
INTRODUCTION
Base excision repair (BER) is a critical and highly conserved mechanism for the repair of damage induced by alkylation and oxidation of DNA, including by chemotherapy and ionising radiation [1]. Abasic sites (also known as apurinic/apyrimidinic or AP sites) are cytotoxic obligate repair intermediates generated during BER and processed by human apurinic/apyrimidinic endonuclease 1 (APE1). APE1 cleaves the phosphodiester DNA backbone 5’ to the AP site prior to further processing via either the short patch or the long patch BER pathway. Unrepaired AP sites generate single strand breaks, which stall replication fork progression and induce DNA double strand breaks (DSBs) that are toxic to the cell at high density [2].
APE1 is a multifunctional protein [1, 3]. In addition to BER functions, it possesses N-terminus redox activity, which can activate pro-angiogenic and pro-survival transcription factors. APE1 also has roles in acetylation-mediated gene regulation and RNA quality control [4]. SiRNA-mediated APE1 downregulation induces AP site accumulation and is associated with hypersensitivity to DNA damaging agents, including alkylators and ionising radiation [1]. Overexpression of APE1 confers resistance to these agents, both in vitro and in vivo [1]. Furthermore, exposure to alkylating agents causes upregulation of endogenous APE1 levels, suggesting a role in the development of treatment resistance [5]. APE1 expression in human tumours may have prognostic or predictive significance in patients [1].
In light of the evidence presented above, APE1 is an emerging anti-cancer drug target. [1, 3]. We have initiated drug development programmes to identify novel inhibitors of APE1 DNA repair function [6-11]. Several of these compounds have shown promising preclinical activity, including the potentiation of the cytotoxicity of the alkylating agent temozolomide in cancer cell lines. More recently, we have demonstrated synthetic lethality of APE1 inhibition in BRCA-deficient cell systems [12], analagous to results observed with PARP inhibitors currently under development for treatment of HR-deficient cancer [13, 14].
Phosphatase and tensin homolog (PTEN) is a negative regulator of the anti-apoptotic PI3K/Akt pathway [15]. PTEN mutation is reported in 5-20% of primary melanomas, although PTEN mutation is more frequently seen in melanoma cell lines (30-50%) [16, 17]. Furthermore, transcriptional and translational repression of PTEN function has been reported in up to 65% of melanomas [18]. In addition to its inositol phosphatase function, PTEN has recently been implicated in the maintenance of genomic integrity [19-21]. PTEN-null cells are associated with centromere-clustered chromosome breakages, possibly due to interaction with the centromeric protein CENP-C, which is vital for centromeric stability during mitosis. Additionally, PTEN might function as a transcriptional regulator of the critical homologous recombination (HR) protein RAD51 via the transcription factor Egr-1 [19-21]. Alternatively, PTEN loss may be associated with altered expression of the RAD51 paralogs [22] or impaired HR factor recruitment to DNA damage due to cell cycle checkpoint defects [20]. SUMOylation may be essential for PTEN DNA repair functions by directing nuclear PTEN localisation, with PTEN-null or non-SUMOylatable mutant cells exhibiting enhanced sensitivity to DNA damaging agents [23]. In keeping with these findings, PTEN -/- cells have been demonstrated to possess a HR defect that is associated with synthetic lethality following PARP inhibitor exposure [24]. However, although an association between RAD51 deficiency, impaired HR and PTEN deficiency has been demonstrated in colorectal cancer cells [24] and endometrial cancer cells [25], the association was not demonstrated in prostate cancer models [22].
Loss of PTEN may promote melanoma development [26], possibly as a cooperating mutation with BRAF V600E [27]. Oncogenic BRAF V600 driver mutations have recently emerged as a key therapeutic target [28], leading to the development of vemurafanib [29]. Despite BRAF-targeted therapy, many patients eventually progress and succumb to the disease. Interestingly, a recent study has suggested that PTEN loss may contribute to BRAF inhibitor resistance in melanoma [30]. Therefore, development of therapeutic strategies targeting PTEN deficiency is highly desirable. In the current study, we hypothesised a synthetic lethal relationship between PTEN and APE1 in melanoma. We have measured mRNA expression of PTEN and APE1 in 191 human melanomas and correlated this with clinical and pathological factors. We have confirmed the utility of APE1 inhibitors in the presence of PTEN deficiency in melanoma cell lines.
