
Introduction
The molecular mechanisms underlying the pathogenesis of acute myeloid leukemia (AML) have been extensively studied, and are known to involve members of the HOX family of transcription factors, both as partners in chimeric fusion proteins, and also in their wild type form [1]. Whilst these roles are reflected in the relationship between the expression of individual HOX genes and clinicopathological factors such as disease subtype and patient survival [2], the role of HOX proteins in the survival of AML cells has proved difficult to assess as many have redundant functions, which makes a conventional knock down experiment difficult to interpret. For example, knocking down the expression of either HOXA6 or HOXA9 alone has little effect on AML cells, but their double knock-down induces cell death and also increases their sensitivity to cytarabine [3]. An alternative strategy to targeting HOX proteins is to inhibit their interaction with the PBX co-factor, which can be achieved using a short, cell-penetrating peptide (HXR9) that mimics the conserved hexapeptide in HOX proteins responsible for PBX binding [4]. HXR9 has been shown to induce apoptosis in a range of solid cancers, both in vitro and in vivo, including those of the lung [5], breast [6] and prostate [7], and melanoma [8]. It has also been shown to be cytotoxic for malignant B cells [9], and a number of AML cell lines [10]. However, the mechanism of HXR9 induced-cell death is not well understood in hematological malignancies. In this study we evaluated the relationship between HOX gene expression and overall survival, and the mechanism by which HXR9 causes cell death in AML. Our findings indicate that HXR9 induces necroptosis, rather than apoptosis, and that its cytotoxicity can be greatly enhanced by inhibition of protein kinase C (PKC).
Results
Despite the public availability of large datasets relating HOX gene expression to survival in AML, relatively little has been reported on the relationship between the expression of individual HOX genes and survival. We therefore analyzed the relationship between survival and expression of HOX genes that encode proteins capable of binding to the HXR9 target, PBX, amongst a cohort of 269 patients from the Gene Expression Omnibus (GEO) database [11]. This revealed that a number of HOX genes were significantly related to survival in AML, including HOXA5 (p = 0.03), HOXB2 (p = 0.002), HOXB4 (p = 0.037), HOXB9 (p = 0.001), and HOXC9 (p = 0.007) (Figure 1), whilst HOXA4 (p = 0.067) and HOXA9 (p = 0.06) showed borderline significance. In contrast, the expression of a number of other HOX genes including HOXA1 (p = 0.242), HOXA2 (p = 0.595), HOXA3 (p = 0.407), HOXA7 (p = 0.529), HOXB3 (p = 0.783), HOXB5 (p = 0.979), HOXB6 (p = 0.246), HOXB7 (p = 0.996), HOXB8 (p = 0.74), and HOXC6 (p = 0.876) were not related to patient survival (data not shown).
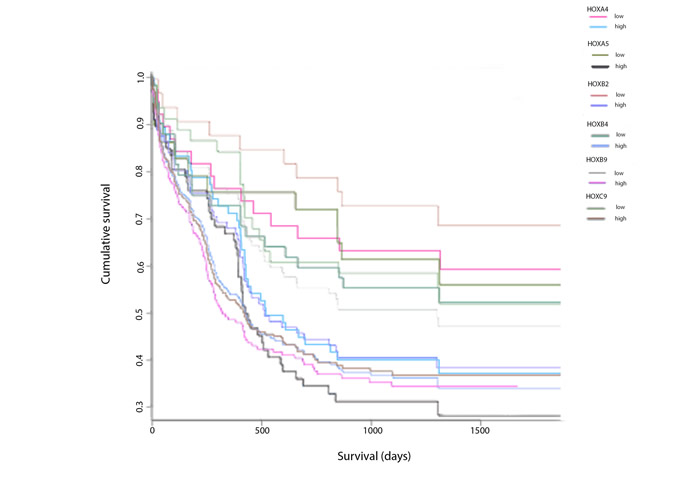
Figure 1: Association of expression of HOX genes in combination with AML patient survival data. Kaplan-Meier plots of the cumulative proportion of patients surviving in the AML dataset (n = 269) from the Gene Expression Omnibus database GSE23312 in patients with a low level and a high level of expression of each specified HOX gene.
In order to evaluate the molecular mechanisms underlying the cytotoxicity of HXR9 in AML cells, we determined the sensitivity of a number of AML-derived cell lines and primary AML cells. Three of the cell lines were derived from primary AML (KG1, HEL 92.1.7, and HL-60) and 2 from secondary AML (KU812F, and K562). The IC50s of cell killing by HXR9, as determined using an LDH assay, were 4.5, 6.1, 16.9, 9.1, and 10.4 µM, respectively (Figure 2A). None of these cell lines were sensitive to CXR9, an inactive variant of HXR9 that differs from it by only a single amino acid [7]. In order to test the effect of HXR9 on primary AML cells we isolated cells from the peripheral blood of AML patients and used a proliferation assay to evaluate the response to HOX/PBX inhibition. This revealed that HXR9 can significantly reduce the proliferation of primary AML cells at a concentration < 1 µM (Figure 2B), which is considerably lower than for other primary cancer cells isolated from solid malignancies [8].
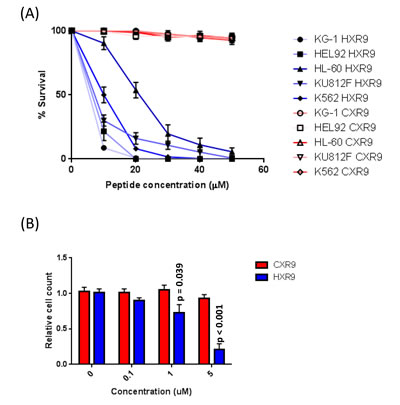
Figure 2: A. IC50 survival curves for AML-derived cell lines treated with HXR9 or CXR9. B. Proliferation of primary AML cells treated with varying concentrations of HXR9 or CXR9. Each value is the mean of 3 independent repeats, error bars show the SEM.
