
INTRODUCTION
The Dedicator of cytokinesis (DOCK) family protein is a novel class of guanine nucleotide exchange factors (GEF) for Rac GTPases of the Rho family, [1] and is known to involved in the regulation of actin cytoskeleton, cell motility, [2] as well as cell cycle, survival, gene expression, and tumorigenesis [1]. There are 11 DOCK proteins (DOCK1 to DOCK11) in mammals, which are further classified into four subgroups (denoted as A to D) according to the sequence similarity and domain organization. The proteins in DOCK-A and B subgroups contain a terminal domain that allows binding of the adaptor protein ELMO (Engulfment and Motility) to promote efficient GEF activity [2]. Knocking down ELMO1 impairs long-term expansion of leukemic cell lines and depletion of ELMO1 in human CD34+ cells inhibit proliferation, possibly through the inhibition of ELMO-Rac axis [3]. The DOCK proteins are involved in several diseases including cancers, and disorders in the immune and central nervous systems [1]. Among them, DOCK2 plays a pivotal role in lymphocyte migration and activation as well as differentiation of T cells. A recent study identified DOCK2 as one of the FLT3 interacting protein [4]. DOCK4 participates in erythroid maturation [5]. The only known function of DOCK5 so far is the connection with osteoclasts [6]. Deletion of DOCK8 in human results in a combined immune deficiency syndrome [7]. Deregulation of the remaining DOCK proteins may contribute to neurodegenerative diseases and central nervous system defects [8]. As a prototype member in DOCK family, DOCK1 is involved in migration and invasion of various cancer cells including breast cancer, ovarian cancer and glioblastoma multiforme [9–11]. Despite the protean functions of DOCK proteins in cancers and hematopoiesis, their roles in acute myeloid leukemia (AML) have not been explored. In this study, we first screened the expression levels of DOCK family genes in a cohort of AML patients. The members with at least modest expression in AML were selected to test their prognostic significance in ours and the other two independent cohorts. We found that expression level of DOCK1 but not the other members harbored prominent prognostic significance in both ours and other independent cohorts of AML patients. Higher expression of DOCK1 served as an independent unfavorable prognostic marker. The prediction power of this single gene expression even outperformed several published gene signatures in AML prognostication. Bioinformatics analyses were performed to identify potential pathway and function of DOCK1 in AML.
RESULTS
Expression of DOCK family members in AML
To obtain a comprehensive expression landscape of DOCK family genes in hematopoietic system, we profiled gene expression microarrays of the 347 AML samples (NTUH cohort) and reprocessed several public expression datasets of AML and/or normal samples. In the TCGA RNA-Seq data of AML patients (n=179), expression levels of DOCK genes were quite diverse (Figure 1A). DOCK2 and DOCK8 were among the top 10% of genome-wide genes (median RPKM, 60.8 and 50.3), while DOCK3, DOCK4, DOCK6, and DOCK9 seemed silent (all median RPKM values<2), and DOCK5, DOCK10, and DOCK11 showed modest expression. DOCK1 had low but measurable expression levels among the AML patients. To investigate the involvement of DOCK genes in hematopoiesis, we analyzed another microarray dataset of samples across various hematopoietic cell stages (GSE24759, n=211). DOCK1 seemed to be exclusively abundant in hematopoietic stem cells (HSC) (average z-value=1.17; one-sample t-test p<0.001; Figure 1B). In erythroid cells, expression levels of DOCK2 and DOCK10 were especially low (average z, -1.53 and -1.36; both p<0.001). DOCK3, DOCK9, and DOCK10 formed a cluster of high expression in T cells (all p<0.001; Figure 1B).
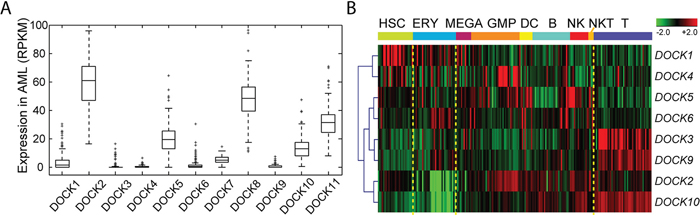
Figure 1: Expression profile of DOCK family genes in AML and normal samples. (A) Expression levels of DOCK family genes in AML. Samples of 179 AML patients were profiled with Illumina RNA-Seq by TCGA. RPKM, reads per kilobase of transcript per million mapped reads. (B) Heatmap of DOCK genes across hematopoietic cell states in GSE24759 (n=211). Expression levels were z-transformed in each probe, and multiple probes representing the same gene were averaged. Genes were clustered using hierarchical clustering. hematopoietic stem cells (HSC), erythroid cells (ERY), megakaryocytes (MEGA), granulocyte/monocyte progenitors (GMP), dendritic cells (DC), B cells (B), natural killer cells (NK), natural killer T cells (NKT), and T cells (T).