RESULTS
Prognostic significance of PTEN mRNA and APE1 mRNA expression in human melanomas
Patient demographics of the 191 cases are summarized in Supplementary Table S1. The clinicopathological association data are summarised in Supplementary Table S2. Relapse free and overall survival data are summarized in Supplementary Table S3. Low PTEN and high APE1 mRNA expression associated with presence of vascular invasion (p=0.05) and high mitotic rate (p=0.4), respectively. In the whole cohort (n=191), low PTEN mRNA expression was significantly associated with poor relapse free survival and overall survival (Supplementary Table S3 and Figure 1A). High APE1 mRNA expression was also significantly associated with poor relapse free survival and overall survival (supplementary Table S3 and Figure 1B) in the whole cohort. When PTEN and APE1 are considered together, patients with tumours that exhibit high PTEN and low APE1 mRNA expression have a significantly better prognosis compared to tumours that have low PTEN/high APE1 mRNA expression or low PTEN/low APE1 mRNA expression or high PTEN/high APE1 mRNA expression (Figure 1C).
Given the evidence in murine models that low PTEN and BRAF V600 mutations results in development of metastatic melanoma [27], we conducted an exploratory analysis based on PTEN mRNA expression and BRAF/NRAS status. None of the patients with BRAF V600 mutation had received vemurafanib (BRAF inhibitor) therapy. In tumours that had no BRAF or NRAS mutation, low PTEN was significantly associated with poor overall survival [HR 95%CI)=0.27 (0.12, 0.64), P=0.003] (Figure 1D). In tumours that have BRAF mutation (Figure 1B) [HR (95%CI)=0.81 (0.46, 1.43), p=0.47] and in NRAS mutants, low PTEN level did not significantly influence prognosis (Figure 1C) [HR 95%CI)=0.88 (0.29, 2.69), p=0.82]. Taken together, the data suggest that PTEN deficiency is a promising prognostic marker in BRAF wild type melanomas.
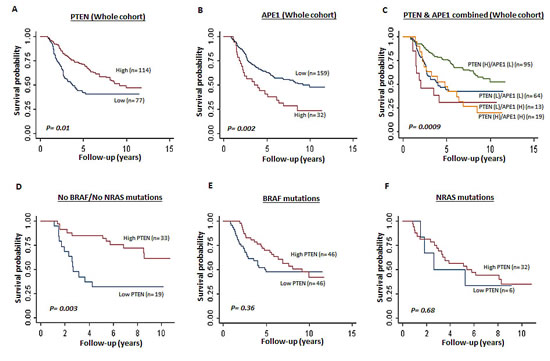
Figure 1: Kaplan Meier curves showing overall survival in melanoma. A. Whole cohort (PTEN mRNA high and low, p=0.01). B. Whole cohort (APE1 mRNA high and low, p=0.002). C. Whole cohort (PTEN and APE1 m RNA combined high and low, p=0.0009). D. Tumours with no BRAF/NRAS mutations (PTEN mRNA high and low, p=0.003). E. Tumours with BRAF V600E mutation (PTEN mRNA high and low, p=0.36). F. Tumours with NRAS mutations (PTEN mRNA high and low, p=0.68).
PTEN-deficient melanoma cell lines exhibit altered DSB repair protein levels
The emerging role of PTEN in DNA repair [23] and the association between low PTEN and impaired DNA DSB repair [19-21] implies that PTEN- deficient melanoma cells with defective DSB repair may be more reliant upon BER to maintain genomic stability. To investigate the hypothesis that PTEN loss is associated with impaired HR, DSB repair protein levels were examined in melanoma cell lines. MeWo (BRAF wildtype, NRAS wildtype), SkMel28 (BRAF V600E mutant, NRAS wildtype), HT144 (BRAF V600E mutant, NRAS wildtype) and UACC62 (BRAF V600E mutant, NRAS wildtype) melanoma cell lines were screened for PTEN protein. As shown in Figure 2A and 2B, western blot analysis confirmed high PTEN protein levels in MeWo cells. UACC62 and HT144 cells demonstrate an almost complete absence of PTEN protein; SkMel28 exhibits an intermediate level. All four cell lines are proficient in APE1 expression. In keeping with previous studies [21, 24], PTEN-deficient HT144 and UACC62 melanoma cells were also RAD51-deficient. RAD51 level was also lower in SkMel28 cells compared to MeWo. However, Fraser et al. did not observe RAD51 deficiency in PTEN-deficient prostate cancer cells [22]. Therefore, for additional clarification, we explored protein levels of other key DSB repair factors. As shown in Figure 2A and 2B, PTEN-deficient cells exhibited consistently lower levels of a number of DSB repair proteins compared to PTEN-proficient MeWo.