We investigated whether these cells underwent apoptosis after HXR9 treatment. Although changes in the plasma membrane consistent with apoptosis were apparent in all of these cell lines (Figure 3), which concurs with previous findings [10], this is not in itself an absolute indication that cells have undergone apoptosis, as these membrane changes can also occur during necrosis [12]. Indeed, further studies with the K562 and HL-60 cell lines revealed no evidence of other, more definitive changes associated with apoptosis including caspase-3 activation (Figure 4A) or nuclear fragmentation (Figure 4B). We also measured the expression of a number of genes involved in apoptosis using qRT-PCR (Figure 4C). This analysis revealed that HXR9 did not cause transcriptional changes in the initiator caspase-9 or its partner Apaf1 that together form an apoptosome complex with cytochrome C (cyt C) [13], nor in the executioner caspases-3, -6 and -7, which are key mediators of apoptosis [14], nor the pro-apoptotic members of the Bcl-2 family including Bad, Bax, Bak1 and Bid, nor the anti-apoptotic member Bcl-2, which mediate mitochondrial cell death [15]. In addition, neither the transcription of PARP1 nor XIAP was affected by HXR9 treatment. Furthermore, cell death was not dependent on ATP (Figure 4D), and could not be reversed by the pan-caspase inhibitor z-VAD-FMK (Figure 4E). We therefore explored the possibility of necrotic cell death. K562 cells could be rescued from HXR9-mediated cell killing using CsA (Figure 4F), an inhibitor of mitochondrial necrosis that targets the CypD protein, although only a limited rescue was achieved in HL-60 cells (Figure 4G) [16]. However, inhibition of the RIP1 kinase using its inhibitor Nec-1 [17] resulted in a significant rescue of K562 and HL-60 cells from HXR9-mediated cytotoxicity (Figure 5A). RIP1 is a central component of the necroptosis pathway [18], suggesting that this might play a key role in HXR9-induced cell death. We also explored further molecular pathways that might influence necroptosis. Inhibition of signaling through p38 (Figure 5B) and MEK/ERK (Figure 5C) had no effect on HXR9 cytotoxicity, nor did the inhibition of the p53 tumor suppressor protein (Figure 5D), although HXR9 treatment of both K562 and HL-60 cells resulted in a significant increase in expression of the p21 tumor suppressor gene at both the RNA (Figure 5E) and protein (Figure 5F) level. Furthermore, inhibition of PKC also significantly sensitized cells to HXR9 (Figure 6).
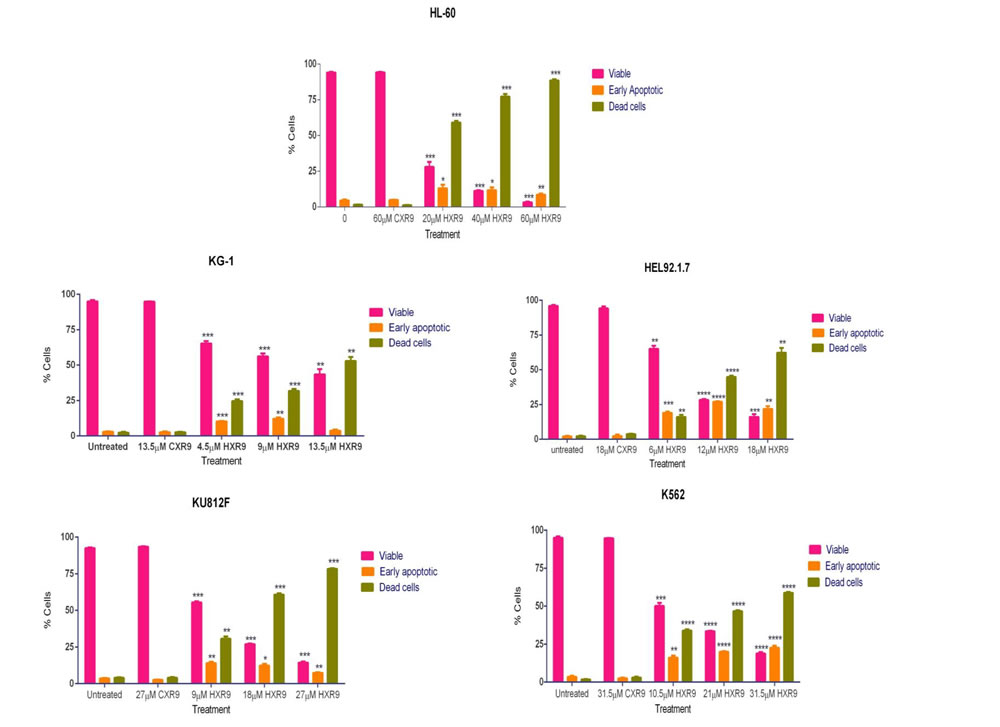
Figure 3: Annexin assay for detection of cell death in AML-derived cell lines. Cells were treated with the IC50, 2xIC50, or 3xIC50 of HXR9, and with a concentration of CXR9 equivalent to the highest concentration of HXR9 for 2 hours. Cells were then stained with annexin V and 7-AAD and analysed by flow cytometry. The number of early apoptotic and dead cells increased with increasing HXR9 concentrations. Results are expressed as the mean of 3 separate experiments and error bars show the SEM. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 with respect to untreated cells.
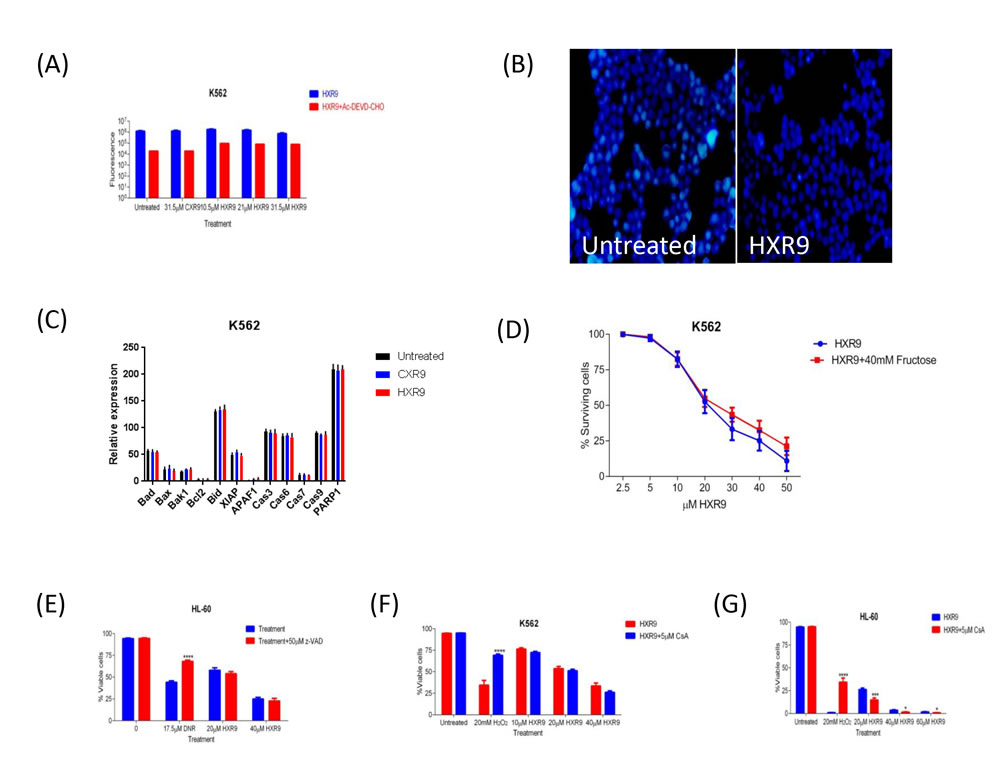
Figure 4: A. Inhibition of apoptosis by Ac-DEVD-CHO in AML K562 cells using Z-DEVD-R110. Cells were treated with IC50, 2xIC50, 3xIC50 of HXR9, with an equivalent CXR9 to the highest concentration of HXR9 or untreated (control) for 2 hours. The cells were then lysed and the Z-DEVD-R110 substrate was added. The results were normalised to lysis buffer alone. There was no statistical difference in caspase-3 activity between both HXR9- or CXR9-treated cells and untreated control cells. Graphs show the mean of 3 independent experiments and error bars show the SEM. B. Nuclear staining of K562 cells either untreated or incubated for 2 hours with 10 µM HXR9. C. Analysis of transcriptional changes of several pro- and anti-apoptotic genes upon HXR9 treatment. K562 cells were treated with the IC50 of HXR9, CXR9 or untreated (control) for 2 hours and gene expression was analysed using RT-PCR. There was no significant change in any of the analysed genes. Results