The prognostic significance of gene expression of DOCK family members in AML
Among the 347 AML patients in the NTUH cohort, 227 who received standard chemotherapy were dichotomized into two groups by the median expression levels of individual DOCK members according to the array data. With a median follow up of 57 months, we observed that a higher DOCK1 expression level was associated with worse OS (median 20 vs. 116.8 months, p<0.001) and DFS (median 11.0 vs. 101.7months, p<0.001) (Figure 2A and 2B). Convincingly, these observations could be validated by two independent cohorts TCGA (N=186) and GSE12417 (N=162) (Figure 2C and 2D). The similar prognostic impact of DOCK1 expression was also present in the patients with AML other than acute promyelocytic leukemia (Figure 2E) as well as cytogenetically normal AML (Figure 2F) in our cohort. Overall, these data confirmed that DOCK1 expression levels harbored a significant impact on survival in AML patients. On the contrary, higher expression of DOCK2 was associated with a favorable outcome in our patients (median OS 68.0 months vs. 24.9 months, p=0.020), however, this result was not able to be validated by TCGA or GSE12417 cohort (Supplementary Figure 1). The expression of other DOCK genes failed to demonstrate impacts on survival in our AML patients. We thus focused on DOCK1 gene expression in subsequent analyses.
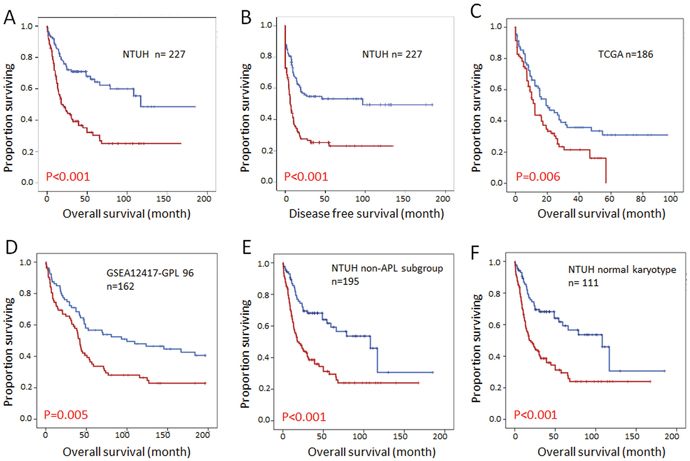
Figure 2: Kaplan Meier survival curves for AML patients stratified by DOCK1 expression levels. (A) Overall survival in the NTUH cohort. (B) Disease free survival in the NTUH cohort. (C) Overall survival in the TCGA cohort. (D) Overall survival in the GSEA12417-GPL96 cohort. (E) Overall survival in non-APL patients from the NTUH cohort. (F) Overall survival in AML patients with normal karyotype from the NTUH cohort. Red line: higher DOCK1 expression; blue line: lower DOCK1 expression.
For the 227 patients who received standard chemotherapy, 61% of those with higher DOCK1 levels achieved complete remission (CR), compared to 84% in the lower expression group (p<0.001, Table 1). Univariate Cox analysis revealed that higher DOCK1 expression (p<0.001), unfavorable karyotype (p=0.001), older age (p=0.003), higher WBC count (p=0.015), CD 34 expression (p=0.018), FLT3-ITD (p= 0.001), MLL-PTD (p=0.001), and mutations in TP53 (p<0.001), RUNX1 (p=0.040) and WT1 (p=0.020) were all associated with shorter OS (Supplementary Table 1) while similar parameters, except WT1 and DNMT3A mutations, adversely affected DFS (Supplementary Table 2). Presence of CEBPA double mutations correlated with better OS and DFS. Multivariate analysis confirmed higher DOCK1 expression, along with older age, higher WBC count, unfavorable karyotype, absence of CEBPA double mutations and TP53 mutation, as an independent unfavorable prognostic factor for both overall survival and disease free survival, p=0.005 (Table 2) and p<0.001 (Supplementary Table 3) respectively.
