
INTRODUCTION
Ovarian cancer is the fifth leading cause of cancer-related female deaths and was reported to be responsible for approximately 152,000 deaths worldwide in 2012 [1]. Although up to 80% of patients with primary disease respond to first-line chemotherapy, relapse and resistance to further treatment is prevalent, leading to lack of long-term benefit from treatment [2]. Mucinous and clear cell histological subtypes do not respond well to current chemotherapy strategies and are clinically challenging to treat [3, 4]. Different strategies have been developed to treat the recurrent and/or resistant disease, including targeted therapies such as angiogenesis inhibitors and poly(ADP-ribose) polymerase (PARP) inhibitors [5].
The inhibition of PARP enzymatic activity hinders DNA repair via the base excision repair (BER) pathway. For cells deficient in homologous DNA recombination repair (HRR) this results in multiple lethal DNA double-strand breaks normally repaired by the HRR pathway. The tumor cells with BRCA1/2 mutation or other HRR defective status cannot efficiently repair these double-strand breaks, leading to cell death [6–8]. Another mode of action for PARP inhibitors is to trap PARP proteins at the sites of DNA damage, which is highly toxic to cells due to blockade of DNA replication and induction of a replication stress response. PARP inhibitors proficiently result in synthetic lethality in tumor cells with BRCA1/2 or other HRR deficiencies, more than in normal DNA repair proficient cells [9, 10]. Rucaparib is one of a series of tricyclic benzimidazole carboxamide PARP inhibitors with a Ki of 1.4 nM for PARP1 in a cell-free assay. It is a poly(ADP-ribose) polymerase (PARP) inhibitor successfully granted a license by the FDA and indicated as a monotherapy for the treatment of patients with a deleterious BRCA mutation (germline and/or somatic) associated advanced ovarian cancer who have been treated with two or more chemotherapies [11].
Reactivation of wild-type p53 by preventing the protein-protein binding interaction between p53 and its negative regulator MDM2 induces the growth inhibitory and/or pro-apoptotic functions of p53, and has been demonstrated to have potential as a therapeutic strategy for non-genotoxic activation of p53. Nutlin-3 provided the mechanistic proof-of-concept for small molecule inhibition of the MDM2-p53 interaction and continues to be a useful reference tool compound; however, its potency and pharmacological properties are suboptimal for clinical use [12, 13]. RG7388, a second generation MDM2 inhibitor, was subsequently developed with superior potency, selectivity and oral bioavailability suitable for clinical development, with a cell-free IC50 value of 6 nM [14]. These compounds target a small hydrophobic pocket on MDM2, to which p53 normally binds, leading to p53 stabilization and upregulation of p53 downstream transcriptional targets involved in cell cycle arrest and/or apoptosis [15, 16].
Up to 50% to 60% of epithelial ovarian cancer is estimated to be deficient in HRR and hence likely to respond to PARP inhibitors [17]. The approximately 34% of ovarian cancer patients with tumors harboring wild-type TP53 may benefit from MDM2 inhibitor treatment [16]. Combination chemotherapy for cancer treatment has a long established history, particularly for agents having different mechanism of action and non-overlapping toxicities. Utilizing targeted cancer therapeutic agents in combination is starting to be explored, although it has substantial complexity [18].
In the current study it was hypothesized that combination treatment of Nutlin-3/RG7388 with rucaparib further activates the p53 pathway by inhibition of PARP and results in enhanced induction and stabilization of p53 via Nutlin-3/RG7388 treatment to increase growth arrest and/or apoptosis in wild-type TP53 ovarian cancer cell lines.
RESULTS
The growth inhibitory response of ovarian cancer cell lines to Nutlin-3/RG7388 and rucaparib
A sulforhodamine-B (SRB) assay was used to investigate growth inhibition by Nutlin-3/RG7388 or rucaparib for a panel of wild-type and mutant TP53 ovarian cancer cell lines derived from tumors of different histological subtypes [19–22] (Figure 1 and Table 1). The GI50 values, required concentration of each compound leading to 50% growth inhibition, showed that wild-type TP53 ovarian cancer cell lines were significantly more sensitive to Nutlin-3/RG7388 compared to mutant, which is consistent with their mechanism of action (p<0.0001 Mann-Whitney test) (Figure 1A & 1B). The GI50 values for wild-type TP53 cell lines for RG7388 and Nutlin-3 were in the nanomolar range ( 253.3 ± 73.1 (SEM) nM) and micromolar range (1.76 ± 0.51 (SEM) μM) respectively. In contrast, TP53 mutant cell lines had GI50 values greater than 10 μM (17.8 ± 2.9 (SEM) μM) for RG7388 and range 21.2->30 μM for Nutlin-3 (Table 1).
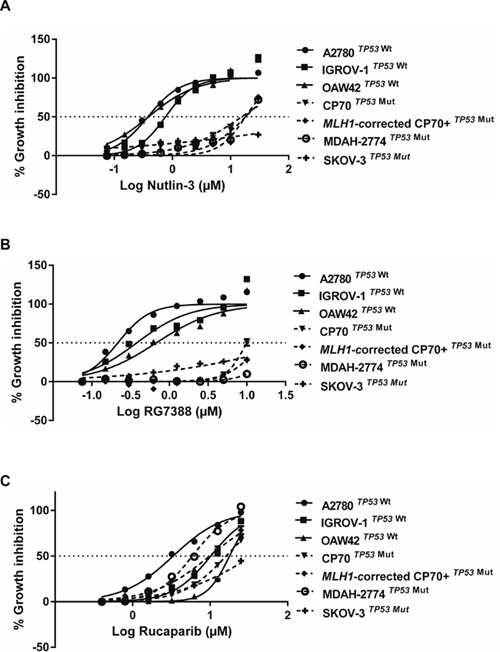
Figure 1: The sensitivity to MDM2 antagonists, Nutlin-3 and RG7388, and rucaparib in a panel of wild-type and mutant TP53 ovarian cancer cell lines. Wild-type TP53 cell lines are significantly more sensitive to growth inhibition by (A) Nutlin-3 (Mann Whitney test, p< 0.0001) and (B) RG7388 (Mann Whitney test, p< 0.0001) treatment for 72 hours compared to mutant TP53 cell lines. (C) The sensitivity to rucaparib is p53-independent. Data shown are the average of at least three independent experiments and error bars represent SEM.
Table 1: GI50 concentrations of rucaparib, Nutlin-3 and RG7388 for the panel of ovarian cancer cell lines of varying TP53 status
Cell line |
TP53 status |
Histotype |
Rucaparib (μM) |
Nutlin-3 (μM) |
RG7388 (μM) |
|---|---|---|---|---|---|
A2780 |
Wild-type |
Undifferentiated [19] |
3.26 ± 0.47 |
1.23 ± 0.23 |
0.11 ± 0.01 |
IGROV-1 |
Wild-type |
Mixed, EC with CCC/UD [21] |
11.34 ± 0.05 |
2.8 ± 0.48 |
0.35 ± 0.04 |
OAW42 |
Wild-type |
Serous Cystadenocarcinoma [20] |
19.00 ± 0.58 |
1.3 ± 0.1 |
0.31 ± 0.04 |
CP70 |
Mutant (Heterozygous) c.514 G->T; p.Val172Phe |
Undifferentiated [19] |
17.00 ± 0.64 |
21.2 ± 2.5 |
11.7 ± 1.81 |
MLH1-Corrected CP70+ |
Mutant (Heterozygous) c.514 G->T; .Val172Phe |
Undifferentiated [19] |
14.00 ± 2.84 |
21.2 ± 1.22 |
14.5 ± 1.09 |
MDAH-2774 |
Mutant (Homozygous) c.818G->A; p.Arg273His |
Endometrioid carcinoma [22] |
6.92 ± 1.48 |
21.4 ± 0.9 |
20.7 ± 1.43 |
SKOV-3 |
Mutant (Homozygous) 265delC; p.Pro89fsX33 |
Adenocarcinoma [20] |
>25 |
> 30 |
24.6 ± 1.54 |
Data represent the mean of at least 3 independent experiments ± SEM. EC, endometrioid carcinoma; CCC, clear cell carcinoma; UD, undifferentiated.