are presented as a ratio with β-actin (x10 000). Statistical analysis was performed using student’s t-test by comparing the relative expression of a gene of interest in HXR9 or CXR9 treated cells to its counterpart in untreated cells. Graphs show the mean of 3 independent experiments and error bars show the SEM. D. The effect of ATP depletion on HXR9 cytotoxicity. Cells were pre-incubated with or without 40 mM fructose and then with a range of HXR9 concentrations (2.5 μM - 50 μM) for 2 hours prepared in media either with or without 40 mM fructose, red and blue curves, respectively. HXR9 cytotoxicity was then measured by assessing LDH enzyme activity in cell-free supernatants. ATP depletion did not inhibit HXR9-induced death. Graphs show the mean of 3 independent experiments and error bars show the SEM. E. General inhibition of caspase activity in HXR9 treated AML cell lines by z-VAD-FMK. Cells were pre-treated either with or without 50μM z-VAD-FMK for 1 hour, and then treated with the IC50, or 2xIC50 of HXR9 for 2 hours or with 17.5 μM DNR for 24 hours, which was used as a positive control. There was no statistical difference in terms of sensitivity to HXR9 between pre-treated cells with or without 50μM z-VAD-FMK. Graphs show the mean of 3 independent experiments and error bars show the SEM. ****p < 0.0001 with respect to z-VAD-FMK untreated cells. F. and G. Effect of CsA on HXR9 cytotoxicity. Cells were pre-treated with or without 5μM CsA for 1 hour, and then with the IC50, 2xIC50, 3xIC50 of HXR9, 20 mM H2O2 (positive control), or were left untreated (negative control) for 2 hours with or without 5 μM CsA. The cells were then stained with annexin V and 7-AAD and analysed by flow cytometry. There was no statistical difference in the proportion of viable cells between K562 cells F. incubated with or without 5μM CsA, while incubation with 5μM CsA led to a significant decrease in proportion of viable HL-60 cells G.. Graphs show the mean of 3 independent experiments and error bars show the SEM. *p < 0.05, ***p < 0.001 and ****p < 0.0001 with respect to 5 μM CsA untreated cells.
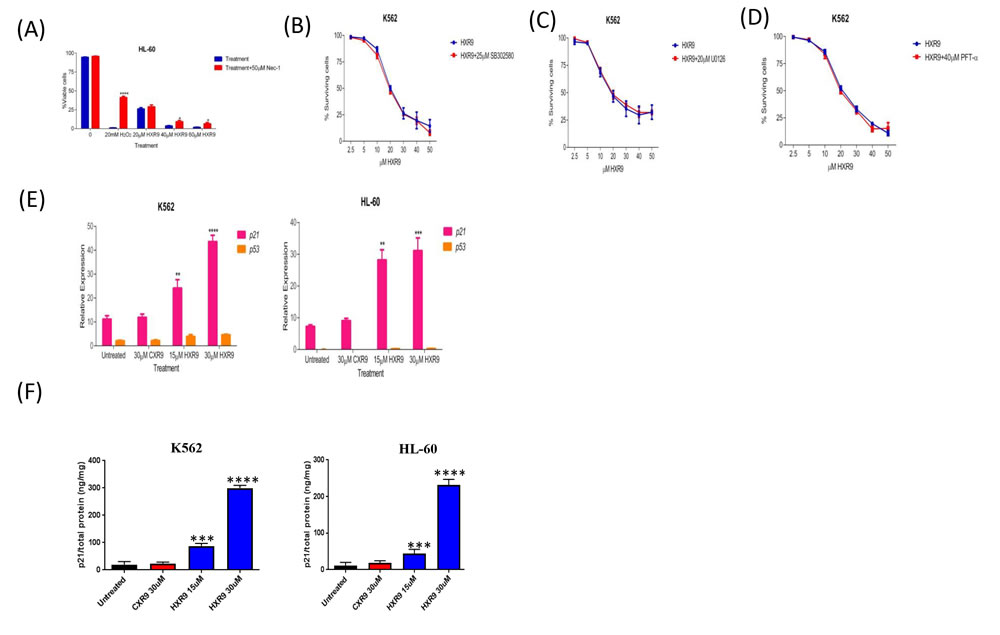
Figure 5: Evaluation of potential cell death pathways. A. Inhibition of RIP1 rescues HL-60 cells from HXR9-mediated cell killing. RIP1 was inhibited using 50 µM Nec1. Hydrogen peroxide was used as a positive control as it has previously been shown to induce RIP1-dependent cell killing. B. Cells were pre-treated with or without 25 μM of the p38 inhibitor SB302580 for 1 hour, and then with a titration of HXR9 prepared in media with or without 25 μM SB302580, red and blue curves, respectively. The cytotoxicity of HXR9 was analysed by measuring LDH enzyme activity in cell-free supernatants. Pre- and co-treatment of cells with 25 μM SB302580 did not affect the cytotoxicity of HXR9. Statistical significance was tested using the student’s t-test. Graphs show the mean of 3 independent experiments and error bars show the SEM. C. Cells were pre-treated with or without 20 μM of the MEK/ERK inhibitor U0126 for 1 hour, and then with increasing concentrations of HXR9 prepared in media with or without 20 μM U0126, red and blue curves, respectively. The proportion of surviving cells was then measured using the LDH assay. Pre- and co-treatment of cells with 20μM U0126 did not prevent HXR9 cytotoxicity. Graphs show the mean of three independent experiments and error bars show the SEM. D. The effect of blocking p53 protein on HXR9 cytotoxicity. Cells were pre-treated with or without 40 μM PFT-α for 1 hour, and then for 2 hours with a titration of HXR9 prepared in media supplemented with or without 40 μM PFT-α, red and blue curves, respectively. Graphs show the mean of 3 independent experiments and error bars show the SEM. E. HXR9 treatment of K562 and HL-60 cells leads to a significant increase in expression of the p21 tumor suppressor gene. p21 expression was measured using quantitative PCR after 2 hours of incubation with the IC50 concentration of HXR9 for each cell type. F. p21 protein expression was measured using an ELISA with the same concentrations of HXR9.