Table 1: Comparison of clinical manifestations between AML patients with higher and lower BM DOCK1 expression
Variables |
Total (n=347) |
DOCK1 expression |
P value |
|
|---|---|---|---|---|
Higher (n=174) |
Lower (n=173) |
|||
Sex† |
0.253 |
|||
Male |
196 |
93 |
103 |
|
Female |
151 |
81 |
70 |
|
Age (year)‡ |
62 (15-89) |
53 (16-91) |
0.010 |
|
Lab data |
||||
WBC (/μL)‡ |
24415 (890-423000) |
18770 (380-417500) |
0.188 |
|
Hemoglobin (g/dL)‡ |
8.1 (3.3-14.0) |
8.1 (3.7-16.2) |
0.441 |
|
Platelet (×1,000/μL)‡ |
60 (7-655) |
39 (2-493) |
<0.001 |
|
Blast (/μL)‡ |
12325 (0-345964) |
5080 (0-369070) |
0.005 |
|
LDH (U/L)‡ |
897 (202-9097) |
931 (242-13130) |
0.412 |
|
FAB† |
174 |
173 |
<0.001 |
|
M0 |
9 |
4 |
2 |
0.414 |
M1 |
67 |
35 |
32 |
0.703 |
M2 |
109 |
54 |
55 |
0.879 |
M3 |
28 |
1 |
27 |
<0.001 |
M4 |
103 |
62 |
41 |
0.015 |
M5 |
20 |
11 |
9 |
0.655 |
M6 |
8 |
4 |
4 |
0.993 |
Undetermined |
6 |
3 |
3 |
0.994 |
Induction response† |
227 |
114 |
113 |
<0.001 |
CR† |
165 (72.7%) |
70 (61.4%) |
95 (84.1%) |
<0.001 |
PR+ refractory† |
47 (20.7%) |
34 (29.8%) |
13 (11.5%) |
<0.001 |
Induction death† |
15 (6.6%) |
10 (8.8%) |
5 (4.4%) |
0.187 |
Relapse† |
72 (31.7%) |
40 (35.1%) |
32 (28.3%) |
0.273 |
†Number of patients (%).
‡Median (range).
WBC, white blood cell count; FAB, French-American-British classification; CR, complete remission; PR, partial remission.
Table 2: Multivariate analysis (Cox regression) on the overall survival*
Variables |
Overall survival |
|||
|---|---|---|---|---|
HR |
95% CI |
P value |
||
Lower |
Upper |
|||
Total cohort (n=227) |
||||
Age |
1.014 |
1.001 |
1.027 |
0.036 |
WBC |
1.000 |
1.000 |
1.000 |
0.036 |
Unfavorable cytogenetics |
2.672 |
1.484 |
4.809 |
0.001 |
FLT3-ITD |
1.320 |
0.840 |
2.073 |
0.229 |
CEBPAdouble mutation |
0.331 |
0.131 |
0.837 |
0.019 |
RUNX1 mutation |
1.279 |
0.718 |
2.275 |
0.403 |
MLL-PTD |
1.636 |
0.729 |
3.620 |
0.225 |
WT1 mutation |
1.827 |
1.022 |
3.267 |
0.042 |
TP53 mutation |
3.838 |
1.416 |
10.402 |
0.008 |
DOCK1 higher expression |
1.501 |
1.127 |
1.998 |
0.005 |
*The model was generated from a stepwise Cox regression model that included age, WBC, unfavorable cytogenetics, gene mutations of FLT3, WT1, CEBPA, RUNX1, MLL, TP53 and expression level of DOCK1.
HR, hazard ratio; CI, confidence interval; WBC, white blood cell count.
Association of expression levels of DOCK1 with clinical and biological parameters
Patient with higher DOCK1 expression were older (p=0.010) and had higher platelet (p<0.001) and peripheral blast counts (p=0.005) at diagnosis than the lower expression group (Table 1). In addition, higher DOCK1 expression level was closely associated with intermediate-risk cytogenetics (p=0.003) and normal karyotype (p=0.021), but inversely correlated with favorable karyotype, including t(8;21) and t(15:17) (all p<0.001) (Supplementary Table 4). Moreover, mutations in CEBPA, KIT, and IDH2 occurred less often whilst FLT3-ITD and mutations in PTPN11, MLL, NPM-1, RUNX1, ASXL1 and DNMT3A appeared more frequently in patients with higher DOCK1 expression than in those with lower expression (Supplementary Table 5).