For rucaparib, the cells were treated with a wide range of concentrations (0.4-25 μM) for 72 hours to construct growth inhibition curves and calculate the GI50 values. The GI50 values significantly varied showing a range of responses, with A2780 (3.26±0.47 μM) and SKOV-3 (> 25 μM) as the most sensitive and resistant cell lines respectively. Although CP70 cells are derived from A2780, they have genetic alterations in addition to the difference in TP53 genomic status. In particular, CP70 cells are mismatch repair (MMR)-deficient due to loss of the MLH1 gene and they are resistant to rucaparib even though A2780 cells are MMR-competent and sensitive to rucaparib. The CP70+ cell line is MLH1-corrected by chromosome 3 transfer into the CP70 cells and retains the heterozygous TP53 mutation. The A2780 and derived cell lines differ in growth rate, TP53 status and MLH1 status and show differences in response to cisplatin and rucaparib, they are nevertheless all derived from the same tumor and thus overall are genetically closely related. The results of this study showed no relationship between the status of TP53 and response to rucaparib (Mann-Whitney, p>0.05) (Figure 1C).
Nutlin-3/RG7388 synergizes with rucaparib for growth inhibition of wild-type TP53 ovarian cancer cell lines
The effect of rucaparib in combination with Nutlin-3/RG7388 was investigated for three wild-type TP53 ovarian cancer cell lines using median-effect analysis. The sensitivity of the wild-type TP53 cell lines to growth inhibition during 72 hours exposure to rucaparib and Nutlin-3/RG7388 was determined as single agents, and in combination at 5 equipotent concentrations between 0.25× and 4× their respective GI50 concentrations for the A2780 cell line. Owing to the high GI50 for IGROV-1 and OAW42 in response to rucaparib, 3 equipotent concentrations between 0.25× and 1x their respective GI50 concentrations were used to evaluate the combination effect of rucaparib with Nutlin-3/RG7388. The effect of combined treatment was cell type dependent. The combination of Nutlin-3/RG7388 with rucaparib at all concentrations led to greater growth inhibition compared to either agent alone for the A2780, IGROV-1 and OAW42 cell lines, although the increase was smaller for the OAW42 cells (Figure 2). Combination treatment of rucaparib with Nutlin-3/RG7388 at concentrations equal and lower than the individual 1x GI50 dose resulted in more growth arrest compared to doses higher than 1x GI50 for the A2780 cell line.
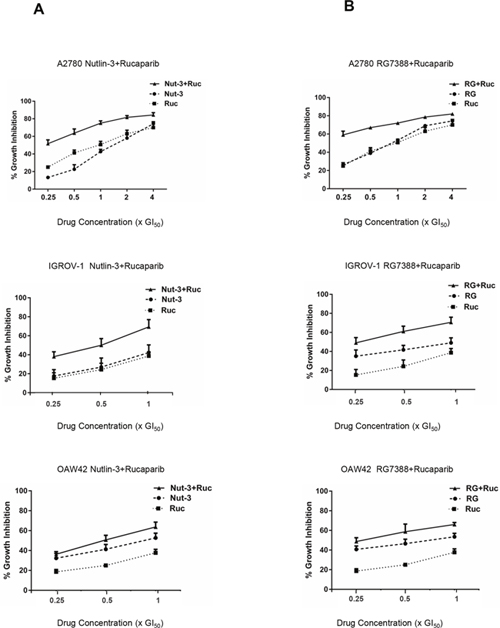
Figure 2: Nutlin-3/RG7388 synergizes with rucaparib in wild-type TP53 ovarian cancer cells. (A) Growth inhibition curves of three wild-type TP53 cell lines exposed to Nutlin-3 and rucaparib alone, and in combination at constant 1:1 ratios of 0.25X, 0.5X, 1X, 2X and 4X (A2780) or 0.25X, 0.5X and 1X (IGROV-1 & OAW42) their respective GI50 concentration for 72 hours. (B) Growth inhibition curves of three wild-type TP53 cell lines exposed to RG7388 and rucaparib alone, and in combination at constant 1:1 ratios of 0.25X, 0.5X, 1X, 2X and 4X (A2780) or 0.25X, 0.5X and 1X (IGROV-1 & OAW42) their respective GI50 concentrations for 72 hours. Nut-3, Nutlin-3; RG, RG7388; Ruc, rucaparib.
To determine whether the observed differences in growth inhibition were additive, synergistic or antagonistic, the data were analyzed using median-effect analysis and Combination Index (CI) values calculated. CI values for each constant ratio combination and at effect levels of ED50, ED75 and ED90 were computed and the average of CI values at ED50, ED75 and ED90 was also determined (Figure 3 & Table 2). Combined treatment of rucaparib with Nutlin-3/RG7388 ranged from additive to strong synergism based on the CI at ED50 and overall CI for A2780 and IGROV-1 cell lines, whereas only slight synergism to antagonism was observed for the OAW42 cell line (Figure 3 & Table 2). Interestingly, rucaparib, Nutlin-3 and RG7388 had favorable Dose Reduction Index (DRI) values for combined treatment with all experimental values ranging from 1.2-fold to 7.8-fold dose reduction (Table 3).
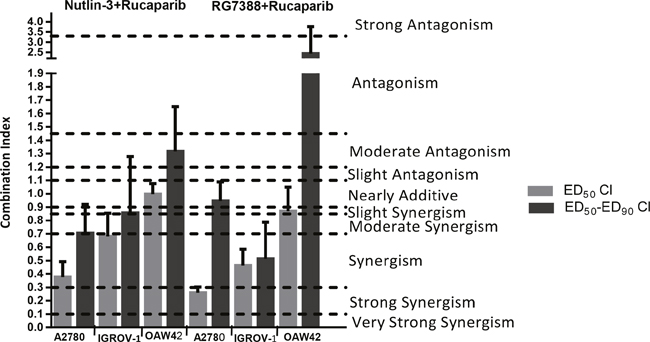
Figure 3: The CI values for Nutlin-3/RG7388 in combination with rucaparib at ED50 and the average of CI values at effect levels ED50, ED75 and ED90 in three wild-type TP53 ovarian cancer cell lines. Data are shown as the average of at least 3 independent experiments and error bars represent SEM. CI, Combination Index; ED, Effective Dose.
