
INTRODUCTION
Type I interferons (IFNs) are pleiotropic immunomodulatory cytokines that were originally described based on their ability to interfere in the viral infection cycle [1], that is to say, activate protective antiviral machinery in infected cells, their neighbors and, on a systemic scale, in antigen presenting cells (APCs) and T lymphocytes [2]. The IFN family is sub-divided into three types of cytokines — type I, type II and type III — which differ in their protein sequence, function, producer cell and cognate receptor. In humans, type I IFNs contain 18 distinct members (13 subtypes of IFN-α and one for each IFN-β, IFN-ε, IFN-κ, IFN-τ and IFN-ω) that, interestingly, all bind to the same cognate receptor, composed of the IFN-α/β receptor 1 (IFNAR1) and IFNAR2 subunits [3, 4]. Unlike the other type I IFNs, IFN-α and IFN-β have much more established and known roles in immunity and our review will focus on them. There is only one type II IFN — identified as IFN-γ — which binds to the IFN-γ receptor 1 (IFNGR1) and IFNGR2 subunits and is mainly produced by CD4+ helper T lymphocytes and natural killer (NK) cells. Type III IFNs consist of IFN-λ1, IFN-λ2, IFN-λ3 and IFN-λ4, which bind to the IFN-λ heterodimeric receptor 1 (IFNLR1) and Interleukin-10 (IL-10) receptor subunit β [3, 4].
During the past decades, a growing body of evidence clearly indicates that type I IFNs also play a pivotal role in naturally occurring and therapy induced immune responses to cancer [5]. This conclusion is based on two key observations: First, Ifnar1 knockout (KO) mice are more tumor-prone upon exposure to the carcinogen methylcholanthrene (MCA) in comparison with mice that have functional type I IFN signaling. Second, tumors that arise from this IFN-α/β deficient context were more immunogenic (i.e., immune rejected when transplanted into a immunocompetent, naïve, syngeneic host) than when they were originated in the wild type background, thus demonstrating a significant role for type I IFNs in immune surveillance during carcinogenesis and tumor progression [6].
The ability of the immune system to eliminate nascent transformed cells, control and sculpt the immunogenicity of developing tumors while in a state of equilibrium, and upon escape of the immune control, exert pro-tumor functions, are all contemplated in the cancer immunoediting hypothesis [7, 8]. And among the cellular and molecular pathways identified so far, type I IFNs seem to be critical components for the host immune response against tumor, more specifically for the dendritic cell (DC) compartment [6, 9].
First identified by Steinman and Cohn [10, 11] DCs are professional APCs that act as central regulators of the antitumor immune cycle [12]. While in the steady state, DCs are present in their immature form, characterized by high capacity to capture antigens, but low secretion of cytokines and expression of co-stimulatory molecules (e.g., CD80, CD40, CD86). Yet, in the face of tissue injury, cell death or microbial infection, DCs are activated and migrate to the draining lymph nodes (LN) where they acquire fully mature phenotype (i.e, high expression of major histocompatibility complex (MHC) molecules and co-stimulatory signaling potential). DCs will then pass on the message received in the microenvironment where the antigen was encountered, delivering both antigenic (through MHC-I and MHC-II, due to their cross-presentation ability) and co-stimulatory signals, via membrane and secreted molecules, such as CD80, CD86 and IL-12, respectively, to prime naïve T cells [13, 14]. Interestingly, spontaneous immune responses to tumor cells have been shown to depend on the activation of DCs by type I IFNs [15] and as a result, one of the first cancer immunotherapies ever to be approved by the US Food and Drug Administration (FDA) consisted of high doses of recombinant IFN-α2b for melanoma and renal cell carcinoma [16]. Since then, numerous other antitumor strategies have exploited the immunomodulatory properties of type I IFNs to bring the full force of the immune system to the cancer fighting arena. For these reasons, in this review we will discuss the pleiotropic effects of type I IFNs on cancer and immunity and some of the therapeutic opportunities based on this critical interaction.
DENDRITIC CELL SUBSETS IN CANCER
All DCs originate from bone marrow hematopoietic stem cells through sequential steps of differentiation that first form a common progenitor of macrophages/DCs and, secondly, give rise to two lineage specific precursors, one for monocytes and the other for DCs. The latter finally branches out into two major subsets, plasmacytoid DCs (pDCs) and conventional DCs (cDCs), which are further divided into cDC1 and cDC2, based on the transcription factors that drive the development process, cell surface markers and functions [17]. It is important to stress that much of the following nomenclature was obtained from studies of the mouse immune system and not all data from murine models perfectly match with their human counterpart.
In the mouse, cDC1s are negative for the CD11b marker, dependent on the inhibitor of DNA binding 2 (ID2), interferon regulatory factor 8 (IRF8) or basic leucine zipper ATF-like transcription factor 3 (BATF3) transcriptional factors, express the X-C Motif chemokine receptor 1 (XCR1) and display a remarkable capacity to cross-present antigens on MHC-I to activate CD8+ T cell responses. Among the BAFT3 driven DCs, CD8α+ DCs are localized in lymphoid organs, such as spleen and LN, thus not found in the non-lymphoid organs, whereas CD103+ cDCs are found in non-lymphoid organs [18, 19]. Importantly, Baft3 KO mice, which lack both CD103+ and CD8α+ cDCs, when transplanted with highly immunogenic tumors (i.e., spontaneously regresses after being inoculated in immunocompetent mice) are not able to reject them [20] and even more critically, do not respond to checkpoint blockade immunotherapy [21]. On the other hand, cDC2s that can induce innate lymphoid cells (ILCs) and a Th2 immune response against multicellular parasites are a CD11b+ heterogeneous population, dependent on the IRF4 and zinc finger E-box binding homeobox 2 (ZEB2) transcription factors, express the signal regulatory protein alpha (SIRPα/CD172a) transmembrane protein and present MHC-II antigens to CD4+ T cells [15, 18]. Yet, their role in cancer remains unclear [15, 18, 19].
In humans, equivalent BAFT3 dependent DCs are identified by the expression of CD11c, CLEC9A, XCR1 and CD141 [22], have been found in different tumor types and, just as the murine DC, seem to be relevant in anti-tumor responses, since their presence correlates with a superior outcome in melanoma patients [14, 23]. Key insights on why these CD103+/CD141+ DCs display such unique function came from a recent work that used a mouse model of cancer to question which of the different tumor associated APCs (resident CD11b+, migratory CD11b+, CD8α+, CD103+ and macrophages) could phagocytose ovalbumin (OVA) and m-cherry from tumor cells, migrate to the draining LN and still be positive for m-cherry fluorescence, indicating the presence of the intact antigen. Remarkably, only in the CD103+ DCs subset could m-cherry fluorescence be detected. Upon isolation of APCs from the draining LN, once again, just CD103+ DCs were able to drive T cell responses against the OVA antigen. Furthermore, it was shown that C-C motif chemokine receptor 7 (CCR7) is required for CD 103+ DCs to traffic tumor antigens to the LN and that CCR7 levels correlate with T cell infiltrate and patient survival [24].
The other important subset is comprised of pDCs, whose development is driven by the E2-2 transcription factor and, curiously, morphologically resemble plasma cells. The pDCs specialize in producing and secreting large amounts of IFN-α after pathogen stimulation of toll-like receptor 7 (TLR7, detects single stranded viral RNA) and TLR9 (double stranded DNA), thus having a relevant role in the innate immune response against viruses [15, 18, 19]. In mice, pDCs are mostly found in blood and spleen, categorized by the expression of B220, Ly6C and the plasmacytoid dendritic cell antigen-1 (PDCA-1) markers and in humans, while negative for T, B and NK cell markers, pDCs are positive for CD4, CD123 (IL-3R), CD303 (BDCA-2), immunoglobulin-like transcript 3 (ILT3), and ILT7] [25]. Though the available data do not show a clear role for them in antigen presentation and initiation of the adaptive immune response, they are able to present antigens in the context of MHC-II molecules [15, 18, 19]. Indeed, upon activation by viruses, cytokines such as IL-3, CD40L or CpG oligonucleotide pDCs differentiate into full DC morphology and activate CD8+and CD4+ T cell responses [26, 27]. Interestingly, in different types of tumors, including melanoma and prostate cancer, pDCs have been shown to be present in reduced frequency in the circulation, as they express multiple chemokine receptors, such as CXCR4 and ChemR23, that determine their tropism for sites undergoing pathological processes [28, 29], a subject thoroughly reviewed in [27] and [28].
However, in spite of suggestions that they may be involved in the initiation of the response by producing IFN-α in the tumor microenvironment, their role is still not convincing and, actually, some human studies have even associated pDC infiltration of tumors with poor survival [14, 30]. Accordingly, for reasons not fully understood, tumor-associated pDCs display a reduced responsiveness to TLR9 stimulation, become defective in IFN-α production and secrete immunosuppressive factors (e.g., IL-10) that along with regulatory T cells participate in immune surveillance escape, hence, favor tumor progression [27, 31]. Along the same lines, Le Mercier and colleagues used an orthotopic murine mammary tumor model to show that depletion of pDCs retarded tumor growth, evidencing their pro-tumor role. Remarkably, instead of TLR9, intratumoral administration of TLR7 ligand activated tumor associated pDCs and provoked a strong tumor regression effect [32]. Depletion of pDC and neutralization of type I IFNs prevented this outcome. Nevertheless, production of IFN-α by human pDCs can also be negatively regulated through the receptors BDCA2, NKp44 and ILT7, although only ILT7 has a known ligand, BST2 found on the cell membrane upon exposure to type I IFNs and also in a fraction of melanomas [27, 33].
Monocyte derived DCs (moDCs), or inflammatory DCs, originate from circulating monocytes that are thought to be drawn to the inflammatory cancer microenvironment since they are not present in a steady state [34]. moDCs express the Ly6C marker in mice or CD14high in humans, but since in the mouse they are also positive for MHC-II, CD11b, CD11c and F4/80, it is hard to discriminate them from other CD11b+ populations or macrophages. Currently, CD64 (high-affinity IgG receptor gamma chain FcγRI) and MAR-1 (high-affinity IgE receptor FcεRIα chain) staining is used to discriminate moDCs from CD11b+ cDCs [14, 15, 34]. The capacity of moDCs to activate näive T cells requires further elucidation, since depending of the context and cytokines that are present, Ly6C+ monocytes can give rise to both macrophages and DCs [15]. Additionally, in tumors, Ly6Chigh monocytes can remain as a heterogeneous population called myeloid-derived suppressor cells (MDSC) and through up-regulation of nitric-oxide, arginase, prostaglandin-E2 and production of transforming growth factor β2 (TGF-β2) can deeply impair function of effector immune cells [14, 35].
Regarding the infiltration of other DC subtypes into the tumor mass, data indicates that cDCs represent a scarce population, and as such are likely competing with other more abundant myeloid populations, such as macrophages and monocytes, for antigen capture and priming of T cells [36, 37]. In mouse models of cancer, the localization of most DC subtypes have been shown to be mostly localized in the tumor margins, with limited infiltration into the center [38]. In humans, owning to the difficulty to characterize the DCs in situ, their scarcity as well as cancer related heterogeneity, the localization of DCs remains poorly studied. Even so, in melanoma, peritumoral DCs have been observed which a more mature phenotype than infiltrating DCs’ [39, 40]. However, in a recent breakthrough, Lavin and collaborators aiming to determine the immune landscape of early lung adenocarcinoma lesions, used a multiscale immune profiling strategy based on mass cytometry by time of-flight (CyTOF) combined with single-cell transcriptomics and multiplex tissue imaging and observed that CD141+ DC (categorized by the high levels of CD207, CLEC9A, and XCR1) are significantly depleted in comparison with non-lung adenocarcinoma tissue, whereas CD1c+ DCs (expressing CD1c, CX3CR1 and IRF4) were observed more frequently [41]. However, the impact of tumor infiltrating DCs on clinical outcome needs further investigation, as other cells present in the tumor stroma are also playing a role, a topic that was thoroughly discussed in [40].
TYPE I INTERFERONS SIGNALING PATHWAYS
In humans, the type I IFN family includes proteins encoded by at least 13 IFN-α genes (IFN-α1, -α2, -α4, -α5, -α6, -α7, -α8, -α10, -α13, -α14, -α16, -α17 and -α21) and one gene each for IFN-β, IFN-ε, IFN-κ, IFN-ω and IFN-τ [42]. Interestingly, IFN-α genes share 70–80% sequence homology and have about 35% identity with the IFN-β gene [43]. All type I IFN genes lack introns and are located on the short arm of chromosome 9 in humans and chromosome 4 in mice. IFNs-α and IFN-β have 186–190 amino acids, but they have a peptide that signals cleavage resulting in secreted proteins of 165 or 166 amino acids with the amino terminal domain being important for biological activity [44].
The various type I IFNs have differential tissue expression and although they bind to the same receptor (IFNAR1/IFNAR2) and signal through similar mechanisms [4, 45], they have different binding affinities and, consequently, give rise to different antiviral, antiproliferative, and immunomodulatory outcomes [46–48]. IFN-β has a ~50-fold higher receptor-binding affinity to IFNAR1 than IFN-α [49], resulting in a more potent antiproliferative and perhaps distinct immunoregulatory action [47]. Interestingly, only IFN-β, but not IFN-α, stimulation enables the co-immunoprecipitation of IFNAR1 and IFNAR2 subunits [50]. Also, the IFNAR2 subunit binds type I IFNs with relatively higher affinity than IFNAR1, but the latter is absolutely required for signal transduction [42, 51, 52].
The IFNAR1/IFNAR2 receptor consists of transmembrane proteins which lack intrinsic kinase domains. They associate with a family of nonreceptor cytoplasmic tyrosine kinases, the Janus kinases (JAK1 and TYK2), so they can phosphorylate specific tyrosine residues of signal transducer and activator of transcription (STAT) proteins [53]. TYK2 associates with IFNAR1 while JAK1 acts in association with IFNAR2 [52, 54] on the inner side of the membrane, thus providing stability to the receptors and facilitating their cell surface localization, while serving as key components of signaling complexes [55, 56]. These complexes phosphorylate Tyr701 in STAT1α and in its spliced variant STAT1β, and Tyr690 in STAT2, enabling p-STAT to form heterodimers via their Src homology 2 (SH2) domains (Figure 1) [57, 58].
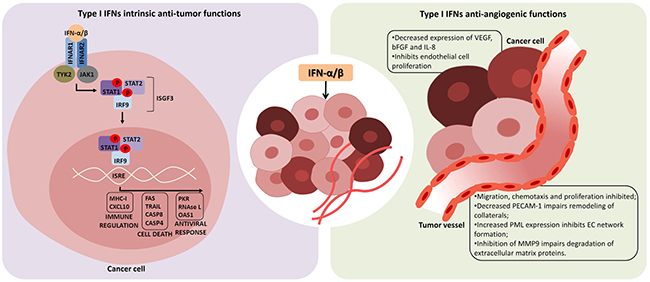
Figure 1: Intrinsic anti-tumor and anti-angiogenic functions of type I interferons. Interferon-α/β (IFN-α/β) has direct effects in tumor cells inducing growth arrest and cell death (left panel). After binding to the heterodimeric IFN-α/β receptor 1 and 2 (IFNAR1/IFNAR2), type I IFNs induce a cascade of intracellular events which culminates in expression of genes whose promoters contain the IFN-stimulated response element (ISRE). In this way, several immuno-regulatory cytokines, cell death factors and proteins related to antiviral response are produced, as well as more IFN-α/β, which in turn affects neighboring cells. In addition, anti-tumor effects of type I IFN may also be a consequence of its anti-angiogenic function, impairing tumor vessel formation and leading to death of tumors by lack of oxygen and nutrients (right panel). IFN-α/β can inhibit the production of angiogenic factors by tumor cells, and also directly affects endothelial cells (EC), inhibiting their proliferation and secretion of factors responsible for EC chemotaxis and remodeling of extracellular matrix. Tyrosine kinase 2 (TYK2), Janus kinase 1 (JAK1), signal transducer and activator of transcription (STAT), IFN-regulatory factor 9 (IRF9), IFN-stimulated gene factor 3 (ISGF3), vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), interleukin-8 (IL-8), platelet endothelial cell adhesion molecule-1 (PECAM-1), promyelocytic leukemia protein (PML), matrix metalloproteinase 9 (MMP9).
Once phosphorylated, the heterodimer STAT1/STAT2 binds to IRF9, forming the IFN-stimulated gene factor 3 (ISGF3) transcription factor complex [59]. The ISGF3 complex translocates to the nucleus and binds to cis-acting IFN-stimulated response elements (ISREs) in the promoters of IFN-stimulated genes (ISGs). While STAT1 and STAT2 require phosphorylation to be active, IRF9 functions as a DNA adapter molecule independently of its posttranslational modification induced by IFNα/β [60, 61].
Expression of type I IFNs is intimately connected and influenced by IRFs, a family of nine transcription factors with a similar DNA binding domain in their N-termini. IRF1 is expressed constitutively and also in response to IFN-γ, like IRF8, and it may determine which species of IFNs are induced by TLR activation. IFN-β induces synthesis of IRF7, which amplifies the synthesis of IFNs together with the constitutive expressed IRF3 [62, 63], and induces transcription of IFN-α genes [64].
In the type II IFN-γ signaling pathway, the homodimer of tyrosine-phosphorylated STAT1 binds to Gamma-Activated Sites (GAS) in ISG promoters [65]. Type I IFNs can also induce this pathway, triggering expression of genes with GAS in their promoters. Also, STAT1 can form heterodimers with other STATs leading to activation of other pathways (Figure 1) [66].
Differences between IFN-α and IFN-β and their antitumor effects
Type I IFNs can induce expression of different genes depending on their concentration. Some genes are highly sensitive and require low picomolar concentrations, while other genes require 100-fold higher IFN-α/β concentrations for activation. Microarray analysis of expressed genes revealed that antiviral activity genes (such as Mx dynamin like GTPase 1 - Mx1, protein kinase R - PKR and 2′-5′-oligoadenylate synthetase 2 - OAS2) are induced by low amounts of IFN-α/β, whereas genes related to cell proliferation, chemokine activity and inflammation (like IL-6, C-X-C motif chemokine ligand 11 - CXCL11, and tumor necrosis factor related apoptosis inducing ligand - TRAIL) need a stronger IFN-α/β signal in order to be activated [47, 48, 67, 68]. The difference between antiviral and antiproliferative activities for IFN-α2 was 1,000-fold, while for IFN-β it was only 50-fold in WISH cells [51].
IFN-β binds to IFNAR1/IFNAR2 with higher affinity and thus forms more stable ternary complexes than IFN-α [47, 48, 69]. Because of this, IFN-β regulates cellular functions at concentrations that are orders of magnitude lower than any IFN-α subtype. Yet, all type I IFNs can induce antiviral responses at picomolar concentrations [69, 70].
In a study performed by Jaitin and collaborators, the authors engineered an IFN-α2 triple mutant (with H57A, E58A and Q61A mutations) that binds IFNAR1 with a 30-fold higher affinity than the wild-type protein and thus comparable to the binding affinity of IFN-β to IFNAR1. They observed that the HEQ mutant exhibited several functional characteristics of IFN-β, like similar patterns of gene induction and therefore substantially increased antiproliferative activity, without altering antiviral activity and ISGF3 formation. In this way, they indicated that functional differences between IFN-α2 and IFN-β are mainly due to their different binding affinities for IFNAR1. Therefore, the differential phenotypes of IFN-α2 and IFN-β are not qualitative, an observation that applies to both biological activities and gene induction patterns [47].
Also, the concentration of IFNAR1/IFNAR2 in the plasma membrane is critical to determine the cell sensitivity to type I IFNs. While IFN-β is more potent than IFN-α2 to induce antiproliferative activity in cells with native receptor numbers, IFN-α2 was equally able in cells with highly increased receptor numbers [46]. This may explain why IFN-β, but not IFN-α2, provides long-term signaling [70]. In Daudi cells, picomolar concentrations of type I IFNs are already enough to cause growth arrest because these cells have higher levels of the interferon receptors [51].
A recent report has suggested that tumor cells have lower interferon receptor levels, making them resistant to the antiproliferative activity of type I IFNs [71]. At low IFNAR1 concentrations, the complex is formed only with IFN-β, which binds IFNAR1 tightly, but not with IFN-α2 [72]. The duration of IFN stimulus is also critical to the decision between the antiviral state or antiproliferative response. While induction of antiviral activity requires a few hours of IFN-α or IFN-β exposure, antiproliferative activity requires days of constant IFN-α/β binding [70].
Alterations in the type I interferon pathway in cancer
Since type I IFNs play a central role in the tumor microenvironment, especially with regard to anti-tumor activities, genetic alterations in this pathway are expected to be detrimental to prognosis and responses to therapy. The interferon gene cluster, found on human chromosome 9p21, encodes nearly all of the IFN genes and pseudogenes [43]. Interestingly, the cyclin dependent kinase inhibitor 2A (CDKN2A) gene also resides in this same region, encoding the p14ARF (alternate reading frame, a functional partner of p53) and p16INK4a (inhibitor of CDK4/6, thus an activator of retinoblastoma - Rb) proteins [73]. As such, 9p21 deletions could impact the p53, Rb and interferon pathways together or individually, depending on the exact nature of the deletion.
Homozygous deletion of IFN-α/β has been reported in leukemias, such as chronic myeloid leukemia (CML) and acute lymphoblastic leukemia (ALL) [74–78], and may be associated with resistance to IFN-α [79]. However, other studies indicate that a 400kb deletion including p16INK4a, but not the IFN gene cluster, is critical in lymphoblastic leukemias [80]. Deletion in the gene encoding IFN-α has also been correlated with post-transplant lymphoproliferative disorder [81]. Homozygous deletion of IFN-α contributes to the recurrence of head and neck squamous cell carcinoma (HNSC) [82]. The loss of the gene encoding IFN-β has been observed in glioma [83–85]. While for melanoma, hemizygotic deletion of 9p21 has been reported, observed in 12 of 14 cases [86], but this may reflect loss of CDKN2A alone or in combination with IFN-β [85]. Studies in a variety of cancer cell lines also support the notion that loss of the genes encoding IFN-α/β is a frequent event, though co-deletion of p16INK4a and/or p14ARF must be specifically examined [87–89].