We proceeded to generate a doxycycline inducible PTEN-knockdown, BRAF-proficient MeWo cell line. Figure 2C and 2D demonstrates time-dependent knockdown of PTEN protein expression following doxycycline induction of lentiviral shRNA transduced into MeWo cells. By day 7, PTEN protein level was reduced to ~50% baseline, reaching >90% by day 14. We confirmed reduced PTEN transcript production at day 21 by qRT-PCR (Figure 2D). Western blot examination confirmed that levels of a number of DSB repair proteins were reduced in PTEN-knockdown cells compared to non-induced controls (Figure 2E and 2F). Taken together, these data confirm that PTEN loss is associated with reduced DSB repair protein levels in melanoma cells.
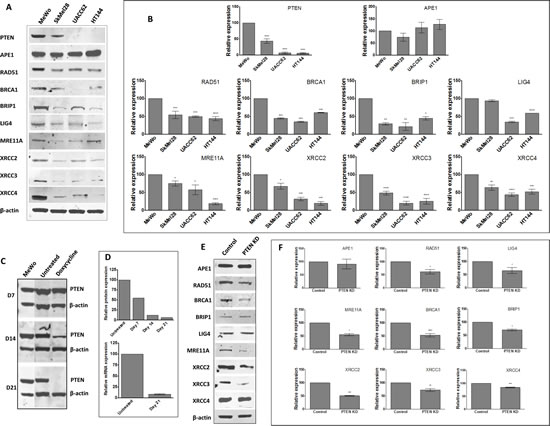
Figure 2: A. Representative Western blots of PTEN and selected DNA repair proteins in human melanoma cell lines. B. Quantification of PTEN and selected DNA repair factor protein levels in melanoma cell lines. C. Generation of doxycycline-inducible PTEN knockdown MeWo cells. Following lentiviral transduction, continuous doxycycline exposure induced PTEN shRNA expression. Whole cell lysates were examined for PTEN protein levels every 7 days, with almost complete absence observed by day 21. This was confirmed by mRNA analysis by qRT-PCR (D). E. Representative Western blots and quantification (F) of PTEN and selected DNA repair proteins in control and PTEN knockdown (KD) cell line. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 compared to control cells.
PTEN-deficient melanoma cells have dysregulated DNA repair mRNA expression
To investigate DNA repair expression at the mRNA level, we profiled a panel of 88 DNA repair genes in MeWo, PTEN-knockdown MeWo, SkMel28, HT144 and UACC62 melanoma cells using the RT2 Profiler DNA Repair PCR array. Significantly reduced mRNA expression of several HR repair genes was observed in PTEN-deficient cells (summarised in Supplementary Table S5 and Supplementary Figure S1). This included several repair factors that have previously been implicated in PTEN deficiency, including RAD51 [21, 24, 37], MRE11 [22], and the RAD51 paralogs RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3 [38]. We also observed low expression of a number of genes involved in non-homologous end joining(NHEJ), as well as nucleotide excision repair(NER) and mismatch repair(MMR). Interestingly, significantly increased mRNA expression of ATM, CCNH and DDB2 was also observed. Taken together, the data suggest that PTEN-deficient melanoma cells have complex patterns of DNA repair dysregulation with consistent downregulation of genes involved in DSB repair.
PTEN-deficient melanoma cells are sensitive to APE1 inhibitors
As PTEN-deficient cells exhibited differential expression of HR factors, blockade of BER through APE1 inhibition could lead to synthetic lethality. To test this hypothesis, we conducted studies in clinically relevant PTEN-deficient and PTEN-proficient melanoma cells using a panel of APE1 inhibitors. Four prototypical APE1 inhibitors were evaluated (chemical structures are shown in Supplementary Figure S2) in MeWo, SKMel28, UACC62, HT144 and PTEN-knockdown MeWo cells. We first confirmed target inhibition using the ARP assay. A significant accumulation of AP sites in genomic DNA was demonstrated following 2, 4 and 8 hours exposure to inhibitor 1, inhibitor 2, inhibitor 3 or inhibitor 4 (Figures 3A-D). We then evaluated whether these inhibitors could induce selective cytotoxicity in PTEN-deficient cells using clonogenic survival assays. Treatment with each inhibitor resulted in reduced survival of PTEN-deficient SKMel28, UACC62, and HT144 cells in comparison to PTEN-proficient MeWo cells (Figure 3E-3H). Similarly, PTEN-knockdown MeWo cells exhibited significantly increased sensitivity to APE1 inhibitor treatment compared to control cells (Figures 4A-4D).