Table 2: Growth inhibition CI values for RG7388/Nutlin-3 in combination with rucaparib for the wild-type TP53 ovarian cancer cell lines
Cell Line |
Combination |
CI |
CI ED50 |
CI ED75 |
CI ED90 |
CI Ave ED50-90 |
||||
|---|---|---|---|---|---|---|---|---|---|---|
XGI50 |
||||||||||
0.25 |
0.5 |
1 |
2 |
4 |
||||||
A2780 |
Nutlin-3+Rucaparib |
0.4 |
0.5 |
0.5 |
0.6 |
1.0 |
0.4 |
0.6 |
1.1 |
0.7 |
RG7388+Rucaparib |
0.4 |
0.4 |
0.6 |
0.7 |
1.2 |
0.3 |
0.7 |
1.9 |
0.9 |
|
IGROV-1 |
Nutlin-3+Rucaparib |
0.6 |
0.7 |
0.6 |
ND |
ND |
0.7 |
0.8 |
1.1 |
0.9 |
RG7388+Rucaparib |
0.4 |
0.4 |
0.3 |
ND |
ND |
0.5 |
0.4 |
0.7 |
0.5 |
|
OAW42 |
Nutlin-3+Rucaparib |
0.9 |
0.7 |
0.9 |
ND |
ND |
1.0 |
1.3 |
1.7 |
1.3 |
RG7388+Rucaparib |
0.9 |
1.0 |
1.4 |
ND |
ND |
1.2 |
3.2 |
4.6 |
2.4 |
|
The combined treatment was performed at the indicated fixed 1:1 ratios relative to their respective GI50 concentrations. CI values were calculated for each constant ratio combination and at effect levels ED50, ED75 and ED90 from the average of at least three independent experiments. CI Ave ED50-90 represents the average of CI values at effect levels of ED50, ED75 and ED90. CI range: < 0.1 very strong synergism; 0.1-0.3 strong synergism; 0.3-0.7 synergism; 0.7-0.85 moderate synergism; 0.85-0.9 slight synergism; 0.9-1.1 nearly additive; 1.1-1.2 slight antagonism; 1.2-1.45 moderate antagonism; 1.45-3.3 antagonism; 3.3-10 strong antagonism; > 10 very strong antagonism. Synergistic combinations are highlighted in bold font. CI, Combination Index; ED, Effective Dose.
Table 3: DRI values for growth inhibition by RG7388/Nutlin-3 in combination with rucaparib for the wild-type TP53 ovarian cancer cell lines
Cell Line |
Combination |
Component |
DRI |
||||
|---|---|---|---|---|---|---|---|
XGI50 |
|||||||
0.25 |
0.5 |
1 |
2 |
4 |
|||
A2780 |
Nutlin-3+Rucaparib |
Rucaparib |
3.1 |
2.7 |
3.0 |
2.5 |
1.6 |
Nutlin-3 |
5.00 |
3.9 |
3.6 |
2.6 |
1.6 |
||
RG7388+Rucaparib |
Rucaparib |
5.4 |
4.0 |
3.1 |
2.5 |
1.4 |
|
RG7388 |
5.1 |
4.4 |
3.0 |
2.4 |
1.6 |
||
IGROV-1 |
Nutlin-3+Rucaparib |
Rucaparib |
3.5 |
3.0 |
3.9 |
ND |
ND |
Nutlin-3 |
3.5 |
2.8 |
3.3 |
ND |
ND |
||
RG7388+Rucaparib |
Rucaparib |
5.7 |
5.0 |
3.7 |
ND |
ND |
|
RG7388 |
2.7 |
3.9 |
7.8 |
ND |
ND |
||
OAW42 |
Nutlin-3+Rucaparib |
Rucaparib |
2.3 |
1.6 |
1.9 |
ND |
ND |
Nutlin-3 |
1.9 |
1.2 |
1.2 |
ND |
ND |
||
RG7388+Rucaparib |
Rucaparib |
4.3 |
4.0 |
3.0 |
ND |
ND |
|
RG7388 |
2.0 |
2.1 |
1.8 |
ND |
ND |
||
The combined treatment was performed at the indicated fixed 1:1 ratios relative to their respective GI50 concentrations. DRI values were calculated for each constant ratio combination from the average of at least three independent experiments. Favorable DRI values (>1.0) are highlighted in bold font. DRI, Dose Reduction Index.
The effect of combination treatment with rucaparib and Nutlin-3/RG7388 on activation of the p53 pathway
Western blotting was used to investigate the effect of combination treatment on the p53 molecular pathway. Wild-type TP53 cell lines were treated with rucaparib, Nutlin-3 or RG7388 alone, and in combination at constant 1:1 ratios of 1/2x and 1x their respective GI50 concentrations for 4 hours. They were also treated with rucaparib, Nutlin-3 or RG7388 alone, and in combination at constant 1:1 ratios of 1x their respective GI50 concentrations for 24 hours, to test the induction of PUMA (BBC3) as a TP53-related pro-apoptotic gene. Western blot analysis showed that rucaparib treatment as a single agent had no effect on p53 stabilization, upregulation of p21WAF1, MDM2 or PUMA compared to DMSO control, nor generally did rucaparib increase the effect of the MDM2 inhibitors on the p53 pathway (Figure 4 & Figure 5). Although rucaparib in combination with Nutlin-3/RG7388 appeared to increase stabilization of p53 and its downstream transcriptional targets in some cases in IGROV-1, otherwise there were no convincing differences. No evidence of synergy was observed at the molecular level to indicate that the mechanism of synergy for growth inhibition and apoptosis by rucaparib involved enhancement of the p53 pathway activation by MDM2 inhibitors (Figure 4 & Figure 5).
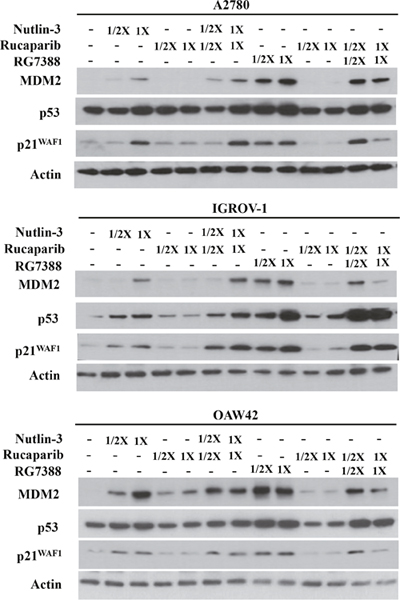
Figure 4: Combination of Nutlin-3/RG7388 with rucaparib increased stabilization of p53 and upregulation of its downstream targets, MDM2 and p21WAF1 compared to rucaparib on its own but not compared to Nutlin-3/RG7388. Total levels of p53, p21WAF1, MDM2 4 hours after the commencement of treatment with Nutlin-3 and RG7388 alone, and in combination with rucaparib at constant 1:1 ratios of 1/2X and 1X their respective GI50 concentration analyzed by western blot in three wild-type TP53 ovarian cancer cell lines.
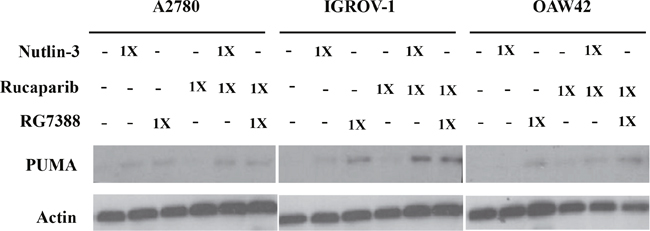
Figure 5: Combination of Nutlin-3/RG7388 with rucaparib increased upregulation of TP53 downstream target, PUMA compared to rucaparib on its own but not compared to Nutlin-3/RG7388. Total levels of PUMA 24 hours after the commencement of treatment with Nutlin-3 and RG7388 alone, and in combination with rucaparib at constant 1:1 ratios of 1X their respective GI50 concentration analyzed by western blot in three wild-type TP53 ovarian cancer cell lines.
Rucaparib in combination with Nutlin-3/RG7388 induces cell cycle distribution changes and/or apoptosis in wild-type TP53 ovarian cancer cell lines
Wild-type TP53 cell lines were treated with rucaparib and Nutlin-3/RG7388, alone and in simultaneous combination at constant 1:1 ratios of 1/2x and 1x their respective GI50 concentrations for 24, 48 and 72 hours. Then, they were analyzed by flow cytometry for cell cycle phase distribution changes and evidence of apoptosis in response to treatment.