Comparisons of prognostic significance of DOCK1 expression with published prognostic gene signatures
To further evaluate the potential of DOCK1 expression as a prognostic marker for AML, we compared the survival impact of DOCK1 expression with other published gene expression-based prognostic signatures. We performed pairwise multivariate Cox analysis of DOCK1 expression with each of the published 3-gene, [12] 7-gene, [13] 11-gene, [14] and 24-gene predictors [15]. Notably, DOCK1 expression remained a prognostic factor independent to all these composite signatures (with multivariate Cox p<0.05) and achieved even higher significance in most of the comparison settings (10 out of 12; Table 3). Considering that these signatures were originally developed by diverse study designs in multiple AML cohorts, our data suggested DOCK1 expression as a simple, powerful, and widely applicable prognostic marker.
Table 3: Comparisons of DOCK1 expression to published prognostic gene signatures
Predictor |
NTUH (n=227) |
TCGA (n=186) |
GSE12417 (n=162) |
|---|---|---|---|
DOCK1 |
<0.001* (1.50;1.27-1.76)** |
0.001 (1.73;1.26-2.37) |
0.001 (1.34;1.12-1.60) |
3-gene score (Wilop et al.) |
0.010 (1.34;1.07-1.68) |
0.003 (1.37;1.11-1.69) |
0.194 (1.15;0.93-1.42) |
DOCK1 |
<0.001 (1.43;1.20-1.70) |
0.003 (1.66;1.19-2.31) |
0.008 (1.28;1.07-1.55) |
7-gene score (Marcucci et al.) |
0.003 (1.19;1.06-1.34) |
0.092 (1.10;0.98-1.22) |
0.031 (1.19;1.02-1.38) |
DOCK1 |
0.001 (1.39;1.15-1.67) |
0.005 (1.68;1.17-2.41) |
0.146 (1.17;0.95-1.44) |
11-gene score (Chuang et al.) |
0.007 (1.05;1.01-1.09) |
0.337 (1.02;0.98-1.07) |
0.004 (1.06;1.02-1.11) |
DOCK1 |
0.004 (1.36;1.11-1.68) |
0.034 (1.43;1.03-2.00) |
0.023 (1.32;1.04-1.68) |
24-gene score (Li et al.) |
0.029 (1.07;1.01-1.13) |
<0.001 (1.11;1.05-1.18) |
0.493 (1.02;0.97-1.07) |
*Pairwise multivariate P-value (**hazard ratio; 95% confidence interval of hazard ratio).
Functional enrichment analysis of DOCK1
We conducted functional enrichment analysis to gain biological insights into the underlying mechanisms of unfavorable prognosis related to DOCK1 over-expression. The up-regulation of DOCK1 in HSC cells (Figure 1B) implied its role in stem cell biology in AML. To test this hypothesis, we curated two published stem cell gene signatures [16, 17]. GSEA analysis showed concordant up-regulation of HSC and leukemic stem cell (LSC)-associated genes (Supplementary Tables 6 and 7) in patients with higher DOCK1 expression (GSEA p<0.001 and p=0.004, respectively; Figure 3A). Furthermore, GSEA revealed a concordant enrichment of homeobox genes (Supplementary Table 8, p<0.001; Figure 3A), indicating the association of DOCK1 in stem cell functions as homeobox genes are well-known central player in determining stem cell fate [18].
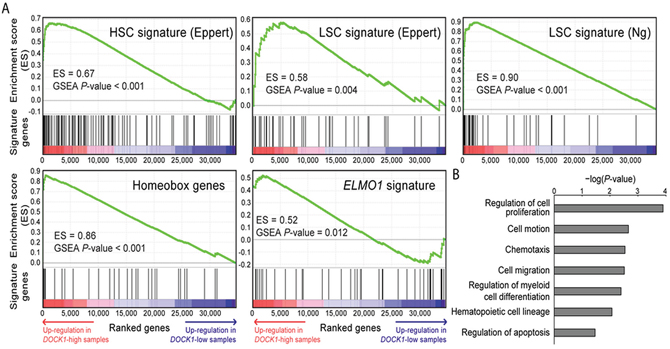
Figure 3: Functional enrichment analysis of DOCK1 in AML. (A) GSEA enrichment plots. GSEA sorted genome-wide genes by the significance of differential expression between patients with higher and lower DOCK1 expressions (denoted by red and indigo arrows) in the NTUH cohort. Each set of genes was tested for an enrichment (measured by an enrichment score and permutation-based p-value) at either side of the list. Four gene sets were analyzed, including gene signatures of HSC and LSC, described previously by Eppert et al [16] and Ng et al [17], respectively and our manually curated ELMO1-interacting genes and Homeobox genes. All of these gene sets showed significant enrichment in samples with DOCK1 up regulation. (B) Functions and pathways of DOCK1 in the NTUH cohort. We used the DAVID web tool to analyze associated functions and pathways of 321 probes differentially expressed between DOCK1-high and -low patients. DAVID-generated modified Fisher’s exact test p-values of selected terms are represented in log-10 scale.
