antiestrogen in mammary tissue, but showing estrogenic activity in the endometrium [18]. Currently, it is estimated that up to 40% of all endometrial cancers could be related to obesity [13]. Since the prevalence of obesity is increasing, the incidence of obesity-related endometrial cancer is also on the rise [19]. Additional risk factors for the development of endometrial cancer include: polycystic ovarian syndrome, skipping menstrual periods, being nulliparous, late menopause onset, and the use of unopposed estrogen as hormone substitution therapy [20]. Hence, enhanced estrogen signaling for prolonged periods of time, also as the result of insufficient progesterone levels, represents the main risk factor in endometrial cancer.
Progesterone antagonizes the proliferative activity of estrogen by inducing epithelial and stromal cell differentiation in the endometrium [21] [22]. In fact, although progesterone (in the form of medroxyprogesterone acetate (MPA)) can be used for the palliative treatment of well-advanced and recurrent endometrial cancer, this treatment has a modest response rate (15-25%) [23]. In contrast, when progesterone, is employed as a primary treatment (e.g. in pre-menopausal women suffering from well-differentiated endometrial cancer and determined to preserve fertility), response rates are considerably improved (up to 60% or more) [24] [25].
Progesterone inhibition of estrogen-induced proliferation
The mechanisms by which progesterone induces its anti-proliferative effect on the endometrium are still largely unknown. Several studies have shown a direct effect of progesterone on estrogen receptor signaling and expression levels [26], while others show that progesterone affects the availability of bioactive estrogens [27] [28] [29]. Furthermore, differential expression of the progesterone receptor (PR) isoforms (PRA and PRB) and the relative expression levels of ERα compared to ERβ could also play a role in controlling endometrial proliferation during the menstrual cycle [30] [27].
In order to investigate in more detail the molecular mechanism(s) underlying progesterone-driven inhibition of estrogen signaling, expression profiling analysis was conducted on endometrial tissue specimens obtained from postmenopausal women treated for three weeks with estrogens (E-only) or estrogens+progestagens (E+P) and compared to untreated controls [21] [22]. The endometrial gene profiles obtained from this study were also compared with endometrial gene expression levels of untreated women during the different stages of the menstrual cycle [4]. Upon reviewing regulation of expression of the twenty most significantly estrogen-regulated genes (Fig. 1), it was observed that most of them were completely counterbalanced by concurrent administration of progestagens (E+P). However, estrogen-induced upregulation of specific genes (e.g. IGF1 and IGFBP5) is only partly counterbalanced by progestagens, whereas in other cases (e.g. SCGB1D2) the increase of expression level upon estrogen signaling is not at all counterbalanced by progestagens. Notably, these genes also behave differently during the menstrual cycle: IGF1 and IGFB5 are upregulated during the proliferative, early and mid secretory phases of the menstrual cycle, whereas SCGB1D2 is upregulated exclusively during the early and mid secretory phases (Fig. 1).
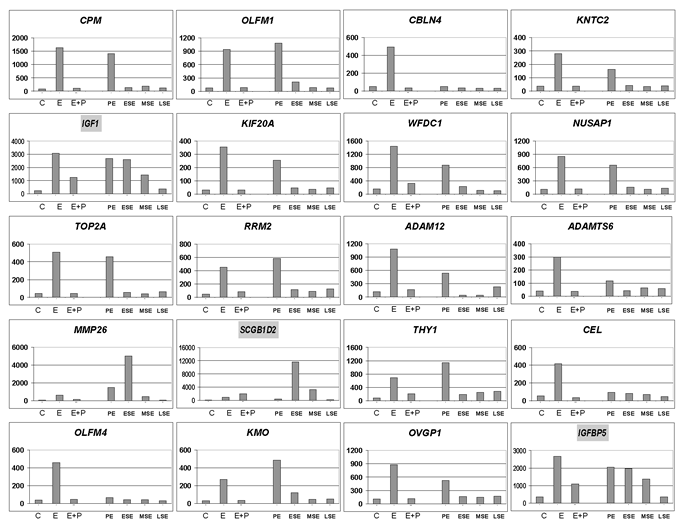
Figure 1: Inhibition of estrogen-induced genes by progesterone. The data employed in this figure were obtained by combining endometrial gene expression profiles from postmenopausal women after no treatment (C), 21-day treatment with estradiol (E), or estradiol + medroxyprogesterone acetate (E+P) [22], with gene expression profiles from the proliferative endometrium (PE), early secretory endometrium (ESE), mid secretory endometrium (MSE), or late secretory endometrium (LSE) [4]. The bars represent the relative expression levels of the indicated genes under the indicated conditions. For this figure the twenty most significantly estrogen regulated genes are shown. The three highlighted gene-names are those discussed in depth in the text.
Next to gene profiling studies, immunohistochemical analysis of PRA/PRB and ERα expression were performed together with measurements of estrogen availability [21]. These investigations revealed that besides a small increase in PRA/PRB levels in glandular cells upon treatment with E-only, no pronounced differences in ERα and PR expression levels were observed between the E-only and the E+P group [21]. Furthermore, in order to fully asses estrogen signaling, next to the biologically most potent estrogen, estradiol (E2), the less active estradiol precursor, estrone (E1), as well as its inactivated form, sulfated estrone (E1S), were measured [28] [31] [32]. It was observed that both in sera and in uterine tissues, estradiol, estrone and sulfated estrone were increased as a result of E-only and E+P treatment, though with no significant difference between the treatment groups [32] These studies seem to indicate that progestagenic regulation of estrogen signaling at the receptor level and at the level of ligand availability is not the major mechanism through which progesterone counterbalances estrogen signaling during the menstrual cycle, and additional molecular mechanisms are likely to play a role.