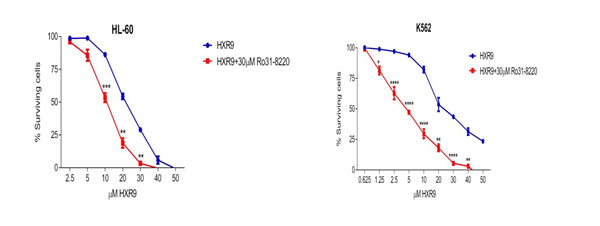
Figure 6: Inhibition of PKC sensitizes cells to killing by HXR9. PKC was inhibited using Ro31-8220, reducing the IC50 of HXR9 to 5 µM for K562 cells, and to 10.7 µM for HL-60 cells.
In order to test these findings in vivo, we used a previously described murine model of AML based on a xenograft flank tumor model using K562 cells. Treatment with HXR9 significantly reduced tumor growth compared to vehicle (PBS) alone or CXR9, and combining HXR9 with the PKC inhibitor Ro31 resulted in a significantly longer delay in tumor growth (Figure 7).
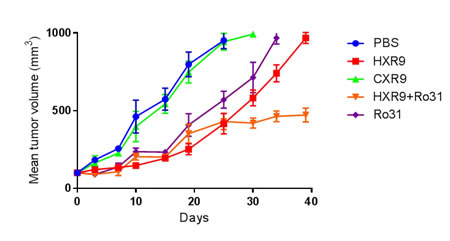
Figure 7: Inhibition of K562 tumor growth in a mouse xenograft model. Tumors were initiated by subcutaneous injection of K562 cells in to the flank of each mouse and were treated intratumourally with HXR9 or CXR9 at a dose of 50 mg/Kg, or Ro31 at a dose of 10 mg/Kg, or both HXR9 and Ro31, every 3 days.
Discussion
The HOX genes have previously been shown to have a key role in the development of a malignant phenotype in AML, although this has principally been related to their incorporation in chimeric oncogenes formed by chromosomal breakage and rearrangement [1]. Less is known of their role in their wild type from, although the overexpression of a number of HOX genes is important for cell survival and drug resistance [3]. In this study, we examined the relationship between overall survival and the expression of individual HOX genes in AML. It was previously shown that high HOXA9 expression was associated with a poor outcome in this malignancy [2], although in the larger cohort examined here this relationship was only of borderline significance. The reason for this difference is unclear, although it may reflect the high level of functional redundancy amongst HOX genes with respect to proliferation and survival. Indeed, we found that the expression of a number of other HOX genes in AML showed a strong association with survival, including HOXA5, which has been shown to be upregulated by the H3K79 methyltransferase hDOT1L in MLL-AF10-mediated leukaemogenesis [19].
The strong relationships between the elevated expression of multiple HOX genes in AML and patient survival support the possibility that HOX proteins could be therapeutic targets in this malignancy, and this is supported further by our findings that inhibition of HOX/PBX dimers cause cell death in AML-derived cell lines and primary AML cells. Taken together, our findings point to a mechanism of HXR9-mediated cell death that depends not on apoptosis, but instead on necroptosis, and which can be blocked by PKC signaling. Necroptosis is considered to be a regulated form of necrosis that in some respects parallels apoptosis, as it can be triggered by the same external stimuli [18]. It is a major mechanism of cell death in chronic lymphocytic leukaemia (CLL) [20], induced by drugs that target Bcl-2 [21] and the proteasome [22]. In addition, the induction of necroptosis in AML cells has been used as a strategy to overcome resistance to apoptosis, through the combined use of a Smac mimetic that inhibits XIAP and agents that promote DNA demthylation [23]. It has also been shown to mediate the killing of AML cells by a diphtheria toxin-GMCSF conjugate [24]. Despite this, the lack of any apoptosis following HXR9 treatment in these AML-derived cell lines is curious, as some solid malignancies have been shown to undergo apoptosis and can, for example, be rescued by the inhibition of caspase activity [8, 25].
PKC has not previously been show to directly interact with the necroptosis pathway, although signaling through PKC is known to promote the transcription of pro-survival genes, and to be a potential target in CLL [26]. Furthermore, the delta-PKC isotype was previously identified as a critical regulator of TNF signalling in adherent neutrophils. Delta PKC associates with tumour necrosis factor receptor-1 (TNFR-1) in response to TNF binding whereupon it promotes receptor serine phosphorylation and consequently enhances binding of TNFR-1-associated death domain protein (TRADD) to TNFR-1 and formation of a TNFR1-TRADD-RIP1 complex [27]. The formation of this complex in turn results in the polyubiquitination of RIP1 and activation of NF-kB signalling, which promotes cell survival. Conversely, disruption of this complex leads to RIP deubiquitination, and its subsequent recruitment to a complex with either Caspase 8, leading to apoptosis, or its phosphorylation by RIP1 kinase followed by binding to RIP3 and the induction of necroptosis (Figure 8) [28]. Our findings indicate that the latter pathway is important as an inhibitor of RIP1 kinase, but not a global caspase inhibitor, significantly reduced the sensitivity of AML cells to killing by HXR9.
In addition to inhibiting necroptosis, our findings indicate that HOX/PBX dimers block p21 expression in AML cells at both the RNA and protein level. The relationship between p21 and apoptosis is complex and varies between cell types: p21 can promote apoptosis in some contexts but prevent apoptosis in others [29]. The lack of apoptosis after HXR9 treatment might be a result of increased p21 expression (Figure 8), although further dissection of this complex molecular pathway is needed in order to confirm this.
The synergistic interaction between HXR9 and PKC-mediated signaling is particularly noteworthy, as it points to possible combinatorial approaches when targeting AML using HOX/PBX inhibitors, and indeed this was supported by results obtained using a murine model. Furthermore, the strong relationship between survival in AML and the expression of a number of HOX genes indicates that targeting HOX function via the disruption of HOX/PBX dimers could be a useful therapeutic target in this disease, and that HOX gene expression might also allow patients to be stratified according to their likely response to such a treatment. We hope that further work will help us to refine this approach to establish the selective activation of necroptosis as a therapeutic target in AML.