Alterations in the interferon gene cluster are certainly not the only mechanism by which the type I IFN pathway may be disrupted. At least in cell lines, loss of STAT2 is associated with reduced apoptosis in response to IFN-α treatment [90]. Interestingly, methylthioadenosine phosphorylase (MTAP) also resides on chromosome 9p21 and can be epigenetically silenced in melanoma, resulting in impaired STAT1 signaling and serving as a marker of response to IFN-α therapy [91]. Infection with human papillomavirus (HPV), whether of high or low risk subtypes, has been correlated with resistance to IFN-α [92, 93]. Stimulator of interferon genes (STING) acts in the endoplasmic reticulum and promotes the transcriptional functions of nuclear factor-ĸB (NF-ĸB) and IRF3, thus playing a major role in anti-viral response [94]. In established melanoma and colorectal cancer cell lines, STING signaling is repressed due, typically, to epigenetic silencing of cyclic GMP-AMP synthase (cGAS) or STING itself. Loss of STING and/or cGAS was confirmed in some 54% of human colorectal cancer (Stage II-IV) while loss of both was seen in 41.7% of advanced stage human melanoma samples [95, 96].
Type I IFN and its receptor (IFNAR1/2) also contribute to the immunosuppressive tumor microenvironment. The paradox between inflammation and immunosuppression, especially with regard to type I IFN, was reviewed recently [97]. Expression of type I IFN is expected to promote the immune response, but it can also lead to the expression of indoleamine 2,3-dioxygenase (IDO), programmed cell death-ligand 1 (PD-L1) and IL-10, culminating in immunosuppression, as discussed here and in the review by Snell et al. [97]. In a recent report, Katlinski et al. [98] showed that IFNAR1 expression is reduced in colorectal cancer microenvironment, specifically in cytotoxic T lymphocytes, contributing to an immune-privileged niche that supports tumor growth. Restoration of IFNAR1 expression in the T cells was associated with renewed control over tumor growth [98]. The magnitude and duration of type I IFN signaling may be critical points to consider when designing therapies aimed at this complex pathway since sustained inflammation may lead to immunosuppression.
ANTITUMOR FUNCTIONS OF TYPE I INTERFERONS
Inhibition of cell growth and induction of apoptosis
The effects of type I IFNs on tumor cells may vary depending on the type of tumor and even more so when considering the specific cell in question. For example, IFN-β has a stronger antitumor effect than IFN-α in the early stage hepatocelular carcinoma (HCC) in patients with chronic hepatitis C. While IFN-α has been shown to induce apoptosis in HCC cell lines [99–101], Murata and co-authors showed that IFN-β had a superior antiproliferative effect as compared to IFN-α on three HCC cell lines, inducing cell cycle change and apoptosis, and more strongly upregulating ISGs, like Fas antigen and human leukocyte antigen (HLA)-class I molecules [102].
A time course study in WM9 melanoma cells with IFN-β (500 units/ml) identified more than 30% terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive cells at 96 h, while IFN-α2 did not result in any positive staining. Other melanoma cell lines revealed similar sensitivity, like FemX cells, while Guilliams cells were partially sensitive and A375 cells were relatively resistant to either IFN-α2 or IFNβ, even at higher doses (up to 1000 units/ml) [103]. In another study with melanoma cells, IFN-β potency was also greater than IFN-α2 for induction of ISGs, like cytomegalovirus-induced gene 5 (CIG5/viperin), CIG49, ISG54, TLR3, CXCL10, TRAIL, as seen by microarray analysis of both WM9 and WM35 cell lines, while for the IFN-β-sensitive WM9 cell line, IFN-β also induced expression of SP100, tumor necrosis factor-stimulated gene 6 (TSG6), augmented in prostate carcinoma gene (AIPC), Cyclin-E, ubiquitin E2-like (UBEL-2) and ubiquitin-specific protease (USP18), as seen by RT-qPCR [104].
SK-MEL-2 and SK-MEL-24 cell lines were also more sensitive to the anti-proliferation effects of IFN-β than those of IFN-α2b in vitro. Matrigel invasion of SK-MEL-24 was significantly inhibited by both IFN-α2b and IFN-β and treatment of SK-MEL-24 with IFN-α2b or IFN-β decreased vascular endothelial growth factor-C (VEGF-C) and VEGF receptor-3 (VEGFR-3) protein expression. In a human melanoma xenograft model, SK-MEL-24 cells were injected intradermally in mice and tumor growth was reduced after IFN-α2b or IFN-β treatment. LN metastases were more frequent in mice treated with IFN-β than with IFN-α2b. One of six mice showed LN metastasis in the IFN-α2b group compared to three of six mice in the IFN-β group. Tumors were evaluated and revealed that both IFN-α2b and IFN-β decreased cell proliferation and increased the number of apoptotic cells, yet these effects were superior in IFN-β treated tumors. Also, VEGF-C/VEGFR-3 levels were reduced in tumors treated with IFN-α2b or IFN-β, but LYVE-1 was decreased only in IFN-α2b treated tumors, representing less intratumoral and peritumoral lymphatic vessels [105].
Cell lines that were relatively resistant to inhibition of cell growth by IFNs, including U937 (histiocytic lymphoma), HeLa (HPV-infected cervical adenocarcinoma), and T47D (ductal breast carcinoma) were also not TUNEL-positive in response to IFN-α2 or IFN-β. Other cell lines, like ACHN (renal cell carcinoma), Minors (melanoma), NIHOVCAR3 (ovarian carcinoma) and MCF-7 (breast carcinoma) had an increase in TUNEL-positive cells in response to IFN-β but not IFN-α2 [103].
Chen and colleagues showed that both IFN-α and IFN-β induced apoptosis in U266, RPMI-8266, and NCI-H929 multiple myeloma cell lines and plasma cells from 10 patients with multiple myeloma [106]. Expression of TRAIL, which contains 2 IFN-stimulated regulatory elements in its promoter [107], seems to be the main event for cell death induction, followed by caspase-8 activation, Bid cleavage, cyt c release and caspase-3 activation [106].
In a study performed by Rozera and co-authors, TS/A adenocarcinoma cells were injected with retrovirus encoding IFN-α or IFN-β and these cells were inoculated in BALB/c mice. More host-infiltrating cells were observed in TS/A-IFN-α and TS/A-IFN-β than in parental TS/A tumors (macrophages, granulocytes, and lymphocytes, being CD8+ T cells more numerous than CD4+ T cells). Also, fewer blood vessels were observed in TS/A-IFN-α or TS/A-IFN-β tumors as compared with parental TS/A tumors, being the vasculature of TS/A-IFN-β tumors scarcer than TS/A-IFN-α tumors, even when no differences in the expression of angiogenic factors (VEGF and basic fibroblast growth factor - bFGF) were found. However, the expression of pro-inflammatory cytokines, such as IL-1β, IFN-γ and tumor necrosis factor-α (TNF-α), was higher in TS/A-IFN-α than TS/A-IFN-β tumors and absent parental TS/A tumor. Finally, survival of TS/A-IFN-β mice that produced higher levels of IFN-β or TS/A-IFN-α mice was three- to four fold longer than the control group, while metastatic ability of TS/A cells was reduced in mice injected with either TS/A IFN-α or IFN-β cells [108].
Inhibition of angiogenesis
Angiogenesis is an important antitumor therapeutic target because it is required for tumor growth [109, 110] for the delivery of oxygen and nutrients to the fast-growing tumor cells [111]. As shown in Figure 1, the anti-proliferative and cell death inducing functions of type I IFNs also inhibit angiogenesis [112, 113]. For example, after IFN-α/β treatment, tumor vessels undergo necrosis [113]. IFN-α/β also prevents tumor cell production of angiogenic growth factors, like bFGF [114–116], VEGF [117, 118], and IL-8 [119, 120]. Interestingly, when MBT-2 (murine transitional carcinoma of the bladder) or L1210R (murine leukemia resistant to the antiproliferative effects of IFN) cells were treated in vitro with IFN-β and then inoculated intracutaneously in C3H/He or Swiss mice, respectively, the inhibition of angiogenesis was noted within 24 hours of tumor cell inoculation, even before their antiproliferative effects on tumor cells [112]. Thus, inhibition of angiogenesis can be counted among type I IFN’s anti-tumor benefits.
McCarty and collaborators showed that endogenous type I IFN signaling is involved in the regulation of angiogenesis. These authors implanted sponges filled with proangiogenic molecules (bFGF, VEGF, and TGF-α) in mice deficient for IFN-α/β receptor and observed superior vascularization when compared to control mice with functional type I IFN signaling. Moreover, the antiangiogenic effects of type I IFNs resulted in inhibition of tumor growth in animal models [121].
It is known that in inflamed tissues, the release of IFN-α by leukocytes trigger macrophage activation, which then causes the release of TNFα. Both IFN-α and TNF-α induce accumulation of promyelocytic leukemia protein (PML) in HUVECs and in microvascular endothelial cells (HMVECs). PML was shown to be indispensable for TNF-α and IFN-α-mediated inhibition of EC network formation, but no significant differences in apoptosis of HUVEC treated with TNF-α, IFN-α or vehicle were detected [122]. PML is highly expressed in normal vascular endothelium and inflamed tissues [123] and known as an ISG [124] through STAT1 induction, since knockdown of STAT1 significantly impairs PML expression in these cells. The authors have shown PML suppressed integrin β1 (ITGB1) expression in both HUVECs and HMVECs, an important protein which regulates ischemic neovascularization [125], cell-to-cell and cell-to-extracellular matrix adhesion and cell migration [126].
Spaapen and others have observed that IFN-β–secreting B16 cells injected subcutaneously in mice had impaired growth, yet when implanted into mice lacking IFNAR1, tumors grew progressively. This shows that the antitumor effect of IFN-β was dependent on signaling via host cells and did not act directly on tumor cells, a situation that was also observed with IFN-α. This effect was also independent of adaptive immunity, since tumor regression was also observed in Rag2−/−γc−/− mice (deficient in T, B, and NK cells). However, a diminution of blood vessel density was observed in the IFN-β-secreting tumors. In the work, the authors showed that IFN-β has a direct effect on nonhematopoietic Tie2+ cells, that is to say, vascular ECs, causing inhibition of angiogenesis [127].
While IFN-β may reduce the number of tumor vessels, it can also contribute to vessel maturation. Dickson and others showed that treatment of human xenografts in immunodeficient mice with an adeno-associated virus (AAV)-vector encoding the human IFN-β gene resulted in maturation of the intra-tumor vasculature, yet inhibition of angiogenesis [128]. Also, treatment of tumors with IFN-β, encoded by an adenoviral vector, promotes an increase in inducible nitric oxide synthase (iNOS) and a decrease in bFGF and TGF-β1 levels [129], resulting in inhibition of tumor growth.
Indirectly, IFN-β inhibits matrix metalloproteinase 9 (MMP9) gene expression, which is responsible, together with MMP2, for degradation of extracellular matrix proteins collagen and elastin, a process required for initiating the enlargement of collateral vessels [130]. This is in accordance with Nelissen and others (2002), who have shown that IFN-β inhibited expression of MMP9 in monocytic and peripheral blood mononuclear cells [131] (Figure 1).
The mechanisms by which IFN-β inhibits tumor growth and angiogenesis may also involve tumor-infiltrating neutrophils. Jablonska and others showed that IFN-β-deficient mice presented faster tumor growth of injected tumor cells and larger tumors compared to wild-type mice. This was associated with an increase in tumor angiogenesis and tumor-infiltrating CD11b+Gr1+ neutrophils, which are responsible for expression of proangiogenic and homing factors. After treatment with IFN-β, these neutrophils had reduced gene expression of VEGF, MMP9, CX-C chemokine receptor type 4 (CXCR4) and the receptor for stromal-derived factor-1 (SDF-1), contributing to limiting tumor angiogenesis. Also, when neutrophils obtained from IFNAR-deficient mice were injected in wild-type mice, tumor growth was increased and accompanied by more mature vessels when compared with neutrophils obtained from WT mice [132].
Type I IFN may affect endothelial cell (EC) survival [133] and function, as indicated by inhibition of migration, chemotaxis and proliferation of HUVEC (human umbilical vein endothelial cells) [134], but as said before, although these cytokines share the same receptors, they exert different effects on tumor viability and angiogenesis. While there are several studies demonstrating the antiangiogenic effects of type I IFNs, there are few studies comparing the different effects of IFN-α and IFN-β, and even less including other members of the type I IFN family. In a study comparing type I IFNs, IFN-α2b inhibited in vitro vessel formation of HUVEC by 20%, whereas inhibition due to IFN-β was around 80%. In vivo, IFN-α2 inhibited vessel growth by 30% in SK-MEL-1 tumors, whereas IFN-β inhibited vessel formation by 80%. While both IFN-α2b and IFN-β inhibited HUVEC proliferation, neither of them was able to induce apoptosis. Genes induced by IFN-β in HUVECs are p56, CXCL11, ISG20, melanoma differentiation-associated-5 (MDA-5), HECT and RLD domain containing E3 ubiquitin protein ligase family member 6 (HERC6), CXCL10, SAM and HD domain containing deoxynucleoside triphosphate triphosphohydrolase 1 (SAMHD1), p60, Sp100B and monocyte chemoattractant protein 2 (MCP-2) [135]. This effect was also observed by Erdmann and others who described that IFN-β inhibited cell cycle and proliferation of human micro and macrovascular ECs, but did not induce apoptosis [136]. Interestingly, VEGF is responsible for phosphorylation of IFNAR1, followed by ubiquitination induced by the protein kinase D2 (PKD2), which results in the degradation of IFNAR1 and promotion of angiogenesis [137].
Albini and co-authors have compared the effects of retroviral vector-packaging cell lines encoding IFN-α or IFN-β cDNAs (α1Am12 and βAm12) upon ECs. In this study, both conditioned media from α1Am12 or βAm12 decreased chemotaxis and invasion of ECs, however only βAm12 inhibited EC differentiation into capillary-like structures on Matrigel. Also, IFN-β’s superiority in inhibiting angiogenesis was confirmed in an in vivo model, in which sponges containing a very potent angiogenic cocktail co-injected along with α1Am12 cells in C57BL/6 mice produced a limited inhibitory effect on angiogenesis, while βAm12 cells markedly impaired vessel formation. This was also observed in nude mice, confirming the effect of IFN-β on ECs independent of a T-cell response [138].
As will be discussed below, the first antitumor efforts involving type I IFNs were developed using IFN-α, but IFN-β later gained importance in this field as a result of studies showing its increased antitumor and antiangiogenic effects, although further investigation is needed to support the notion that IFN-β provides superior antiangiogenic activity. Studies comparing the gene expression profiles of ECs treated with different type I IFNs could be very enlightening as to the different effects of these cytokines on angiogenesis. Also, in vivo and in vitro treatments of ECs with different type I IFNs and posterior analysis of angiogenesis could reveal type I IFN’s functional effects. Finally, knockout models specific for each type I IFN could be used to confirm their individual functions, thus providing evidence based on endogenous proteins.
Immunomodulatory and regulatory effects of type I interferons
So far, we have exposed several anti-neoplastic functions attributed to type I IFN, but its main function is most often associated with immune modulation. Depending on the stimulus, both IFN-α/β can be produced by almost any cell type, including fibroblasts and leucocytes [139]. Such inducers act through pattern recognition receptors (PRRs) that sense pathogen-derived and non-pathogenic components, for example: double-stranded RNA (dsRNA) from RNA viruses detected by TLR3, both cytosolic DNA and second messenger cyclic di-GMP24 from bacteria by STING [5, 139], as well as danger-associated molecular patterns (DAMPs) released upon cellular stress or therapy induced cell death. Heil and Land suggest dividing mammalian DAMPs into five classes according to different PRRs. Class I DAMPs, like HMGB1 or heat shock proteins, are sensed by TLRs and trigger the MAPK signaling cascade. Class II DAMPs are perceived indirectly by the NOD-like receptor family protein 3 (NLRP3) inflammasome and comprise ROS, monosodium ureate, eATP and dsDNA. Both classes I and II are involved in maturation of DCs. Class III comprises stress-induced soluble major histocompatibility complex class I-related chains A/B (MIC-A/B) and UL-binding proteins (ULBPs) and are sensed by receptors such as NKG2D, expressed by innate lymphocytes, like NK cells, and innate-like T-lymphocytes, like gamma delta T-cells. Class IV represents neoantigens, such as non-muscle myosin-II (NMHC-II), actin cytoskeleton and oxidized phospholipids that, together with IgM antibodies, bind to classical lectin receptors and trigger activation of the complement cascade and alternative pathways. Finally, class V DAMPs are called Dyshomeostasis – Associated Molecular Patterns, which comprise altered pattern of molecules resulted from perturbations in the steady-state of the intra- and/or extracellular microenvironment, like hypoxia, changes in acidity or osmolarity, and metabolic stress [140]. Thus, type I IFNs may be released as part of the natural evolution of a disease or as a consequence of therapeutic interventions. However, in cancers, an important difference between these two scenarios is that the local immune suppressive environment modifies the expected “physiological” response, either counteracting or amplifying the immunomodulatory functions of type I IFNs (Figure 2).
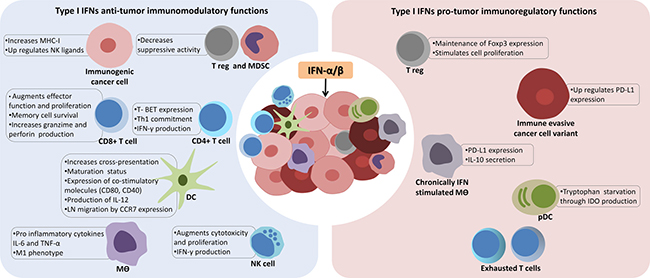
Figure 2: The context dependent and complex role of type I interferons in cancer immunity. Activation or delivery of interferon-α/β (IFN-α/β) into the tumor microenvironment can result in immunomodulatory and regulatory functions. In the first scenario (left panel), in order to unleash an effective immune attack against cancer cells, type I IFNs modulate innate and adaptive compartments through multiple mechanisms to provide a pro-inflammatory context suitable for antigen recognition by tumor associated dendritic cells (DCs) and priming of T lymphocytes. Importantly, as type I IFNs enhance co-stimulatory molecules of DCs, they also increase the unique ability of DCs to cross-present phagocytized tumor antigens to CD8+ T cells. Additionally, immunogenic tumor clones (represented in pink) have their antigenicity increased by up regulating major histocompatibility complex class I (MHC-I) molecules. However, type I IFNs can also favor tumor progression and escape from immune control (right panel), especially under chronic exposure conditions, since they can induce macrophages (mϴ) to produce Interleukin-10 (IL-10), that along with tryptophan starvation mediated by indoleamine 2,3-dioxygenase (IDO) and expression of programmed death-ligand 1 (PD-L1) by immune evasive tumor cells (dark red), greatly impairs T cells functions. Tumor necrosis factor–α (TNF-α), plamacytoid DCs (pDCs), myeloid-derived suppressor cells (MDSC), regulatory T cell (T reg), C-C chemokine receptor type 7 (CCR7), natural killer cells (NK).
Immunomodulatory effects triggered by type I IFNs can act on both innate and adaptive immune compartments [141]. In a temporal scale, macrophages can be considered as early producers of type I IFNs that act on nearby macrophages and other innate cells, such as NK, to provide a pro-inflammatory context (release of cytokines IL-6, TNF-α) suitable for antigen capture and presentation by tumor associated APCs and priming of immune effector cells [2, 142]. Macrophages, similarly to DCs, produce both type I and II IFNs, and upon activation display high levels of MHC class I and II in order to boost a T cell response, thus working as a link between innate and adaptive immunity [143]. Type I and II IFNs have also been show to polarize macrophages into an M1 immunostimulatory phenotype with anti-tumor functions, rather than an M2 phenotype, which may have pro-tumor activities [143, 144].
However, as part of an adaptive immune resistance mechanism that takes place after an inflammatory response, negative regulators of the immune response are induced, aiming to limit both duration and specificity of the immune attack [145]. Regulatory mechanisms induced after IFN-α/β production include secretion of IL-10 and expression of PD-L1 [146]. Both IL-10 and PD-L1 are well known inhibitors of CD8+ T function and, as shown by Shaabani and collaborators, upon infection with lymphocytic choriomeningitis virus (LCMV), type I IFNs are produced in high amounts by CD169+ macrophages to combat the virus, but as a consequence leads to up-regulation of PD-L1 and therefore CD8+ T cell exhaustion [147]. Lack of this CD169+ macrophage population, known for their unique distribution in secondary lymphoid organs and antigen handling capacity to prime CD8+ T cells, impairs viral control, IFN-α production, and eventually mice succumb, thus confirming the regulatory function type I IFNs on macrophages and CD8+ responses [147, 148]. Interestingly, in a more general view of the tumor microenvironment context, PD-L1 expressing tumor-infiltrating immune cells, including macrophages and DCs, in head and neck cancer patients have been shown to result in a more favorable prognosis than when PD-L1 is expressed on tumor cells [149]. Indeed, in a recent study by Noguchi and colleagues, IFN-γ was responsible for up regulating PD-L1 in tumor cells, contributing to immune escape, but unexpectedly, the majority of PD-L1 molecules were expressed by the host immune system, especially in the macrophage compartment, suggesting a mechanism in trans to impair T cells function. Intriguingly, after antibody mediated blockade of IFN-γ, levels of PD-L1 on tumor cells drastically decreased, but remained elevated on tumor associated macrophages, suggesting an additional mechanism not dependent on IFN-γ to induce PD-L1 [150]. The exact role for type I IFNs in mediating PD-L1 immune escape remains to be elucidated, but it is tempting to speculate that early production of IFN-α by APCs augments IFN-γ production by CD4+ T lymphocytes [151] and NK cells [152], and thus leads to PD-L1 up regulation in the tumor microenvironment.
Within the DC compartment, type I IFNs act as strong maturation signals, increasing expression of co-stimulatory proteins CD40, CD80, CD86 and MHC molecules [153], enhancing their unique properties to process and present apoptotic cell antigens through their scavenger receptor lectin-like oxidized-LDL receptor-1 (LOX-1) [154], and stimulating migration to the draining LNs by up regulating the CCR7 chemokine receptor [155]. Notably, together with the production of IL-12 and IL-23, all of these actions support Th1 and Th17 cytotoxic T lymphocyte (CTL) responses, including increased survival of CD8+ memory T cells and expression of granzyme B and perforin 1 [2, 5].