In literature there are a number of reports implicating an important role for Wnt/β-catenin signaling in regulating endometrial proliferation and differentiation. For example, the activity of Wnt/β-catenin signaling was observed to change between different stages of the menstrual cycle [33] and plays an important role in preparing the endometrium for embryo implantation [34] and subsequent placental formation [35]. Furthermore, Wnt/β-catenin signaling is often found activated in endometrial cancer [14] [36] and, upon reviewing regulation of estrogen and progesterone regulated genes [21] [22] [4], it was also observed that a significant number of downstream targets and components of the Wnt/β-catenin signaling pathway were regulated [37]. Moreover, some of the sex hormone-regulated downstream targets and components of the Wnt/β-catenin signaling pathway have also been implicated in endometrial carcinogenesis: both FOXO1 and CDH1 (E-cadherin) expression is decreased in endometrial cancer and is induced by progesterone [38] [39] while Survivin (BIRC5) is expressed at high levels in endometrial cancer and upregulated by estradiol [40]. Based on these observations, a role for Wnt/β-catenin signaling downstream from sex hormone signaling in the endometrium was hypothesized and will be discussed in more detail in the next paragraphs.
The relationship between sex hormone and Wnt/β-catenin signaling in the endometrium
Central in canonical Wnt signaling is the destruction complex, a multi-protein complex consisting of the scaffold proteins AXIN1 and AXIN2 (conductin), β-catenin (CTNNB1), the tumor suppressor APC (adenomatosis polyposis coli) and the Ser-Thr kinases CK1 (casein kinase I) and GSK3β (glycogen synthase kinase 3 beta) [41]. In the absence of Wnt ligands, formation of the destruction complex triggers Thr/Ser-phosphorylation of β-catenin by CK1 and GSK3β, and its subsequent ubiquitination and proteasomal degradation. Upon Wnt signaling, the formation of the destruction complex is inhibited thus leading to cytoplasmic accumulation of β-catenin and its nuclear translocation [42]. Once in the nucleus, β-catenin interacts with members of the TCF/LEF transcription factor family, thus regulating the expression of a broad spectrum of Wnt downstream target genes [42] [43] [44]. The latter include genes encoding for proteins with a central role in cell proliferation and survival such as cyclin D1 (CCND1) and c-MYC, in addition to a broad spectrum of other cellular functions i.e. cellular migration (e.g. CD44), cell adhesion (CDH1), extracellular matrix (MMP7) and many others [42] [45].
The central role of Wnt/β-catenin signaling in the regulation of tissue homeostasis has been extensively investigated for the gut [42] [46]. Along the intestinal tract, stem cells are located at the bottom of the crypts of Lieberkuhn where they give rise to new stem cells and to proliferating progenitor cells (transient amplifying, TA, cells) [47]. These progenitor cells actively divide and produce new cells that are pushed up along the flank of the crypt towards the villus and eventually differentiate into Goblet cells, enteroendocrine cells and absorptive epithelial cells [48]. A somewhat similar process seems to take place in the endometrium, where estrogen receptor activation in the basalis stimulates endometrial cell proliferation during the first two weeks of the menstrual cycle, thus giving rise to the functionalis. In week three and four of the menstrual cycle, the corpus luteum will produce progesterone, which reduces estrogens-driven proliferation and induces differentiation of the functionalis thus preparing the endometrium for implantation of the fertilized oocyte around day 20 - 22 of the menstrual cycle.
Wnt/β-catenin signaling activity along the crypt-villus axis of the intestine follows a decreasing gradient from the stem cell (SC) and proliferative (TA) compartment to the more differentiated compartment [49]. In the endometrium Wnt/β-catenin signaling has also been suggested to play a role in regulating proliferation and differentiation during the menstrual cycle. Nei et al. [33] observed that in the human endometrium, nuclear β-catenin was clearly enhanced during the proliferative phase of the menstrual cycle, while it was mostly found in the cytoplasm and at the cell membrane during the secretory phase. Recently, it was also observed that estradiol induces stabilization of intracellular β-catenin in the endometrium, and, upon inhibition of Wnt signaling (by using adenoviral SFRP2), estradiol-induced proliferation was abolished [50]. In two other studies [51] [52], LiCl was administered through the drinking water of mice to inhibit GSK3β activity and thereby activate Wnt signaling. LiCl-treated animals were characterized by increased proliferation and hyperplasia of the endometrium, thus mimicking sustained estrogen signaling.
In summary, it seems likely that, during the menstrual cycle, sex hormones can modulate Wnt/β-catenin signaling to maintain the balance between proliferation and differentiation.
Estradiol regulation of Wnt/β-catenin signaling
The putative mechanisms underlying estrogen-mediated Wnt/β-catenin activation in the uterus are at present poorly understood. A direct effect of ERα as a transcription factor on the expression of Wnt ligands, modulators and targets has been described by many authors: the ligands Wnt4, Wnt5A and Wnt7A have been shown to be induced by estradiol [53] [54] [50] [55] [56]; the Wnt inhibitor DKK1 was shown to be inhibited by estrogens in bone forming osteoblasts [57] and in the hippocampal CA1 region [58] and last, the Wnt-target gene WISP2 (Wnt-1 induced signaling pathway protein 2) was shown to be upregulated through direct interaction of activated ERα with its promoter region in human breast cancer cells [59].