Combination with Nutlin-3
Rucaparib on its own slightly increased the proportion of cells in G2/M phase and the number of SubG1 events in a dose and time-dependent manner. Combination treatment of rucaparib with Nutlin-3 resulted in an increased percentage of cells in the G2/M cell cycle phase compared to either agent alone, in a treatment time and dose-dependent manner for A2780 and IGROV-1 cell lines (Figure 6A & 6B). For the OAW42 cell line after 24 hours, the combination treatment led to an increased proportion of the cell population in the G2/M phase of the cell cycle compared to Nutlin-3 as a single agent. After 48 and 72 hours, there was little change in the cell cycle distribution following combination of rucaparib with Nutlin-3 compared to either agent alone (Figure 6C).
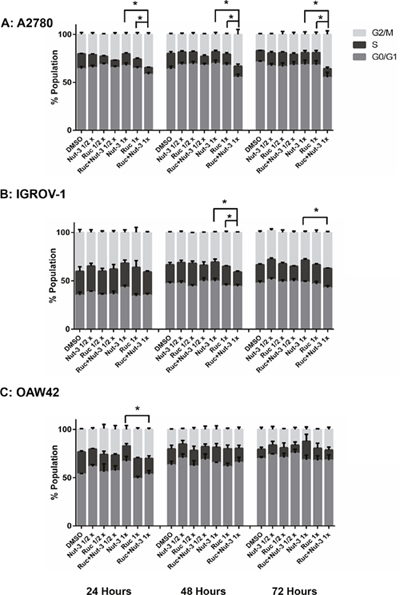
Figure 6: Combination of Nutlin-3 with rucaparib affects the cell cycle distribution. Wild-type TP53 ovarian cancer cells were treated for 24, 48 and 72 hours with Nutlin-3 or rucaparib alone and at constant 1:1 combination ratios of 1/2 X & 1X their respective GI50 concentrations. (A) A2780 cell line, (B) IGROV-1 cell line, (C) OAW42 cell line. Nut-3, Nutlin-3; Ruc, rucaparib; *, p < 0.05. Data are shown as the average of at least 3 independent experiments and error bars represent SEM.
The basal levels of apoptosis, indicated by SubG1 signals on FACS analysis, differed markedly between the cell lines, with much higher basal levels evident with the IGROV1 cells and least for the OAW42 cells. However, across all 3 cell lines, the trend was for combination treatments to induce an increased number of SubG1 signals in a concentration and time-dependent manner (Figure 7).
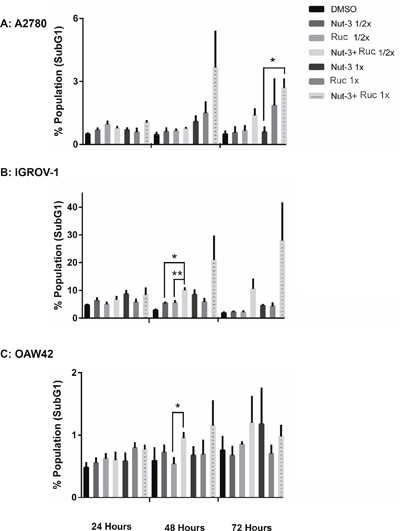
Figure 7: Combination of Nutlin-3 with rucaparib affects the apoptotic endpoints. FACS analysis for SubG1 events. Wild-type TP53 ovarian cancer cells were treated for 24, 48 and 72 hours with Nutlin-3 or rucaparib alone and at constant 1:1 combination ratios of 1/2 X & 1X their respective GI50 concentrations. (A) A2780 cell line, (B) IGROV-1 cell line, (C) OAW42 cell line. Nut-3, Nutlin-3; Ruc, rucaparib; *, p <0.05; **, P < 0.01. Data are shown as the average of at least 3 independent experiments and error bars represent SEM.
Combination with RG7388
For A2780 cells, the combination of rucaparib with RG7388 increased the proportion of cells in G2/M phase and SubG1 signals compared to either agent alone, in a treatment time and dose-dependent manner. The combination treatment also decreased the proportion of cells in S-phase compared to either agent alone (Figure 8A & 9A). In terms of the proportional distribution of IGROV-1 cells in G0/G1 or G2/M, the effect of rucaparib combination with RG7388 was time dependent. Combined treatment for 24 and 48 hours led to proportionally more cells in G0/G1 compared to the effect of rucaparib on its own and a higher proportion of cells in G2/M compared to the effect of RG7388 alone. After 72 hours treatment, the combination of rucaparib with RG7388 resulted in increased G2/M cell cycle arrest compared to RG7388 on its own, with little change in the percentage of cells in G0/G1 cell cycle compared to rucaparib alone. Combined treatments also increased the percentage of SubG1 signals compared to either agent alone, in a time and dose-dependent manner (Figure 8B & 9B). For OAW42 cells, combination of rucaparib with RG7388 led to proportionally more G2/M cells compared to either agent alone. It also decreased the percentage of cells in S-phase compared to either agent alone after 24 and 48 hours (Figure 8C & 9C).
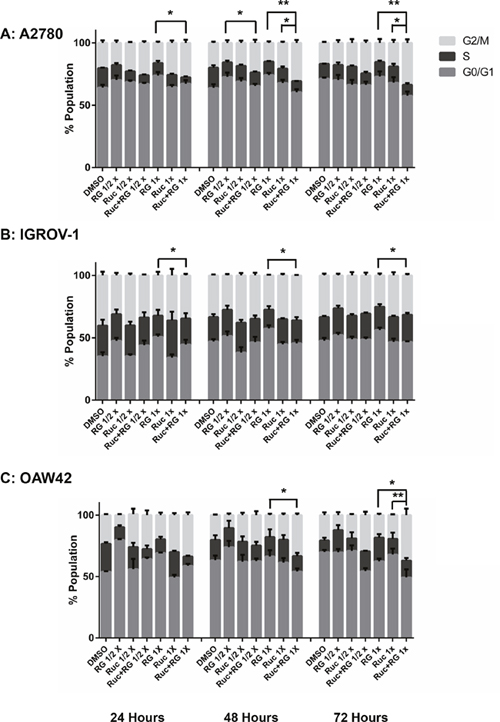
Figure 8: Combination of RG7388 with rucaparib affects the cell cycle distribution. Wild-type TP53 ovarian cancer cells were treated for 24, 48 and 72 hours with RG7388 or rucaparib alone and at constant 1:1 combination ratios of 1/2 X & 1X their respective GI50 concentrations. (A) A2780 cell line, (B) IGROV-1 cell line, (C) OAW42 cell line. RG, RG7388; Ruc, rucaparib; *, p <0.05; **, P < 0.01. Data are shown as the average of at least 3 independent experiments and error bars represent SEM.