Among the different DC subsets, using bone marrow chimera experiments, Diamond and collaborators have demonstrated that Ifnar1−/− CD8α+ DCs lose their capability to cross present tumor antigens and as a consequence, when regressor tumors are transplanted, the host is no longer able to immune reject them [9]. Cross-presentation is defined as the presentation of internalized antigens in the context of MHC-I molecules to CD8+ T cells, instead of the MHC-II context [5]. This data implies that this DC subset must be endowed with mechanisms to recognize stress signals from dying tumor cells, capture their antigens and present to CD8+ T cells, in order to trigger CTL responses. A plausible mechanism for the re-routing of capture antigens has been made by Reis and Sousa, investigating the C-type lectin domain family 9 member A (CLEC9A), a plasmatic receptor for necrotic cells that is highly expressed in CD8α DCs. They found that instead of activating these cells, CLEC9A was directing necrotic cell cargo into a recycling endosomal compartment, favoring cross-presentation to CD8+ T cells. Along the same lines, CLEC9A deficiency in CD8α+ DCs impaired their capability to prime CD8+ T cells but not CD4+ T cells, indicating that this plasmatic receptor was promoting cross-presentation of dead cell–associated antigens on MHC class I molecules. However the role of CLEC9A on the cancer immunity cycle needs further investigation [156]. Along the same lines, Tmen173−/− DCs, that lack the STING pattern recognition receptor gene, have also been found to be unable to prime CD8+ T cells [5, 157].
Taken together these data indicate that type I IFNs are involved in DCs antigen preseting functions, although immune regulatory mechanism are also triggered in order to limit the magnitude of the inflammatory response. Evidence of such regulatory mechanisms can be observed in pDCs, which have an unclear role in cancer, but are known to be a major source of type I IFNs. pDCs that infiltrate breast cancers have been shown to be defective in producing IFN-α and to co-localize with T regulatory cells (T-reg, CD3+CD4+CD25+ FOXP3+) [158], suggesting that either T-reg cells may be inhibiting IFN-α production or that pDCs somehow support the proliferation of T-regs. Indeed, mature pDCs can orchestrate tolerogenic immune responses through the induction of IL-10 IDO [159, 160], which catabolizes the essential amino acid tryptophan into a more stable metabolite, kynurenine and, as a consequence, stimulates T-reg proliferation while inducing CD8+ T-cell dysfunction, anergy and apoptosis [160, 161]. Catabolism of tryptophan in cancer is being recognized as a powerful suppressor of antitumor immunity since several tumor types were found to over-express IDO [162] and this was recently implicated as a critical mechanism of resistance to checkpoint blockade immunotherapy targeting the cytotoxic T lymphocyte antigen-4 (CTLA-4) pathway [163]. Interestingly, the IDO promoter contains transcriptional targets of both IFN-γ and IFN-α/β and in the case of pDC, both type I and type II IFNs were shown to be equipotent and exert additive effects on the induction of IDO [164].
In the lymphocyte compartment, type I IFNs have been show to act on both CD4+ and CD8+ T cells to dictate a Th1 immune response through the activation of STAT4 and T-bet expression, which is a T-box transcription factor expressed in CD4+ T lymphocytes committed to Th1 development [165, 166]. Interestingly, type I IFNs can also reverse the commitment of a Th2 humoral response by suppressing the GATA3 transcription factor, but when compared to IL-12, another Th1 inducing cytokine, it was shown that type I IFN cannot sustain T-bet expression by itself, needing other cytokines, such as IL-1β, to maintain this phenotype [166]. In terms of cancer, Th1 cells are known for orchestrating CTL responses that are implicated in the destruction of a tissue during autoimmune responses as well in antitumor responses [167], as demonstrated by a mechanistic degree of similarity shared between them [167, 168] and increased survival rate observed in patients with a CTL tumor infiltrate [169].
Another critical immunomodulatory mechanism induced by type I IFNs that directly affects T cell responses is the positive regulation of tumor antigens that are presented on cancer cells by MHC-I molecules, allowing the immune system to detect the tumor and distinguish it from a normal cell [170, 171]. In fact, up regulation of MHC-I by type I IFNs [172], has the potential to counteract the frequent down regulation of MHC-I found in human tumors resistant to immunotherapies [173]. Indeed, as recently demonstrated in a mouse model using tumors resistant to PD-L1 checkpoint blockade immunotherapy, high dose radiation directly upon the tumor mass induces a systemic increase of IFN-β levels and restores therapeutic efficacy by up-regulating MHC-I molecules in the tumor cells. Antibody mediated blockade of IFNAR1 completely abrogated this effect [174].
Furthermore, the regulation of immunity in cancer by IFN-α/β also involves increased cytotoxic functions of NK cells [146], which are lymphoid cells that through a balance of activating (e.g., NKG2D) and inhibitory receptors (e.g, killer-cell immunoglobulin-like, KIR) can effectively kill tumor cells [175]. Activation of the NK response was demonstrated to positively sustain an M1 macrophage phenotype and to edit tumor immunogenicity in a process independent of T cells [176]. And, curiously, type I IFNs inhibit the elimination of CD8+ T cells by NK cells [177], a phenomenon observed in conditions where NK cells assume a regulatory function over the adaptive immune response in order to prevent chronic inflammation and generation of auto-immune reactions [178].
Finally, in support of defining the appropriate context for an effective CTL response, type I IFNs were also shown to inhibit immune-suppressive actions of T-reg cells and MDSCs [144]. Yet in inflammatory conditions, it has been shown that type I IFNs were required for maintenance of Foxp3 expression and immune suppressive functions of T-regs, since transfer of T-regs obtained from Ifnar1 KO mice were not able to inhibit the induction of the T-cell mediated colitis, as seen for T-regs obtained from wild-type mice. Indeed, administration of recombinant IFN-α reduced T cell-mediated colitis by increasing the number of T-regs and their suppressive functions [179].
As discussed here, the role of type I IFNs in immunity is complex and context dependent, assuming either antitumor or pro-tumor functions determined by the exposure to type I IFNs pre or post antigen encounter, if produced acutely or chronically, in low or high levels. For example, using an LCMV model, it was observed that early and transient production of IFN-α by pDCS exerts minimal effects on CD8+ T cell responses, but administration of recombinant IFN-α5 and IFN-β on days that coincide with endogenous type I IFNs decline, hence providing sustained stimulation, can prevent CD8+ T cell exhaustion and viral persistence [180]. Furthermore, this complex role of type I IFNs was evidenced in two complementary works both exploring a model of chronic LCMV infection, where IFN-α was transiently produced, yet ISG expression was prolonged. In the first work, it was shown that genetic or antibody mediated blockade of IFN-α signaling prior to infection leads to increased viral replication and loss of infection control, thus confirming the antiviral role of IFN-α [181]. Whereas, in the second, after establishment of the chronic infection, IFN-α blockade acted by reducing IL-10 and PD-L1 levels and, as a result, ameliorated T cell exhaustion and, even though it took 2 months, resulted in significantly lower virus titers [182]. Therefore, there seems to be a paradoxical function of type I IFNs: early (i.e., prior to antigen encounter) antiviral effects of type I IFNs are critical for host protection, promoting immune activation by stimulating an NK cell attack, enhancing DC antigen presenting function and favoring T cell proliferation, but after this adaptive immune response has been unleashed, chronic stimulation of the type I IFN pathway can result in immunoregulatory mechanisms that aim to shut down long lasting and unresolved immune responses, although as discussed below, therapeutically induced IFN-α/β can restart or reinvigorate a new immunity cycle. Examples of such duality are also observed during hepatitis C virus infection, in which strong IFN-α/β signature correlates with poor responses to therapy, as well as in chronic HIV infections and on latent Mycobacterium tuberculosis [183].
In cancer, the opposing role IFNs is better characterized with IFN-γ, especially on the induction of PD-L1 on cancer cells. As recently demonstrated by Benci and colleagues, prolonged exposure of tumors to IFN-γ induces a STAT-1 epigenetic signature as well as ligands for inhibitory receptors that results in PD-L1 dependent and independent mechanisms of resistance to checkpoint blockade immunotherapy [184]. Intriguingly, type I IFNs were also shown to be required for maintenance, not induction, of the PD-L1 independent resistance phenotype, but the precise contribution was not thoroughly explored [184]. Moreover, disrupting IFN-γ driven resistance with ruxolitinib, a JAK1/JAK2 inhibitor, renders CTLA-4 checkpoint blockade resistant tumors sensitive again. Accordingly, the work also provides clinical evidence that high expression of ISG and IFN-γ signaling is associated with tumor progression after PD-L1 therapy [184]. The mechanism that is behind the complex and opposing functions of IFNs is likely mediated by a qualitative and quantitative difference of regulators. For example, in a mouse melanoma model, therapeutic efficacy of high-doses of intratumoral IFN-α/β appears not to be T cell dependent, but rather relies on their anti-angiogenic properties, acting directly on the tumor vasculature [127]. Further studies that can dissect the molecular basis of this complex mechanism, specifically the influence of the producing cell, timing and magnitude, are surely needed.
TYPE I INTERFERONS IN CANCER THERAPY
IFN-α/β therapy for melanoma
Type I IFN therapy as treatment for melanoma utilizes the recombinant protein itself or as a complex with polyethylene glycol (PEG) in order to improve protein stability [185]. High dose IFN-α2b has been approved as an adjuvant therapy after surgical resection of cutaneous melanoma in patients with a high risk of death from recurrence. This approach is beneficial for improving disease-free survival, but the therapy itself is not well tolerated. Here we will address some of the progress and pitfalls of IFN-α/β therapy.
When caught early, surgical excision of melanoma can be curative. However, once disseminated, the treatment of melanoma is quite inefficient and survival rates are quite dismal [186]. The use of IFN-α2b was approved as an adjuvant therapy by the FDA in 1996 based on clinical findings that showed high-dose treatment was beneficial for prolonging relapse free survival and overall survival [187]. Unfortunately, the high-dose treatment is associated with severe adverse effects, including fatigue, myalgia, pyrexia and depression. While lower doses may decrease the adverse effects, they do not offer the same benefit in relapse free survival [188]. The use of PEG-IFN-α2b has been shown to reduce some of the fatigue and flu-like symptoms seen with the non-pegylated protein [189, 190]. In comparison, adjuvant therapy with IFN-β is standard practice in Japan where low-dose administration has been reported as beneficial for maintenance therapy [191].
High-dose IFN-α treatment has also been tested as a neoadjuvant for patients with locally advanced disease with the intention of reducing T-regs and improving CD8+ T cell memory [192]. Clinically, neoadjuvant IFN-α therapy was associated with increased intratumoral DC in 11/20 patients who showed objective clinical response [193]. For the treatment of disseminated melanoma, higher doses or continuous administration of IFN-β were met with limited efficacy and toxicity [194, 195]. Association of IFN-α with dacarbazine or other chemotherapies was not beneficial [192].
The results from several long term and large cohort trials exploring melanoma patient populations and treatment regimens have been published in the past few years. For example, in the Sunbelt Melanoma Trial, started in 1997, treatment of patients with sentinel lymph node involvement were treated with high dose IFN-α2b (HDI) with or without complete lymph node dissection and clinical progress was compared to patients who did not receive HDI. In this trial involving more than 900 patients, no clinical benefit was associated with the use of HDI [196]. Long term follow up of the EORTC 18952 trial was recently reported, revealing that a 13 month IFN-α2b treatment regimen was inferior to a 25 month regimen in patients with stage IIB-III melanoma, however the difference was marginal. Interestingly, ulceration of the primary tumor was associated with increased sensitivity to IFN-α2b [197]. Final analysis of the Dermatologic Cooperative Oncology Group Trial was reported in 2015, showing benefit of IFN-α2b treatment for relapse free survival, but not overall survival [198].
The study of melanoma treatment using recombinant type I IFN is ongoing and aims to identify patient populations that will benefit from this and other adjuvant approaches, including ipilimumab and vemurafenib [186, 197, 199–201]. Even so, the use of type I IFN for the treatment of cancer is certainly not limited to melanoma. For example, treatment of prostate carcinoma [202] and myeloproliferative disorders [203] with type I IFN has revealed some benefit, yet concerns over decline in quality of life and the toxicity of the treatment continue. As evidenced by the large number of clinical trials involving type I interferon for the treatment of cancer (more than 450 listed on https://clinicaltrials.gov, including some 50 trials that are recruiting patients at this time), study of this approach continues in order to better develop delivery methods, treatment regimens and identify those patients who are most likely to benefit.
Inducers of endogenous type I IFNs
The main purpose of cancer immunotherapy is to induce immune cells to effectively eliminate tumors, overcoming the immunosuppressive tumor microenvironment [204] and, as discussed here, the induction of type I IFNs may be a critical step towards this end. Indeed, in contrast with traditional vaccine adjuvants, such as aluminum compounds, that mostly stimulate humoral immune responses, targeting of DAMPs and/or PAMPs receptors to induce IFN-α/β is a very effective strategy for cell-mediated immunity and therefore an alternative as adjuvant in cancer vaccines (Figure 3). Here we explore the induction of endogenous type I IFN both as an adjuvant and as an immunotherapy on its own.
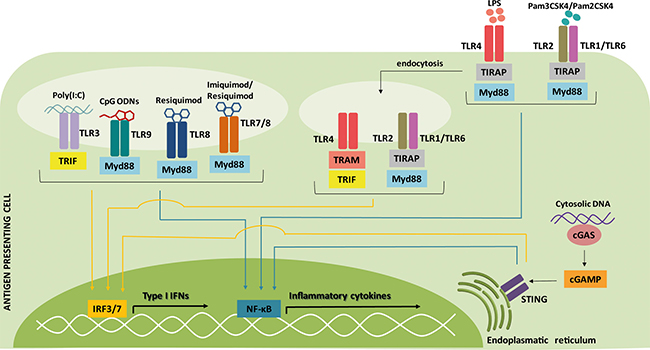
Figure 3: Signaling pathways of type I IFN inducers commonly used as adjuvants for cancer therapy. Nearly all cells are capable of producing type I IFNs after sensing pathogen-associated microbial patterns (PAMPs) and danger-associated molecular patterns (DAMPs). This strategy is used to improve therapeutic cancer vaccines, increasing the immunologic response. The activation of pattern-recognition receptors (PRRs) leads to signaling through the adaptor molecules toll/interleukin-1 receptor domain-containing adaptor protein inducing interferon-β (TRIF) and/or Myd88, which culminates in the activation of IFN-regulatory factor 3/7 (IRF3/7) (yellow arrows) or nuclear factor-ĸB (NF-ĸB) (blue arrows) transcription factors and, consequently, in the expression of type I IFNs or inflammatory cytokines. Regarding the dendritic cell (DC) subtypes, the PRRs toll-like receptor (TLR)1, TLR2, TLR3, TLR4, TLR6 and TLR8 are expressed by monocyte-derived DCs and myeloid DCs, while TLR7 and TLR9 are only expressed by plasmacytoid DC. Lipopolysaccharide (LPS), triacylated lipopeptides (Pam3CSK4), diacylated lipopeptides (Pam2CSK4), polyribosinic-polyribocytidylic acid [Poly(I:C)], oligodeoxynucleotide (CpG ODN), TIR domain-containing adaptor protein (TIRAP), TRIF-related adaptor molecule (TRAM), 2′-3′-cyclic GMP-AMP (cGAMP), cGAMP synthase (cGAS), stimulator of interferon genes (STING).
Poly(I:C)
Polyribosinic-polyribocytidylic acid [Poly(I:C)] is a synthetic analog of double-stranded RNA, a ligand of the TRIF-dependent toll-like receptor-3 (TLR3) [205]. TLR3 is highly expressed in several tumors [206] and in immature myeloid DC, NK cells, T cells and macrophages [207, 208]. Poly-ICLC is a derivative of Poly(I:C) stabilized with poly-lysine (Hiltonol®, Oncovir Inc.) and indeed was shown to be 5- to 10-fold more resistant to hydrolysis [209]. Both are included in the National Cancer Institute’s ranking of immunotherapeutic agents with the highest potential of improving the cancer immunotherapy response [205].
After stimulation, TLR3 recruits adaptor protein TRIF and signals through activation of IRF3, NF-κB and activator protein-1 (AP-1), stimulating activation of the antiviral and pro-inflammatory responses [210].
In vitro studies have shown that Poly(I:C) induces maturation and activation of DCs, including enhancing cross-presentation [211], stimulates T cells [212] and NK cells [213], and induces secretion of pro-inflammatory cytokines by tumor and immune cells [214]. Poly(I:C) can also directly affect tumor growth and induce apoptosis of tumor cells [215], resulting in the availability of tumor-associated antigens (TAAs) for uptake by APCs.
In healthy volunteers, Poly-ICLC was shown to upregulate genes involved in the innate immune response including IFN-α, IFN-β, IFN-γ, the complement system and the inflammasome [216]. Poly-ICLC stimulated Th1 cytokines, increasing the Th1/Th2 ratio [217] and driving T cells toward a Th1 response. Sabbatini and colleagues have shown that 91% of patients with ovarian cancer in a poly-ICLC-vaccine cohort showed functional CD8+ T cell responses, compared to 25–62% of patients in non-poly-ICLC groups. Similarly, CD4+ T cell responses were stronger in patients treated with poly-ICLC-containing vaccines [218].
Interestingly, lymphocytes extracted from patients with persistent HPV infection were exposed to HPV16 virus-like particles (VLP) and then treated with Poly-ICLC, resulting in increased MHC class I and II, CD40, CD80, and CD86 expression and inducing HPV16 E7-specific CD8+ T cell responses in vitro [219].
Another version of Poly(I:C), Poly(I:C12U) (Ampligen®, Hemispherx Biopharma), has been used for chronic fatigue syndrome treatment [220] and activated moDCs, increasing the expression of surface MHC class I and II, CD83, CCR7, CD86, CD40 and IL-12 [221].
Lipopolysaccharide
Lipopolysaccharides (LPS) from Gram-negative bacteria are TLR4 agonists. After LPS binding, TLR4 dimerizes and this is sensed by an adaptor molecule called toll/interleukin-1 receptor domain-containing adapter protein (TIRAP) [222]. Then, TIRAP recruits the signaling adaptor MyD88 and several interleukin-1 receptor-associated kinase (IRAK) family members [223], which activates inflammatory transcription factors such as AP-1 and NF-κB [224]. At the same time that TLR4 signaling is induced, several events take place to promote the TLR4 endocytosis. Upon delivery to endosomes, TLR4 recruits TRIF-related adaptor molecule (TRAM) and TRIF, which signals through a cascade of activated proteins in the cytosol and culminates in the induction of and IRF3 [225]. In this way, TLR4 engagement promotes the expression of pro-inflammatory cytokines and type I IFNs.
Several reports show that DCs generated from mobilized monocytes pulsed in vitro with TAAs and stimulated with LPS, with or without IFN-γ, were able to express IL-12 and CXCL10, polarize a Th1 immune response and may be useful for DC-based immunotherapy [226–230]. Interestingly, other studies have demonstrated that DCs generated using LPS are capable of inhibiting suppression mediated by CD4+CD25+Foxp3+ regulatory T cells [231] and that they restored CD4+ and CD8+ T cell proliferation, while DCs matured with a conventional cocktail (IL-1, IL-6, TNF-α, prostaglandin E2 - PGE2) did not fully restore T cell proliferation [232]. Also, the 3-O-deacylated monophosphoryl lipid A (MPLA) is a less toxic LPS derived from Salmonella minnesota R595 and used with alum in a prophylactic vaccine against human papillomavirus 16 and 18 (Cervarix®, GSK Vaccines) [233].
Imidazoquinoline-like molecules
Imiquimod and Resiquimod are imidazoquinoline-like molecules that have been identified as TLR7/8 agonists based on their ability to induce DC maturation. TLR7 is mainly expressed in pDCs and, to some extent, in B cells and monocytes/macrophages [234], while TLR8 is primarily expressed in monocytes/macrophages and myeloid DCs [235]. Therefore, Imiquimod is used specifically to activate pDCs, inducing expression of IFN-α, IL-6, IL-8, IL-12 and TNF-α and stimulating a Th1 immune response; it has been approved by the FDA for treating basal cell skin cancer (Aldara®, 3M Pharmaceuticals) [236].
TLR7 also recognizes single-stranded RNA (ssRNA) derived from RNA viruses (like vesicular stomatitis virus, influenza A virus and human immunodeficiency virus) [237], synthetic poly(U) RNA and certain small interfering RNAs [238], thus pDCs are able to produce large amounts of type I IFN and cytokines in response to virus infection [237]. TLR8 recognizes Resiquimod and viral ssRNA and is upregulated after bacterial infection, having its highest expression in monocytes, although is expressed in several tissues [239].
These sensors utilize the universal adapter protein MyD88, which in turn activates the expression of IRF7 and NF-κB, thereby stimulating transcription of type I and III IFNs, inflammatory cytokines and chemokines [240], especially IFN-α, TNF-α and IL-12 [241]. Upon activation, pDCs also expresses the co-stimulatory molecules CD40, CD80 and CD86 and gain the ability to cross-present antigens in the context of MHC [242].
The use of Imiquimod brought to light intriguing observations regarding DC functions that are not usually considered. Drobits and co-workers showed that Imiquimod treatment promoted the secretion of both TRAIL and granzyme B resulting in pDC-mediated tumor killing [243]. Also, pDCs stimulated with agonists for TLR7 and 9 upregulated the surface expression of TRAIL in a type I IFN-dependent manner, causing the lysis of Jurkat cells and melanoma cell lines SKMel2 and WM793 [244].
The topical treatment of basal cell carcinoma, perianal Bowen's disease and superficial malignant melanomas with Imiquimod led to an increase in activated-pDC infiltration and to a reduction in neoplastic cells with complete regression in some cases [245–248].
Also, Imiquimod may have direct antitumor effects inducing apoptosis via modulation of the expression of Bcl-2/Bax [249–251] and autophagy [252, 253] in several cancer cells, as well as antiangiogenic properties, based on its induction of interferons, IL-10, and IL-12 [254], which end up inhibiting angiogenesis independently of their immunomodulatory functions. In fact, Imiquimod has been successfully used as an antiangiogenic agent to treat vascular proliferative lesions, such as infantile haemangioma, pyogenic granuloma and Kaposi’s sarcoma [254–256]. And, in a patient with melanoma, treatment with Imiquimod induced gene expression of angiogenesis or MMP inhibitors, like IFN-α, KiSS1, TIMP metallopeptidase inhibitor 1 (TIMP1), and thrombospondin 1 (THBS1), while decreasing expression of bFGF and MMP9, as shown by quantitative PCR of cutaneous melanoma metastasis biopsies performed before and after treatment [257].