ERα, however, can also function as a transcriptional modulator without directly binding to DNA sequences in the promoter region of the affected genes. For example, Shi et al [60] could show that EZH2 (the polycomb group protein enhancer of zeste homolog 2) physically interacts with ERα and β-catenin. Hence, estradiol can affect the transcription of Wnt/β-catenin target genes without directly interacting with estrogen response elements at the DNA level.
Furthermore, ERα has also been observed to associate with important growth factor pathways such as the PI3K pathway thus indirectly cross-talking with canonical Wnt signaling [61] [62]. Binding of ERα to p85 (PIK3R2), the regulatory subunit of PI3K, activates AKT1 which results in inhibition of GSK-3β. This, in turn, prevents N-terminal Ser/Thr phosphorylation of β-catenin, enhances its intracellular stabilization and eventual allowes for translocation to the nucleus, where it complexes with members of the TCF/LEF transcription factor family and lead to the activation of Wnt target genes [63] [64] (Fig. 2A and Fig 3).
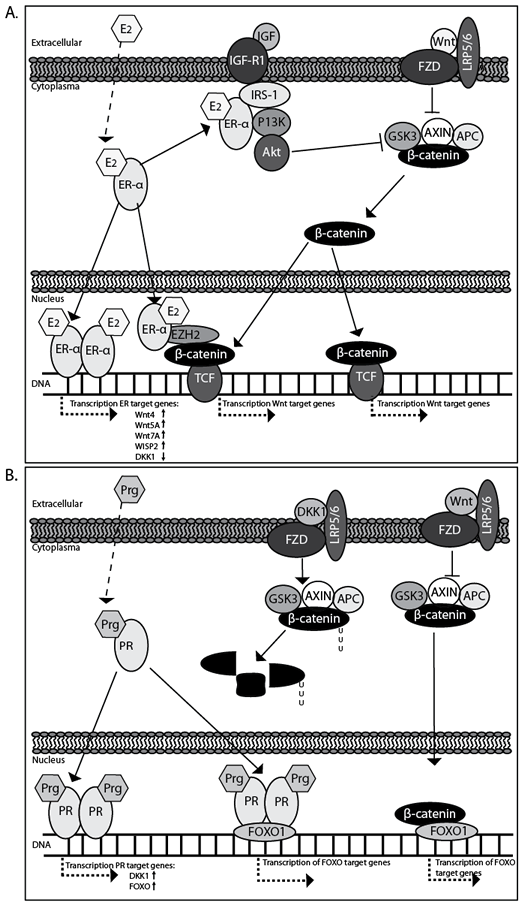
Figure 2: Estrogen (A) and progestagen (B) regulation of Wnt/β-catenin signaling in the endometrium.
Progesterone regulation of Wnt/β-catenin signaling
A role for progesterone in regulating Wnt/β-catenin signaling was suggested by Kao et al. [65] and by Tulac et al. [66] who observed a profound progesterone-specific increase in expression of the Wnt/β-catenin signaling inhibitor DKK1 in endometrial stroma cells during the secretory phase of the menstrual cycle. Using the antiprogestagen mefipristone (RU486), Catalano et al. [67] could indeed confirm progestagen regulation of many Wnt/β-catenin pathway components. Furthermore, Kane at al. [68] subsequently showed that TGFβ 1 attenuates both the expression of PR and DKK1 in differentiated endometrial stromal cells corroborating a close link between progesterone and Wnt/β-catenin signaling.
Recently, our group has followed up on these observations [37] and could show that progesterone efficiently inhibited the expression of a Wnt/β-catenin signaling reporter plasmid (TOP/FOPflash) in the Ishikawa endometrial cancer cell line by induction of the Wnt/β-catenin inhibitors DKK1 [66] and FOXO1 [38]. Furthermore, when induction of both DKK1 and FOXO1 was prevented, progesterone inhibition of Wnt signaling was also partly circumvented. FOXO1, in this respect, is an interesting molecule, as it has been shown to physically interact with the progesterone receptor to coordinate cell cycle regulation and differentiation of human endometrial stromal cells [69]. Furthermore, FOXO1 is also able to interact with β-catenin [70] [71], thus possibly directly inhibiting Wnt/β-catenin signaling [72].
Another pathway likely to play a role in regulating the interplay between progesterone and Wnt/β-catenin signaling is Hedgehog (Hh). Combined endometrial microarray data from different stages of the menstrual cycle [4] and from E-only or E+P treated patients [22] indicated profound sex hormone regulation of Hh signaling. The Hedgehog ligand IHH (Indian hedgehog), its receptor PTCH, and the transcription factor and target gene GLI1 are all up-regulated upon estrogen signaling and downregulated by progesterone during the menstrual cycle. Furthermore, it has been shown that when IHH expression is impaired, the downstream effects of progesterone are lost in the uterus [73]. As indicated, progesterone itself downregulates Hh signaling in the uterus and it has been shown that Wnt/β-catenin signaling may act downstream to Indian hedgehog signaling [74].