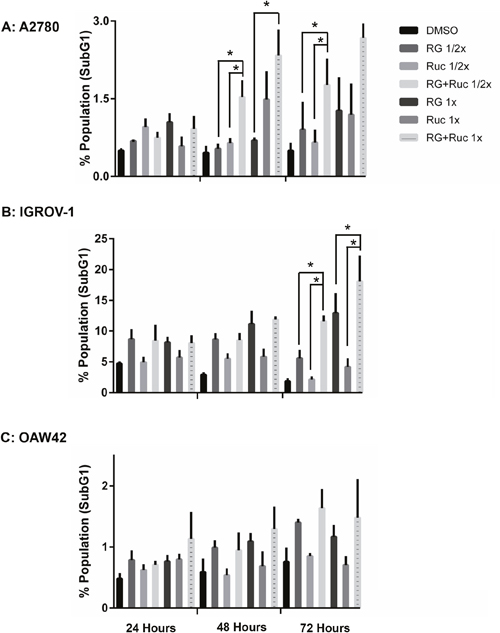
Figure 9: Combination of RG7388 with rucaparib affects the apoptotic endpoints. FACS analysis for SubG1 events. Wild-type TP53 ovarian cancer cells were treated for 24, 48 and 72 hours with RG7388 or rucaparib alone and at constant 1:1 combination ratios of 1/2 X & 1X their respective GI50 concentrations. (A) A2780 cell line, (B) IGROV-1 cell line, (C) OAW42 cell line. RG, RG7388; Ruc, rucaparib; *, p <0.05. Data are shown as the average of at least 3 independent experiments and error bars represent SEM.
Caspase 3/7 enzymatic activity was assessed to evaluate induction of apoptosis. Wild-type TP53 ovarian cancer cell lines were treated for 24 hours with 1x their respective Nutlin-3/RG7388 GI50 concentrations as a single agent and in combination with rucaparib (Figure 10). For A2780 and OAW42 there was no significant increase in the caspase 3/7 activity in response to Nutlin-3/RG7388 alone. Also no significant increase was observed for the combination of Nutlin-3/RG7388 with rucaparib compared with the effect of either agent alone in the A2780 and OAW42 cells. In contrast, there was a significant increase in caspase 3/7 activity in response to both Nutlin-3 and RG7388 as single agents in IGROV-1. Furthermore, the combination of Nutlin-3/RG7388 with rucaparib resulted in more caspase 3/7 activity in IGROV-1 compared to either agent alone (Figure 10).
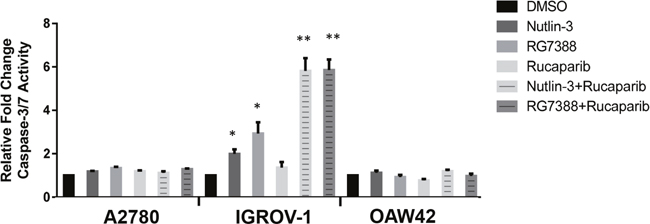
Figure 10: Combinations of Nutlin-3 or RG7388 with rucaparib affect caspase3/7 activity. The wild-type TP53 ovarian cancer cells treated at constant 1:1 ratios of 1x their respective GI50 concentrations of Nutlin-3, RG7388 or rucaparib alone, and in combination for 24 hours. Caspase 3/7 activity is represented as fold change relative to DMSO solvent control. *, The single star represents a significant increase in the caspase3/7 activity compared to DMSO control (p<0.05); **, The double stars represent a significant increase in the caspase3/7 activity compared to Nutlin-3, RG7388 or rucaparib alone (p<0.01).
DISCUSSION
PARP inhibitors have been developed over a number of decades and in more recent years have been shown to have efficacy as single agents against tumor cells with intrinsic deficiencies in DNA repair. They currently have been undergoing clinical trials in different types of cancers, including ovarian cancer [23–25]. Encouraging clinical trial results for the use of PARP inhibitors have been reported for ovarian cancer [11, 26]. The promising results showing activity of MDM2-p53 antagonists against wild-type TP53 ovarian cancer cell lines [16, 27] have prompted this investigation of combination treatment with rucaparib and Nutlin-3/RG7388 in a panel of wild-type TP53 ovarian cancer cell lines. Mechanistically it was of interest to investigate the combined effect of PARP inhibition and MDM2-p53 binding antagonists on the p53 pathway activation. This study evaluates for the first time the effect of the MDM2-p53 binding antagonists Nutlin-3 and RG7388 in combination with rucaparib in wild-type TP53 ovarian cancer cell lines and explores the interplay between the p53 pathway and PARP dependent pathways modulated by rucaparib.
Within the panel of ovarian cancer cell lines studied, wild-type TP53 cell lines were significantly more sensitive to Nutlin-3 and the more potent RG7388 compared to mutant TP53 cell lines, which is consistent with their mechanism of action [28]. Following the scheme suggested by Mukhopadhyay et al, the 10 μM cut-off value was used to categorize cell lines into sensitive and resistant to PARP inhibitor [17]. Among the individual cell lines, A2780 and MDAH-2774 were sensitive (GI50 < 10 μM) and other cell lines (IGROV-1, OAW42, CP70, MLH1-corrected CP70+ and SKOV-3) were resistant (GI50 > 10 μM). No relationship was found between the TP53 status of cells and response to rucaparib. These results are in line with those of previous studies reporting the sensitivity of A2780 and resistance of OAW42, IGROV-1, CP70 and SKOV-3 in response to rucaparib [29, 30]. Overall, the GI50 values for rucaparib across all cell lines were much higher than the concentration needed to reduce the cell-free enzymatic activity by half, Ki=1.4 nM [31]. However, they were in the range of rucaparib concentrations achievable in vivo and used in clinical trials [10, 11, 26, 32].
To explore the potential mechanism involved in the sensitivity of A2780 and MDAH-2774 and resistance of other cell lines to rucaparib, the status of genes implicated as biomarkers in response to PARP inhibitors were studied [29, 33–37]. The most interesting finding is that the A2780 cell line was the most sensitive cell line even though among the relevant genes previously implicated in the sensitivity to rucaparib it has only a heterozygous mutation in PTEN (Deletion-In frame, heterozygous, (c.380_388delGAAAGGGAC) (89682879_89682887delAGGGACGAA) (Supplementary Table 1). MDAH-2774 is an ovarian endometrioid tumor with mutant TP53 and KRAS [22].
Overall, the reasons for the greater sensitivity of A2780 and MDAH-2774 to rucaparib are not clear. Further research is needed to identify reliable biomarkers to stratify patients who will benefit from treatment with PARP inhibitors.
Resistance to MDM2-p53 binding antagonists has been suggested to be acquired by prolonged exposure of cells to sub-lethal doses through de novo inactivating TP53 mutations or selection of pre-existing subclones of TP53 mutant cells that might be present as a result of cancer cell genomic instability and tumor heterogeneity [38, 39]. Development of resistance to PARP inhibitors occurs through different mechanisms. For example, acquired resistance to PARP inhibitors resulting from a secondary mutation of the BRCA gene has been confirmed to occur in patients [10]. Combination therapy is suggested to delay or prevent drug resistance. Other major benefits of combination therapy are the potential for a synergistic therapeutic effect, or at least the possibility of dose and toxicity reduction [40, 41].
This study set out with the aim of assessing the effect of combination treatment of Nutlin-3 or RG7388 with rucaparib in a panel of ovarian cancer cell lines of known TP53 status as combined targeted therapeutics. Updated results from the ARIEL2 clinical trial of rucaparib in 152 patients with wild-type BRCA1/2 who were sensitive to platinum reported a response rate of 36% in those with BRCA-like DNA repair deficiency status. Less common mutations involved in Homologous Recombination Deficiency (HRD) and predictive of response to platinum may be present in almost one third (33%) of ovarian cancer patients [42]. Almost 30% of ovarian cancers include histological subtypes other than high grade serous and these are mostly TP53 wild-type. Thus nearly 10% of ovarian cancer patients (one third of the 30% not high grade serous) who are likely to be sensitive to both MDM2 inhibitors and rucaparib may benefit from combination treatment with these agents. In addition, almost 4% of high grade serous ovarian cancers nevertheless have wild-type TP53 and these mostly have BRCA1/2 mutation or other HRD status [42], rendering them sensitive to both MDM2 inhibitors and rucaparib and therefore likely to benefit from the combined treatment. Furthermore, those ovarian tumours which include a histological mixture of high grade serous with clear cell, mucinous, endometrioid or low grade serous might gain benefit from this combination treatment to target the different components.