CpG ODNs
The unmethylated CpG ODN (oligodeoxynucleotide) TLR9 agonists are powerful adjuvants for the activation of pDCs. In humans, TLR9 is expressed only in pDCs and B cells [258] and recognizes unmethylated CpG motifs that are found in bacterial and viral genomes [239]. CpG ODNs are divided into four classes depending on the differences in their structure and immunoreactivity. Of these classes, almost all the CpG ODNs used in clinical trials have been class-B CpG ODNs (also known as K-type ODNs), however type I IFN is weakly induced by CpG-B ODNs. Class-A CpG ODNs (also known as D-type ODNs) have also been used but in fewer clinical trials. Class A and C CpG ODNs enter the lysosome compartments of pDCs and B cells to stimulate IFN-α production, while class B CpG ODNs enter the endosomal compartments of pDCs to induce their maturation [259].
CpG ODNs activate TLR9–MyD88–IRF7 and TLR9–MyD88–NF-κB signaling pathways of pDC to induce expression of MHC and costimulatory molecules such as CD40, CD80, and CD86, which results in CD4+ and CD8+ T cell maturation [260] and secretion of type I IFN [261] and IL-6, IL-12 and TNF-α [262]. Additionally, type I IFN and TNF-α secreted from pDCs activate NK and NKT cells [263].
A prospective Phase I trial with stage II–IV metastatic melanoma patients vaccinated with melanoma-associated antigen recognized by T cells-1 (MART-1) peptide, Montadine® ISA-51 (an agonist made of mineral oil and surfactant from mannide monnooleate family [264]) and CpG 7909 showed that in the presence of CpG ODN there was 10-fold more MART-1 specific T cells induced in patients [265].
MelQbG10, which is G10 CpG ODN and the tumor peptides MART-1 coated with bacteriophage protein, was used in combination with Montanide® ISA-51 and topical 5% Imiquimod cream in stage III/IV malignant melanoma patients. Patients vaccinated with MelQbG10 plus Montanide® ISA-51 had significantly higher T cell induction versus MelQbG10 alone, but there were no significant differences in clinical outcome among the different treatment groups [266].
Bacterial triacylated or diacylated lipopeptides
The fatty acid groups of triacylated lipopeptides are recognized by TLR2/TLR1 heterodimers [267], while the fatty acid groups of diacylated lipopeptides are ligands for TLR2/TLR6 heterodimers [268]. Previously, it was thought that TLR2/TLR1 and TLR2/TLR6 engagement elicited the pro-inflammatory pathway, but not the type I IFN responses [269]. It was shown that administration of TLR2 agonists can enhance effector and memory T cell responses, culminating in improved tumor rejection [270, 271]. TLR2 agonists can also increase expression of costimulatory molecules in B cell lymphoma, enhancing its sensitivity to NK and CD8+ T cells [272], or inducing caspase 8-dependent apoptosis [273].
However, recent studies have shown that bacterial ligands can induce type I IFN responses through TLR2 binding. After stimulation, the TLR2 heterodimers are internalized into endolysosomal vesicles, from which they induce IFN-β via MyD88 and IRF1/IRF7 [274] and this pathway requires TRAM [275], but is yet to be fully elucidated. In this way, TRAM acts as adaptor molecule for both TLR4 and TLR2, inducing IRF1 and IRF7 signaling from the endosome.
Dietrich and colleagues observed that stimulation of bone marrow derived macrophages (BMDMs) with Pam3CSK4 (synthetic triacylated lipopeptides TLR2/TLR1 agonist) and Pam2CSK4 (synthetic diacylated lipopeptides TLR2/TLR6 ligand) induced not just pro-inflammatory cytokines like TNF-α and IL-12, but also type I IFN-inducible genes, such as CXCL-10, Mx2, IL-6 and iNOS [274].
SUP3 is a TLR2 agonist based on the structure of Pam3CSK4, but with a chemically more stable structure. SUP3 was shown to enhance cross-presentation by CD8+ cDCs in vitro, up-regulate the expression of CD40 and CD86 co-stimulatory molecules and induce production of IL-6 and TNFα in DC, culminating in an antigen-specific CD8+ T cell response and increased immunization against tumor challenge. SUP3 also induced antigen-specific antibodies such as IgM, total IgG and high affinity IgG [276].
2′-3′-Cyclic GMP-AMP
Several authors have shown that 2′-3′-cyclic GMP-AMP (cGAMP) can be used directly as an adjuvant for antitumor therapy [277]. cGAS is a major sensor of cytosolic DNA, irrespective of the DNA sequence [278, 279]. Cytosolic DNA can trigger strong production of type I IFNs and other inflammatory cytokines in immune and non-immune cells. After DNA binding, cGAS undergoes a conformational change that promotes the conversion of GTP and ATP into cGAMP [280]. cGAMP then acts as a second messenger that activates the adaptor protein STING, at the endoplasmic reticulum membrane [279]. STING in turn activates the proteins inhibitor of nuclear factor-κB kinase (IKK) and TANK binding kinase 1 (TBK1), which activate NF-κB and IRF3, respectively, inducing production of cytokines and type I IFN [281].
DCs can also activate the cGAS–STING pathway after DCs phagocytose tumor cells and some of the tumor DNA escapes to the cytoplasm. Woo and co-workers (2014) showed that mice deficient for Myd88, TRIF, the purinergic receptor P2X7 (P2XR7), mitochondrial antiviral-signaling protein (MAVS) or retinoic acid inducible gene I (RIG) had no defect in priming of CD8+ T cells. Strikingly, in both STING-deficient and IRF3-deficient mice, there was a substantially diminished CD8+ T cell response against tumor-associated antigens and, in wild-type mice, transfer of tumor DNA to host APCs resulted in TBK1 and IRF3 phosphorylation and, as a consequence, production of IFN-β [282]. ECs are also producers of type I IFN in response to STING activation. Demaria and collaborators showed that intratumoral injection of exogenous cGAMP enhanced STING activation in the tumor microenvironment, resulting in stimulation of type I IFN response and antitumor CD8+ T cells, leading to growth inhibition of injected and contralateral tumors. Interestingly, this effect resulted mainly from tumor ECs, which were the main producers of IFN-β in response to cGAMP injection in both mouse and human [283].
In a study performed by Wang and co-authors, PD-L1 antibody was administered in a mouse model of melanoma and they observed that cGAS-deficient mice are refractory to the antitumor effects of a PD-L1 antibody. They showed a large increase of tumor-infiltrating leukocytes in wild-type mice after PD-L1 antibody treatment, but not in cGAS- or STING-deficient mice. This may be due to tumor cell killing caused by PD-L1 antibody treatment, which exposes tumor-associated antigens and DNA that are taken up by DCs. Then, tumor DNA escapes to the cytoplasm of DCs and activates the cGAS–STING pathway, inducing the production of type I IFN and the co-stimulatory molecule CD86, and activating a Th1 response. When they applied cGAMP intramuscularly, this caused inhibition of tumor growth and prolonged mouse survival after PD-L1 antibody treatment [284].
Previous studies have shown that intratumor injection of cGAMP and its analogs also induced antitumor effects. However, some authors suggest that STING activation may induce a suppressive tumor microenvironment and contribute to tumor growth and metastasis [285]. Metastatic brain cancer cells generate cGAMP, which is transferred by gap junctions to astrocytes, activating the STING pathway in these cells and producing proinflamatory cytokines, which in turn activate STAT1 and NF-κB pathways in the metastatic cells, thus supporting tumor growth [286].
Immunogenic cell death
During the last decade, the newly defined concept of immunogenic cell death (ICD) induced a thorough revision to the previously accepted, classic point of view cell death as a dichotomized phenomenon as either apoptotic, associated with a tolerogenic immune response that maintains tissue homeostasis or, in contrast, necrotic, a promoter of the inflammatory response [287]. Undeniably, along with the success of checkpoint blockade immunotherapy [288], ICD helped to cement the importance of the immune system during cancer treatment, especially pertinent when selecting which chemotherapy to administer since unsuccessful approaches are often immunosuppressive and unable to activate antitumor immunity.
ICD was originally demonstrated as a cellular and molecular response of cancer cells to anthracyclines that involves the exposure and secretion of immunogenic DAMPS, in a defined temporal sequence, providing both antigenic and stimulatory signals for the DC compartment to generate an effective CD8+ T cell attack against remaining tumor cells [287]. The first study that unveiled this mechanism showed that ex vivo treatment of MCA205 sarcoma cells with doxorubicin and subsequent inoculation of these dying tumor cells into naïve syngeneic mice protected them against a subsequent tumor challenge. Remarkably, this protection was not seen when cells were treated with mitomycin C (another chemotherapeutic agent) or when caspase-3 activity was blocked, showing that a specific property of cell death induced by doxorubicin was mediating immune stimulation. Furthermore, intratumoral application of doxorubicin in subcutaneous established tumors only exhibited therapeutic efficacy when treatment was performed in immunocompetent mice, whereas treatment in the nude background (lacking mature T cells) or in animals depleted of DCs abolished the immune response against the tumor cells [289].
Key mechanistic insights came later when Obeid and colleagues used a large panel of apoptosis inducers and identified changes in the plasma membrane proteome that were exclusively present in anthracycline treated cells and not in the presence of the pan-caspase inhibitor. Comparison of two-dimensional electrophoresis, followed by mass spectroscopic analyses, led to the identification of the endoplasmic reticulum chaperone calreticulin (CRT). Accordingly, knockdown of CRT negatively affected phagocytic uptake of dying tumor cells by DCs and abrogated the immune protection effect. Therefore, CRT release (ecto-CRT) was the first key feature identified as a determinant of the interaction of DCs and dying immunogenic cells and consequently the anticancer immune response [290].
Furthermore, as revealed in subsequent studies, other ICD determinants have been identified. This way, the proposed mechanism postulates that: (i) in response to lethal insult from doxorubicin treatment, dying tumor cells activate autophagy machinery and secrete ATP that, in turn, is recognized by purinergic receptors (P2RY2 and P2RX7) of DCs, promoting DC recruitment and activation; (ii) exposition of Annexin A1 mediates DC contact with the dying tumor cell; (iii) secretion of CRT, as a consequence of endoplasmic reticulum stress, acts on antigen uptake by DCs; (iv) release of the alarmin high-mobility group box 1 (HMGB1) from the nucleolus, which then binds to TLR4 of DCs to induce full maturation status, secretion of IL-1β and eventually leading to priming of T cells with complete cytotoxic capacity [291].
Interestingly, all of these processes regarding release and secretion of ICD markers alone or in combination cannot predict with certainty if the cell death process will truly be immunogenic, which suggests that additional, unknown factors remain to be identified [292]. Along these same lines, recently a novel mechanism was uncovered: the cancer cell–autonomous secretion of type I IFNs [293].
The role of type I IFNs in ICD was revealed by analyzing immunologically relevant transcriptional changes induced in sarcomas upon intratumoral treatment with doxorubicin. Among the modulated pathways, transcripts associated with response to viral infections were indicated by type I IFN stimulated-genes, pointing to a type I IFN fingerprint in the cancer cells. Indeed, antibody mediated blockade of IFNAR1 or IFN-α/β neutralization markedly inhibited doxorubicin’s antitumor effects. Sarcoma tumors derived from an Ifnar2−/− background did not respond to doxorubicin when transplanted into a wild-type host, suggesting that the IFN-β produced during ICD was most likely affecting the tumor cells, not the host immune system. As further demonstrated, Ifnar1−/− tumor cells failed to secrete CXCL10 in response to doxorubicin, just as was also seen in tumor cells derived from Tlr3 KO or its adaptor Trif. These data demonstrated that ICD inducers act by stimulating IFN-β secretion through an autocrine and paracrine mechanism that takes place upon TLR3 recognition of self RNA from dying cells, activating the CXCL10-CXCR3 signaling axis to attract effector immune cells [293].
In accordance with these findings, radiation therapy (RT), a known ICD inducer, was also reported to depend on IFN-β signaling. In the work of Burnette and collaborators, local ablative RT of B16-SIY tumors resulted in striking tumor regression and local production of IFN-β by tumor-infiltrating CD45+ cells. In agreement, tumor associated DCs presented higher levels of maturation markers (CD40, CD80 and MHC-I and II molecules) after RT, yet if treatment was performed in an Ifnar1 KO background, therapeutic control was completely lost. Additionally, to determinate the host compartment in which IFN-β was necessary, bone marrow transplants from Ifnar1 KO mice to a WT host revealed a requirement for the hematopoietic cells, more specifically the CD11c+ and CD11c- myeloid populations, that in a context lacking IFN-β lost their cross-priming capacity within the tumor microenvironment [294].
Another ICD inducer that relies of type I IFN activity is the oncolytic Newcastle Disease Virus (NDV) [295], which is an anticancer virotherapy strategy, and as such, was expected to be impaired by the antiviral properties of IFNs, thus negatively affecting therapy outcome. However, as demonstrated in the work of Zamarin and colleagues, intratumoral applications of the oncolytic NDV accompanied with CTLA-4 checkpoint blockade promoted complete regression of B16 tumors in both primary and non-treated secondary sites, showing a remarkable systemic immune protection. Additionally, the authors found that this immune protection was mediated by both NK and CD8+ T cells, and, unexpectedly, if treatment was performed in a Ifnar1−/− host, therapeutic efficacy was abrogated in the injected tumors as well as in the contralateral challenge, even when combined NDV plus CTLA-4 treatment was applied [296].
Three pathways of innate immune sensing can lead to Ifnb gene transcription: (i) TLR stimulation signals through MyD88 and TRIF adaptors, (ii) RIG-I senses cytosolic RNA and signals through the adaptor protein IPS-1, and (iii) STING senses cytosolic DNA and promotes type I IFN expression [297, 298]. Consequently, induction of type I IFN in the tumor microenvironment correlates with T cell infiltration. Gajewski demonstrated, through melanoma gene expression profiling, that tumors infiltrated with CD8+ T cells also exhibited a type I IFN transcriptional signature [299], a suggestion that type I IFN signaling might participate in innate recognition of tumors [157]. Also, as said before, mice deficient in type I IFN response showed decreased spontaneous T cell priming in transplantable tumor models and increased tumor induction using methylcholanthrene [6].
Overall, the findings presented here highlight the critical role of integrated innate and adaptive immune responses in order to achieve full therapeutic efficacy and, most importantly, revealing type I IFN signaling as an indispensable propeller of the cancer immunity cycle. Yet, it remains to be determined which tumors are likely to benefit from ICD treatment, for example, one report indicates that spontaneous mammary tumors in (MMTV)-NeuT transgenic female mice can successfully respond to immunogenic chemotherapy even in the absence of the immune system [300]. But, as shown by Pfirschke and collaborators, pretreatment with the combination of oxaliplatin-cyclophosphamide can increase T cell infiltration in resistant Kras/Trp53 mutant tumors, rendering them sensitive to anti-PD-1 and CTLA-4 blockade [301], suggesting the potential benefit of associating chemo and immunotherapy. However, inducing ICD using multiple inflammatory immunotherapeutic agents may not boost immune attack, since negative regulatory mechanisms will likely be stimulated and, thus, impede the immune response.
TYPE I IFNS IN CANCER GENE THERAPY: TARGETING TUMOR AND DENDRITIC CELLS
According to the Journal of Gene Medicine, 2409 clinical trials making use of gene transfer approaches have been included in their online database since 1989, of which 1554 (approximately 64.5% of the total) were aimed at treating cancer. Cancer gene therapy is expected to remediate faulty gene function in order to kill tumor cells or render them susceptible to killing by chemo/radiotherapy or immune attack. Such approaches may include the silencing of oncogenes [302], the transfer of tumor suppressor genes, generally aiming to trigger mechanisms of cell death [303], and the transfer of immune modulating genes in order to elicit antitumor systemic responses [304, 305]. As will be discussed further, the gene therapy approach may not target the tumor cell directly, but can be used to trigger an anti-tumor immune response, such as in the case of vaccines based on modified DCs.
Many vectors have been employed for the transfer of type I IFN genes, including for modification of tumor cells or DCs (Table 1). Among these vectors, adenovirus, AAV and, more recently, non-viral liposome mediated gene transfer have been used in both basic and clinical research protocols [306–311]. Specifically in the case of IFN-α gene transfer, adenovirus is one of the most commonly employed vectors for in situ treatment models, yet lentivirus is better suited for ex vivo creation of IFN-α secreting cells.
Table 1: Properties of the main vectors used in gene therapy protocols
Vector |
Integrative |
in vitro delivery efficiency |
in vivo delivery Efficiency |
Capacity to trigger immune response |
Efficient production |
|---|---|---|---|---|---|
Adenoviral |
No |
High |
High |
High |
Yes |
Lentiviral |
Yes |
High |
High |
Low |
No |
Retroviral |
Yes |
High |
High |
Low |
No |
AAV |
No |
High |
High |
High |
No |
Liposomes |
No |
High |
Low |
Low |
Yes |
Abbreviations: AAV, Adeno-associated virus.
In preclinical models, as expected from its antitumor functions, IFN-α gene transfer induced: (i) cell cycle arrest, (ii) apoptotic cell death mechanisms [312–314], (iii) decreased hemoglobin index and microvessel density, and (iv) necrotic ischemia in tumor tissue [312, 313, 315]. Regarding the immunomodulatory property of IFN-α, it was also observed that the gene transfer, alone or combined with other therapy approaches, lead to: (i) increased presence of infiltrating CD8+ and CD4+ T cells and decreased Foxp3+ cells in the tumor parenchyma and also augmented MHC-I expression on tumor cells [316]; (ii) enhancement of NK cell cytolytic activity [312] and (iii) increased presence of CD11c+ cells in regional lymph nodes [317]. In this way, the immunosuppressive features of the tumor microenvironment can be overcome upon IFN-α gene transfer and indications of a systemic antitumor immune response are uncovered.
More recently, the effect of the IFN-α on metastasis was investigated. In this approach, hematopoietic stem cells (HSCs) were modified with a lentiviral vector in order to generate Tie2+ macrophages/monocytes that constitutively express IFN-α. The natural homing of these cells to tumor sites was observed, leading to a reduction in hepatic metastases from colorectal cancer, without exerting a negative influence on the homeostasis of hematopoiesis [318, 319].
In light of the successful results obtained from IFN-α gene transfer to cancer in basic-research models, a phase-I clinical trial was carried out using a recombinant adenoviral vector to treat non-muscle invasive bladder cancer in which 17 patients were enrolled. The subjects received escalating doses of the vector and the gene transfer efficacy was assessed by examining cytokine levels in the urine. Even at the highest dose, the treatment was well tolerated, with only mild adverse events being observed. Regarding the treatment efficacy, it was reported that 7 patients achieved complete response at 3 months [320].
In a similar way, in basic research protocols aiming to assess the effects of IFN-β gene transfer in both in vitro and in vivo models, it was observed that the treatment: (i) reduced tumor cell proliferation [133, 321–323]; (ii) decreased cell viability in both monolayer and spheroids cultures [308, 322]; (iii) increased long-term survival with reduced tumor burden [311, 323–327] and (iv) reduced tumor volume without notable toxicity, yet an increase in apoptotic cells and areas of necrosis in tumor tissue [133, 321, 324]. In this way, IFN-β gene transfer may offer an advantage, localized high concentrations of this protein, which cannot be achieved with biochemotherapy.
Delving into the effect of gene transfer on the microenvironment and immune system, in preclinical models after treatment it was reported that: (i) the cells presented downregulation of genes associated with angiogenesis, such as bFGF, MMP9, VEGF-A and IL-8 [311, 322]; (ii) there was decreased quantity and density of blood vessels and diminished levels of hemoglobin in the tumor [311, 327]; (iii) treated animals were less prone to develop spontaneous metastasis, became resistant to a second tumor challenge or to the establishment of induced metastases [323, 326, 327]; (iv) increased infiltrating CD8+ T lymphocytes [323, 326, 328], activated NK cells [326, 329] and macrophages [133, 324, 329] as well as increased levels of MHC-I on the tumor cells [326]. The efficacy of IFN-β gene therapy was considerably decreased only in animals depleted of CD8+ T cells, indicating that this class of lymphocytes play a critical role in the immunomodulation stimulated by IFN-β [328].
Although many vectors are being used for gene transfer, armed oncolytics carrying IFN-β has recently gained ground. Approaches using the Vesicular Stomatitis Virus (VSV) encoding IFN-β have been shown to elicit a strong antitumor immune response, decreasing infiltrating T-reg cells and increasing CD8+ cells, and also stimulating the expression of PD-L1 on tumor cells [330]. Since VSV-IFN-β offers increased capacity to elicit both innate and adaptive immune responses as well as preferential replication in tumor cells, it is safer and more effective as compared to VSV with no transgene [331, 332], features that led to the establishment of a phase I clinical trial in 2012, that is still ongoing with estimated primary completion date in June 2017 (https://clinicaltrials.gov/ct2/show/study/NCT01628640).
Despite the positive responses seen in preclinical studies, only a few clinical protocols using recombinant vectors for the delivery of IFN-β in cancer patients have been carried out [333–335]. As an example, in a phase I clinical trial using a recombinant adenoviral vector, 11 patients with recurrent malignant glioma received different doses of the vector by stereotactic injection in the tumor site. The first injection was performed approximately one week before the scheduled tumor resection surgery and a second right after the procedure. After the treatment regimen it was clear that the vector treatment reproduced some data from the preclinical models, i.e, induction of apoptosis and the presence of necrotic areas in the treated tissue. Although a treatment-related dose limiting toxicity was seen in one patient enrolled in the highest dose cohort, IFN-β gene therapy was shown to be a safe and potentially effective approach [336]. On the other hand, despite its proven safety, it was also seen in another clinical trial that repeated doses of the adenoviral vector did not considerably improve the clinical outcome in patients with mesothelioma due to the fast development of neutralizing antibodies against the vector, an obstacle that, perhaps, could be circumvented by using a non-immunogenic vector or by combining gene transfer with additional therapeutic approaches [332].