DOCK1 is known to interact physically with ELMO1, a mediator of chemotaxis in AML, with an implication in cell migration [2, 3]. We reasoned that the adverse effect of higher DOCK1 expression in prognosis might be mediated by this axis. To verify this, we manually curated a signature of ELMO1-interacting genes (Supplementary Table 9) to measure its activity and ran another GSEA analysis. Indeed, the ELMO1 signature was positively associated with DOCK1 overexpression (p=0.012; Figure 3A). DAVID analysis confirmed the association between 321 differentially expressed probes between DOCK1-high and DOCK1-low patients (Supplementary Table 10) (with t-test p<0.05 and >1.5-fold change between DOCK1-high and -low patients) and functions involved in chemotaxis and cell migration-related functions, cell proliferation, and apoptosis (modified Fisher’s exact test p-values<0.05; Figure 3B). Overall, our data implied the involvement of DOCK1 in chemotaxis and migration in hematopoietic cells, at least partially accounting for unfavorable survival in AML patients with higher DOCK1 expression. Further biological investigations are needed to delineate the underlying mechanisms.
DISCUSSION
While DOCK proteins have pleiotropic functions in immunity, neurology, cytokinesis, and motility, their functions in hematopoietic system and prognostic significance in AML are totally unknown. In this study, we addressed these questions by analyzing the expression patterns of the 11 DOCK member genes in normal hematopoietic and AML cells. The data were extracted from ours and other public databases. Our analyses focused on DOCK1 since it was the only one in the family with prognostic significance. The finding can be validated in other independent cohorts.
DOCK1 protein is involved in the regulation of actin cytoskeleton and cell motility [2]. The dysregulation of these molecules may be responsible for invasive and metastatic properties of cancer cells [9, 10, 19]. DOCK1 gene expression carries prognostic significance in breast cancer, ovarian cancer and glioblastoma multiforme through activation of c-JUN, STAT3, and Rac1 [9, 10, 19]. Given these findings in solid cancers, the role of DOCK1 protein in AML remains unexplored. To the best of our knowledge, the current study is the first to report the clinical implication of DOCK gene expression in AML. DOCK1 gene seems to be a special DOCK member because of its unique enrichment in HSC and adverse prognostic impact in AML.
The mechanisms underlying poor prognostic implication of higher DOCK1 expression in AML remain to be explored. Our bioinformatics approach revealed DOCK1 overexpression coincided with some of its known functions such as cell migration, motion, and chemotaxis in AML cells (Supplementary Table 10). HIF-1α [20, 21] and CXCR4, two factors related to cell motion and chemotaxis, have been reported as unfavorable prognostic markers for AML [22, 23]. The analysis of microarray data exhibited significant correlation of the expression of DOCK1 with that of HIF-1α and CXCR4 in our cohort (data not shown). This was consistent with previous study in which knock down of ELMO1, the partner gene of DOCK1, resulted in reduced chemotaxis of leukemia cells [3].
We showed that DOCK1 was associated with both HSC and LSC signatures. Since stemness is an established property pertaining to drug resistance and poor prognosis in cancer patients [24], these data provided an explanation for the unfavorable prognostic impact of DOCK1 over-expression in AML. We compared the prognostic significance of DOCK1 expression to several published multi-gene signatures originally derived from a wide range of molecular mechanisms (e.g., epigenetic changes and chemoresistance genes) and cohorts with different cytogenetic subtypes and races. The comparisons were performed among datasets from both cytogenetically normal (GSE12417) and cytogenetically heterogeneous AML patients (NTUH and TCGA) (Table 3). The promising performance of DOCK1 expression reinforces its potential as a simple and powerful prognostic marker that seems to be independent of cytogenetic abnormalities, but further studies are necessary to confirm it.
We noted that the three datasets analyzed in this study were derived from different profiling platforms. Probe-wise z-transformation was thus performed to each dataset in order to eliminate cross-platform and cross-cohort biases. For simplicity, in this study we adopted a median of DOCK1 expression as a cutoff level. For clinical practice, a receiver operating characteristic (ROC) analysis of 2-year survival in the NTUH dataset suggested an optimal cutoff at 0.625 (z-transformed expression of DOCK1, or, if multiple probes exist, average of z-transformed expression of all probes of DOCK1). The cutoff achieved significant differences in survival curves in the three datasets (all p-values<0.001) and warrants further testing by a large prospective study.