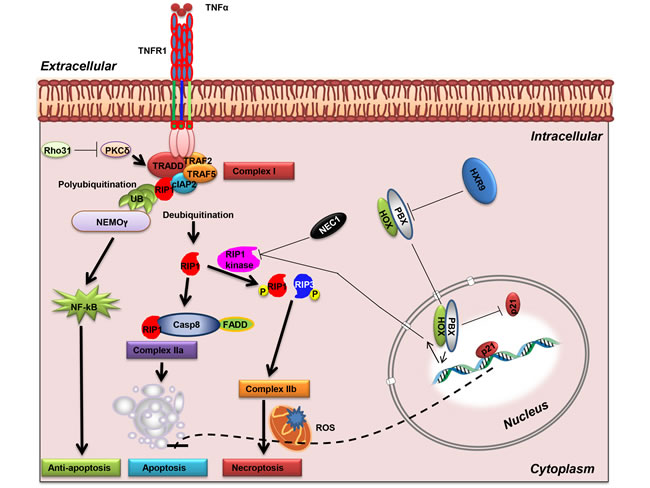
Figure 8: Activation of RIP1-mediated necroptosis. TNF-α binding to TNFR1 can activate NF-κB activation, apoptosis, or necroptosis. Phosphorylation of TNFR1 favours TRADD recruitment that in turn leads to RIP1 ubiquitination (complex I), and the subsequent activation of the NF-κB pathway via NEMO. Inhibition of PKC delta leads to the deubiquitination of RIP1 and the formation of either complex IIa with FADD and caspase-8, or, if RIP1 is phosphorylated, the formation of a necrosome with phosphorylated RIP3 (complex IIb), leading to necroptosis. A dimer between HOX and PBX proteins represses the necroptosis pathway, possibly through inhibition of RIP kinase. HOX/PBX dimers also repress p21 expression, which might in turn block apoptosis (dotted line). Abbreviations: PKC, protein kinase C; NEMO, NF-kappa-B essential modulator; RIP1, Receptor-interacting protein 1; cIAP2, Cellular inhibitor of apoptosis-1; TRAF, TNF receptor associated factors; FADD, Fas-associated protein with death domain; TRADD, Tumor necrosis factor receptor type 1-associated death domain protein.
materials and Methods
Analysis of AML microarray data
The bioinformaritcs analysis was performed in R. The GSE23312 microarray data set [30] was downloaded from the GEO database [11] and pre-processed in R/Bioconductor [31]. The patients were stratified equally into 2 groups based on the expression levels of each HOX gene. The association between the expression level and patient survival was assessed using Kaplan-Meier plots in R using the survival package.
Primary AML cell isolation
Primary AML cells were obtained after patient’s informed consent and approval of the study by the ethics committee (SGUL). Cells were isolated from bone marrow or peripheral blood using density gradient centrifugation and cultured in IMDM medium (Life Technologies, Carlsbad, CA, USA) with 10% foetal bovine serum (FBS; Biochrom GmbH, Berlin, Germany), 10% horse serum (HS; Gibco, Invitrogen, Carlsbad, USA), and 1 µM hydrocortisone (Sigma-Aldrich, St. Louis, MO).
Proliferation assays
For adaptation to oxygen content, primary AML cells were cultured for three days under hypoxic conditions and then seeded for proliferation assays. The cells were subsequently incubated with different concentrations of HXR9 or CXR9 ranging from 100 nM to 5000 nM. All assays were done in triplicate. Cell numbers were determined on day 3 after HXR9/CXR9 treatment using the cell viability analyzer Vi-Cell XR (Beckman Coulter, Brea, CA, USA). Each assay was performed at least twice.
P21 ELISA
P21 protein was measured using the p21 simplestep ELISA kit (Abcam, Cambridge, UK) according to the manufacturer’s instructions.
AML derived cell lines
In this study we used three primary AML cell lines derived from patients with de novo mutations: KG-1, a cell line derived from an erythroleukaemia [32], HEL 92.1.7, an erythroleukaemia cell line [33], and HL-60, a cell line obtained from acute promyelocytic leukaemia (APML) [34]. The two other lines, K562 (26) and KU812F (27) were derived from secondary AML (chronic myeloid leukaemia cells in blast crisis).
HXR9 and CXR9 synthesis
The synthesis of these peptides has been described previously [35].
Tissue culture
K562, KU812F and HEL92.1.7 cells were grown in RPMI-1640 medium complemented with 10% FBS, 5% L-glutamine and 5% antibiotics (1000 units of penicillin/ml and 10 mg/ml streptomycin, P/S). KG-1 and HL-60 cells were cultured in IMDM supplemented with 20% FBS and 5% antibiotics. All cell lines were cultured in T-75 sterile cell culture flasks at 37°C in a humidified environment containing 5% CO2 and 95% air.
Gene expression analysis by RT-PCR
Gene expression analysis using SYBR greenTM was carried out in order to measure the changes in expression of genes of interest. The primer sequences for the pro- and anti-apoptotic genes are listed in Table 1. Each gene of interest was tested in triplicate. The housekeeping gene β-actin was used as an endogenous control. The thermal profiles employed were: heating once for 10 minutes at 95°C, followed by 30 seconds at 95°C, 1 minute at 60°C, 30 seconds at 72°C for 40 cycles. The mean of the cycle threshold values of the triplicate repeats for each gene was calculated and used for measuring gene expression.