Overall, the effect of combined treatment was cell type and exposure time dependent, with a higher synergistic effect for the combination of Nutlin-3/RG7388 with rucaparib for A2780 and IGROV-1 cell lines. Interestingly, although both IGROV-1 and OAW42 were resistant to rucaparib, there was nevertheless a synergistic effect for the combination of Nutlin-3/RG7388 with rucaparib. A possible explanation for this is that the defects conferring sensitivity to PARP inhibitors as single agents may be different from those which play an important role in the response to the combination treatment. For example, serious deficiencies in HRR may affect the sensitivity to PARP inhibitors as single agents, while mild defects in HRR may have no effect on response to PARP inhibitors alone but may nevertheless influence the effect of combination treatments [8, 29]. Another possibility is that different p53-dependent off-target effects of these MDM2 inhibitors [43, 44] and/or off-target effects of rucaparib with respect to PARP1 and PARP2 may influence the growth inhibitory effect of combined treatment compared to rucaparib as a single agent [45, 46].
An important and clinically relevant finding from the data is the favorable DRI values in both combination treatment of Nutlin-3 or RG7388 with rucaparib. Additive and even mildly antagonistic results of combined treatment can nevertheless be of potential clinical use due to favorable DRI values [47, 48]. These favorable DRI values demonstrate that combination of Nutlin-3 or RG7388 with rucaparib has the potential to reduce the dose of drugs in most cases to achieve the same overall level of effect, indicating a potential clinical benefit of combining these therapeutic agents, particularly when they have differing dose limiting toxicities.
Rucaparib on its own had no effect on p53 stabilization and upregulation of its downstream targets p21WAF1, MDM2 and PUMA across all three cell lines. Combination treatment of Nutlin-3 or RG7388 with rucaparib induced stabilization of p53 and upregulation of p21WAF1, MDM2 and PUMA compared to rucaparib on its own, whereas rucaparib had no enhancement of the p53 activation by MDM2 inhibitors alone. These results demonstrate that the synergistic effect on growth inhibition observed for the combination of rucaparib and Nutlin-3/RG7388 is not the result of an accentuated p53 response and hence not related to the p53 molecular pathway. The interplay between PARP and p53 is controversial which may be related to the type of DNA damage, type of PARP inhibitors and intensity of replicative stress [49, 50].
Individually, Nutlin-3 and RG7388 induced cell cycle arrest in wild-type TP53 ovarian cancer cell lines in a time and dose-dependent manner. Rucaparib had little effect on the cell cycle distribution for IGROV-1 and OAW42 cell lines, which is in agreement with the results obtained by Porcelli et al. [51] that indicated no effect of rucaparib on the cell cycle progression of pancreatic cancer cells. However, in the current study rucaparib significantly decreased the proportion of cells in S-phase in A2780 cells, consistent with a recent study indicating a robust decrease in the percentage of cells in S-phase following treatment of U2OSDR-GFP cells with olaparib [50]. Furthermore, there was only a slight increase in the SubG1 cell subpopulation across all cell lines treated with rucaparib compared to DMSO control, suggesting that cells are not undergoing apoptosis. These results are in line with those of Jelinic and Levine who observed low SubG1 events in cancer cells treated with olaparib or veliparib [49].
Across all three cell lines, combined treatment of Nutlin-3/RG7388 with rucaparib increased the proportion of cells in the G2/M phase of the cell cycle, which was marked for A2780 and IGROV-1. This can be explained by induction of p21WAF1 expression in response to MDM2 inhibitors and its critical role in cell cycle arrest in G2/M by inhibiting mitotic Cdk complexes and Rb phosphorylation [52]. Also, p53-mediated repression of FoxM1 results in the maintenance of a stable G2/M arrest which is partially p21 dependent [53]. The increased proportion of cells in G2/M phase following PARP inhibitor treatments may reflect their mechanism of action during DNA replication in S-phase, which is to trap PARP-1 and -2 to DNA and induce a replicative stress response [50]. Combination treatment with Nutlin-3 or RG7388 and rucaparib also led to more SubG1 events and/or higher levels of caspase 3/7 activity compared to either agent alone in a cell type dependent manner which was marked for IGROV-1 cells.
In conclusion, the present study demonstrates that combination treatment with MDM2 inhibitors and rucaparib has synergistic and/or dose reduction potential dependent on cell genotype and compound and merits further investigation. Monitoring HRR status, cell cycle markers, PARP expression and PARP activity are likely to provide additional useful information to assess the effectiveness of PARP inhibitors. This information may be helpful to stratify the patients who might benefit from PARP inhibitors and their potential combination with the emerging class of MDM2 inhibitors.
MATERIALS AND METHODS
Chemicals and antibodies
Nutlin-3, a 1:1 mixture of the active enantiomer Nutlin-3a and the inactive enantiomer Nutlin-3b, was purchased from NewChem (Newcastle, UK) and RG7388 (Idasanutlin) was synthesized by Ruth Bawn and Amy Heptinstall within the Newcastle Drug Discovery Group. Both were dissolved in dimethyl sulfoxide (DMSO). Rucaparib was kindly supplied by Clovis Oncology, and prepared in 10mM stocks solubilized in DMSO.
Cell lines
The ovarian cancer cell lines used in this study, their TP53 status and histological subtype are listed in Table 1. All cell lines were sourced from the NICR authenticated cell bank and regularly tested for Mycoplasma. A2780, IGROV-1, OAW42 and CP70 were cultured in RPMI-1640 supplemented with 10% (v/v) FBS and 5% (v/v) penicillin/streptomycin. The CP70 cell line harbors a heterozygous TP53 mutation (c.514 G->T, p.Val172Phe) [54]. The MLH1-corrected CP70+ cell line was grown in RPMI-1640 supplemented with 10% (v/v) FBS and Hygromycin B (200 μg/ml: Life Technologies, Inc.) [54]. This cell line has the heterozygous TP53 mutation (c.514 G->T, p.Val172Phe). SKOV-3 and MDAH-2774 cell lines were cultured in DMEM supplemented with 10% and 5% (v/v) FBS and penicillin/streptomycin respectively. As information on the TP53 status of SKOV-3 and IGROV-1 in the literature was contradictory, sequencing was performed. The results of PCR-based Sanger sequencing of TP53 exon 5 and exon 4 confirmed the wild-type TP53 status of the IGROV-1 cell line (Supplementary Figure 1). For the SKOV-3 cell line, frame shift deletion (c.265delC, p.Pro89fsX33) was confirmed in TP53 exon 4 and no substitution mutation (c.179 A->G) was detected in TP53 exon 5 (Supplementary Figure 2). The MDAH-2774 cells were confirmed to harbor a TP53 mutation located in exon 8 (c.818G->A, p.Arg273His) [55].
Growth inhibition assays and median-effect analysis
The GI50 values, the required concentrations of each compound leading to 50% growth inhibition, were determined by Sulforhodamine B (SRB) growth inhibition assays for drug exposure over 72 hours and the absorbance of the re-dissolved SRB protein stain was measured at 570 nm using a 96-well plate spectrophotometer (Spectramax 250 Molecular Devices) [56]. Growth curves were constructed using GraphPad Prism statistical analysis software version 5.04. For combination treatment of Nutlin-3 or RG7388 with rucaparib, the wild-type TP53 cell lines were treated for 72 hours with each agent alone and in combination simultaneously at constant 1:1 ratios of 0.25x, 0.5x, 1x, 2x, and 4x their respective GI50 concentrations. Median-effect analysis was used to calculate Combination Index (CI) and Dose Reduction Index (DRI) values [57] using CalcuSyn software v2 (Biosoft, Cambridge, UK).