In summary, our study concluded that the overexpression of DOCK1 as an unfavorable prognostic marker in acute myeloid leukemia possibly through its correlation with stemness, cell proliferation, motility and chemotaxis. Current study focused on the clinical and bioinformatics analyses on the prognostic and biological impacts of DOCK1 in AML. Prospective studies are warranted to confirm our observation. Moreover, further mechanistic studies are necessary to delineate how DOCK1 participates in the stem cell biology of hematopoietic lineages and its role in modulating unfavorable prognostic impact in AML.
MATERIALS AND METHODS
Patients
We recruited 347 adult patients with newly diagnosed AML at the National Taiwan University Hospital (NTUH) from 1995 to 2011. Among them, 227 patients underwent standard intensive chemotherapy as described previously [25]. We obtained written informed consent from each patient in accordance with Declaration of Helsinki. This study was approved by the Research Ethics Committee of the National Taiwan University Hospital.
Cytogenetic and mutation analyses
Cytogenetic risk was defined according to the European Leukemia Net (ELN) guideline [26]. Analyses of genetic mutations including FTL3-ITD, FTL3-TKD, NPM1, MLL-PTD, NRAS, KRAS, PTPN11, KIT, JAK2, WTI, CEBPA, RUNX1, ASXL1, IDH1, IDH2, TET2 and DNTM3A mutations were carried out in bone marrow mononuclear cells obtained at diagnosis as described previously [27–31].
High-throughput mRNA assay and data analysis
Illumina HumanHT-12 v4 Expression BeadChips (Illumina, Inc., San Diego, CA) were utilized to profile global gene expression of bone marrow cells from the 347 AML patients as previously described (GEO accession numbers: GSE68469 and GSE71014) [14, 32, 33]. Expression of DOCK1 was analyzed in binomial manner with a median cutoff to ensure balance comparison, as in previous literatures [13, 15, 32]. To confirm the prognostic implication of DOCK1 in our cohort, we incorporated publicly available gene expression microarray datasets from two independent AML cohorts, The Cancer Genome Atlas (TCGA; n=186) [34] and GSE12417 (n=162) [35]. The RNA-Seq dataset of the TCGA cohort (n=179) was also utilized in this study which was represented in RPKM (reads per kilobase of transcript per million mapped reads) counts. All microarray data were converted to the log-2 scale if not performed originally. Probe expression levels were transformed to z-values (subtraction of probe mean and division by probe standard deviation) and averaged into gene-level data.
Functional enrichment analysis
Differentially expressed genes associated with the gene of interest (i.e., DOCK1) were analyzed by the Database for Annotation, Visualization and Integrated Discovery (DAVID) [36, 37] for related functions, such as Gene Ontology terms and biological pathways, with default settings. We also utilized the Gene Set Enrichment Analysis (GSEA) [38] to investigate the association between expression profile of the gene and other biological functions or gene signatures. Briefly, previously published or our manually curated gene signatures were collected and tested for enrichment in a genome-wide gene list, which was sorted by the significance of differential expression between patients with highest (>3rd quartile) and lowest (<1st quartile) expression of the gene. We adopted default GSEA settings and the significance of an enrichment score was evaluated by a permutation test with respect to genes.
Statistical analysis
Statistical analysis was generated using SPSS software 21.0. We first examine the normality of array parameter with Shapiro-Wilk test. Non-parametric variables were analyzed with Mann-Whitney test while nominal parameters were compared by Chi-square test. The overall survival (OS) was calculated and plotted with the use of Kaplan-Meier curves from the date of first diagnosis to date of last follow-up or death from any cause. The disease free survival (DFS) was measured from the date of primary treatment to the date of disease relapse or death from any cause. Log-rank test was utilized to determine statistical significance. Whole patient cohort (n=347) was included in the analysis of association between DOCK expression levels and clinical characteristics yet only the 227 of them who received standard chemotherapy were included for survival analyses. The prognostic significance of parameters such as age, white blood cell counts, CD34 expression, mutation status of the afore-mentioned 16 genes and expression levels of DOCK1 were first evaluated individually in univariate analysis and the significant factors were then examined together with multivariate analysis using Cox regression. Level of significance was set at p<0.05.
Author contributions
Conception and design: WC Chou, HF Tien, YC Chiu.
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): CC Lin, HA Hou, YC Chiu, YH Li.
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): SH Lee, YC Chiu.
Writing, review, and/or revision of the manuscript: WC Chou, HF Tien, SH Lee, YC Chiu.
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): CC Lin, SH Lee, YC Chiu, YH Li.
Study supervision: WC Chou, HF Tien.