Recently the link between Hedgehog and Wnt/β-catenin signaling has also been confirmed for atypical endometrial hyperplasia and endometrial cancer: in hyperplasia and in well differentiated endometrial cancers GLI1 overexpression overlaps with β-catenin nuclear immunoreactivity [75]. Because our own data on staining for the Wnt target gene CD44 indicated that progesterone can also act as a profound inhibitor of Wnt/β-catenin signaling in vivo in hyperplasia as well as in well differentiated endometrial cancer, a physiological and functional link between progesterone, hedgehog and Wnt/β-catenin signaling seems plausible. (Fig. 2B and Fig. 3)
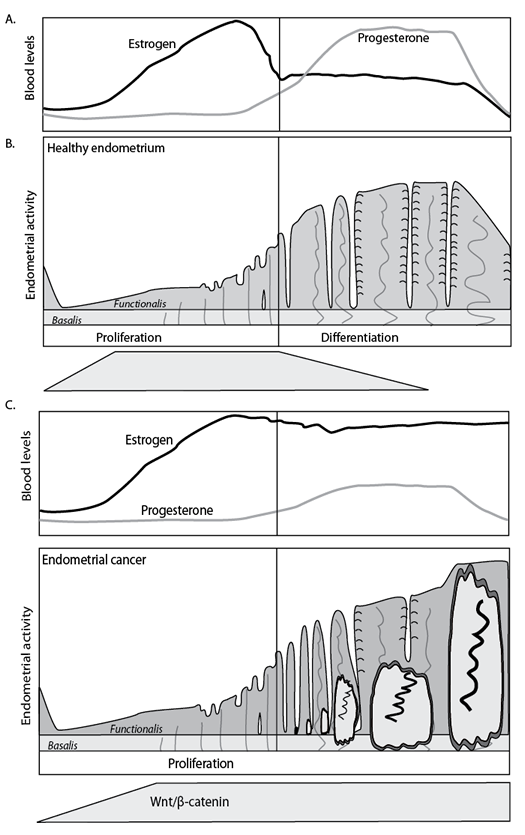
Figure 3: Hormonal and morphological changes during the normal menstrual cycle (A) and in case of increased estrogen activity (B). In case of enhanced or unopposed estrogen signaling, the endometrium may develop hyperplasia, which may eventually develop further into endometrial cancer.
Role of Wnt signaling in endometrial cancer
Gene mutations leading to constitutive activation of canonical Wnt signaling have been found in many different cancer types (e.g. breast, colon, stomach, liver, ovary skin etc). Also in the case of endometrial cancer, activation of Wnt/β-catenin signaling is likely to play an important role in early tumorigenesis [14] [36]. A substantial fraction of well differentiated endometriod carcinomas (Type I) cases (31%: [76]; 85%: [77]) show nuclear β-catenin staining (Fig. 4). Accordingly, loss- and gain-of-function mutations in members of the Wnt/β-catenin signaling pathway known to act as tumor suppressors (APC) and oncogenes (β-catenin) have also been identified. β-catenin activating mutations at its GSK-3β binding consensus site located within exon 3 have been identified in 15-40 % of endometrial tumors [78] [79], whereas LOH at the APC locus was observed in 24 % of the cases with nuclear β-catenin staining [80]. APC mutation analysis showed truncating mutations in 10% of all endometrial cancers [81]. Moreover, the APC A1 promoter was found to be hypermethylated in 46.6% of endometrial cancers with nuclear β-catenin [80], often in correlation with micro-satellite instability [82]. Notably, these somatic mutations in members of the canonical Wnt pathway were preferentially found in Type I endometrial cancer, which accounts for about 85% of the total number of endometrial cancer cases. Enhanced estrogen signaling over a prolonged period of time is believed to be a causative factor for Type I endometrial cancer [9]. Hence, although continuous estrogen induced Wnt/β-catenin signaling may represent an early step in endometrial tumorigenesis, tumor progression and malignant transformation seem to additionally require somatic mutations leading to the constitutive activation of the pathway (Fig. 3).
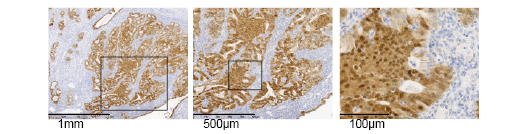
Figure 4: Nuclear β-catenin accumulation in well differentiated endometrial cancer. Immunohistochemical staining for β-catenin of type I endometrial cancer showing areas with nuclear β-catenin accumulation.
Current investigations into the role of Wnt/β-catenin signaling in endometrial carcinogenesis.
Enhanced or unopposed estrogen signaling is the most important risk factor for endometrial hyperplasia and endometrial cancer. In view of the observations according to which i. Wnt/β-catenin signaling plays a central role in endometrial homeostasis, ii. it possibly represents one of the downstream effectors of estrogen signaling, and iii. its constitutive activation is likely to underlie malignant transformation in the uterus, it is important to assess whether canonical Wnt can trigger endometrial hyperplasia and cancer in the absence of enhanced estrogen signaling. In other words, among the allegedly broad spectrum of downstream effectors of estrogen signaling, does constitutive Wnt/β-catenin activation play a rate-limiting role for endometrial malignancies?