Western blotting
Lysis buffer (12.5 ml Tris HCL, 2g SDS, 10 ml Glycerol, 67.5 ml Distilled Water) was used to harvest whole-cell lysates, followed by sonication. The concentration of protein in the cell lysates was estimated by using a bicinchoninic acid (BCA) assay. Novex® 4-20% Tris-Glycine 12-well polyacrylamide gradient gels (Invitrogen, UK) were used to separate proteins. The separated proteins were transferred by perpendicular electrophoresis to a nitrocellulose Hybond™ C membrane (Amersham, Buckinghamshire, UK). Monoclonal Mouse Anti-Human primary antibodies Actin 1:1000 (#: A4700, Sigma-Aldrich), MDM2 1:300 (#: OP46-100UG, Merck Millipore), p21WAF1 1:100 (#: OP64, Calbiochem), PUMA 1:1000 (#dd716, Santa Cruz Biotechnology) and p53 1:500 (#: NCL-L-p53-DO7, Leica Microsystems Ltd.) were used. Secondary goat anti-mouse HRP-conjugated antibodies (#: P0447/P0448, Dako) were used at 1:1000. All antibodies were diluted in 5% milk/1XTBS-Tween (w/v). Enhanced chemiluminescence (GE Life Sciences) and X-ray film (Fujifilm) were used to visualize the proteins.
Flow cytometry
Cells were treated with Nutlin-3, RG7388 and rucaparib alone and with combinations of Nutlin-3 or RG7388 with rucaparib simultaneously at constant 1:1 ratios of 0.5x and 1x their respective GI50 concentrations for 24, 48 and 72 hours. Harvested cells, both floating and adherent, were washed with PBS and resuspended in 500 μL PBS with 1mg/mL sodium citrate (Sigma, St Louis, MO), 100 μg/mL propidium iodide (Sigma), 200 μg/mL RNAse A (Sigma) and 0.3% Triton-X (Sigma). Samples were analyzed on a FACSCalibur™ flow cytometer using CellQuest Pro software (Becton Dickinson, Oxford, UK). Cell cycle distribution was determined using Cyflogic (CyFlo Ltd, Turku, Finland).
Caspase 3/7 activity assay
Caspase 3/7 activity was measured using a Caspase- Glo 3/7 assay following the manufacturer’s instructions (Promega, Southampton, UK).
Statistical analysis
All statistical tests presented were carried out using GraphPad Prism version 5.04 software. A p-value of <0.05 was considered to be statistically significant based on at least n=3 experimental repeats.
ACKNOWLEDGMENTS
The authors would like to thank Professor Herbie Newell, members of Drug Discovery in the NICR, Newcastle University and Clovis Oncology for the provision of rucaparib.
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
GRANT SUPPORT
The authors gratefully acknowledge the funding bodies that allowed us to conduct these studies, including Cancer Research UK (C240/A1571; JL & C27826/A11524; JL & RJE), Northern Cancer Care & Research Society (RJE), Newcastle University & Northern Institute for Cancer Research (C0190R4011; MZ).
REFERENCES
1. Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray, F. GLOBOCAN 2012 v1.0. Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. Lyon, France: International Agency for Research on Cancer; 2013.
2. Kim A, Ueda Y, Naka T, Enomoto T. Therapeutic strategies in epithelial ovarian cancer. J Exp Clin Cancer Res. 2012; 31:14-14.
3. Köbel M, Kalloger SE, Boyd N, McKinney S, Mehl E, Palmer C, Leung S, Bowen NJ, Ionescu DN, Rajput A, Prentice LM, Miller D, Santos J, et al. Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med. 2008; 5:e232.
4. Makii C, Oda K, Ikeda Y, Sone K, Hasegawa K, Uehara Y, Nishijima A, Asada K, Koso T, Fukuda T, Inaba K, Oki S, Machino H, et al. MDM2 is a potential therapeutic target and prognostic factor for ovarian clear cell carcinomas with wild type TP53. Oncotarget. 2016; 7:75328-75338. https://doi.org/10.18632/oncotarget.12175.
5. Luvero D, Milani A, Ledermann JA. Treatment options in recurrent ovarian cancer: latest evidence and clinical potential. Ther Adv Med Oncol. 2014; 6:229-239.
6. Lupo B, Trusolino L. Inhibition of poly(ADP-ribosyl)ation in cancer: Old and new paradigms revisited. Biochim BiophysActa. 2014; 1846:201-215.
7. Michels J, Vitale I, Saparbaev M, Castedo M, Kroemer G. Predictive biomarkers for cancer therapy with PARP inhibitors. Oncogene. 2014; 33:3894-3907.
8. Turner N, Ashworth A. Biomarkers of PARP inhibitor sensitivity. Breast Cancer Res Treat. 2011; 127:283-286.
9. Underhill C, Toulmonde M, Bonnefoi H. A review of PARP inhibitors: from bench to bedside. Ann Oncol. 2010; 22:268-79.
10. Weil MK, Chen A. PARP Inhibitor Treatment in Ovarian and Breast Cancer. Curr Probl Cancer. 2011; 35:7-50.
11. Brown JS, Kaye SB, Yap TA. PARP inhibitors: the race is on. Br J Cancer. 2016; 114:713-715.
12. Vu B, Wovkulich P, Pizzolato G, Lovey A, Ding Q, Jiang N, Liu JJ, Zhao C, Glenn K, Wen Y, Tovar C, Packman K, Vassilev L, Graves B. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med Chem Lett. 2013; 4:466-469.
13. Ribeiro C, Rodrigues C, Moreira R, Santos M. Chemical variations on the p53 reactivation theme. Pharmaceuticals. 2016; 9:25.
14. Lu M, Xia L, Li Y, Wang X, Hoffman R. The orally bioavailable MDM2 antagonist RG7112 and pegylated interferon alpha 2a target JAK2V617F-positive progenitor and stem cells. Blood. 2014; 124:771-779.
15. Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J. MDMX Overexpression Prevents p53 Activation by the MDM2 Inhibitor Nutlin. J Biol Chem. 2006; 281:33030-33035.
16. Mir R, Tortosa A, Martinez-Soler F, Vidal A, Condom E, Perez-Perarnau A, Ruiz-Larroya T, Gil J, Gimenez-Bonafe P. Mdm2 antagonists induce apoptosis and synergize with cisplatin overcoming chemoresistance in TP53 wild-type ovarian cancer cells. Int J Cancer. 2013; 132:1525-1536.
17. Mukhopadhyay A, Plummer ER, Elattar A, Soohoo S, Uzir B, Quinn JE, McCluggage WG, Maxwell P, Aneke H, Curtin NJ, Edmondson RJ. Clinicopathological features of homologous recombination–deficient epithelial ovarian cancers: sensitivity to PARP inhibitors, platinum, and survival. Cancer Res. 2012; 72:5675-5682.
18. Kummar S, Chen HX, Wright J, Holbeck S, Millin MD, Tomaszewski J, Zweibel J, Collins J, Doroshow JH. Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements. Nat Rev Drug Discov. 2010; 9:843-856.
19. Pizao PE, Lyaruu DM, Peters GJ, van Ark-Otte J, Winograd B, Giaccone G, Pinedo HM. Growth, morphology and chemosensitivity studies on postconfluent cells cultured in 'V'-bottomed microtiter plates. Brit J Cancer. 1992; 66:660-665.