Modified dendritic cells expressing type I interferons: crossroad between cancer vaccines and gene therapy
In order to induce a host immune response against tumor cells, genetically modified DCs have been used in vaccination protocols. In spite of the clear rationale, DC-based vaccination faces some technical obstacles, such as the best condition for their activation such that the antitumor immune response is efficiently induced [337]. Given the influence of type I IFNs on DCs, it is reasonable that IFN-α/β gene transfer be used to activate DCs and positive results have been reported. For example, mice bearing GL261 glioma tumors were treated with one intratumoral injection of an adenoviral vector encoding IFN-α followed by implantation of syngeneic bone marrow-derived DCs resulting in increased survival due to an antitumor immune response dependent on CD8+ T cells, and it was shown that the animals acquired a certain level of resistance against a second tumor challenge [338]. In another model the vaccination protocol consisted of intratumoral delivery of DCs previously modified with a recombinant adenoviral vector encoding IFN-α in combination with irradiated tumor cells engineered to express IL-4 or GM-CSF, resulting in stronger tumor specific CTL responses in the cervical lymph node and increased survival [339].
Specifically regarding the genetic modification of DCs, many recombinant vectors have been used, such as those derived from adenovirus, lentivirus, retrovirus, AAV and Sendai virus [340]. In spite of the many types of vectors available for the genetic modification of DCs, adenoviral vectors are most commonly used since they provide highly efficient gene transfer and expression, easy handling and high-titer preparations. Even so, adenoviral vectors present some disadvantages, e.g. pre-existing neutralizing antibodies and transductional dependency on the coxsackievirus and adenovirus receptor (CAR) [341]. However, the former should not present a barrier when virus is applied to DCs ex vivo and the latter can be overcome with the use of modified adenoviral vectors that no longer depend on CAR. Still, adenoviral vectors trigger molecular mechanisms leading to the maturation of DCs and, as a consequence, a more consistent antitumor immune response [342, 343].
In addition to adenovirus, other vectors have been gaining space in the field of genetically modified DCs. For example, non-viral methods are being more frequently used, including mRNA transfection, due to their lower manufacturing costs, comparable levels of expression, and relative transfection efficacy, especially with the use of electroporation [344]. Another emerging viral vector is the Sendai virus due to its high transduction efficiency ex vivo and in vivo, augmented capacity to trigger antitumor immune responses and elicit DC maturation [345, 346]. Along the same lines, exploring the properties of IFN-α produced by pDCs, infection of pDCs with a replication-deficient herpes simplex virus 1 (HSV-1) d106S vaccine strain showed a robust cytotoxic effect against various melanoma cell lines that was equivalent or superior to the effects induced by synthetic TLR7 and TLR9 agonists [347].
p19Arf and interferon-β combined gene transfer
As presented above, gene transfer of IFN-α/β directly into the tumor mass or into dendritic cells presents promising results that merit further development for clinical application. However, if one aims to increase the intrinsic antitumor and immunomodulatory properties of type I IFNs, which strategy should be used? More specifically, using a gene transfer method, how can we assure that most of the transduced tumors cells would die? Since type I IFNs are already considered sufficiently immunogenic, can we enhance their immune stimulation?
In light of these questions, our lab has previously developed a set of unique adenoviral vectors which utilize a p53 responsive promoter, called PGTxβ [348] to direct the expression of the cDNAs for p19Arf (p19Arf for mouse and p14ARF for humans) and for IFN-β. Arf is a tumor suppressor protein that is encoded by the CDKN2A locus (also encoding the p16INK4a protein) [349] and is mainly known for being a functional partner of p53, since after oncogenic stress Arf associates with MDM2 and prevents MDM2 mediated ubiquitination of p53 for posterior degradation [350]. Thus, Arf can enable p53 to trigger growth arrest, apoptosis and also acts in a p53 independent manner by inhibiting ribosomal RNA processing, promoting apoptosis and regulating autophagy [351, 352].
By combining Arf and IFN-β along with the p53 responsive promoter, we hoped to create interplay between: (i) transgene control, (ii) p53/Arf pro-apoptotic functions and (iii) IFN-β antiviral and immunomodulatory activities. Indeed, other studies have already pointed potential benefits of targeting the p53/Arf/IFN-β pathways, but never explored its therapeutic application. For example, Takaoka and collaborators have shown that IFN-α/β activates p53 transcription and stabilizes its protein levels [353]. Interestingly, they showed that p53 and type I IFNs work cooperatively to potentiate the apoptotic machinery and mediate tumor suppression and viral control functions. Furthermore, Sandoval and colleagues showed that apoptosis induced by type I IFNs requires p14Arf, but not p53, since human sarcoma cells null for p14ARF undergo apoptosis when p14ARF is reintroduced in the presence of IFN-α/β, but the same observation is not seen with p53 [354].
Based on this evidence, we decided to explore the murine B16F10 (B16) melanoma cell line as a model since it harbors p53 in its wild type form, as seen in 90% of human melanoma cases [355] and is a well-known model for immunotherapies. Remarkably, in our initial observations we noticed that combined gene transfer of p19Arf and IFN-β, but not the either single treatment, provoked massive cell death while up-regulating p53 target genes p21Waf1, Mdm2 and Puma [356].
Evidence for superior immune stimulation came from two distinct immunization contexts. In the first, mice were vaccinated prophylactically with ex vivo transduced B16 cells that while dying were inoculated in naïve syngeneic C57BL/6 mice and seven days later, mice were challenged with fresh B16 cells in the contralateral flank. Tumor formation was completely abrogated at the vaccine site in hosts with competent NK cell compartment due to the up regulation of Il-15, Ulbp1 NK cell receptor, Killer/Dr5 and Fas/Apo-1 death receptors on the treated cell, thus providing a safety benefit for the combination. At the challenge site, a dramatic decrease in tumor progression was observed and was dependent on tumor-infiltrating CD4+ and CD8+ T lymphocytes. Unexpectedly, in this prophylactic model, IFN-β alone or in combination with p19Arf showed similar protection and T cells presented similar killing capabilities and levels of IFN-γ and TNF-α secretion. This would argue that there was no evident immunological superiority for the combination. However, when exploring a therapeutic tumor model, where the tumor challenge was made before the immunization step, only p19Arf/IFN-β vaccinated mice displayed reduction in tumor progression [357].
In support of this evidence, now in the second immunization context, mice bearing heterotopic (s.c) lewis lung carcinoma (LLC1) tumors were treated with four rounds of adenoviral injections directly into the tumor mass and subsequently challenged with fresh LLC1 cells in the opposite flank. Remarkably, mice that had their primary tumor treated with the p19Arf/IFN-β combination showed improved tumor control at the challenge site even when compared to IFN-β single treatment, showing superior immune protection by the combined p19Arf and IFN-β in situ gene therapy [358].
Furthermore, seeking to gain mechanistic insights on how the combination could induce cell death and immune stimulation, we evaluated the transcriptional profile of critical protein pathways, revealing that only the p19Arf/IFN-β combination induced genes related to both the p53 pathway and apoptosis as well as IFN-β immune response and antiviral functions [359]. We also noted that the use of the adenoviral vector was a critical component for inducing cell death, reinforcing the antiviral aspect of the response. Intriguingly, the p19Arf/IFN-β combination promoted cell death by a different mechanism than that seen for the individual treatments, since inhibition of capase-3/7 increased the levels of cell death upon the individual treatment with p19Arf or IFN-β, but did not affect the p19Arf/IFN-β group, suggesting that a caspase-independent mechanism of cell death was induced by the combined treatment. Of the three groups, the combination showed the lowest caspase 3 activity, while displaying features of necroptotic death, as revealed by the increase of Rip-3 (key mediator of necroptosis) and TNF receptor (Tnfrsf1A, an activator of the necrosome complex). Moreover, consistent with the recent demonstration that necroptosis can promote ICD [360], only the combined gene transfer of p19Arf and IFN-β was accompanied by the exposition of calreticulin, ATP secretion and HMGB1 release, providing mechanistic support for the immunomodulatory superiority of the combination [359].
Taken together, we believe that our data provide functional and mechanistic evidence to classify our p19Arf and IFN-β combined gene transfer as a novel agent for cancer immunotherapy. In fact, to the best of knowledge, no other gene transfer strategy employing non-replicating viral vectors has been shown to unleash ICD. Although we have identified NK cells, CD4+ and CD8+ T lymphocytes as critical cell mediators, we do not fully understand the mechanism by which these cells cooperate to bring about the anititumor immune response, most importantly we have not yet analyzed how DCs are being affected. Since NK cells can assume a helper phenotype to modulate DC priming function [178], it will be interesting to investigate how the NK cells activated in the vaccine site are interacting with DCs and promoting antigen uptake in our approach. Along the same lines, we expect that the p19Arf/IFN-β combination will provide not only an IFN-β immunomodulatory stimulus, but also immunogenic DAMPs unleashed during the ICD process, which together may provide an ideal stimulus for DC maturation, especially in the immunosuppressive tumor microenvironment. Another possibility regarding DCs would be to generate ex vivo derived DCs and use treated dying tumor cells as adjuvant as well as a source of antigen, an application that was successfully demonstrated in a pre-clinical model of high-grade glioma tumors [361]. Nevertheless, despite the long road ahead, IFN-β gene transfer and its combination with Arf holds a promising position in the cancer immunotherapy field.
CONCLUSIONS
Type I IFNs certainly play a critical role in the anti-cancer immune response and represent attractive strategy for therapy. Indeed, considering immunomodulation of the tumor microenvironment and its components, including stroma, immune and tumor cells, type I IFNs can be exploited by several strategies, such as inducers of ICD, agonists of TLRs, gene therapy and recombinant protein for the treatment of cancer (summarized in Figure 4). However, as discussed above, owing to their complex regulatory mechanisms, depending on the model of study, therapeutically induced type I IFNs have been shown to be required for either tumor cells or for infiltrating immune cells, especially DCs, to affect an anti-tumor response. Studies that can obtain deeper mechanistic insights are surely needed to clarify this dual requirement. For example, using a therapeutic model of systemic poly A:U application, Nocera and colleagues have visualized IFN-β in the tumor microenvironment, identifying the CD11c+ population as the main host source of IFN-β, but not the only one [362]. In this model, host type I IFN signaling was absolutely required for therapeutic efficacy and for poly A:U induced antitumor immunity. Moreover, using the same IFN-β luciferase reporter mouse, Lienenklaus and collaborators have previously revealed tissue-specific expression of IFN-β following infection with influenza or La Crosse virus, but most importantly, that IFN-β is constitutively expressed in low amounts by several tissues, including thymic epithelial cells, to maintain an activated state prepared for infection by pathogens [363].
Figure 4: Harnessing type I interferons in cancer therapy. During the last decades several strategies have been developed in order to exploit the antitumor properties of type I interferons (IFNs) in the tumor microenvironment. Indeed, diverse strategies range from the stimulation of tumor cells to produce their own IFN-α/β [e.g., inducers of immunogenic cell death, agonists of toll like receptors (TLRs) and gene therapy] or to deliver it to the cancer microenvironment, for example recombinant protein or dendritic cells (DCs) modified ex vivo. Though there is no consensus on which strategy is likely to provide the best results and much more remains to be understood concerning type I IFN’s pleiotropic functions, its combination with other treatment modalities, such as checkpoint blockade immunotherapy, is expected to unleash the full force of the immune system against cancer. Newcastle disease virus (NDV), antibody (Ab), oligodeoxynucleotide (CpG ODNs), polyribosinic-polyribocytidylic acid [Poly(I:C)].
Nevertheless, having in mind the central position of DCs in the cancer immunity cycle and the profound immunomodulation exerted by type I IFNs, even in the scenario that type IFNs are mainly impacting tumor cells, it would be reasonable to expect that antigen presenting functions of DCs are also being affected. Moreover, the dynamics and expression levels are also important factors to be considered, for example, should a tumor that already displays an ISG signature or a CD8+ T cell infiltrate be treated with the same amount of type I IFNs as compared to tumors that do not? Along the same lines, taking in to consideration the adaptive resistance mechanisms observed both in tumor and host cells, would an IFN based treatment in an inflamed tumor simply favor immunosuppression [184]? Some of the other major hurdles that must be overcome include the toxicity seen when high-dose recombinant protein is administered systemically as well as the relatively bland response encountered when IFN-α/β are applied as single agent gene therapies. We and others propose that more sophisticated ways to deliver a more localized concentration of type I IFN along with a tighter control over expression dynamics would alleviate adverse effects while still providing the desired biological effect. Gene therapy continues to be a promising method for the delivery of IFN-α/β in such manner, though we have strong evidence that the delivery of a second factor may be critical to releasing the full force of ICD. Even so, more experimentation is necessary to identify novel partners for IFN-α/β, including their pairing with chemo/radiotherapy and checkpoint blockade. Key regulators of the interplay between IFN-α/β, DCs and immune activation are still being revealed and, we propose, will continue to play an ever more critical role in cancer therapy.
ACKNOWLEDGMENTS AND FUNDING
We are grateful to Dr. Ramon Kaneno for encouraging us to write this review and for the São Paulo Research Foundation for grants (BES) 13/25167-5 and 15/26580-9; and fellowships (RFVM) 13/09474-5, (AH) 11/10656-5.
CONFLICTS OF INTEREST
The authors declare that there is no conflicts of interest regarding the publication of this article.
REFERENCES
1. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. 1957; 147:258–67.
2. Sin WX, Li P, Yeong JP, Chin KC. Activation and regulation of interferon-β in immune responses. Immunol Res. 2012; 53:25–40.
3. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005; 5:375–86.
4. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004; 202:8–32.
5. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015; 15:405–14.
6. Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, Diamond MS, Koebel CM, Arthur C, White JM, Schreiber RD. A critical function for type I interferons in cancer immunoediting. Nat Immunol. 2005; 6:722–9.
7. Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006; 6:836–48.
8. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011; 331:1565–70.
9. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011; 208:1989–2003.
10. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973; 137:1142–62.
11. Steinman RM, Kaplan G, Witmer MD, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. V. Purification of spleen dendritic cells, new surface markers, and maintenance in vitro. J Exp Med. 1979; 149:1–16.
12. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013; 39:1–10.
13. Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol. 2007; 7:19–30.
14. Veglia F, Gabrilovich DI. Dendritic cells in cancer: the role revisited. Curr Opin Immunol. 2017; 45:43–51.
15. Gardner A, Ruffell B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016; 37:855–65.
16. Belardelli F, Ferrantini M, Proietti E, Kirkwood JM. Interferon-alpha in tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002; 13:119–34.
17. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. 2014; 14:571–8.
18. Durai V, Murphy KM. Functions of Murine Dendritic Cells. Immunity. 2016; 45:719–36.
19. Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseño CG, Iwata A, Kretzer NM, Durai V, Murphy KM. Transcriptional Control of Dendritic Cell Development. Annu Rev Immunol. 2016; 34:93–119.
20. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008; 322:1097–100.
21. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, Chakarov S, Rivera C, Hogstad B, et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity. 2016; 44:924–38.
22. Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, Keller AM, Joffre O, Zelenay S, Nye E, Le Moine A, Faure F, Donckier V, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med. 2010; 207:1261–71.
23. Sluijter BJ, van den Hout MF, Koster BD, van Leeuwen PA, Schneiders FL, van de Ven R, Molenkamp BG, Vosslamber S, Verweij CL, van den Tol MP, van den Eertwegh AJ, Scheper RJ, de Gruijl TD. Arming the Melanoma Sentinel Lymph Node through Local Administration of CpG-B, GM-CSF: Recruitment and Activation of BDCA3/CD141(+) Dendritic Cells and Enhanced Cross-Presentation. Cancer Immunol Res. 2015; 3:495–505.
24. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, Krummel MF. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell. 2016; 30:324–36.
25. Cao W. Molecular characterization of human plasmacytoid dendritic cells. J Clin Immunol. 2009; 29:257–64.
26. Guiducci C, Ott G, Chan JH, Damon E, Calacsan C, Matray T, Lee KD, Coffman RL, Barrat FJ. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J Exp Med. 2006; 203:1999–2008.
27. Vermi W, Soncini M, Melocchi L, Sozzani S, Facchetti F. Plasmacytoid dendritic cells and cancer. J Leukoc Biol. 2011; 90:681–90.
28. Sozzani S, Vermi W, Del Prete A, Facchetti F. Trafficking properties of plasmacytoid dendritic cells in health and disease. Trends Immunol. 2010; 31:270–7.
29. Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. 1999; 5:919–23.
30. Saadeh D, Kurban M, Abbas O. Plasmacytoid dendritic cell role in cutaneous malignancies. J Dermatol Sci. 2016; 83:3–9.
31. Hartmann E, Wollenberg B, Rothenfusser S, Wagner M, Wellisch D, Mack B, Giese T, Gires O, Endres S, Hartmann G. Identification and functional analysis of tumor-infiltrating plasmacytoid dendritic cells in head and neck cancer. Cancer Res. 2003; 63:6478–87.
32. Le Mercier I, Poujol D, Sanlaville A, Sisirak V, Gobert M, Durand I, Dubois B, Treilleux I, Marvel J, Vlach J, Blay JY, Bendriss-Vermare N, Caux C, et al. Tumor promotion by intratumoral plasmacytoid dendritic cells is reversed by TLR7 ligand treatment. Cancer Res. 2013; 73:4629–40.
33. Cao W, Bover L. Signaling and ligand interaction of ILT7: receptor-mediated regulatory mechanisms for plasmacytoid dendritic cells. Immunol Rev. 2010; 234:163–76.
34. Segura E, Amigorena S. Inflammatory dendritic cells in mice and humans. Trends Immunol. 2013; 34:440–5.
35. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016; 37:208–20.
36. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, Amigorena S, Van't Veer LJ, Sperling AI, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 2014; 26:638–52.
37. Keirsse J, Van Damme H, Van Ginderachter JA, Laoui D. Exploiting tumor-associated dendritic cell heterogeneity for novel cancer therapies. J Leukoc Biol. 2017 Apr 7. [Epub ahead of print].
38. Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, Egeblad M, Krummel MF. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell. 2012; 21:402–17.
39. Klarquist JS, Janssen EM. Melanoma-infiltrating dendritic cells: Limitations and opportunities of mouse models. OncoImmunology. 2012; 1:1584–93.
40. Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL. Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol. 2015; 194:2985–91.
41. Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, Meinhof K, Chow A, Kim-Shulze S, et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell. 2017; 169:750–65.e17.
42. Uzé G, Schreiber G, Piehler J, Pellegrini S. The receptor of the type I interferon family. Curr Top Microbiol Immunol. 2007; 316:71–95.
43. Díaz MO, Pomykala HM, Bohlander SK, Maltepe E, Malik K, Brownstein B, Olopade OI. Structure of the human type-I interferon gene cluster determined from a YAC clone contig. Genomics. 1994; 22:540–52.
44. Pestka S. The human interferon-alpha species and hybrid proteins. Semin Oncol. 1997; 24:S9–4–S9–17.
45. Viscomi GC. Structure-activity of type I interferons. Biotherapy. 1997; 10:59–86.
46. Moraga I, Harari D, Schreiber G, Uzé G, Pellegrini S. Receptor density is key to the alpha2/beta interferon differential activities. Mol Cell Biol. 2009; 29:4778–87.
47. Jaitin DA, Roisman LC, Jaks E, Gavutis M, Piehler J, Van der Heyden J, Uze G, Schreiber G. Inquiring into the differential action of interferons (IFNs): an IFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol Cell Biol. 2006; 26:1888–97.
48. Kalie E, Jaitin DA, Podoplelova Y, Piehler J, Schreiber G. The stability of the ternary interferon-receptor complex rather than the affinity to the individual subunits dictates differential biological activities. J Biol Chem. 2008; 283:32925–36.
49. Gavutis M, Lata S, Lamken P, Müller P, Piehler J. Lateral ligand-receptor interactions on membranes probed by simultaneous fluorescence-interference detection. Biophys J. 2005; 88:4289–302.
50. Platanias LC, Uddin S, Domanski P, Colamonici OR. Differences in interferon alpha and beta signaling. Interferon beta selectively induces the interaction of the alpha and betaL subunits of the type I interferon receptor. J Biol Chem. 1996; 271:23630–3.
51. Cohen B, Novick D, Barak S, Rubinstein M. Ligand-induced association of the type I interferon receptor components. Mol Cell Biol. 1995; 15:4208–14.
52. Domanski P, Fish E, Nadeau OW, Witte M, Platanias LC, Yan H, Krolewski J, Pitha P, Colamonici OR. A region of the beta subunit of the interferon alpha receptor different from box 1 interacts with Jak1 and is sufficient to activate the Jak-Stat pathway and induce an antiviral state. J Biol Chem. 1997; 272:26388–93.
53. Stark GR. How cells respond to interferons revisited: from early history to current complexity. Cytokine Growth Factor Rev. 2007; 18:419–23.
54. Colamonici OR, Uyttendaele H, Domanski P, Yan H, Krolewski JJ. p135tyk2, an interferon-alpha-activated tyrosine kinase, is physically associated with an interferon-alpha receptor. J Biol Chem. 1994; 269:3518–22.
55. Ragimbeau J, Dondi E, Alcover A, Eid P, Uzé G, Pellegrini S. The tyrosine kinase Tyk2 controls IFNAR1 cell surface expression. EMBO J. 2003; 22:537–47.
56. Gauzzi MC, Barbieri G, Richter MF, Uzé G, Ling L, Fellous M, Pellegrini S. The amino-terminal region of Tyk2 sustains the level of interferon alpha receptor 1, a component of the interferon alpha/beta receptor. Proc Natl Acad Sci USA. 1997; 94:11839–44.
57. Schindler C, Shuai K, Prezioso VR, Darnell JE. Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science. 1992; 257:809–13.
58. Shuai K, Horvath CM, Huang LH, Qureshi SA, Cowburn D, Darnell JE. Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell. 1994; 76:821–8.
59. Mowen K, David M. Role of the STAT1-SH2 domain and STAT2 in the activation and nuclear translocation of STAT1. J Biol Chem. 1998; 273:30073–6.
60. Levy DE, Kessler DS, Pine R, Darnell JE. Cytoplasmic activation of ISGF3, the positive regulator of interferon-alpha-stimulated transcription, reconstituted in vitro. Genes Dev. 1989; 3:1362–71.
61. Levy DE, Lew DJ, Decker T, Kessler DS, Darnell JE. Synergistic interaction between interferon-alpha and interferon-gamma through induced synthesis of one subunit of the transcription factor ISGF3. EMBO J. 1990; 9:1105–11.
62. Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006; 6:644–58.
63. Novick D, Orchansky P, Revel M, Rubinstein M. The human interferon-gamma receptor. Purification, characterization, and preparation of antibodies. J Biol Chem. 1987; 262:8483–7.
64. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006; 25:349–60.
65. Durbin RK, Kotenko SV, Durbin JE. Interferon induction and function at the mucosal surface. Immunol Rev. 2013; 255:25–39.
66. van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006; 25:361–72.
67. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. Corrigendum: A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2015; 525:144.
68. Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, Hertzog PJ. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013; 41:D1040–6.
69. Jaks E, Gavutis M, Uzé G, Martal J, Piehler J. Differential receptor subunit affinities of type I interferons govern differential signal activation. J Mol Biol. 2007; 366:525–39.
70. Piehler J, Thomas C, Garcia KC, Schreiber G. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol Rev. 2012; 250:317–34.
71. Wagner TC, Velichko S, Chesney SK, Biroc S, Harde D, Vogel D, Croze E. Interferon receptor expression regulates the antiproliferative effects of interferons on cancer cells and solid tumors. Int J Cancer. 2004; 111:32–42.
72. Whitty A, Raskin N, Olson DL, Borysenko CW, Ambrose CM, Benjamin CD, Burkly LC. Interaction affinity between cytokine receptor components on the cell surface. Proc Natl Acad Sci USA. 1998; 95:13165–70.
73. Chin L, Pomerantz J, DePinho RA. The INK4a/ARF tumor suppressor: one gene—two products—two pathways. Trends Biochem Sci. 1998; 23:291–6.
74. Diaz MO, Ziemin S, Le Beau MM, Pitha P, Smith SD, Chilcote RR, Rowley JD. Homozygous deletion of the alpha- and beta 1-interferon genes in human leukemia and derived cell lines. Proc Natl Acad Sci USA. 1988; 85:5259–63.
75. Neubauer A, Neubauer B, Liu E. Polymerase chain reaction based assay to detect allelic loss in human DNA: loss of beta-interferon gene in chronic myelogenous leukemia. Nucleic Acids Res. 1990; 18:993–8.
76. Einhorn S, Grander D, Bjork O, Brondum-Nielsen K, Soderhall S. Deletion of alpha-, beta-, and omega-interferon genes in malignant cells from children with acute lymphocytic leukemia. Cancer Res. 1990; 50:7781–5.
77. Grander D, Heyman M, Brondum-Nielsen K, Liu Y, Lundgren E, Soderhall S, Einhorn S. Interferon system in primary acute lymphocytic leukemia cells with or without deletions of the alpha-/beta-interferon genes. Blood. 1992; 79:2076–83.
78. Heyman M, Grander D, Brondum-Nielsen K, Liu Y, Soderhall S, Einhorn S. Deletions of the short arm of chromosome 9, including the interferon-alpha/-beta genes, in acute lymphocytic leukemia. Studies on loss of heterozygosity, parental origin of deleted genes and prognosis. Int J Cancer. 1993; 54:748–53.
79. Colamonici OR, Domanski P, Platanias LC, Diaz MO. Correlation between interferon (IFN) alpha resistance and deletion of the IFN alpha/beta genes in acute leukemia cell lines suggests selection against the IFN system. Blood. 1992; 80:744–9.
80. Aguiar RC, Sill H, Goldman JM, Cross NC. The commonly deleted region at 9p21-22 in lymphoblastic leukemias spans at least 400 kb and includes p16 but not p15 or the IFN gene cluster. Leukemia. 1997; 11:233–8.
81. Wood A, Angus B, Kestevan P, Dark J, Notarianni G, Miller S, Howard M, Proctor S, Middleton P. Alpha interferon gene deletions in post-transplant lymphoma. Br J Haematol. 1997; 98:1002–3.
82. Lydiatt WM, Davidson BJ, Schantz SP, Caruana S, Chaganti RS. 9p21 deletion correlates with recurrence in head and neck cancer. Head Neck. 1998; 20:113–8.
83. Sugawa N, Ekstrand AJ, Ueda S, Collins VP. Frequency of IFN beta 1 gene loss in 47 primary human gliomas. Noshuyo Byori. 1993; 10:161–3.
84. James CD, He J, Collins VP, Allalunis-Turner MJ, Day RS 3rd. Localization of chromosome 9p homozygous deletions in glioma cell lines with markers constituting a continuous linkage group. Cancer Res. 1993; 53:3674–6.
85. Coleman A, Fountain JW, Nobori T, Olopade OI, Robertson G, Housman DE, Lugo TG. Distinct deletions of chromosome 9p associated with melanoma versus glioma, lung cancer, and leukemia. Cancer Res. 1994; 54:344–8.
86. Fountain JW, Karayiorgou M, Ernstoff MS, Kirkwood JM, Vlock DR, Titus-Ernstoff L, Bouchard B, Vijayasaradhi S, Houghton AN, Lahti J. Homozygous deletions within human chromosome band 9p21 in melanoma. Proc Natl Acad Sci USA. 1992; 89:10557–61.
87. Heyman M, Grander D, Brondum-Nielsen K, Cederblad B, Liu Y, Xu B, Einhorn S. Interferon system defects in malignant T-cells. Leukemia. 1994; 8:425–34.
88. Zhang H, Chen ZH, Savarese TM. Codeletion of the genes for p16INK4, methylthioadenosine phosphorylase, interferon-alpha1, interferon-beta1, and other 9p21 markers in human malignant cell lines. Cancer Genet Cytogenet. 1996; 86:22–8.
89. Chen ZH, Zhang H, Savarese TM. Gene deletion chemoselectivity: codeletion of the genes for p16(INK4), methylthioadenosine phosphorylase, and the alpha- and beta-interferons in human pancreatic cell carcinoma lines and its implications for chemotherapy. Cancer Res. 1996; 56:1083–90.
90. Romero-Weaver AL, Wang HW, Steen HC, Scarzello AJ, Hall VL, Sheikh F, Donnelly RP, Gamero AM. Resistance to IFN-alpha-induced apoptosis is linked to a loss of STAT2. Mol Cancer Res. 2010; 8:80–92.
91. Meyer S, Wild PJ, Vogt T, Bataille F, Ehret C, Gantner S, Landthaler M, Klinkhammer-Schalke M, Hofstaedter F, Bosserhoff AK. Methylthioadenosine phosphorylase represents a predictive marker for response to adjuvant interferon therapy in patients with malignant melanoma. Exp Dermatol. 2010; 19:e251–7.
92. Barnard P, Payne E, McMillan NA. The human papillomavirus E7 protein is able to inhibit the antiviral and anti-growth functions of interferon-alpha. Virology. 2000; 277:411–9.
93. Beglin M, Melar-New M, Laimins L. Human papillomaviruses and the interferon response. J Interferon Cytokine Res. 2009; 29:629–35.
94. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009; 461:788–92.
95. Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Reports. 2016; 14:282–97.
96. Xia T, Konno H, Barber GN. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016; 76:6747–59.
97. Snell LM, McGaha TL, Brooks DG. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017; 38:542–557.
98. Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, Carbone CJ, Beiting DP, Girondo MA, Peck AR, Pure E, Chatterji P, Rustgi AK, et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell. 2017; 31:194–207.
99. Yano H, Iemura A, Haramaki M, Ogasawara S, Takayama A, Akiba J, Kojiro M. Interferon alfa receptor expression and growth inhibition by interferon alfa in human liver cancer cell lines. Hepatology. 1999; 29:1708–17.
100. Yano H, Ogasawara S, Momosaki S, Akiba J, Kojiro S, Fukahori S, Ishizaki H, Kuratomi K, Basaki Y, Oie S, Kuwano M, Kojiro M. Growth inhibitory effects of pegylated IFN alpha-2b on human liver cancer cells in vitro and in vivo. Liver Int. 2006; 26:964–75.
101. Murphy D, Detjen KM, Welzel M, Wiedenmann B, Rosewicz S. Interferon-alpha delays S-phase progression in human hepatocellular carcinoma cells via inhibition of specific cyclin-dependent kinases. Hepatology. 2001; 33:346–56.
102. Murata M, Nabeshima S, Kikuchi K, Yamaji K, Furusyo N, Hayashi J. A comparison of the antitumor effects of interferon-alpha and beta on human hepatocellular carcinoma cell lines. Cytokine. 2006; 33:121–8.
103. Chawla-Sarkar M, Leaman DW, Borden EC. Preferential induction of apoptosis by interferon (IFN)-beta compared with IFN-alpha2: correlation with TRAIL/Apo2L induction in melanoma cell lines. Clin Cancer Res. 2001; 7:1821–31.
104. Leaman DW, Chawla-Sarkar M, Jacobs B, Vyas K, Sun Y, Ozdemir A, Yi T, Williams BR, Borden EC. Novel growth and death related interferon-stimulated genes (ISGs) in melanoma: greater potency of IFN-beta compared with IFN-alpha2. J Interferon Cytokine Res. 2003; 23:745–56.
105. Roh MR, Zheng Z, Kim HS, Jeung HC, Rha SY, Chung KY. Difference of interferon-α and interferon-β on melanoma growth and lymph node metastasis in mice. Melanoma Res. 2013; 23:114–24.
106. Chen Q, Gong B, Mahmoud-Ahmed AS, Zhou A, Hsi ED, Hussein M, Almasan A. Apo2L/TRAIL and Bcl-2-related proteins regulate type I interferon-induced apoptosis in multiple myeloma. Blood. 2001; 98:2183–92.
107. Gong B, Almasan A. Genomic organization and transcriptional regulation of human Apo2/TRAIL gene. Biochem Biophys Res Commun. 2000; 278:747–52.
108. Rozera C, Carlei D, Lollini PL, De Giovanni C, Musiani P, Di Carlo E, Belardelli F, Ferrantini M. Interferon (IFN)-beta gene transfer into TS/A adenocarcinoma cells and comparison with IFN-alpha: differential effects on tumorigenicity and host response. Am J Pathol. 1999; 154:1211–22.
109. Folkman J, Klagsbrun M. Angiogenic factors. Science. 1987; 235:442–7.
110. Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990; 82:4–6.
111. Fidler IJ, Yano S, Zhang RD, Fujimaki T, Bucana CD. The seed and soil hypothesis: vascularisation and brain metastases. Lancet Oncol. 2002; 3:53–7.
112. Sidky YA, Borden EC. Inhibition of angiogenesis by interferons: effects on tumor- and lymphocyte-induced vascular responses. Cancer Res. 1987; 47:5155–61.
113. Dvorak HF, Gresser I. Microvascular injury in pathogenesis of interferon-induced necrosis of subcutaneous tumors in mice. J Natl Cancer Inst. 1989; 81:497–502.
114. Singh RK, Gutman M, Bucana CD, Sanchez R, Llansa N, Fidler IJ. Interferons alpha and beta down-regulate the expression of basic fibroblast growth factor in human carcinomas. Proc Natl Acad Sci USA. 1995; 92:4562–6.
115. Dinney CP, Bielenberg DR, Perrotte P, Reich R, Eve BY, Bucana CD, Fidler IJ. Inhibition of basic fibroblast growth factor expression, angiogenesis, and growth of human bladder carcinoma in mice by systemic interferon-alpha administration. Cancer Res. 1998; 58:808–14.
116. Huang S, Bucana CD, Van Arsdall M, Fidler IJ. Stat1 negatively regulates angiogenesis, tumorigenicity and metastasis of tumor cells. Oncogene. 2002; 21:2504–12.
117. von Marschall Z, Scholz A, Cramer T, Schäfer G, Schirner M, Oberg K, Wiedenmann B, Höcker M, Rosewicz S. Effects of interferon alpha on vascular endothelial growth factor gene transcription and tumor angiogenesis. J Natl Cancer Inst. 2003; 95:437–48.
118. Rosewicz S, Detjen K, Scholz A, von Marschall Z. Interferon-alpha: regulatory effects on cell cycle and angiogenesis. Neuroendocrinology. 2004; 80:85–93.
119. Singh RK, Gutman M, Llansa N, Fidler IJ. Interferon-beta prevents the upregulation of interleukin-8 expression in human melanoma cells. J Interferon Cytokine Res. 1996; 16:577–84.
120. Reznikov LL, Puren AJ, Fantuzzi G, Mühl H, Shapiro L, Yoon DY, Cutler DL, Dinarello CA. Spontaneous and inducible cytokine responses in healthy humans receiving a single dose of IFN-alpha2b: increased production of interleukin-1 receptor antagonist and suppression of IL-1-induced IL-8. J Interferon Cytokine Res. 1998; 18:897–903.
121. McCarty MF, Bielenberg D, Donawho C, Bucana CD, Fidler IJ. Evidence for the causal role of endogenous interferon-alpha/beta in the regulation of angiogenesis, tumorigenicity, and metastasis of cutaneous neoplasms. Clin Exp Metastasis. 2002; 19:609–15.
122. Cheng X, Liu Y, Chu H, Kao HY. Promyelocytic leukemia protein (PML) regulates endothelial cell network formation and migration in response to tumor necrosis factor α (TNFα) and interferon α (IFNα). J Biol Chem. 2012; 287:23356–67.
123. Terris B, Baldin V, Dubois S, Degott C, Flejou JF, Hénin D, Dejean A. PML nuclear bodies are general targets for inflammation and cell proliferation. Cancer Res. 1995; 55:1590–7.
124. Stadler M, Chelbi-Alix MK, Koken MH, Venturini L, Lee C, Saïb A, Quignon F, Pelicano L, Guillemin MC, Schindler C. Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene. 1995; 11:2565–73.
125. Caporali A, Pani E, Horrevoets AJ, Kraenkel N, Oikawa A, Sala-Newby GB, Meloni M, Cristofaro B, Graiani G, Leroyer AS, Boulanger CM, Spinetti G, Yoon SO, et al. Neurotrophin p75 receptor (p75NTR) promotes endothelial cell apoptosis and inhibits angiogenesis: implications for diabetes-induced impaired neovascularization in ischemic limb muscles. Circ Res. 2008; 103:e15–26.
126. Reineke EL, Liu Y, Kao HY. Promyelocytic leukemia protein controls cell migration in response to hydrogen peroxide and insulin-like growth factor-1. J Biol Chem. 2010; 285:9485–92.
127. Spaapen RM, Leung MY, Fuertes MB, Kline JP, Zhang L, Zheng Y, Fu YX, Luo X, Cohen KS, Gajewski TF. Therapeutic activity of high-dose intratumoral IFN-β requires direct effect on the tumor vasculature. J Immunol. 2014; 193:4254–60.
128. Dickson PV, Hamner JB, Streck CJ, Ng CY, McCarville MB, Calabrese C, Gilbertson RJ, Stewart CF, Wilson CM, Gaber MW, Pfeffer LM, Skapek SX, Nathwani AC, et al. Continuous delivery of IFN-beta promotes sustained maturation of intratumoral vasculature. Mol Cancer Res. 2007; 5:531–42.
129. Cao G, Su J, Lu W, Zhang F, Zhao G, Marteralli D, Dong Z. Adenovirus-mediated interferon-beta gene therapy suppresses growth and metastasis of human prostate cancer in nude mice. Cancer Gene Ther. 2001; 8:497–505.
130. Ma Z, Qin H, Benveniste EN. Transcriptional suppression of matrix metalloproteinase-9 gene expression by IFN-gamma and IFN-beta: critical role of STAT-1alpha. J Immunol. 2001; 167:5150–9.
131. Nelissen I, Ronsse I, Van Damme J, Opdenakker G. Regulation of gelatinase B in human monocytic and endothelial cells by PECAM-1 ligation and its modulation by interferon-beta. J Leukoc Biol. 2002; 71:89–98.
132. Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest. 2010; 120:1151–64.
133. Izawa JI, Sweeney P, Perrotte P, Kedar D, Dong Z, Slaton JW, Karashima T, Inoue K, Benedict WF, Dinney CP. Inhibition of tumorigenicity and metastasis of human bladder cancer growing in athymic mice by interferon-beta gene therapy results partially from various antiangiogenic effects including endothelial cell apoptosis. Clin Cancer Res. 2002; 8:1258–70.
134. Park JA, Joe YA, Kim TG, Hong YK. Potentiation of antiglioma effect with combined temozolomide and interferon-beta. Oncol Rep. 2006; 16:1253–60.
135. Taylor KL, Leaman DW, Grane R, Mechti N, Borden EC, Lindner DJ. Identification of interferon-beta-stimulated genes that inhibit angiogenesis in vitro. J Interferon Cytokine Res. 2008; 28:733–40.
136. Erdmann J, Vitale G, van Koetsveld PM, Croze E, Sprij-Mooij DM, Hofland LJ, van Eijck CH. Effects of interferons α/β on the proliferation of human micro- and macrovascular endothelial cells. J Interferon Cytokine Res. 2011; 31:451–8.
137. Zheng H, Qian J, Carbone CJ, Leu NA, Baker DP, Fuchs SY. Vascular endothelial growth factor-induced elimination of the type 1 interferon receptor is required for efficient angiogenesis. Blood. 2011; 118:4003–6.
138. Albini A, Marchisone C, Del Grosso F, Benelli R, Masiello L, Tacchetti C, Bono M, Ferrantini M, Rozera C, Truini M, Belardelli F, Santi L, Noonan DM. Inhibition of angiogenesis and vascular tumor growth by interferon-producing cells: A gene therapy approach. Am J Pathol. 2000; 156:1381–93.
139. Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998; 67:227–64.
140. Heil M, Land WG. Danger signals - damaged-self recognition across the tree of life. Front Plant Sci. 2014; 5: 578.
141. Le Bon A, Tough DF. Links between innate and adaptive immunity via type I interferon. Curr Opin Immunol. 2002; 14:432–6.
142. Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, Tomasello F, Gambuzza M, Macrì G, Ruggeri A, Leanderson T, Teti G. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol. 2007; 178:3126–33.
143. Siveen KS, Kuttan G. Role of macrophages in tumour progression. Immunol Lett. 2009; 123:97–102.
144. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016; 16:131–44.
145. Ribas A. Adaptive Immune Resistance: How Cancer Protects from Immune Attack. Cancer Discov. 2015; 5:915–9.
146. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015; 15:87–103.
147. Shaabani N, Duhan V, Khairnar V, Gassa A, Ferrer-Tur R, Häussinger D, Recher M, Zelinskyy G, Liu J, Dittmer U, Trilling M, Scheu S, Hardt C, et al. CD169(+) macrophages regulate PD-L1 expression via type I interferon and thereby prevent severe immunopathology after LCMV infection. Cell Death Dis. 2016; 7:e2446.
148. Martinez-Pomares L, Gordon S. CD169+ macrophages at the crossroads of antigen presentation. Trends Immunol. 2012; 33:66–70.
149. Kim HR, Ha SJ, Hong MH, Heo SJ, Koh YW, Choi EC, Kim EK, Pyo KH, Jung I, Seo D, Choi J, Cho BC, Yoon SO. PD-L1 expression on immune cells, but not on tumor cells, is a favorable prognostic factor for head and neck cancer patients. Sci Rep. 2016; 6:36956.
150. Noguchi T, Ward JP, Gubin MM, Arthur CD, Lee SH, Hundal J, Selby MJ, Graziano RF, Mardis ER, Korman AJ, Schreiber RD. Temporally Distinct PD-L1 Expression by Tumor and Host Cells Contributes to Immune Escape. Cancer Immunol Res. 2017; 5:106–17.
151. Brinkmann V, Geiger T, Alkan S, Heusser CH. Interferon alpha increases the frequency of interferon gamma-producing human CD4+ T cells. J Exp Med. 1993; 178:1655–63.
152. Hunter CA, Gabriel KE, Radzanowski T, Neyer LE, Remington JS. Type I interferons enhance production of IFN-gamma by NK cells. Immunol Lett. 1997; 59:1–5.
153. Gessani S, Conti L, Del Cornò M, Belardelli F. Type I interferons as regulators of human antigen presenting cell functions. Toxins (Basel). 2014; 6:1696–723.
154. Parlato S, Romagnoli G, Spadaro F, Canini I, Sirabella P, Borghi P, Ramoni C, Filesi I, Biocca S, Gabriele L, Belardelli F. LOX-1 as a natural IFN-alpha-mediated signal for apoptotic cell uptake and antigen presentation in dendritic cells. Blood. 2010; 115:1554–63.
155. Parlato S, Santini SM, Lapenta C, Di Pucchio T, Logozzi M, Spada M, Giammarioli AM, Malorni W, Fais S, Belardelli F. Expression of CCR-7, MIP-3beta, and Th-1 chemokines in type I IFN-induced monocyte-derived dendritic cells: importance for the rapid acquisition of potent migratory and functional activities. Blood. 2001; 98:3022–9.
156. Zelenay S, Keller AM, Whitney PG, Schraml BU, Deddouche S, Rogers NC, Schulz O, Sancho D, Reis e Sousa C. The dendritic cell receptor DNGR-1 controls endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J Clin Invest. 2012; 122:1615–27.
157. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013; 14:1014–22.
158. Sisirak V, Faget J, Gobert M, Goutagny N, Vey N, Treilleux I, Renaudineau S, Poyet G, Labidi-Galy SI, Goddard-Leon S, Durand I, Le Mercier I, Bajard A, et al. Impaired IFN-α production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. 2012; 72:5188–97.
159. Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, Messina JL, Chandler P, Koni PA, Mellor AL. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. 2004; 114:280–90.
160. Lippens C, Duraes FV, Dubrot J, Brighouse D, Lacroix M, Irla M, Aubry-Lachainaye JP, Reith W, Mandl JN, Hugues S. IDO-orchestrated crosstalk between pDCs and Tregs inhibits autoimmunity. J Autoimmun. 2016; 75:39–49.
161. Harden JL, Egilmez NK. Indoleamine 2,3-dioxygenase and dendritic cell tolerogenicity. Immunol Invest. 2012; 41: 738–64.
162. Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, Muller AJ. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. 2014; 63:721–35.
163. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013; 210:1389–402.
164. Puccetti P. On watching the watchers: IDO and type I/II IFN. Eur J Immunol. 2007; 37:876–9.
165. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000; 100:655–69.
166. Huber JP, Farrar JD. Regulation of effector and memory T-cell functions by type I interferon. Immunology. 2011; 132:466–74.
167. Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005; 54:721–8.
168. Caspi RR. Immunotherapy of autoimmunity and cancer: the penalty for success. Nat Rev Immunol. 2008; 8:970–6.
169. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012; 12:298–306.
170. Schiavoni G, Mattei F, Gabriele L. Type I Interferons as Stimulators of DC-Mediated Cross-Priming: Impact on Anti-Tumor Response. Front Immunol. 2013; 4:483.
171. Greiner JW, Hand PH, Noguchi P, Fisher PB, Pestka S, Schlom J. Enhanced expression of surface tumor-associated antigens on human breast and colon tumor cells after recombinant human leukocyte alpha-interferon treatment. Cancer Res. 1984; 44:3208–14.