To this aim, several genetically engineered mouse models have been employed. Tanwar et al. [83] investigated the effects of conditional Wnt signaling activation in the uterus by Amhr2-Cre-driven oncogenic activation of β-catenin (Ctnnb1tm1Mmt/+). The Amhr2 gene is expressed as of embryonic day 12.5 onwards in mesenchymal cells surrounding the Mullerian duct and in adult mice in the myometrium. The corresponding Amhr2Cre/+;Ctnnb1tm1Mmt/+ mice develop myometrial hyperplasia and mesenchymal tumors (similar to leiomyomas) and endometrial sarcomas. Furthermore, hyperplasia of endometrial glands was occasionally observed in the uterus, suggesting that mesenchymal activation of Wnt/β-catenin signaling plays a role in the early events of epithelial tumorigenesis in the endometrium. Jeong et al. [84] employed Pgr-Cre to drive oncogenic activation of β-catenin (PgrCre/+;Ctnnb1f(Ex3)/+) and of canonical Wnt signaling in a broad spectrum of uterine cells (endometrium + myometrium). These authors could show that activation of Wnt/β-catenin signaling in the uterus resulted in enhanced proliferation of glandular epithelial cells, endometrial hyperplasia at 6 weeks of age, and in defective estrogen signaling, though not in endometrial cancer. These results suggest that constitutive activation of Wnt/β-catenin signaling on its own is insufficient for endometrial cancer onset and possibly that the synergistic action of additional downstream effectors of estrogen signaling are necessary for full-blown malignant transformation. However, as predicted by the ”just-right” signaling model [85] [86], different levels of pathway activation may differently trigger tumorigenesis in distinct tissues. Therefore, it would be of interest to investigate the consequences of hypomorphic mutations in members of the Wnt pathway other than β-catenin whose oncogenic activation invariably leads to extremely high Wnt signaling levels. By inducing Apc mutations in the myometrium [87], we recently observed defects in the myometrial layer of the uterus, where a significant loss of muscle fibers was apparent [Wang et al, submitted). Likewise, we also employed Pgr-Cre to drive loss of Apc function in epithelial cells of the endometrium and in myometrial cells. Also in this case, muscular defects were apparent with invasion of endometrial glands and stroma into the muscular layer. Notably, we also observed hyperplasia and early stage endometrial cancer [manuscript in preparation). Hence, it is possible that “just-right” levels of Wnt/β-catenin signaling are sufficient to trigger tumor initiation in the endometrium in a dosage- and context-dependent fashion.
Conclusions
In this review we discussed the role of estrogens and progestagens in the regulation of Wnt/β-catenin signaling as well as the involvement of activated Wnt/β-catenin signaling in the development of endometrial cancer. Evidence indicates that estrogens can induce Wnt/β-catenin signaling and that enhanced or unopposed estrogen signaling, as well as activated Wnt/β-catenin signaling may underlie endometrial hyperplasia and cancer. Furthermore, progesterone was shown to be a strong inhibitor of Wnt/β-catenin signaling. The latter is of clinical relevance as it is well known that progesterone can inhibit endometrial hyperplasia and well differentiated endometrial cancer growth.
These observations place Wnt/β-catenin signaling at the center of physiological regulation of the menstrual cycle and point at a central functional role for its uncontrolled activation in endometrial carcinogenesis. Although Wnt/β-catenin signaling inhibitors (e.g. CGP049090, PKF115-584 [88] and XAV939 [89]) have not yet been introduced into clinical practice and likewise have not been considered for endometrial cancer treatment, the experimental data here summarized may lay the basis for future tailor-made therapies based on members of the canonical Wnt pathway or its downstream targets.
ACKNOWLEDGEMENTS
We would like to thank Liesbeth Kuhne (BSc) for IHC shown in Figure 4.
The work of RF is supported by grants from the Dutch Cancer Society, the BSIK program of the Dutch Government grant 03038 (www.stemcells.nl), and the EU FP6 and FP7 consortia Migrating Cancer Stem Cells program (MCSCs; www.mcscs.eu) and TuMIC (integrated concept of tumour metastasis (http://itgmv1.fzk.de/www/tumic/tumic_main.htm ).
The work of LJB is supported by a grant from the Dutch Cancer Society.
CONFLICT OF INTEREST STATEMENT
No conflict of interest to declare
REFERENCES
1. Jabbour HN, Kelly RW, Fraser HM, Critchley HO. Endocrine regulation of menstruation. Endocr Rev. 2006; 27: 17-46.
2. Padykula HA, Coles LG, Okulicz WC, Rapaport SI, McCracken JA, King NW, Jr., et al. The basalis of the primate endometrium: a bifunctional germinal compartment. Biol Reprod. 1989; 40: 681-90.
3. Vienonen A, Miettinen S, Blauer M, Martikainen PM, Tomas E, Heinonen PK, et al. Expression of nuclear receptors and cofactors in human endometrium and myometrium. J Soc Gynecol Investig. 2004; 11: 104-12.
4. Talbi S, Hamilton AE, Vo KC, Tulac S, Overgaard MT, Dosiou C, et al. Molecular phenotyping of human endometrium distinguishes menstrual cycle phases and underlying biological processes in normo-ovulatory women. Endocrinology. 2006; 147: 1097-121.
5. Markee JE. Physiology of reproduction. Annu Rev Physiol. 1951; 13: 367-96.
6. Farage MA, Neill S, MacLean AB. Physiological changes associated with the menstrual cycle: a review. Obstet Gynecol Surv. 2009; 64: 58-72.
7. Maruyama T, Yoshimura Y. Molecular and cellular mechanisms for differentiation and regeneration of the uterine endometrium. Endocr J. 2008; 55: 795-810.
8. Gambrell RD, Jr. Estrogens, progestogens and endometrial cancer. J Reprod Med. 1977; 18: 301-6.
9. Schindler AE. Progestogen deficiency and endometrial cancer risk. Maturitas. 2009; 62: 334-7.
10. Parkin DM, Pisani P, Ferlay J. Global cancer statistics. CA Cancer J Clin. 1999; 49: 33-64.
11. Cramer DW, Knapp RC. Review of epidemiologic studies of endometrial cancer and exogenous estrogen. Obstet Gynecol. 1979; 54: 521-6.
12. Amant F, Moerman P, Neven P, Timmerman D, Van Limbergen E, Vergote I. Endometrial cancer. Lancet. 2005; 366: 491-505.