Table 1: Pro- and anti-apoptotic and β-actin gene primers used for PCR amplification
Gene |
Forward Sequence 5’-3’ |
Reverse Sequence 5’-3’ |
Amplicon Length (bp) |
Bad |
CGGAGGATGAGTGACGAGTT |
GATGTGGAGCGAAGGTCACT |
180 |
Bak1 |
TTTTCCGCAGCTACGTTTTT |
GGTGGCAATCTTGGTGAAGT |
248 |
Bax |
TTTGCTTCAGGGTTTCATCC |
CAGTTGAAGTTGCCGTCAGE |
246 |
Bcl2 |
GAGGATTGTGGCCTTCTTTG |
ACAGTTCCACAAAGGCATCC |
170 |
Bid |
CTGCAGGCCTACCCTAGAGA |
ACTGTCCGTTCAGTCCATCC |
195 |
XIAP |
GGGGTTCAGTTTCAAGGACA |
CGCCTTAGCTGCTCTTCAGT |
182 |
Apaf1 |
TTCTGATGCTTCGCAAACAC |
CTGGCAAATCTGCCTTCTTC |
237 |
Caspase-3 |
TTTTTCAGAGGGGATCGTTG |
CGGCCTCCACTGGTATTTTA |
151 |
Caspase-6 |
ATCCTCACCGGGAAACTGTG |
AATTGCACTTGGGTCTTTGC |
161 |
Caspase-7 |
AGTGACAGGTATGGGCGTTC |
CGGCATTTGTATGGTCCTCT |
164 |
Caspase-9 |
CTAGTTTGCCCACACCCAGT |
GCATTAGCGACCCTAAGCAG |
172 |
PARP1 |
GCTCCTGAACAATGCAGACA |
CATTGTGTGTGGTTGCATGA |
233 |
c-FOS |
CCAACCTGCTGAAGGAGAAG |
GCTGCTGATGCTCTTGACAG |
232 |
p21 |
GACACCACTGGAGGGTGACT |
CAGGTCCACATGGTCTTCCT |
171 |
p53 |
GTGGAAGGAAATTTGCGTG |
CCAGTGTGATGATGGTGAGG |
183 |
β-actin |
ATGTACCCTGGCATTGCCGAC |
GACTCGTCATACTCCTGCTTG |
227 |
Lactate dehydrogenase (LDH) assay
Cells were suspended in 5% FBS of the appropriate culture medium at a concentration of 5×105 cell/ml and seeded in 96-well flat-bottom plates at a concentration of 5×104 cell/well. The cells were then treated with 100 μl of a serial dilution of HXR9 or its negative control CXR9 in 5% FBS for 2 hours. Each plate included untreated cells (negative control), 2% triton-treated cells (positive control), and 5% FBS medium only (background control). LDH enzymatic activity was determined according to the manufacturer’s instructions.
Annexin V- PE assay
The annexin V-PE assay was used to quantify the percentage of the apoptotic cells and evaluate the mechanism of cell death. Cells were suspended in 5% FBS of the appropriate culture medium at a concentration of 1×106 cell/ml and seeded in 96-well flat-bottom plates at a concentration of 1×105 cell/well. The percentage of the different cell populations, viable, early apoptotic and dead cells, were plotted by GraphPad Prism software (California, USA) on column graphs to allow the comparison of treated and untreated negative control cells. Each experiment was repeated at least 3 times independently.
DAPI staining
Cells were suspended in 5% FBS culture medium at a concentration of 1×106 cell/ml and seeded in 24-well plates at a concentration of 5×105 cell/well. Then cells were treated either with HXR9 at the IC50 or double the IC50, in duplicate. Each plate included untreated negative control cells.
Cyclosporin A (CsA) protection assay
CsA attenuates mitochondrial permeability transition pore (mPTP) formation by binding to cyclophilin D (cypD), which is required for mPTP formation in necrotic mitochondrial cell death [36-38]. In this assay cells were pre- and co-treated with or without 5 μM CsA and hydrogen peroxide (H2O2) treatment was used as a positive control. Cells were treated in triplicate with HXR9 at the IC50, double or triple the IC50 or with 20 mM H2O2. Cell pellets were prepared and stained with annexinV-PE as described for the annexinV-PE assay. The proportion of positively stained cells was determined using flow cytometry.
Necrostatin-1 (Nec-1) protection assay
Nec-1 is a selective inhibitor of receptor interacting protein 1 (RIP1) [39]. RIP1 inhibition prevents a form of programmed necrosis called necroptosis [40]. Cells were pre- and co-treated with 50 μM Nec-1 using the same method described above for the CsA protection assay.
Fructose protection assay
Fructose is a glycolytic substrate that at high concentrations significantly sequesters phosphate and accordingly intracellular ATP is depleted. This selective depletion of ATP protects cells from apoptosis, but leaves sufficient ATP to rescue cells from spontaneous necrosis [41]. Cells were treated with HXR9 or with 2% triton as a positive control and cell viability was measured using the LDH assay as described above.
Ethilenediaminetetra-acetic acid (EDTA) and HXR9 cytotoxicity
The role of Ca2+ in HXR9-mediated cell death was assessed by two different assays, LDH and annexin V, as described for the fructose protection and CsA protection assays, respectively.
Inhibition of mitogen activated protein kinase (MAPK) pathways
The effect of HXR9 on MAPK pathways was studied using specific inhibitors of extracellular signal-regulated kinase (ERK), Jun N-terminal kinase (JNK), and p38 [42], which were U0126 monoethanolate, SP600125, and SB203580, respectively. Experiments were performed as described for the fructose protection assay.
NADPH oxidase (NOX) inhibition
NOX is a generator of intracellular reactive oxygen species (ROS) [43]. The effects of NOX on the efficiency of HXR9 were assessed using the NOX inhibitor diphenyleneiodonium chloride (DPI) [44]. Cells were pre- and co-treated with or without 60 μM DPI. The assessment of DPI effect on the cytotoxicity of HXR9 was performed as described for the fructose protection assay.
μ-calpain inhibition
Calpains are Ca2+-dependent cysteine proteases that commonly activated in Ca2+ dependent cell death mechanisms [45]. Among them, μ-calpain is one of the most extensively studied in apoptosis/necrosis processes. Calpain inhibitor I (N-Acetyl-Leu-Leu-Norleu-al) was used to inhibit the Ca2+/μ-calpain pathway. Cells were pre- and co-treated with or without 60 μM calpain inhibitor I. The assessment of the role of μ-calpain in the cytotoxicity of HXR9 was performed as described for the fructose protection assay.
PKC inhibition
Ro31-8220 (methanesulfonate salt) was used to inhibit and assess the role of PKC in HXR9 cytotoxicity. Cells were pre- and co-treated with or without 30 μM Ro31-8220, and its effects on the cytotoxicity of HXR9 was assessed as for the fructose protection assay.
Heme oxygenase-1 (HO-1) inhibition
HO-1 is an anti-apoptotic protein [46]. Protoporphyrin IX (PPIX) was used to inhibit HO-1 protein and assess its role in the cytotoxicity of HXR9. Cells were pre- and co-treated with or without 50 μM of PPIX, and the effect of HO-1 inhibition on HXR9 cytotoxicity was determined as described for the fructose protection assay.
p53 inhibition
Pifithrin-α (PFT-α) is an inhibitor of p53-dependent apoptosis and was used to block and assess the role of p53 in the cytotoxicity of HXR9. Cells were pre- and co-treated with or without 40 μM of PFT-α, and the role of p53 in HXR9 cytotoxicity was assessed as described for the fructose protection assay.
In vivo model
Female C57BL/6, nude and severe combined immune-deficient mice, approximately 6 to 8 weeks old, were purchased from Jackson laboratory, Kent, UK. These mice were maintained in pathogen-free cages and fed with irradiated food and acidified water. All experiments were performed under the compliance of Home Office and institutional instructions. The mice were anesthetised using isoflurane and K562 cells (5x106 cell/mouse) were then injected subcutaneously into the flank. Flank tumour sizes were measured three times a week using calliper measurement. Tumour sizes were calculated using the formula: (length x width x width)/2. When the flank tumour became palpable, the mice were divided into two groups with one treated intratumourly with 50 μl of the indicated dose of HXR9, CXR9, Ro31, HXR9 combined with HXR9, or with PBS alone (control group). Mice were sacrificed if the tumour volume was ≥ 1500 mm3.