172. Yang I, Kremen TJ, Giovannone AJ, Paik E, Odesa SK, Prins RM, Liau LM. Modulation of major histocompatibility complex Class I molecules and major histocompatibility complex-bound immunogenic peptides induced by interferon-alpha and interferon-gamma treatment of human glioblastoma multiforme. J Neurosurg. 2004; 100:310–9.
173. Fruci D, Benevolo M, Cifaldi L, Lorenzi S, Lo Monaco E, Tremante E, Giacomini P. Major histocompatibility complex class i and tumour immuno-evasion: how to fool T cells and natural killer cells at one time. Curr Oncol. 2012; 19:39–41.
174. Wang X, Schoenhals JE, Li A, Valdecanas DR, Ye H, Zang F, Tang C, Tang M, Liu CG, Liu X, Krishnan S, Allison JP, Sharma P, et al. Suppression of Type I IFN Signaling in Tumors Mediates Resistance to Anti-PD-1 Treatment That Can Be Overcome by Radiotherapy. Cancer Res. 2017; 77:839–50.
175. Iannello A, Raulet DH. Immune surveillance of unhealthy cells by natural killer cells. Cold Spring Harb Symp Quant Biol. 2013; 78:249–57.
176. O'Sullivan T, Saddawi-Konefka R, Vermi W, Koebel CM, Arthur C, White JM, Uppaluri R, Andrews DM, Ngiow SF, Teng MW, Smyth MJ, Schreiber RD, Bui JD. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J Exp Med. 2012; 209:1869–82.
177. Crouse J, Bedenikovic G, Wiesel M, Ibberson M, Xenarios I, Von Laer D, Kalinke U, Vivier E, Jonjic S, Oxenius A. Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity. 2014; 40:961–73.
178. Schuster IS, Coudert JD, Andoniou CE, Degli-Esposti MA. "Natural Regulators": NK Cells as Modulators of T Cell Immunity. Front Immunol. 2016; 7:235.
179. Lee SE, Li X, Kim JC, Lee J, Gonzalez-Navajas JM, Hong SH, Park IK, Rhee JH, Raz E. Type I interferons maintain Foxp3 expression and T-regulatory cell functions under inflammatory conditions in mice. Gastroenterology. 2012; 143:145–54.
180. Wang Y, Swiecki M, Cella M, Alber G, Schreiber RD, Gilfillan S, Colonna M. Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe. 2012; 11:631–42.
181. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013; 340:202–7.
182. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, Schreiber RD, de la Torre JC, Oldstone MB. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013; 340:207–11.
183. Odorizzi PM, Wherry EJ. Immunology. An interferon paradox. Science. 2013; 340:155–6.
184. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DS, Pauken KE, Huang AC, Gangadhar TC, Amaravadi RK, Schuchter LM, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell. 2016; 167:1540–54.e12.
185. Strauss BE, Costanzi-Strauss E. Gene Therapy for Melanoma: Progress and Perspectives. Recent Advances in the Biology, Therapy and Management of Melanoma. InTech. 2013.
186. Weise AM, Flaherty LE. New options for the adjuvant treatment of cutaneous melanoma? Curr Oncol Rep. 2014; 16: 409.
187. Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 1996; 14:7–17.
188. Kirkwood JM, Ibrahim JG, Sosman JA, Sondak VK, Agarwala SS, Ernstoff MS, Rao U. High-dose interferon alfa-2b significantly prolongs relapse-free and overall survival compared with the GM2-KLH/QS-21 vaccine in patients with resected stage IIB-III melanoma: results of intergroup trial E1694/S9512/C509801. J Clin Oncol. 2001; 19:2370–80.
189. Daud A, Soon C, Dummer R, Eggermont AM, Hwu WJ, Grob JJ, Garbe C, Hauschild A. Management of pegylated interferon alpha toxicity in adjuvant therapy of melanoma. Expert Opin Biol Ther. 2012; 12:1087–99.
190. Kaehler KC, Sondak VK, Schadendorf D, Hauschild A. Pegylated interferons: prospects for the use in the adjuvant and palliative therapy of metastatic melanoma. Eur J Cancer. 2010; 46:41–6.
191. Aoyagi S, Hata H, Homma E, Shimizu H. Sequential local injection of low-dose interferon-beta for maintenance therapy in stage II and III melanoma: a single-institution matched case-control study. Oncology. 2012; 82:139–46.
192. Di Trolio R, Simeone E, Di Lorenzo G, Buonerba C, Ascierto PA. The use of interferon in melanoma patients: a systematic review. Cytokine Growth Factor Rev. 2015; 26:203–12.
193. Moschos SJ, Edington HD, Land SR, Rao UN, Jukic D, Shipe-Spotloe J, Kirkwood JM. Neoadjuvant treatment of regional stage IIIB melanoma with high-dose interferon alfa-2b induces objective tumor regression in association with modulation of tumor infiltrating host cellular immune responses. J Clin Oncol. 2006; 24:3164–71.
194. Borden EC, Jacobs B, Hollovary E, Rybicki L, Elson P, Olencki T, Triozzi P. Gene regulatory and clinical effects of interferon beta in patients with metastatic melanoma: a phase II trial. J Interferon Cytokine Res. 2011; 31:433–40.
195. Voelter-Mahlknecht S, Mahlknecht U, Letzel S, Fierlbeck G. Phase 2 trial of the continuous IV administration of interferon-beta in patients with disseminated malignant melanoma. Skinmed. 2006; 5:271–6.
196. McMasters KM, Egger ME, Edwards MJ, Ross MI, Reintgen DS, Noyes RD, Martin RC, Goydos JS, Beitsch PD, Urist MM, Ariyan S, Sussman JJ, Davidson BS, et al. Final Results of the Sunbelt Melanoma Trial: A Multi-Institutional Prospective Randomized Phase III Study Evaluating the Role of Adjuvant High-Dose Interferon Alfa-2b and Completion Lymph Node Dissection for Patients Staged by Sentinel Lymph Node Biopsy. J Clin Oncol. 2016; 34:1079–86.
197. Eggermont AM, Suciu S, Rutkowski P, Kruit WH, Punt CJ, Dummer R, Salès F, Keilholz U, de Schaetzen G, Testori A, Group EM. Long term follow up of the EORTC 18952 trial of adjuvant therapy in resected stage IIB-III cutaneous melanoma patients comparing intermediate doses of interferon-alpha-2b (IFN) with observation: Ulceration of primary is key determinant for IFN-sensitivity. Eur J Cancer. 2016; 55:111–21.
198. Mohr P, Hauschild A, Trefzer U, Enk A, Tilgen W, Loquai C, Gogas H, Haalck T, Koller J, Dummer R, Gutzmer R, Brockmeyer N, Holzle E, et al. Intermittent High-Dose Intravenous Interferon Alfa-2b for Adjuvant Treatment of Stage III Melanoma: Final Analysis of a Randomized Phase III Dermatologic Cooperative Oncology Group Trial. J Clin Oncol. 2015; 33:4077–84.
199. Tarhini AA, Thalanayar PM. Melanoma adjuvant therapy. Hematol Oncol Clin North Am. 2014; 28:471–89.
200. Gogas H, Abali H, Ascierto PA, Demidov L, Pehamberger H, Robert C, Schachter J, Eggermont AM, Hauschild A, Espinosa E. Who benefits most from adjuvant interferon treatment for melanoma? Am J Ther. 2015; 22:54–60.
201. Minutilli E, Feliciani C. Adjuvant therapy for resected stage III melanoma patients: high-dose interferon-alpha versus ipilimumab combined with kinases inhibitors. Tumori. 2012; 98:185–90.
202. DiPaola RS, Chen YH, Stein M, Vaughn D, Patrick-Miller L, Carducci M, Roth B, White E, Wilding G. A randomized phase II trial of mitoxantrone, estramustine and vinorelbine or bcl-2 modulation with 13-cis retinoic acid, interferon and paclitaxel in patients with metastatic castrate-resistant prostate cancer: ECOG 3899. J Transl Med. 2010; 8: 20.
203. Kiladjian JJ, Giraudier S, Cassinat B. Interferon-alpha for the therapy of myeloproliferative neoplasms: targeting the malignant clone. Leukemia. 2016; 30:776–81.
204. Banday AH, Jeelani S, Hruby VJ. Cancer vaccine adjuvants—recent clinical progress and future perspectives. Immunopharmacol Immunotoxicol. 2015; 37:1–11.
205. Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunol Rev. 2008; 222:357–68.
206. Sato Y, Goto Y, Narita N, Hoon DS. Cancer Cells Expressing Toll-like Receptors and the Tumor Microenvironment. Cancer Microenviron. 2009; 2:205–14.
207. Heinz S, Haehnel V, Karaghiosoff M, Schwarzfischer L, Müller M, Krause SW, Rehli M. Species-specific regulation of Toll-like receptor 3 genes in men and mice. J Biol Chem. 2003; 278:21502–9.
208. Muzio M, Bosisio D, Polentarutti N, D'amico G, Stoppacciaro A, Mancinelli R, van't Veer C, Penton-Rol G, Ruco LP, Allavena P, Mantovani A. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J Immunol. 2000; 164:5998–6004.
209. Levy HB, Baer G, Baron S, Buckler CE, Gibbs CJ, Iadarola MJ, London WT, Rice J. A modified polyriboinosinic-polyribocytidylic acid complex that induces interferon in primates. J Infect Dis. 1975; 132:434–9.
210. Perales-Linares R, Navas-Martin S. Toll-like receptor 3 in viral pathogenesis: friend or foe? Immunology. 2013; 140:153–67.
211. Benwell RK, Hruska JE, Fritsche KL, Lee DR. Double stranded RNA- relative to other TLR ligand-activated dendritic cells induce extremely polarized human Th1 responses. Cell Immunol. 2010; 264:119–26.
212. Salem ML, Diaz-Montero CM, El-Naggar SA, Chen Y, Moussa O, Cole DJ. The TLR3 agonist poly(I:C) targets CD8+ T cells and augments their antigen-specific responses upon their adoptive transfer into naïve recipient mice. Vaccine. 2009; 27:549–57.
213. Huang YK, Zheng Z, Qiu F. Polyinosinic-cytidylic acid as an adjuvant on natural killer- and dendritic cell-mediated antitumor activities. Tumour Biol. 2013; 34:1615–23.
214. McCartney S, Vermi W, Gilfillan S, Cella M, Murphy TL, Schreiber RD, Murphy KM, Colonna M. Distinct and complementary functions of MDA5 and TLR3 in poly(I:C)-mediated activation of mouse NK cells. J Exp Med. 2009; 206:2967–76.
215. Cheng YS, Xu F. Anticancer function of polyinosinic-polycytidylic acid. Cancer Biol Ther. 2010; 10:1219–23.
216. Caskey M, Lefebvre F, Filali-Mouhim A, Cameron MJ, Goulet JP, Haddad EK, Breton G, Trumpfheller C, Pollak S, Shimeliovich I, Duque-Alarcon A, Pan L, Nelkenbaum A, et al. Synthetic double-stranded RNA induces innate immune responses similar to a live viral vaccine in humans. J Exp Med. 2011; 208:2357–66.
217. Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, Nelson SF, Liau LM. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011; 17:1603–15.
218. Sabbatini P, Tsuji T, Ferran L, Ritter E, Sedrak C, Tuballes K, Jungbluth AA, Ritter G, Aghajanian C, Bell-McGuinn K, Hensley ML, Konner J, Tew W, et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin Cancer Res. 2012; 18:6497–508.
219. Da Silva DM, Woodham AW, Skeate JG, Rijkee LK, Taylor JR, Brand HE, Muderspach LI, Roman LD, Yessaian AA, Pham HQ, Matsuo K, Lin YG, McKee GM, et al. Langerhans cells from women with cervical precancerous lesions become functionally responsive against human papillomavirus after activation with stabilized Poly-I:C. Clin Immunol. 2015; 161:197–208.
220. Thompson KA, Strayer DR, Salvato PD, Thompson CE, Klimas N, Molavi A, Hamill AK, Zheng Z, Ventura D, Carter WA. Results of a double-blind placebo-controlled study of the double-stranded RNA drug polyI:polyC12U in the treatment of HIV infection. Eur J Clin Microbiol Infect Dis. 1996; 15:580–7.
221. Navabi H, Jasani B, Reece A, Clayton A, Tabi Z, Donninger C, Mason M, Adams M. A clinical grade poly I:C-analogue (Ampligen) promotes optimal DC maturation and Th1-type T cell responses of healthy donors and cancer patients in vitro. Vaccine. 2009; 27:107–15.
222. Kagan JC, Barton GM. Emerging principles governing signal transduction by pattern-recognition receptors. Cold Spring Harb Perspect Biol. 2014; 7:a016253.
223. Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010; 465:885–90.
224. Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006; 13:816–25.
225. Tan Y, Zanoni I, Cullen TW, Goodman AL, Kagan JC. Mechanisms of Toll-like Receptor 4 Endocytosis Reveal a Common Immune-Evasion Strategy Used by Pathogenic and Commensal Bacteria. Immunity. 2015; 43:909–22.
226. Ueda Y, Itoh T, Fuji N, Harada S, Fujiki H, Shimizu K, Shiozaki A, Iwamoto A, Shimizu T, Mazda O, Kimura T, Sonoda Y, Taniwaki M, et al. Successful induction of clinically competent dendritic cells from granulocyte colony-stimulating factor-mobilized monocytes for cancer vaccine therapy. Cancer Immunol Immunother. 2007; 56:381–9.
227. Dohnal AM, Witt V, Hügel H, Holter W, Gadner H, Felzmann T. Phase I study of tumor Ag-loaded IL-12 secreting semi-mature DC for the treatment of pediatric cancer. Cytotherapy. 2007; 9:755–70.
228. Czerniecki BJ, Koski GK, Koldovsky U, Xu S, Cohen PA, Mick R, Nisenbaum H, Pasha T, Xu M, Fox KR, Weinstein S, Orel SG, Vonderheide R, et al. Targeting HER-2/neu in early breast cancer development using dendritic cells with staged interleukin-12 burst secretion. Cancer Res. 2007; 67:1842–52.
229. Chiang CL, Kandalaft LE, Tanyi J, Hagemann AR, Motz GT, Svoronos N, Montone K, Mantia-Smaldone GM, Smith L, Nisenbaum HL, Levine BL, Kalos M, Czerniecki BJ, et al. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: from bench to bedside. Clin Cancer Res. 2013; 19:4801–15.
230. Chiang CL, Ledermann JA, Aitkens E, Benjamin E, Katz DR, Chain BM. Oxidation of ovarian epithelial cancer cells by hypochlorous acid enhances immunogenicity and stimulates T cells that recognize autologous primary tumor. Clin Cancer Res. 2008; 14:4898–907.
231. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003; 299:1033–6.
232. Lee MK, Xu S, Fitzpatrick EH, Sharma A, Graves HL, Czerniecki BJ. Inhibition of CD4+CD25+ regulatory T cell function and conversion into Th1-like effectors by a Toll-like receptor-activated dendritic cell vaccine. PLoS One. 2013; 8:e74698.
233. Paavonen J, Naud P, Salmerón J, Wheeler CM, Chow SN, Apter D, Kitchener H, Castellsague X, Teixeira JC, Skinner SR, Hedrick J, Jaisamrarn U, Limson G, et al. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): final analysis of a double-blind, randomised study in young women. Lancet. 2009; 374:301–14.
234. Gantier MP, Tong S, Behlke MA, Xu D, Phipps S, Foster PS, Williams BR. TLR7 is involved in sequence-specific sensing of single-stranded RNAs in human macrophages. J Immunol. 2008; 180:2117–24.
235. Alexopoulou L, Desnues B, Demaria O. [Toll-like receptor 8: the awkward TLR]. [Article in French]. Med Sci (Paris). 2012; 28:96–102.
236. Serra-Guillén C, Nagore E, Hueso L, Traves V, Messeguer F, Sanmartín O, Llombart B, Requena C, Botella-Estrada R, Guillén C. A randomized pilot comparative study of topical methyl aminolevulinate photodynamic therapy versus imiquimod 5% versus sequential application of both therapies in immunocompetent patients with actinic keratosis: clinical and histologic outcomes. J Am Acad Dermatol. 2012; 66:e131–7.
237. Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006; 7:131–7.
238. Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, Endres S, Hartmann G. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. 2005; 11:263–70.
239. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010; 11:373–84.
240. Hochrein H, O'Keeffe M, Wagner H. Human and mouse plasmacytoid dendritic cells. Hum Immunol. 2002; 63:1103–10.
241. Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002; 3:196–200.
242. Hellman P, Eriksson H. Early activation markers of human peripheral dendritic cells. Hum Immunol. 2007; 68:324–33.
243. Drobits B, Holcmann M, Amberg N, Swiecki M, Grundtner R, Hammer M, Colonna M, Sibilia M. Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells. J Clin Invest. 2012; 122:575–85.
244. Kalb ML, Glaser A, Stary G, Koszik F, Stingl G. TRAIL(+) human plasmacytoid dendritic cells kill tumor cells in vitro: mechanisms of imiquimod- and IFN-α-mediated antitumor reactivity. J Immunol. 2012; 188:1583–91.
245. Beutner KR, Geisse JK, Helman D, Fox TL, Ginkel A, Owens ML. Therapeutic response of basal cell carcinoma to the immune response modifier imiquimod 5% cream. J Am Acad Dermatol. 1999; 41:1002–7.
246. van Egmond S, Hoedemaker C, Sinclair R. Successful treatment of perianal Bowen's disease with imiquimod. Int J Dermatol. 2007; 46:318–9.
247. Urosevic M, Dummer R, Conrad C, Beyeler M, Laine E, Burg G, Gilliet M. Disease-independent skin recruitment and activation of plasmacytoid predendritic cells following imiquimod treatment. J Natl Cancer Inst. 2005; 97:1143–53.
248. Palamara F, Meindl S, Holcmann M, Lührs P, Stingl G, Sibilia M. Identification and characterization of pDC-like cells in normal mouse skin and melanomas treated with imiquimod. J Immunol. 2004; 173:3051–61.
249. Kauffman EC, Liu H, Schwartz MJ, Scherr DS. Toll-like receptor 7 agonist therapy with imidazoquinoline enhances cancer cell death and increases lymphocytic infiltration and proinflammatory cytokine production in established tumors of a renal cell carcinoma mouse model. J Oncol. 2012; 2012: 103298.
250. van Poelgeest MI, van Seters M, van Beurden M, Kwappenberg KM, Heijmans-Antonissen C, Drijfhout JW, Melief CJ, Kenter GG, Helmerhorst TJ, Offringa R, van der Burg SH. Detection of human papillomavirus (HPV) 16-specific CD4+ T-cell immunity in patients with persistent HPV16-induced vulvar intraepithelial neoplasia in relation to clinical impact of imiquimod treatment. Clin Cancer Res. 2005; 11:5273–80.
251. Ahn MY, Kwon SM, Cheong HH, Park JH, Lee J, Min SK, Ahn SG, Yoon JH. Toll-like receptor 7 agonist, imiquimod, inhibits oral squamous carcinoma cells through apoptosis and necrosis. J Oral Pathol Med. 2012; 41:540–6.
252. Yi JY, Jung YJ, Choi SS, Hwang J, Chung E. Autophagy-mediated anti-tumoral activity of imiquimod in Caco-2 cells. Biochem Biophys Res Commun. 2009; 386:455–8.
253. Cho JH, Lee HJ, Ko HJ, Yoon BI, Choe J, Kim KC, Hahn TW, Han JA, Choi SS, Jung YM, Lee KH, Lee YS, Jung YJ. The TLR7 agonist imiquimod induces anti-cancer effects via autophagic cell death and enhances anti-tumoral and systemic immunity during radiotherapy for melanoma. Oncotarget. 2017; 8:24932–24948. https://doi.org/10.18632/oncotarget.15326.
254. Li VW, Li WW, Talcott KE, Zhai AW. Imiquimod as an antiangiogenic agent. J Drugs Dermatol. 2005; 4:708–17.
255. Janmohamed SR, Madern GC, de Laat PC, Oranje AP. Educational paper: therapy of infantile haemangioma—history and current state (part II). Eur J Pediatr. 2015; 174:259–66.
256. Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013; 68:313–31.
257. Hesling C, D'Incan M, Mansard S, Franck F, Corbin-Duval A, Chèvenet C, Déchelotte P, Madelmont JC, Veyre A, Souteyrand P, Bignon YJ. In vivo and in situ modulation of the expression of genes involved in metastasis and angiogenesis in a patient treated with topical imiquimod for melanoma skin metastases. Br J Dermatol. 2004; 150:761–7.
258. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000; 408:740–5.
259. Krieg AM. Development of TLR9 agonists for cancer therapy. J Clin Invest. 2007; 117:1184–94.
260. Hartmann G, Weiner GJ, Krieg AM. CpG DNA: a potent signal for growth, activation, and maturation of human dendritic cells. Proc Natl Acad Sci USA. 1999; 96:9305–10.
261. Krug A, Rothenfusser S, Hornung V, Jahrsdörfer B, Blackwell S, Ballas ZK, Endres S, Krieg AM, Hartmann G. Identification of CpG oligonucleotide sequences with high induction of IFN-alpha/beta in plasmacytoid dendritic cells. Eur J Immunol. 2001; 31:2154–63.
262. Kumagai Y, Takeuchi O, Akira S. TLR9 as a key receptor for the recognition of DNA. Adv Drug Deliv Rev. 2008; 60:795–804.
263. Ballas ZK, Rasmussen WL, Krieg AM. Induction of NK activity in murine and human cells by CpG motifs in oligodeoxynucleotides and bacterial DNA. J Immunol. 1996; 157:1840–5.
264. Aucouturier J, Dupuis L, Deville S, Ascarateil S, Ganne V. Montanide ISA 720 and 51: a new generation of water in oil emulsions as adjuvants for human vaccines. Expert Rev Vaccines. 2002; 1:111–8.
265. Baumgaertner P, Jandus C, Rivals JP, Derré L, Lövgren T, Baitsch L, Guillaume P, Luescher IF, Berthod G, Matter M, Rufer N, Michielin O, Speiser DE. Vaccination-induced functional competence of circulating human tumor-specific CD8 T-cells. Int J Cancer. 2012; 130:2607–17.
266. Goldinger SM, Dummer R, Baumgaertner P, Mihic-Probst D, Schwarz K, Hammann-Haenni A, Willers J, Geldhof C, Prior JO, Kündig TM, Michielin O, Bachmann MF, Speiser DE. Nano-particle vaccination combined with TLR-7 and -9 ligands triggers memory and effector CD8+ T-cell responses in melanoma patients. Eur J Immunol. 2012; 42:3049–61.
267. Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007; 130:1071–82.
268. Deininger S, Stadelmaier A, von Aulock S, Morath S, Schmidt RR, Hartung T. Definition of structural prerequisites for lipoteichoic acid-inducible cytokine induction by synthetic derivatives. J Immunol. 2003; 170:4134–8.
269. Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol. 2002; 3:392–8.
270. Zhang Y, Luo F, Cai Y, Liu N, Wang L, Xu D, Chu Y. TLR1/TLR2 agonist induces tumor regression by reciprocal modulation of effector and regulatory T cells. J Immunol. 2011; 186:1963–9.
271. Zhang Y, Luo F, Li A, Qian J, Yao Z, Feng X, Chu Y. Systemic injection of TLR1/2 agonist improves adoptive antigen-specific T cell therapy in glioma-bearing mice. Clin Immunol. 2014; 154:26–36.
272. Lee SK, Chwee JY, Ma CA, Le Bert N, Huang CW, Gasser S. Synergistic anticancer effects of Pam3CSK4 and Ara-C on B-cell lymphoma cells. Clin Cancer Res. 2014; 20:3485–95.
273. Rolf N, Kariminia A, Ivison S, Reid GS, Schultz KR. Heterodimer-specific TLR2 stimulation results in divergent functional outcomes in B-cell precursor acute lymphoblastic leukemia. Eur J Immunol. 2015; 45:1980–90.
274. Dietrich N, Lienenklaus S, Weiss S, Gekara NO. Murine toll-like receptor 2 activation induces type I interferon responses from endolysosomal compartments. PLoS One. 2010; 5:e10250.
275. Stack J, Doyle SL, Connolly DJ, Reinert LS, O'Keeffe KM, McLoughlin RM, Paludan SR, Bowie AG. TRAM is required for TLR2 endosomal signaling to type I IFN induction. J Immunol. 2014; 193:6090–102.
276. Guo X, Wu N, Shang Y, Liu X, Wu T, Zhou Y, Huang J, Liao X, Wu L. The Novel Toll-Like Receptor 2 Agonist SUP3 Enhances Antigen Presentation and T Cell Activation by Dendritic Cells. Front Immunol. 2017; 8:158.
277. Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013; 341:1390–4.
278. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013; 339:786–91.
279. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013; 339:826–30.
280. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner KP, Ludwig J, Hornung V. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013; 498:380–4.
281. Ablasser A, Gulen MF. The role of cGAS in innate immunity and beyond. J Mol Med (Berl). 2016; 94:1085–93.
282. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, Alegre ML, Gajewski TF. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014; 41:830–42.
283. Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV, Martinon F, Modlin RL, Speiser DE, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci USA. 2015; 112:15408–13.
284. Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, Chen ZJ. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci USA. 2017; 114:1637–42.
285. Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, Munn D, Mellor AL. STING Promotes the Growth of Tumors Characterized by Low Antigenicity via IDO Activation. Cancer Res. 2016; 76:2076–81.
286. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, Jacob LS, Patwa R, Shah H, Xu K, Cross JR, Massagué J. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016; 533:493–8.
287. Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012; 11:215–33.
288. Adachi K, Tamada K. Immune checkpoint blockade opens an avenue of cancer immunotherapy with a potent clinical efficacy. Cancer Sci. 2015; 106:945–50.
289. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, Coutant F, Métivier D, Pichard E, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005; 202:1691–701.
290. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Métivier D, Larochette N, van Endert P, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007; 13:54–61.
291. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017; 17:97–111.
292. Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, Bracci L, Breckpot K, Brough D, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014; 3: e955691.
293. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, Fend L, Hannani D, Aymeric L, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014; 20:1301–9.
294. Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, Fu YX, Auh SL. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011; 71:2488–96.
295. Koks CA, Garg AD, Ehrhardt M, Riva M, Vandenberk L, Boon L, De Vleeschouwer S, Agostinis P, Graf N, Van Gool SW. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. OncoImmunology. 2015; 136:E313–25.
296. Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014; 6: 226ra32.
297. Gajewski TF, Fuertes MB, Woo SR. Innate immune sensing of cancer: clues from an identified role for type I IFNs. Cancer Immunol Immunother. 2012; 61:1343–7.
298. Barber GN. Cytoplasmic DNA innate immune pathways. Immunol Rev. 2011; 243:99–108.
299. Gajewski TF. Failure at the effector phase: immune barriers at the level of the melanoma tumor microenvironment. Clin Cancer Res. 2007; 13:5256–61.
300. Ciampricotti M, Hau CS, Doornebal CW, Jonkers J, de Visser KE. Chemotherapy response of spontaneous mammary tumors is independent of the adaptive immune system. Nat Med. 2012; 18:344–46.
301. Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, Lin YJ, Wojtkiewicz G, Iwamoto Y, et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity. 2016; 44:343–54.
302. Malek A, Tchernitsa O. Evaluation of targets for ovarian cancer gene silencing therapy: in vitro and in vivo approaches. Methods Mol Biol. 2010; 623:423–36.
303. Bossi G, Sacchi A. Restoration of wild-type p53 function in human cancer: relevance for tumor therapy. Head Neck. 2007; 29:272–84.
304. Schmidt-Wolf GD, Schmidt-Wolf IG. Immunomodulatory gene therapy for haematological malignancies. Br J Haematol. 2002; 117:23–32.
305. Moschos S, Kirkwood JM. Present role and future potential of type I interferons in adjuvant therapy of high-risk operable melanoma. Cytokine Growth Factor Rev. 2007; 18:451–8.
306. Villaverde MS, Gil-Cardeza ML, Glikin GC, Finocchiaro LM. Interferon-β lipofection I. Increased efficacy of chemotherapeutic drugs on human tumor cells derived monolayers and spheroids. Cancer Gene Ther. 2012; 19:508–16.
307. Fondello C, Agnetti L, Villaverde MS, Simian M, Glikin GC, Finocchiaro LM. The combination of bleomycin with suicide or interferon-β gene transfer is able to efficiently eliminate human melanoma tumor initiating cells. Biomed Pharmacother. 2016; 83:290–301.
308. Rossi Ú, Gil-Cardeza ML, Villaverde MS, Finocchiaro LM, Glikin GC. Interferon-β gene transfer induces a strong cytotoxic bystander effect on melanoma cells. Biomed Pharmacother. 2015; 72:44–51.
309. Rossi Ú, Finocchiaro LM, Glikin GC. Interferon-β gene transfer inhibits melanoma cells adhesion and migration. Cytokine. 2017; 89:201–8.
310. Wakabayashi T, Natsume A, Hashizume Y, Fujii M, Mizuno M, Yoshida J. A phase I clinical trial of interferon-beta gene therapy for high-grade glioma: novel findings from gene expression profiling and autopsy. J Gene Med. 2008; 10:329–39.
311. Shih CS, Laurie N, Holzmacher J, Spence Y, Nathwani AC, Davidoff AM, Dyer MA. AAV-mediated local delivery of interferon-beta for the treatment of retinoblastoma in preclinical models. Neuromolecular Med. 2009; 11:43–52.
312. Hara H, Kobayashi A, Yoshida K, Ohashi M, Ohnami S, Uchida E, Higashihara E, Yoshida T, Aoki K. Local interferon-alpha gene therapy elicits systemic immunity in a syngeneic pancreatic cancer model in hamster. Cancer Sci. 2007; 98:455–63.
313. Ohashi M, Yoshida K, Kushida M, Miura Y, Ohnami S, Ikarashi Y, Kitade Y, Yoshida T, Aoki K. Adenovirus-mediated interferon alpha gene transfer induces regional direct cytotoxicity and possible systemic immunity against pancreatic cancer. Br J Cancer. 2005; 93:441–9.
314. Hatanaka K, Suzuki K, Miura Y, Yoshida K, Ohnami S, Kitade Y, Yoshida T, Aoki K. Interferon-alpha and antisense K-ras RNA combination gene therapy against pancreatic cancer. J Gene Med. 2004; 6:1139–48.
315. Indraccolo S, Tisato V, Tosello V, Habeler W, Esposito G, Moserle L, Stievano L, Persano L, Chieco-Bianchi L, Amadori A. Interferon-alpha gene therapy by lentiviral vectors contrasts ovarian cancer growth through angiogenesis inhibition. Hum Gene Ther. 2005; 16:957–70.
316. Aida K, Miyakawa R, Suzuki K, Narumi K, Udagawa T, Yamamoto Y, Chikaraishi T, Yoshida T, Aoki K. Suppression of Tregs by anti-glucocorticoid induced TNF receptor antibody enhances the antitumor immunity of interferon-α gene therapy for pancreatic cancer. Cancer Sci. 2014; 105:159–67.
317. Narumi K, Kondoh A, Udagawa T, Hara H, Goto N, Ikarashi Y, Ohnami S, Okada T, Yamagishi M, Yoshida T, Aoki K. Administration route-dependent induction of antitumor immunity by interferon-alpha gene transfer. Cancer Sci. 2010; 101:1686–94.
318. Catarinella M, Monestiroli A, Escobar G, Fiocchi A, Tran NL, Aiolfi R, Marra P, Esposito A, Cipriani F, Aldrighetti L, Iannacone M, Naldini L, Guidotti LG, et al. IFNα gene/cell therapy curbs colorectal cancer colonization of the liver by acting on the hepatic microenvironment. EMBO Mol Med. 2016; 8:155–70.
319. De Palma M, Mazzieri R, Politi LS, Pucci F, Zonari E, Sitia G, Mazzoleni S, Moi D, Venneri MA, Indraccolo S, Falini A, Guidotti LG, Galli R, et al. Tumor-targeted interferon-alpha delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell. 2008; 14:299–311.
320. Dinney CP, Fisher MB, Navai N, O'Donnell MA, Cutler D, Abraham A, Young S, Hutchins B, Caceres M, Kishnani N, Sode G, Cullen C, Zhang G, et al. Phase I trial of intravesical recombinant adenovirus mediated interferon-α2b formulated in Syn3 for Bacillus Calmette-Guérin failures in nonmuscle invasive bladder cancer. J Urol. 2013; 190:850–6.
321. Ren C, Kumar S, Chanda D, Kallman L, Chen J, Mountz JD, Ponnazhagan S. Cancer gene therapy using mesenchymal stem cells expressing interferon-beta in a mouse prostate cancer lung metastasis model. Gene Ther. 2008; 15:1446–53.
322. Lykhova AA, Kudryavets YI, Strokovska LI, Bezdenezhnykh NA, Semesiuk NI, Adamenko IN, Anopriyenko OV, Vorontsova AL. Suppression of proliferation, tumorigenicity and metastasis of lung cancer cells after their transduction by interferon-beta gene in baculovirus vector. Cytokine. 2015; 71:318–26.
323. Odaka M, Sterman DH, Wiewrodt R, Zhang Y, Kiefer M, Amin KM, Gao GP, Wilson JM, Barsoum J, Kaiser LR, Albelda SM. Eradication of intraperitoneal and distant tumor by adenovirus-mediated interferon-beta gene therapy is attributable to induction of systemic immunity. Cancer Res. 2001; 61:6201–12.
324. GuhaSarkar D, Neiswender J, Su Q, Gao G, Sena-Esteves M. Intracranial AAV-IFN-β gene therapy eliminates invasive xenograft glioblastoma and improves survival in orthotopic syngeneic murine model. Mol Oncol. 2017; 11:180–93.
325. GuhaSarkar D, Su Q, Gao G, Sena-Esteves M. Systemic AAV9-IFNβ gene delivery treats highly invasive glioblastoma. Neuro Oncol. 2016; 18:1508–18.
326. Wilderman MJ, Sun J, Jassar AS, Kapoor V, Khan M, Vachani A, Suzuki E, Kinniry PA, Sterman DH, Kaiser LR, Albelda SM. Intrapulmonary IFN-beta gene therapy using an adenoviral vector is highly effective in a murine orthotopic model of bronchogenic adenocarcinoma of the lung. Cancer Res. 2005; 65:8379–87.
327. Streck CJ, Dickson PV, Ng CY, Zhou J, Gray JT, Nathwani AC, Davidoff AM. Adeno-associated virus vector-mediated systemic delivery of IFN-beta combined with low-dose cyclophosphamide affects tumor regression in murine neuroblastoma models. Clin Cancer Res. 2005; 11:6020–9.
328. Kruklitis RJ, Singhal S, Delong P, Kapoor V, Sterman DH, Kaiser LR, Albelda SM. Immuno-gene therapy with interferon-beta before surgical debulking delays recurrence and improves survival in a murine model of malignant mesothelioma. J Thorac Cardiovasc Surg. 2004; 127: 123–30.
329. Hendren SK, Prabakaran I, Buerk DG, Karakousis G, Feldman M, Spitz F, Menon C, Fraker DL. Interferon-beta gene therapy improves survival in an immunocompetent mouse model of carcinomatosis. Surgery. 2004; 135:427–36.
330. Patel MR, Jacobson BA, Ji Y, Drees J, Tang S, Xiong K, Wang H, Prigge JE, Dash AS, Kratzke AK, Mesev E, Etchison R, Federspiel MJ, et al. Vesicular stomatitis virus expressing interferon-beta is oncolytic and promotes antitumor immune responses in a syngeneic murine model of non-small cell lung cancer. Oncotarget. 2015; 6:33165–77. https://doi.org/10.18632/oncotarget.
331. Kurisetty VV, Heiber J, Myers R, Pereira GS, Goodwin JW, Federspiel MJ, Russell SJ, Peng KW, Barber G, Merchan JR. Preclinical safety and activity of recombinant VSV-IFN-beta in an immunocompetent model of squamous cell carcinoma of the head and neck. Head Neck. 2014; 36:1619–27.
332. Saloura V, Wang LC, Fridlender ZG, Sun J, Cheng G, Kapoor V, Sterman DH, Harty RN, Okumura A, Barber GN, Vile RG, Federspiel MJ, Russell SJ, et al. Evaluation of an attenuated vesicular stomatitis virus vector expressing interferon-beta for use in malignant pleural mesothelioma: heterogeneity in interferon responsiveness defines potential efficacy. Hum Gene Ther. 2010; 21:51–64.
333. Matsumoto K, Kubo H, Murata H, Uhara H, Takata M, Shibata S, Yasue S, Sakakibara A, Tomita Y, Kageshita T, Kawakami Y, Mizuno M, Yoshida J, et al. A pilot study of human interferon beta gene therapy for patients with advanced melanoma by in vivo transduction using cationic liposomes. Jpn J Clin Oncol. 2008; 38:849–56.
334. Sterman DH, Gillespie CT, Carroll RG, Coughlin CM, Lord EM, Sun J, Haas A, Recio A, Kaiser LR, Coukos G, June CH, Albelda SM, Vonderheide RH. Interferon beta adenoviral gene therapy in a patient with ovarian cancer. Nat Clin Pract Oncol. 2006; 3:633–9.
335. Sterman DH, Recio A, Haas AR, Vachani A, Katz SI, Gillespie CT, Cheng G, Sun J, Moon E, Pereira L, Wang X, Heitjan DF, Litzky L, et al. A phase I trial of repeated intrapleural adenoviral-mediated interferon-beta gene transfer for mesothelioma and metastatic pleural effusions. Mol Ther. 2010; 18:852–60.
336. Chiocca EA, Smith KM, McKinney B, Palmer CA, Rosenfeld S, Lillehei K, Hamilton A, DeMasters BK, Judy K, Kirn D. A phase I trial of Ad.hIFN-beta gene therapy for glioma. Mol Ther. 2008; 16:618–26.
337. Berzofsky JA, Terabe M, Oh S, Belyakov IM, Ahlers JD, Janik JE, Morris JC. Progress on new vaccine strategies for the immunotherapy and prevention of cancer. J Clin Invest. 2004; 113:1515–25.
338. Tsugawa T, Kuwashima N, Sato H, Fellows-Mayle WK, Dusak JE, Okada K, Papworth GD, Watkins SC, Gambotto A, Yoshida J, Pollack IF, Okada H. Sequential delivery of interferon-alpha gene and DCs to intracranial gliomas promotes an effective antitumor response. Gene Ther. 2004; 11:1551–8.
339. Kuwashima N, Nishimura F, Eguchi J, Sato H, Hatano M, Tsugawa T, Sakaida T, Dusak JE, Fellows-Mayle WK, Papworth GD, Watkins SC, Gambotto A, Pollack IF, et al. Delivery of dendritic cells engineered to secrete IFN-alpha into central nervous system tumors enhances the efficacy of peripheral tumor cell vaccines: dependence on apoptotic pathways. J Immunol. 2005; 175:2730–40.
340. Smits EL, Anguille S, Cools N, Berneman ZN, Van Tendeloo VF. Dendritic cell-based cancer gene therapy. Hum Gene Ther. 2009; 20:1106–18.
341. Timares L, Douglas JT, Tillman BW, Krasnykh V, Curiel DT. Adenovirus-mediated gene delivery to dendritic cells. Methods Mol Biol. 2004; 246:139–54.
342. Dietz AB, Bulur PA, Brown CA, Pankratz VS, Vuk-Pavlovic S. Maturation of dendritic cells infected by recombinant adenovirus can be delayed without impact on transgene expression. Gene Ther. 2001; 8:419–23.
343. Perreau M, Mennechet F, Serratrice N, Glasgow JN, Curiel DT, Wodrich H, Kremer EJ. Contrasting effects of human, canine, and hybrid adenovirus vectors on the phenotypical and functional maturation of human dendritic cells: implications for clinical efficacy. J Virol. 2007; 81:3272–84.
344. Ponsaerts P, Van Tendeloo VF, Cools N, Van Driessche A, Lardon F, Nijs G, Lenjou M, Mertens G, Van Broeckhoven C, Van Bockstaele DR, Berneman ZN. mRNA-electroporated mature dendritic cells retain transgene expression, phenotypical properties and stimulatory capacity after cryopreservation. Leukemia. 2002; 16:1324–30.
345. Yonemitsu Y, Kitson C, Ferrari S, Farley R, Griesenbach U, Judd D, Steel R, Scheid P, Zhu J, Jeffery PK, Kato A, Hasan MK, Nagai Y, et al. Efficient gene transfer to airway epithelium using recombinant Sendai virus. Nat Biotechnol. 2000; 18:970–3.
346. Yonemitsu Y, Ueda Y, Kinoh H, Hasegawa M. Immunostimulatory virotherapy using recombinant Sendai virus as a new cancer therapeutic regimen. Front Biosci. 2008; 13:1892–8.
347. Thomann S, Boscheinen JB, Vogel K, Knipe DM, DeLuca N, Gross S, Schuler-Thurner B, Schuster P, Schmidt B. Combined cytotoxic activity of an infectious, but non-replicative herpes simplex virus type 1 and plasmacytoid dendritic cells against tumour cells. Immunology. 2015; 146:327–38.
348. Bajgelman MC, Strauss BE. Development of an adenoviral vector with robust expression driven by p53. Virology. 2008; 371:8–13.
349. Sharpless E, Chin L. The INK4a/ARF locus and melanoma. Oncogene. 2003; 22:3092–8.
350. Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci USA. 1998; 95:8292–7.
351. Abida WM, Gu W. p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008; 68:352–7.
352. Ozenne P, Eymin B, Brambilla E, Gazzeri S. The ARF tumor suppressor: structure, functions and status in cancer. Int J Cancer. 2010; 127:2239–47.
353. Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, Sasaki S, Imai K, Shibue T, Honda K, Taniguchi T. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003; 424:516–23.
354. Sandoval R, Xue J, Pilkinton M, Salvi D, Kiyokawa H, Colamonici OR. Different requirements for the cytostatic and apoptotic effects of type I interferons. Induction of apoptosis requires ARF but not p53 in osteosarcoma cell lines. J Biol Chem. 2004; 279:32275–80.
355. Giglia-Mari G, Sarasin A. TP53 mutations in human skin cancers. Hum Mutat. 2003; 21:217–28.
356. Merkel CA, Medrano RF, Barauna VG, Strauss BE. Combined p19Arf and interferon-beta gene transfer enhances cell death of B16 melanoma in vitro and in vivo. Cancer Gene Ther. 2013; 20:317–25.
357. Medrano RF, Catani JP, Ribeiro AH, Tomaz SL, Merkel CA, Costanzi-Strauss E, Strauss BE. Vaccination using melanoma cells treated with p19arf and interferon beta gene transfer in a mouse model: a novel combination for cancer immunotherapy. Cancer Immunol Immunother. 2016; 65:371–82.
358. Catani JP, Medrano RF, Hunger A, Del Valle P, Adjemian S, Zanatta DB, Kroemer G, Costanzi-Strauss E, Strauss BE. Intratumoral Immunization by p19Arf and Interferon-β Gene Transfer in a Heterotopic Mouse Model of Lung Carcinoma. Transl Oncol. 2016; 9:565–74.
359. Hunger A, Medrano RF, Zanatta DB, Del Valle PR, Merkel CA, Salles TA, Ferrari DG, Furuya TK, Bustos SO, de Freitas Saito R, Costanzi-Strauss E, Strauss BE. Reestablishment of p53/Arf and interferon-β pathways mediated by a novel adenoviral vector potentiates antiviral response and immunogenic cell death. Cell Death Dis. 2017; 3:17017.
360. Yang H, Ma Y, Chen G, Zhou H, Yamazaki T, Klein C, Pietrocola F, Vacchelli E, Souquere S, Sauvat A, Zitvogel L, Kepp O, Kroemer G. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. OncoImmunology. 2016; 5:e1149673.
361. Garg AD, Vandenberk L, Koks C, Verschuere T, Boon L, Van Gool SW, Agostinis P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med. 2016; 8: 328ra27.
362. Nocera DA, Roselli E, Araya P, Nuñez NG, Lienenklaus S, Jablonska J, Weiss S, Gatti G, Brinkmann MM, Kröger A, Morón G, Maccioni M. In Vivo Visualizing the IFN-β Response Required for Tumor Growth Control in a Therapeutic Model of Polyadenylic-Polyuridylic Acid Administration. J Immunol. 2016; 196:2860–9.
363. Lienenklaus S, Cornitescu M, Zietara N, Łyszkiewicz M, Gekara N, Jabłónska J, Edenhofer F, Rajewsky K, Bruder D, Hafner M, Staeheli P, Weiss S. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J Immunol. 2009; 183:3229–36.