13. Webb PM. Commentary: weight gain, weight loss, and endometrial cancer. Int J Epidemiol. 2006; 35: 166-8.
14. Fukuchi T, Sakamoto M, Tsuda H, Maruyama K, Nozawa S, Hirohashi S. Beta-catenin mutation in carcinoma of the uterine endometrium. Cancer Res. 1998; 58: 3526-8.
15. Swisher EM, Peiffer-Schneider S, Mutch DG, Herzog TJ, Rader JS, Elbendary A, et al. Differences in patterns of TP53 and KRAS2 mutations in a large series of endometrial carcinomas with or without microsatellite instability. Cancer. 1999; 85: 119-26.
16. Di Cristofano A, Ellenson LH. Endometrial carcinoma. Annu Rev Pathol. 2007; 2: 57-85.
17. Bergman L, Beelen ML, Gallee MP, Hollema H, Benraadt J, van Leeuwen FE. Risk and prognosis of endometrial cancer after tamoxifen for breast cancer. Comprehensive Cancer Centres’ ALERT Group. Assessment of Liver and Endometrial cancer Risk following Tamoxifen. Lancet. 2000; 356: 881-7.
18. Gielen SC, Kuhne LC, Ewing PC, Blok LJ, Burger CW. Tamoxifen treatment for breast cancer enforces a distinct gene-expression profile on the human endometrium: an exploratory study. Endocr Relat Cancer. 2005; 12: 1037-49.
19. Available from: http://info.cancerresearchuk.org.
20. Schindler AE. Non-contraceptive use of hormonal contraceptives. Gynecol Endocrinol. 2008; 24: 235-6.
21. Klaassens AH, van Wijk FH, Hanifi-Moghaddam P, Sijmons B, Ewing PC, Ten Kate-Booij MJ, et al. Histological and immunohistochemical evaluation of postmenopausal endometrium after 3 weeks of treatment with tibolone, estrogen only, or estrogen plus progestagen. Fertil Steril. 2006; 86: 352-61.
22. Hanifi-Moghaddam P, Boers-Sijmons B, Klaassens AH, van Wijk FH, den Bakker MA, Ott MC, et al. Molecular analysis of human endometrium: short-term tibolone signaling differs significantly from estrogen and estrogen + progestagen signaling. J Mol Med. 2007; 85: 471-80.
23. Thigpen JT, Brady MF, Alvarez RD, Adelson MD, Homesley HD, Manetta A, et al. Oral medroxyprogesterone acetate in the treatment of advanced or recurrent endometrial carcinoma: a dose-response study by the Gynecologic Oncology Group. J Clin Oncol. 1999; 17: 1736-44.
24. Kim YB, Holschneider CH, Ghosh K, Nieberg RK, Montz FJ. Progestin alone as primary treatment of endometrial carcinoma in premenopausal women. Report of seven cases and review of the literature. Cancer. 1997; 79: 320-7.
25. Yahata T, Fujita K, Aoki Y, Tanaka K. Long-term conservative therapy for endometrial adenocarcinoma in young women. Hum Reprod. 2006; 21: 1070-5.
26. Kouzmenko AP, Takeyama K, Ito S, Furutani T, Sawatsubashi S, Maki A, et al. Wnt/beta-catenin and estrogen signaling converge in vivo. J Biol Chem. 2004; 279: 40255-8.
27. Bulun SE, Cheng YH, Pavone ME, Xue Q, Attar E, Trukhacheva E, et al. Estrogen receptor-beta, estrogen receptor-alpha, and progesterone resistance in endometriosis. Semin Reprod Med. 2010; 28: 36-43.
28. Dassen H, Punyadeera C, Kamps R, Klomp J, Dunselman G, Dijcks F, et al. Progesterone regulation of implantation-related genes: new insights into the role of oestrogen. Cell Mol Life Sci. 2007; 64: 1009-32.
29. Utsunomiya H, Ito K, Suzuki T, Kitamura T, Kaneko C, Nakata T, et al. Steroid sulfatase and estrogen sulfotransferase in human endometrial carcinoma. Clin Cancer Res. 2004; 10: 5850-6.
30. Mylonas I, Makovitzky J, Friese K, Jeschke U. Immunohistochemical labelling of steroid receptors in normal and malignant human endometrium. Acta Histochem. 2009; 111: 349-59.
31. Pasqualini JR. Estrogen sulfotransferases in breast and endometrial cancers. Ann N Y Acad Sci. 2009; 1155: 88-98.
32. Verheul HA, Blok LJ, Burger CW, Hanifi-Moghaddam P, Kloosterboer HJ. Levels of tibolone and estradiol and their nonsulfated and sulfated metabolites in serum, myometrium, and vagina of postmenopausal women following treatment for 21 days with tibolone, estradiol, or estradiol plus medroxyprogestrone acetate. Reprod Sci. 2007; 14: 160-8.
33. Nei H, Saito T, Yamasaki H, Mizumoto H, Ito E, Kudo R. Nuclear localization of beta-catenin in normal and carcinogenic endometrium. Mol Carcinog. 1999; 25: 207-18.
34. Kao LC, Tulac S, Lobo S, Imani B, Yang JP, Germeyer A, et al. Global gene profiling in human endometrium during the window of implantation. Endocrinology. 2002; 143: 2119-38.
35. Hayashi K, Burghardt RC, Bazer FW, Spencer TE. WNTs in the ovine uterus: potential regulation of periimplantation ovine conceptus development. Endocrinology. 2007; 148: 3496-506.
36. Schlosshauer PW, Pirog EC, Levine RL, Ellenson LH. Mutational analysis of the CTNNB1 and APC genes in uterine endometrioid carcinoma. Mod Pathol. 2000; 13: 1066-71.