20. Hills CA, Kelland LR, Abel G, Siracky J, Wilson AP, Harrap KR. Biological properties of ten human ovarian carcinoma cell lines: calibration in vitro against four platinum complexes. Brit J Cancer. 1989; 59:527-534.
21. Bénard J, Da Silva J, De Blois MC, Boyer P, Duvillard P, Chiric E, Riou G. Characterization of a human ovarian adenocarcinoma line, IGROV1, in tissue culture and in nude mice. Cancer Res. 1985; 45:4970-4979.
22. Gilloteaux J, Lau HL, Gourari I, Neal D, Jamison JM, Summers JL. Apatone® induces endometrioid ovarian carcinoma (MDAH 2774) cells to undergo karyolysis and cell death by autoschizis: A potent and safe anticancer treatment. Trans Res Anat. 2015; 1:25-39.
23. Anwar M, Aslam H, Anwar S. PARP inhibitors. Hereditary Cancer Clin Pract. 2015; 13:4.
24. Benafif S, Hall M. An update on PARP inhibitors for the treatment of cancer. Onco Targets Ther. 2015; 8:519-528.
25. Frey MK, Pothuri B. Targeting DNA repair: poly (ADP-ribose) polymerase inhibitors. Translational Cancer Res. 2015; 4:84-96.
26. Drew Y, Ledermann J, Hall G, Rea D, Glasspool R, Highley M, Jayson G, Sludden J, Murray J, Jamieson D, Halford S, Acton G, Backholer Z, et al. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br J Cancer. 2016; 114:723-730.
27. Zanjirband M, Edmondson RJ, Lunec J. Pre-clinical efficacy and synergistic potential of the MDM2-p53 antagonists, Nutlin-3 and RG7388, as single agents and in combined treatment with cisplatin in ovarian cancer. Oncotarget. 2016; 7:40115-40134. https://doi.org/10.18632/oncotarget.9499.
28. Moll UM, Petrenko O. The MDM2-p53 Interaction. Mol Cancer Res. 2003; 1:1001-1008.
29. Ihnen M, zu Eulenburg C, Kolarova T, Qi JW, Manivong K, Chalukya M, Dering J, Anderson L, Ginther C, Meuter A, Winterhoff B, Jones S, Velculescu VE, et al. Therapeutic potential of the poly(ADP-ribose) polymerase inhibitor rucaparib for the treatment of sporadic human ovarian cancer. Mol Cancer Ther. 2013; 12:1002-1015.
30. McCormick A, Dixon M, Odonnell R, Curtin NJ, Edmondson RJ. Abstract PR06: ovarian cancers harbor defects in nonhomologous end joining resulting in error prone repair and resistance to rucaparib. Clin Cancer Res. 2013; 19:PR06.
31. Thomas HD, Calabrese CR, Batey MA, Canan S, Hostomsky Z, Kyle S, Maegley KA, Newell DR, Skalitzky D, Wang LZ, Webber SE, Curtin NJ. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol Cancer Ther. 2007; 6:945-956.
32. Javle M, Curtin NJ. The potential for poly (ADP-ribose) polymerase inhibitors in cancer therapy. Ther Adv Med Oncol. 2011; 3:257-267.
33. cancer.sanger.ac.uk. In: Institute S, ed.
34. Stordal B, Timms K, Farrelly A, Gallagher D, Busschots S, Renaud M, Thery J, Williams D, Potter J, Tran T, Korpanty G, Cremona M, Carey M, et al. BRCA1/2 mutation analysis in 41 ovarian cell lines reveals only one functionally deleterious BRCA1 mutation. Mol Oncol. 2013; 7:567-579.
35. Lim SL, Smith P, Syed N, Coens C, Wong H, van der Burg M, Szlosarek P, Crook T, Green JA. Promoter hypermethylation of FANCF and outcome in advanced ovarian cancer. Br J Cancer. 2008; 98:1452-1456.
36. Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, Mok SC, D'Andrea AD. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003; 9:568-574.
37. Olopade OI, Wei M. FANCF methylation contributes to chemoselectivity in ovarian cancer. Cancer Cell. 2003; 3:417-420.
38. Aziz MH, Shen H, Maki CG. Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene. 2011; 30:4678-4686.
39. Shen H, Moran DM, Maki CG. Transient Nutlin-3a treatment promotes endoreduplication and the generation of therapy-resistant tetraploid cells. Cancer Res. 2008; 68:8260-8268.
40. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010; 70:440-446.
41. Zhang N, Wu ZM, McGowan E, Shi J, Hong ZB, Ding CW, Xia P, Di W. Arsenic trioxide and cisplatin synergism increase cytotoxicity in human ovarian cancer cells: Therapeutic potential for ovarian cancer. Cancer Sci. 2009; 100:2459-2464.
42. Ledermann JA, Drew Y, Kristeleit RS. Homologous recombination deficiency and ovarian cancer. Eur J Cancer. 2016; 60:49-58.
43. Hoe KK, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014; 13:217-236.
44. Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012; 72:560-567.
45. Murai J, Huang SN, Renaud A, Zhang Y, Ji J, Takeda S, Morris J, Teicher B, Doroshow JH, Pommier Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014; 13:433-443.
46. Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J Pharmacol Exp Ther. 2014; 349.
47. Ohnstad HO, Paulsen EB, Noordhuis P, Berg M, Lothe RA, Vassilev LT, Myklebost O. MDM2 antagonist Nutlin-3a potentiates antitumour activity of cytotoxic drugs in sarcoma cell lines. BMC Cancer. 2011; 11:211-211.
48. Meng G, Wang WEI, Chai K, Yang S, Li F, Jiang KAI. Combination treatment with triptolide and hydroxycamptothecin synergistically enhances apoptosis in A549 lung adenocarcinoma cells through PP2A-regulated ERK, p38 MAPKs and Akt signaling pathways. Int J Oncol. 2015; 46:1007-1017.
49. Bai XT, Moles R, Chaib-Mezrag H, Nicot C. Small PARP inhibitor PJ-34 induces cell cycle arrest and apoptosis of adult T-cell leukemia cells. J Hematol Oncol. 2015; 8:117.
50. Jelinic P, Levine DA. New insights into PARP inhibitors' effect on cell cycle and homology-directed DNA damage repair. Mol Cancer Ther. 2014; 13:1645-1654.
51. Porcelli L, Quatrale AE, Mantuano P, Leo MG, Silvestris N, Rolland JF, Carioggia E, Lioce M, Paradiso A, Azzariti A. Optimize radiochemotherapy in pancreatic cancer: PARP inhibitors a new therapeutic opportunity. Mol Oncol. 2013; 7:308-322.
52. Gire V, Dulić V. Senescence from G2 arrest, revisited. Cell Cycle. 2015; 14:297-304.
53. Barsotti AM, Prives C. Pro-proliferative FoxM1 is a target of p53-mediated repression. Oncogene. 2009; 28:4295-4305.
54. Lu X, Errington J, Curtin NJ, Lunec J, Newell DR. The impact of p53 status on cellular sensitivity to antifolate drugs. Clin Cancer Res. 2001; 7:2114-2123.
55. Dai L, Li C, Shedden KA, Misek DE, Lubman DM. Comparative proteomic study of two closely related ovarian endometrioid adenocarcinoma cell lines using cIEF fractionation and pathway analysis. Electrophoresis. 2009; 30:1119-1131.
56. Houghton P, Fang R, Techatanawat I, Steventon G, Hylands PJ, Lee CC. The sulphorhodamine (SRB) assay and other approaches to testing plant extracts and derived compounds for activities related to reputed anticancer activity. Methods. 2007; 42:377-387.
57. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006; 58:621-681.