CONFLICTS OF INTEREST
All authors have no conflicts of interest to declare.
FUNDING
The study was supported by a National Taiwan University Hospital-National Taiwan University joint research grant (UN103-051), Ministry of Science and Technology of Taiwan (MOST102-2325-B-002-028, 103-2314-B-002-130-MY3, 103-2314-B-002-131MY3 and 104-2923-B-002-001), and Ministry of Health and Welfare of Taiwan (MOHW102-TD-C-111-001 and MOHW103-TD-B-111-04).
REFERENCES
1. Gadea G, Blangy A. Dock-family exchange factors in cell migration and disease. Eur J Cell Biol. 2014; 93:466-477.
2. Cote JF, Vuori K. GEF what? Dock180 and related proteins help Rac to polarize cells in new ways. Trends Cell Biol. 2007; 17:383-393.
3. Capala ME, Vellenga E, Schuringa JJ. ELMO1 is upregulated in AML CD34+ stem/progenitor cells, mediates chemotaxis and predicts poor prognosis in normal karyotype AML. PLoS One. 2014; 9:e111568.
4. Wu M, Hamaker M, Li L, Small D, Duffield AS. DOCK2 interacts with FLT3 and modulates the survival of FLT3-expressing leukemia cells. Leukemia. 2017; 31:688-696.
5. Sundaravel S, Duggan R, Bhagat T, Ebenezer DL, Liu H, Yu Y, Bartenstein M, Unnikrishnan M, Karmakar S, Liu TC, Torregroza I, Quenon T, Anastasi J, et al. Reduced DOCK4 expression leads to erythroid dysplasia in myelodysplastic syndromes. Proc Natl Acad Sci U S A. 2015; 112:E6359-6368.
6. Brazier H, Stephens S, Ory S, Fort P, Morrison N, Blangy A. Expression profile of RhoGTPases and RhoGEFs during RANKL-stimulated osteoclastogenesis: identification of essential genes in osteoclasts. J Bone Miner Res. 2006; 21:1387-1398.
7. Nishikimi A, Kukimoto-Niino M, Yokoyama S, Fukui Y. Immune regulatory functions of DOCK family proteins in health and disease. Exp Cell Res. 2013; 319:2343-2349.
8. Shi L. Dock protein family in brain development and neurological disease. Commun Integr Biol. 2013; 6:e26839.
9. Laurin M, Huber J, Pelletier A, Houalla T, Park M, Fukui Y, Haibe-Kains B, Muller WJ, Cote JF. Rac-specific guanine nucleotide exchange factor DOCK1 is a critical regulator of HER2-mediated breast cancer metastasis. Proc Natl Acad Sci U S A. 2013; 110:7434-7439.
10. Zhao F, Siu MK, Jiang L, Tam KF, Ngan HY, Le XF, Wong OG, Wong ES, Chan HY, Cheung AN. Overexpression of dedicator of cytokinesis I (Dock180) in ovarian cancer correlated with aggressive phenotype and poor patient survival. Histopathology. 2011; 59:1163-1172.
11. Feng H, Hu B, Vuori K, Sarkaria JN, Furnari FB, Cavenee WK, Cheng SY. EGFRvIII stimulates glioma growth and invasion through PKA-dependent serine phosphorylation of Dock180. Oncogene. 2014; 33:2504-2512.
12. Wilop S, Chou WC, Jost E, Crysandt M, Panse J, Chuang MK, Brummendorf TH, Wagner W, Tien HF, Kharabi Masouleh B. A three-gene expression-based risk score can refine the European LeukemiaNet AML classification. J Hematol Oncol. 2016; 9:78.
13. Marcucci G, Yan P, Maharry K, Frankhouser D, Nicolet D, Metzeler KH, Kohlschmidt J, Mrozek K, Wu YZ, Bucci D, Curfman JP, Whitman SP, Eisfeld AK, et al. Epigenetics meets genetics in acute myeloid leukemia: clinical impact of a novel seven-gene score. J Clin Oncol. 2014; 32:548-556.
14. Chuang MK, Chiu YC, Chou WC, Hou HA, Tseng MH, Kuo YY, Chen Y, Chuang EY, Tien HF. An mRNA expression signature for prognostication in de novo acute myeloid leukemia patients with normal karyotype. Oncotarget. 2015; 6:39098-39110.
15. Li Z, Herold T, He C, Valk PJ, Chen P, Jurinovic V, Mansmann U, Radmacher MD, Maharry KS, Sun M, Yang X, Huang H, Jiang X, et al. Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloid leukemia: an international collaborative study. J Clin Oncol. 2013; 31:1172-1181.
16. Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, Canty AJ, Danska JS, Bohlander SK, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011; 17:1086-1093.
17. Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, Arruda A, Popescu A, Gupta V, Schimmer AD, Schuh AC, Yee KW, Bullinger L, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016; 540:433-437.
18. Seifert A, Werheid DF, Knapp SM, Tobiasch E. Role of Hox genes in stem cell differentiation. World J Stem Cells. 2015; 7:583-595.
19. Feng H, Hu B, Liu KW, Li Y, Lu X, Cheng T, Yiin JJ, Lu S, Keezer S, Fenton T, Furnari FB, Hamilton RL, Vuori K, et al. Activation of Rac1 by Src-dependent phosphorylation of Dock180(Y1811) mediates PDGFRalpha-stimulated glioma tumorigenesis in mice and humans. J Clin Invest. 2011; 121:4670-4684.
20. Migliavacca J, Percio S, Valsecchi R, Ferrero E, Spinelli A, Ponzoni M, Tresoldi C, Pattini L, Bernardi R, Coltella N. Hypoxia inducible factor-1a regulates a pro-invasive phenotype in acute monocytic leukemia. Oncotarget. 2016; 7:53540-53557.
21. Deeb G, Vaughan MM, McInnis I, Ford LA, Sait SN, Starostik P, Wetzler M, Mashtare T, Wang ES. Hypoxia-inducible factor-1alpha protein expression is associated with poor survival in normal karyotype adult acute myeloid leukemia. Leuk Res. 2011; 35:579-584.
22. Peled A, Tavor S. Role of CXCR4 in the pathogenesis of acute myeloid leukemia. Theranostics. 2013; 3:34-39.
23. Spoo AC, Lubbert M, Wierda WG, Burger JA. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood. 2007; 109:786-791.
24. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005; 5:275-284.
25. Chou WC, Tang JL, Lin LI, Yao M, Tsay W, Chen CY, Wu SJ, Huang CF, Chiou RJ, Tseng MH, Lin DT, Lin KH, Chen YC, Tien HF. Nucleophosmin mutations in de novo acute myeloid leukemia: the age-dependent incidences and the stability during disease evolution. Cancer Res. 2006; 66:3310-3316.
26. Mrozek K, Marcucci G, Nicolet D, Maharry KS, Becker H, Whitman SP, Metzeler KH, Schwind S, Wu YZ, Kohlschmidt J, Pettenati MJ, Heerema NA, Block AW, et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol. 2012; 30:4515-4523.
27. Hou HA, Chou WC, Kuo YY, Liu CY, Lin LI, Tseng MH, Chiang YC, Liu MC, Liu CW, Tang JL, Yao M, Li CC, Huang SY, et al. TP53 mutations in de novo acute myeloid leukemia patients: longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015; 5:e331.
28. Chou WC, Lei WC, Ko BS, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Wu SJ, Huang SY, Hsu SC, Chen YC, Chang YC, et al. The prognostic impact and stability of Isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia. 2011; 25:246-253.
29. Chou WC, Chou SC, Liu CY, Chen CY, Hou HA, Kuo YY, Lee MC, Ko BS, Tang JL, Yao M, Tsay W, Wu SJ, Huang SY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011; 118:3803-3810.
30. Chou WC, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Ko BS, Wu SJ, Huang SY, Hsu SC, Chen YC, Huang YN, Chang YC, et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood. 2010; 115:2749-2754.
31. Chou WC, Huang HH, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Ko BS, Wu SJ, Huang SY, Hsu SC, Chen YC, Huang YN, et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2010; 116:4086-4094.
32. Chuang MK, Chiu YC, Chou WC, Hou HA, Chuang EY, Tien HF. A 3-microRNA scoring system for prognostication in de novo acute myeloid leukemia patients. Leukemia. 2015; 29:1051-1059.
33. Chiu YC, Tsai MH, Chou WC, Liu YC, Kuo YY, Hou HA, Lu TP, Lai LC, Chen Y, Tien HF, Chuang EY. Prognostic significance of NPM1 mutation-modulated microRNA-mRNA regulation in acute myeloid leukemia. Leukemia. 2016; 30:274-284.
34. Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW, Baty JD, Fulton LL, Fulton R, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013; 368:2059-2074.
35. Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, Heinecke A, Radmacher M, Marcucci G, Whitman SP, Maharry K, Paschka P, Larson RA, et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008; 112:4193-4201.
36. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44-57.
37. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009; 37:1-13.
38. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102:15545-15550.