37. Wang Y, Hanifi-Moghaddam P, Hanekamp EE, Kloosterboer HJ, Franken P, Veldscholte J, et al. Progesterone inhibition of Wnt/beta-catenin signaling in normal endometrium and endometrial cancer. Clin Cancer Res. 2009; 15: 5784-93.
38. Ward RD, Weigel NL. Steroid receptor phosphorylation: Assigning function to site-specific phosphorylation. Biofactors. 2009; 35: 528-36.
39. Moreno-Bueno G, Hardisson D, Sarrio D, Sanchez C, Cassia R, Prat J, et al. Abnormalities of E- and P-cadherin and catenin (beta-, gamma-catenin, and p120ctn) expression in endometrial cancer and endometrial atypical hyperplasia. J Pathol. 2003; 199: 471-8.
40. Erkanli S, Bolat F, Kayaselcuk F, Demirhan B, Kuscu E. COX-2 and survivin are overexpressed and positively correlated in endometrial carcinoma. Gynecol Oncol. 2007; 104: 320-5.
41. Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, et al. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998; 280: 596-9.
42. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006; 127: 469-80.
43. Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R. Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev. 1996; 59: 3-10.
44. Cho EA, Dressler GR. TCF-4 binds beta-catenin and is expressed in distinct regions of the embryonic brain and limbs. Mech Dev. 1998; 77: 9-18.
45. Available from: http://www.stanford.edu/~rnusse/pathways/targets.html.
46. Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001; 1: 55-67.
47. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005; 434: 843-50.
48. Radtke F, Clevers H. Self-renewal and cancer of the gut: two sides of a coin. Science. 2005; 307: 1904-9.
49. van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002; 111: 241-50.
50. Hou YF, Yuan ST, Li HC, Wu J, Lu JS, Liu G, et al. ERbeta exerts multiple stimulative effects on human breast carcinoma cells. Oncogene. 2004; 23: 5799-806.
51. Gunin AG, Emelianov VU, Mironkin IU, Morozov MP, Tolmachev AS. Lithium treatment enhances estradiol-induced proliferation and hyperplasia formation in the uterus of mice. Eur J Obstet Gynecol Reprod Biol. 2004; 114: 83-91.
52. Polotsky AJ, Zhu L, Santoro N, Pollard JW. Lithium chloride treatment induces epithelial cell proliferation in xenografted human endometrium. Hum Reprod. 2009; 24: 1960-7.
53. Katayama S, Ashizawa K, Fukuhara T, Hiroyasu M, Tsuzuki Y, Tatemoto H, et al. Differential expression patterns of Wnt and beta-catenin/TCF target genes in the uterus of immature female rats exposed to 17alpha-ethynyl estradiol. Toxicol Sci. 2006; 91: 419-30.
54. Quintanilla RA, Munoz FJ, Metcalfe MJ, Hitschfeld M, Olivares G, Godoy JA, et al. Trolox and 17beta-estradiol protect against amyloid beta-peptide neurotoxicity by a mechanism that involves modulation of the Wnt signaling pathway. J Biol Chem. 2005; 280: 11615-25.
55. Miyakoshi T, Kajiya H, Miyajima K, Takei M, Tobita M, Takekoshi S, et al. The expression of Wnt4 is regulated by estrogen via an estrogen receptor alpha-dependent pathway in rat pituitary growth hormone-producing cells. Acta Histochem Cytochem. 2009; 42: 205-13.
56. Wagner J, Lehmann L. Estrogens modulate the gene expression of Wnt-7a in cultured endometrial adenocarcinoma cells. Mol Nutr Food Res. 2006; 50: 368-72.
57. Pinzone JJ, Hall BM, Thudi NK, Vonau M, Qiang YW, Rosol TJ, et al. The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood. 2009; 113: 517-25.
58. Zhang QG, Wang R, Khan M, Mahesh V, Brann DW. Role of Dickkopf-1, an antagonist of the Wnt/beta-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J Neurosci. 2008; 28: 8430-41.
59. Fritah A, Redeuilh G, Sabbah M. Molecular cloning and characterization of the human WISP-2/CCN5 gene promoter reveal its upregulation by oestrogens. J Endocrinol. 2006; 191: 613-24.
60. Shi B, Liang J, Yang X, Wang Y, Zhao Y, Wu H, et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol. 2007; 27: 5105-19.
61. Hellyer NJ, Cheng K, Koland JG. ErbB3 (HER3) interaction with the p85 regulatory subunit of phosphoinositide 3-kinase. Biochem J. 1998; 333: 757-63.
62. Cardona-Gomez P, Perez M, Avila J, Garcia-Segura LM, Wandosell F. Estradiol inhibits GSK3 and regulates interaction of estrogen receptors, GSK3, and beta-catenin in the hippocampus. Mol Cell Neurosci. 2004; 25: 363-73.
63. Varea O, Garrido JJ, Dopazo A, Mendez P, Garcia-Segura LM, Wandosell F. Estradiol activates beta-catenin dependent transcription in neurons. PLoS One. 2009; 4: e5153.
64. Varea O, Arevalo MA, Garrido JJ, Garcia-Segura LM, Wandosell F, Mendez P. Interaction of estrogen receptors with insulin-like growth factor-I and Wnt signaling in the nervous system. Steroids. 2010; 75: 565-9.
65. Kao KR, Elinson RP. The legacy of lithium effects on development. Biol Cell. 1998; 90: 585-9.
66. Tulac S, Overgaard MT, Hamilton AE, Jumbe NL, Suchanek E, Giudice LC. Dickkopf-1, an inhibitor of Wnt signaling, is regulated by progesterone in human endometrial stromal cells. J Clin Endocrinol Metab. 2006; 91: 1453-61.