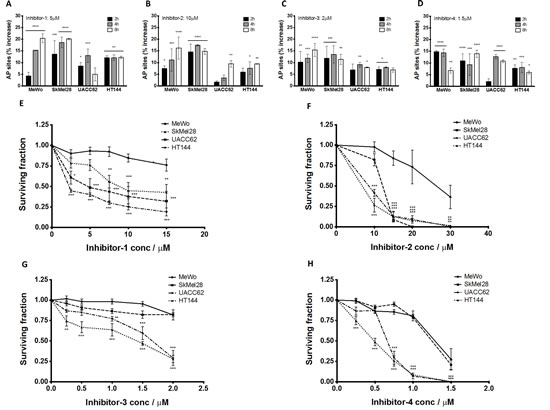
Figure 3: Aldehyde reactive probe assay confirming target inhibition in MeWo, SkMel28, UACC62 and HT144 cells treated with inhibitor -1 (A), inhibitor -2 (B), inhibitor -3 (C) and inhibitor -4 (D). Clonogenic survival assays in MeWo, SkMel28, UACC62 and HT144 cells treated with inhibitor-1 (E), inhibitor-2 (F), inhibitor-3 (G) and inhibitor-4 (H). Inhibitors were added at the indicated concentrations (see methods for details). * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 compared to MeWo (PTEN-wildtype).
To confirm that selective toxicity is due to APE1 inhibtion, we utilised the indirect APE1 inhibitor methoxyamine. Methoxyamine binds irreversibly to AP sites in DNA [36], preventing APE1 from processing the adducted AP lesions. Figure 5E demonstrates that methoxyamine is more lethal to PTEN-deficient SKMel28, UACC62, and HT144 cells than to PTEN-proficient MeWo cells. Similarly, PTEN-knockdown MeWo cells were more sensitive to methoxyamine compared to PTEN-proficient control cells (Figure 4F). To further establish whether this selective cytotoxicity was due to obstruction of BER, we investigated NU1025, a PARP inhibitor that blocks the BER-related single strand break repair (SSBR) pathway [39]. Figure 4G demonstrates the increased toxicity of NU1025 in PTEN-deficient SKMel28, UACC62 and HT144 cells compared to PTEN-proficient MeWo cells. This result was repeated in PTEN-knockdown MeWo cells, which were more sensitive to NU1025 (Figure 4H) than PTEN-proficient MeWo cells. The data is consistent with a previous study in which PTEN-deficient colorectal cells were found to exhibit increased sensitivity to PARP inhibition [24].
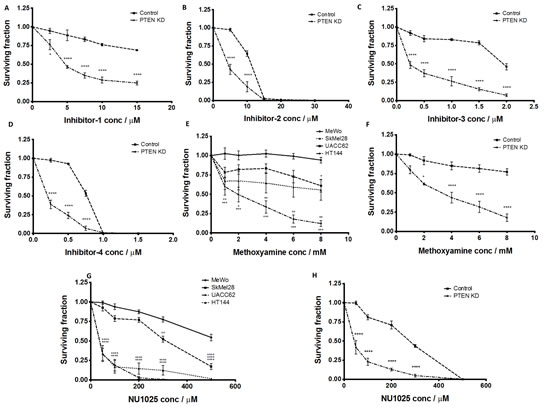
Figure 4: Clonogenic survival assays in control MeWo cells and PTEN knockdown (KD) MeWo cells treated with inhibitor-1 (A), inhibitor-2 (B), inhibitor-3 (C) and inhibitor-4 (D). Clonogenic survival assays in MeWo, SkMel28, UACC62 and HT144 cells treated with methoxyamine (E). Clonogenic survival assays in control MeWo cells and PTEN knockdown (KD) MeWo cells treated with methoxyamine (F). Clonogenic survival assays in MeWo, SkMel28, UACC62 and HT144 cells treated with NU1025 (G). Clonogenic survival assays in control MeWo cells and PTEN knockdown (KD) MeWo cells treated with NU1025 (H). Inhibitors were added at the indicated concentrations (see methods for details). * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 compared to MeWo (PTEN-wildtype).