Statistical analysis
Calcusyn software was used to determine the IC50 of the different drugs for the different cell lines and for assessing whether the interaction between HXR9 with DNR or MTX was synergistic, additive or antagonistic, as described below. Student’s t-test was applied for data analysis of all assays using GraPhpad Prism software. Results are expressed as the mean of 3 separate experiments with error bars to show the SEM. Statistical significance was determined by comparing the different conditions in each assay with each other and considered significant when p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), or p < 0.0001 (****). The IC50 of HXR9, DNR, and MTX for different cell lines were calculated by the median-effect equation of Chou [47] using Calcusyn software (Biosoft, Cambridge, UK). The combination effects of HXR9 with either DNR or MTX were calculated using the combination index (CI) equation [48] with Calcusyn Software. CI > 1, CI = 1, and CI < 1 indicate antagonism, an additive effect and synergism, respectively.
Abbreviations
AML, acute myeloid leukemia; APML, acute promyelocytic leukaemia; CLL, chronic lymphocytic leukaemia; CsA, cyclosporin A; cypD, cyclophilin D; cytC, cytochrome C; DPI, diphenyleneiodonium chloride; EDTA, ethilenediaminetetra-acetic acid; FBS, foetal bovine serum; GEO, gene expression omnibus; HO-1, heme oxygenase-1; HS, horse serum; LDH, lactate dehydrogenase; MAPK, mitogen activated protein kinase; mPTP, mitochondrial permeability transition pore; Nec-1, necrostatin-1; NOX, NADPH oxidase; PFT-α, pifithrin-α; PKC, protein kinase C; PPIX, protoporphyrin IX; RIP1, receptor interacting protein 1; TNFR-1, tumour necrosis factor receptor-1; TRADD, TNFR-1-associated death domain protein.
Author contributions
RA conducted the experiments and analysed the data. RP, KH and HP helped design the study and edited the paper. GRS performed the in vivo experiments. RM designed the study and wrote the paper. KP carried out the statistical analysis on the GEO dataset, and AT helped with the interpretation of the results from this analysis.
Conflicts of interest
Two of the authors (HP and RM) are shareholders in HOX Therapeutics Ltd, a company which is developing novel HOX/PBX binding antagonists, although these reagents were not used in this study.
Funding
This study was funded in part through a grant to RA from the Cultural Bureau of the Kingdom of Saudi Arabia.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Alharbi RA, Pettengell R, Pandha HS, Morgan R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia. 2013; 27: 1000-8. doi: 10.1038/leu.2012.356.
2. Andreeff M, Ruvolo V, Gadgil S, Zeng C, Coombes K, Chen W, Kornblau S, Baron AE, Drabkin HA. HOX expression patterns identify a common signature for favorable AML. Leukemia. 2008; 22: 2041-7. doi: 10.1038/leu.2008.198.
3. Dickson GJ, Liberante FG, Kettyle LM, O’Hagan KA, Finnegan DP, Bullinger L, Geerts D, McMullin MF, Lappin TR, Mills KI, Thompson A. HOXA/PBX3 knockdown impairs growth and sensitizes cytogenetically normal acute myeloid leukemia cells to chemotherapy. Haematologica. 2013; 98: 1216-25. doi: 10.3324/haematol.2012.079012.
4. Morgan R, El-Tanani M, Hunter KD, Harrington KJ, Pandha HS. Targeting HOX/PBX dimers in cancer. Oncotarget. 2017; 8:32322-32331. doi: 10.18632/oncotarget.15971.
5. Plowright L, Harrington KJ, Pandha HS, Morgan R. HOX transcription factors are potential therapeutic targets in non-small-cell lung cancer (targeting HOX genes in lung cancer). Br J Cancer. 2009; 100: 470-5. doi: 10.1038/sj.bjc.6604857.
6. Morgan R, Boxall A, Harrington KJ, Simpson GR, Gillett C, Michael A, Pandha HS. Targeting the HOX/PBX dimer in breast cancer. Breast Cancer Res Treat. 2012; 136: 389-98. doi: 10.1007/s10549-012-2259-2.
7. Morgan R, Boxall A, Harrington KJ, Simpson GR, Michael A, Pandha HS. Targeting HOX transcription factors in prostate cancer. BMC Urol. 2014; 14: 17. doi: 10.1186/1471-2490-14-17.
8. Morgan R, Pirard PM, Shears L, Sohal J, Pettengell R, Pandha HS. Antagonism of HOX/PBX dimer formation blocks the in vivo proliferation of melanoma. Cancer Res. 2007; 67: 5806-13. doi: 10.1158/0008-5472.CAN-06-4231.
9. Daniels TR, Neacato II, Rodriguez JA, Pandha HS, Morgan R, Penichet ML. Disruption of HOX activity leads to cell death that can be enhanced by the interference of iron uptake in malignant B cells. Leukemia. 2010; 24: 1555-65. doi: 10.1038/leu.2010.142.
10. Li Z, Zhang Z, Li Y, Arnovitz S, Chen P, Huang H, Jiang X, Hong GM, Kunjamma RB, Ren H, He C, Wang CZ, Elkahloun AG, et al. PBX3 is an important cofactor of HOXA9 in leukemogenesis. Blood. 2013; 121: 1422-31. doi: 10.1182/blood-2012-07-442004.
11. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30: 207-10.
12. Lecoeur H, de Oliveira-Pinto LM, Gougeon ML. Multiparametric flow cytometric analysis of biochemical and functional events associated with apoptosis and oncosis using the 7-aminoactinomycin D assay. J Immunol Methods. 2002; 265: 81-96.
13. Adrain C, Creagh EM, Martin SJ. Apoptosis-associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. EMBO J. 2001; 20: 6627-36. doi: 10.1093/emboj/20.23.6627.
14. Huerta S, Goulet EJ, Huerta-Yepez S, Livingston EH. Screening and detection of apoptosis. J Surg Res. 2007; 139: 143-56. doi: 10.1016/j.jss.2006.07.034.
15. Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004; 305: 626-9. doi: 10.1126/science.1099320.
16. Dube H, Selwood D, Malouitre S, Capano M, Simone MI, Crompton M. A mitochondrial-targeted cyclosporin A with high binding affinity for cyclophilin D yields improved cytoprotection of cardiomyocytes. Biochem J. 2012; 441: 901-7. doi: 10.1042/BJ20111301.