67. Catalano RD, Critchley HO, Heikinheimo O, Baird DT, Hapangama D, Sherwin JR, et al. Mifepristone induced progesterone withdrawal reveals novel regulatory pathways in human endometrium. Mol Hum Reprod. 2007; 13: 641-54.
68. Kane N, Jones M, Brosens JJ, Saunders PT, Kelly RW, Critchley HO. Transforming growth factor-beta1 attenuates expression of both the progesterone receptor and Dickkopf in differentiated human endometrial stromal cells. Mol Endocrinol. 2008; 22: 716-28.
69. Takano M, Lu Z, Goto T, Fusi L, Higham J, Francis J, et al. Transcriptional cross talk between the forkhead transcription factor forkhead box O1A and the progesterone receptor coordinates cell cycle regulation and differentiation in human endometrial stromal cells. Mol Endocrinol. 2007; 21: 2334-49.
70. Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005; 308: 1181-4.
71. Hoogeboom D, Essers MA, Polderman PE, Voets E, Smits LM, Burgering BM. Interaction of FOXO with beta-catenin inhibits beta-catenin/T cell factor activity. J Biol Chem. 2008; 283: 9224-30.
72. Almeida M, Han L, Martin-Millan M, O’Brien CA, Manolagas SC. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J Biol Chem. 2007; 282: 27298-305.
73. Lee K, Jeong J, Kwak I, Yu CT, Lanske B, Soegiarto DW, et al. Indian hedgehog is a major mediator of progesterone signaling in the mouse uterus. Nat Genet. 2006; 38: 1204-9.
74. Day TF, Yang Y. Wnt and hedgehog signaling pathways in bone development. J Bone Joint Surg Am. 2008; 90: 19-24.
75. Liao X, Siu MK, Au CW, Chan QK, Chan HY, Wong ES, et al. Aberrant activation of hedgehog signaling pathway contributes to endometrial carcinogenesis through beta-catenin. Mod Pathol. 2009; 22: 839-47.
76. Scholten AN, Creutzberg CL, van den Broek LJ, Noordijk EM, Smit VT. Nuclear beta-catenin is a molecular feature of type I endometrial carcinoma. J Pathol. 2003; 201: 460-5.
77. Saegusa M, Okayasu I. Frequent nuclear beta-catenin accumulation and associated mutations in endometrioid-type endometrial and ovarian carcinomas with squamous differentiation. J Pathol. 2001; 194: 59-67.
78. Konopka B, Janiec-Jankowska A, Czapczak D, Paszko Z, Bidzinski M, Olszewski W, et al. Molecular genetic defects in endometrial carcinomas: microsatellite instability, PTEN and beta-catenin (CTNNB1) genes mutations. J Cancer Res Clin Oncol. 2007; 133: 361-71.
79. Hecht JL, Mutter GL. Molecular and pathologic aspects of endometrial carcinogenesis. J Clin Oncol. 2006; 24: 4783-91.
80. Moreno-Bueno G, Hardisson D, Sanchez C, Sarrio D, Cassia R, Garcia-Rostan G, et al. Abnormalities of the APC/beta-catenin pathway in endometrial cancer. Oncogene. 2002; 21: 7981-90.
81. Pijnenborg JM, Kisters N, van Engeland M, Dunselman GA, de Haan J, de Goeij AF, et al. APC, beta-catenin, and E-cadherin and the development of recurrent endometrial carcinoma. Int J Gynecol Cancer. 2004; 14: 947-56.
82. Zysman M, Saka A, Millar A, Knight J, Chapman W, Bapat B. Methylation of adenomatous polyposis coli in endometrial cancer occurs more frequently in tumors with microsatellite instability phenotype. Cancer Res. 2002; 62: 3663-6.
83. Tanwar PS, Lee HJ, Zhang L, Zukerberg LR, Taketo MM, Rueda BR, et al. Constitutive activation of Beta-catenin in uterine stroma and smooth muscle leads to the development of mesenchymal tumors in mice. Biol Reprod. 2009; 81: 545-52.
84. Jeong JW, Lee HS, Franco HL, Broaddus RR, Taketo MM, Tsai SY, et al. beta-catenin mediates glandular formation and dysregulation of beta-catenin induces hyperplasia formation in the murine uterus. Oncogene. 2009; 28: 31-40.
85. Albuquerque C, Breukel C, van der Luijt R, Fidalgo P, Lage P, Slors FJ, et al. The ‘just-right’ signaling model: APC somatic mutations are selected based on a specific level of activation of the beta-catenin signaling cascade. Hum Mol Genet. 2002; 11: 1549-60.
86. Gaspar C, Fodde R. APC dosage effects in tumorigenesis and stem cell differentiation. Int J Dev Biol. 2004; 48: 377-86.
87. Arango NA, Kobayashi A, Wang Y, Jamin SP, Lee HH, Orvis GD, et al. A mesenchymal perspective of Mullerian duct differentiation and regression in Amhr2-lacZ mice. Mol Reprod Dev. 2008; 75: 1154-62.
88. Gandhirajan RK, Staib PA, Minke K, Gehrke I, Plickert G, Schlosser A, et al. Small molecule inhibitors of Wnt/beta-catenin/lef-1 signaling induces apoptosis in chronic lymphocytic leukemia cells in vitro and in vivo. Neoplasia. 2010; 12: 326-35.
89. Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009; 461: 614-20.