Selective sensitivity of PTEN-deficient melanoma cells following APE1 inhibition results from increased DNA damage accumulation
The clonogenic survival data provides compelling evidence for selective toxicity of APE1 inhibitors in PTEN-deficient melanoma cells. To provide mechanistic evidence that APE1 inhibition leads to synthetic lethality in PTEN-deficient cells, we investigated the functional consequence of APE1 inhibitor treatment. The neutral COMET assay detects single and double strand breaks(DSBs) in DNA. Figures 5A-5I summarise the results for MeWo, SKMel28, UACC62, HT144 and PTEN-knockdown MeWo cells treated with APE1 inhibitors. Mean tail moment was increased in all samples after APE1 inhibitor exposure compared to pre-treatment samples, and was significantly higher in PTEN-deficient cells at 2, 4, and 8 hours in comparison to PTEN-proficient cells, with damage persisting to 48 hours. The data demonstrates that PTEN-deficient cells accumulate greater numbers of DNA breaks after exposure to an APE1 inhibitor compared to PTEN-proficient cells.
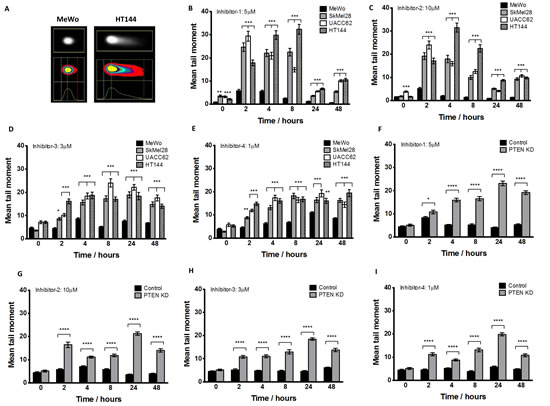
Figure 5: Neutral comet assay was performed at various time points after APE1 inhibitor exposure as indicated in methods. A. Example of increased DNA damage comet ‘tail’ in PTEN-deficient HT144 compared to MeWo cells following 24 hours exposure to inhibitor-1. Significantly increased mean tail moment was observed in PTEN-deficient SkMel28, UACC62 and HT144 cells compared to MeWo cells following treatment with inhibitor-1 (B), inhibitor-2 (C), inhibitor 3 (D) and inhibitor 4 (E). Similarly, significantly increased mean tail moment was observed in PTEN knockdown (KD) MeWo cells compared to control MeWo cells treated with inhibitor-1 (F), inhibitor-2 (G), inhibitor 3 (H) and inhibitor 4 (I). ** p<0.01, *** p<0.001, compared to MeWo (PTEN-wildtype).
Phosphorylation of H2AX at serine 139 (γH2AX) is induced by DSBs, and can be used as a marker of DSB formation. Following inhibitor exposure, γH2AX immunocytochemistry was performed in MeWo, SKMel28, UACC62, HT144 and PTEN-knockdown MeWo cells. As shown in Figures 6A-6I, the percentage of cells with more than six γH2AX foci following inhibitor exposure was significantly higher in PTEN-deficient cells compared to PTEN-proficient control cells. These data provide additional evidence that PTEN-deficient cells accumulate DSBs at an increased rate after exposure to an APE1 inhibitor relative to PTEN-proficient MeWo cells.
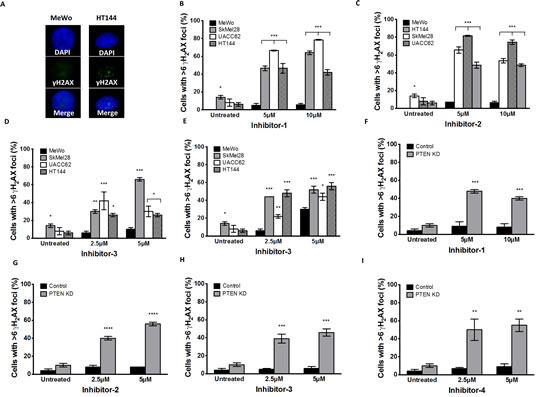
Figure 6: A. γH2AX immunocytochemistry following 24 hours inhibitor-1 exposure demonstrates increased foci in PTEN-deficient HT144 cells compared to PTEN-wildtype MeWo. Significantly increased γH2AX foci was observed in PTEN-deficient SkMel28, UACC62 and HT144 cells compared to MeWo cells following 24 hours treatment with inhibitor-1 (B), inhibitor-2 (C), inhibitor-3 (D) or inhibitor-4 (E). Similar significantly increased γH2AX foci was observed in PTEN knockdown (KD) MeWo cells compared to control MeWo cells following 24 hours treatment with inhibitor-1 (F), inhibitor-2 (G), inhibitor-3 (H) or inhibitor-4 (I). * p<0.05, ** p<0.01, *** p<0.001, compared to MeWo (PTEN-wildtype).