17. Hanus J, Zhang H, Wang Z, Liu Q, Zhou Q, Wang S. Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell Death Dis. 2013; 4: e965. doi: 10.1038/cddis.2013.478.
18. de Almagro MC, Vucic D. Necroptosis: Pathway diversity and characteristics. Semin Cell Dev Biol. 2015; 39:56-62. doi: 10.1016/j.semcdb.2015.02.002.
19. Okada Y, Jiang Q, Lemieux M, Jeannotte L, Su L, Zhang Y. Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L. Nat Cell Biol. 2006; 8: 1017-24. doi: 10.1038/ncb1464.
20. Huang Y, Wu JZ, Li JY, Xu W. Know the enemy as well as the weapons in hand: the aberrant death pathways and therapeutic agents in chronic lymphocytic leukemia. Am J Cancer Res. 2015; 5: 2361-75.
21. Basit F, Cristofanon S, Fulda S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013; 20: 1161-73. doi: 10.1038/cdd.2013.45.
22. Buac D, Shen M, Schmitt S, Kona FR, Deshmukh R, Zhang Z, Neslund-Dudas C, Mitra B, Dou QP. From bortezomib to other inhibitors of the proteasome and beyond. Curr Pharm Des. 2013; 19: 4025-38.
23. Steinhart L, Belz K, Fulda S. Smac mimetic and demethylating agents synergistically trigger cell death in acute myeloid leukemia cells and overcome apoptosis resistance by inducing necroptosis. Cell Death Dis. 2013; 4: e802. doi: 10.1038/cddis.2013.320.
24. Horita H, Frankel AE, Thorburn A. Acute myeloid leukemia-targeted toxin activates both apoptotic and necroptotic death mechanisms. PLoS One. 2008; 3: e3909. doi: 10.1371/journal.pone.0003909.
25. Morgan R, Plowright L, Harrington KJ, Michael A, Pandha HS. Targeting HOX and PBX transcription factors in ovarian cancer. BMC Cancer. 2010; 10: 89. doi: 10.1186/1471-2407-10-89.
26. Kazi JU, Kabir NN, Ronnstrand L. Protein kinase C (PKC) as a drug target in chronic lymphocytic leukemia. Med Oncol. 2013; 30: 757. doi: 10.1007/s12032-013-0757-7.
27. Kilpatrick LE, Sun S, Korchak HM. Selective regulation by delta-PKC and PI 3-kinase in the assembly of the antiapoptotic TNFR-1 signaling complex in neutrophils. Am J Physiol Cell Physiol. 2004; 287: C633-42. doi: 10.1152/ajpcell.00486.2003.
28. Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013; 1833: 3448-59. doi: 10.1016/j.bbamcr.2013.06.001.
29. Karimian A, Ahmadi Y, Yousefi B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst). 2016; 42: 63-71. doi: 10.1016/j.dnarep.2016.04.008.
30. Gaidzik VI, Bullinger L, Schlenk RF, Zimmermann AS, Rock J, Paschka P, Corbacioglu A, Krauter J, Schlegelberger B, Ganser A, Spath D, Kundgen A, Schmidt-Wolf IG, et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol. 2011; 29: 1364-72. doi: 10.1200/JCO.2010.30.7926.
31. Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, Gottardo R, Hahne F, Hansen KD, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods. 2015; 12: 115-21. doi: 10.1038/nmeth.3252.
32. Koeffler HP, Golde DW. Acute myelogenous leukemia: a human cell line responsive to colony-stimulating activity. Science. 1978; 200: 1153-4. doi: 10.1126/science.306682.
33. Martin P, Papayannopoulou T. HEL cells: a new human erythroleukemia cell line with spontaneous and induced globin expression. Science. 1982; 216: 1233-5. doi: 10.1126/science.6177045.
34. Collins SJ, Ruscetti FW, Gallagher RE, Gallo RC. Terminal differentiation of human promyelocytic leukemia cells induced by dimethyl sulfoxide and other polar compounds. Proceedings of the National Academy of Sciences of the United States of America. 1978; 75: 2458-62. doi: 10.1073/pnas.75.5.2458.
35. Morgan R, Pirard PM, Shears L, Sohal J, Pettengell R, Pandha HS. Antagonism of HOX/PBX dimer formation blocks the in vivo proliferation of melanoma. Cancer Research. 2007; 67: 5806-13. doi: 10.1158/0008-5472.can-06-4231.
36. Li Y, Johnson N, Capano M, Edwards M, Crompton M. Cyclophilin-D promotes the mitochondrial permeability transition but has opposite effects on apoptosis and necrosis. Biochem J. 2004; 383: 101-9. doi: 10.1042/bj20040669.
37. Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the Permeability Transition Pore in Mitochondria Devoid of Cyclophilin D. Journal of Biological Chemistry. 2005; 280: 18558-61. doi: 10.1074/jbc.C500089200.
38. Dube H, Selwood D, Malouitre S, Capano M, Simone MI, Crompton M. A mitochondrial-targeted cyclosporin A with high binding affinity for cyclophilin D yields improved cytoprotection of cardiomyocytes. Biochemical Journal. 2012; 441: 901-7. doi: 10.1042/bj20111301.
39. Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nature Chemical Biology. 2008; 4: 313-21. doi: 10.1038/nchembio.83.
40. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005; 1: 112-9.
41. Nieminen AL, Saylor AK, Herman B, Lemasters JJ. ATP depletion rather than mitochondrial depolarization mediates hepatocyte killing after metabolic inhibition. American Journal of Physiology. 1994; 267: C67-C74.
42. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007; 26: 3279-90.
43. Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004; 4: 181-9.
44. Park SE, Song JD, Kim KM, Park YM, Kim ND, Yoo YH, Park YC. Diphenyleneiodonium induces ROS-independent p53 expression and apoptosis in human RPE cells. FEBS letters. 2007; 581: 180-6.
45. Liu XL, Van Vleet T, Schnellmann RG. The role of calpain in oncotic cell death. Annual Review of Pharmacology and Toxicology. 2004; 44: 349-70. doi: 10.1146/annurev.pharmtox.44.101802.121804.
46. Rushworth SA, MacEwan DJ. HO-1 underlies resistance of AML cells to TNF-induced apoptosis. Blood. 2008; 111: 3793-801. doi: 10.1182/blood-2007-07-104042.
47. Chou TC. Relationships between inhibition Constants and Fractional Inhibition in Enzyme-Catalyzed Reactions with Different Numbers of Reactants, Different Reaction Mechanisms, and Different Types and Mechanisms of Inhibition. Molecular Pharmacology. 1974; 10: 235-47.
48. Chou TC. (1991). The median-effect principle and the combination index for quantitation of synergism and antagonism.