DSB accumulation activates a complex cell cycle checkpoint response that may result in eventual induction of apoptosis. Apoptosis detection by FITC-annexin V flow cytometric analysis was therefore performed in MeWo, SKMel28, UACC62, HT144 and PTEN-knockdown MeWo cells following exposure to inhibitor 1, inhibitor 2, inhibitor 3, or inhibitor 4 for 24 or 48 hours. As shown in Figure 7A-7I, the percentage of cells undergoing apoptosis following APE1 inhibitor exposure was significantly higher in PTEN-deficient cells in comparison to PTEN-proficient MeWo cells.
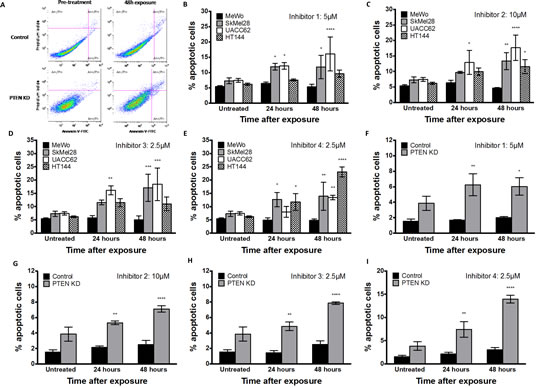
Figure 7: A. Apoptosis detection by FITC-annexin V flow cytometric analysis demonstrates an increased apoptotic fraction in HT144 following 48 hours exposure to inhibitor-1, as observed by increased FITC-Annexin V staining in upper and lower right quadrants. Significantly increased apoptotic fraction was observed in PTEN-deficient SkMel28, UACC62 and HT144 cells compared to MeWo cells following 48 hours treatment with indicated concentrations of inhibitor-1 (B), inhibitor-2 (C), inhibitor-3 (D) or inhibitor-4 (E). Similar significantly increased apoptotic fraction was observed in PTEN knockdown (KD) MeWo cells compared to control MeWo cells following 48 hours treatment with inhibitor-1 (F), inhibitor-2 (G), inhibitor-3 (H) or inhibitor-4 (I). * p<0.05, ** p<0.01, *** p<0.001, compared to MeWo (PTEN-wildtype).
Taken together, these functional studies provide compelling evidence that APE1 inhibition induces a synthetic lethal response in PTEN-deficient cells by causing accumulation of abasic sites and subsequent strand breaks, and ultimately the induction of apoptosis.
DISCUSSION
PTEN is a key regulator of the anti-apoptotic PI3K/Akt pathway [15], and emerging evidence suggests a role in DNA repair [19-21]. The role of PTEN in the regulation of RAD51 remains controversial [24]. However, a number of recent studies provide compelling evidence that nuclear PTEN may have essential roles in DNA repair [40, 41], and that PTEN deficiency may be targeted in a synthetic lethality approach by inhibitors of SSBR [24]. Our primary hypothesis is that this synthetic lethality approach may be applied to PTEN-deficient melanomas using BER inhibitors such as those targeting APE1. We initially investigated the clinicopathological significance of PTEN mRNA and APE1 mRNA in melanoma. Low PTEN was associated with poor survival overall but the effect was absent in BRAF and NRAS-mutated tumours, and only statistically significant in tumours without BRAF or NRAS mutations. This may be related to the complex tumour-suppressing function of PTEN, including its roles as a negative regulator of the anti-apoptotic PI3K/Akt pathway and in DNA repair regulation. That the influence of PTEN expression on prognosis was more pronounced in tumours without BRAF or NRAS mutations is a new observation, and suggests that PTEN loss may be a key biomarker in BRAF-wildtype tumours. Taken together, the clinical data are consistent with pre-clinical study that implicates PTEN loss in melanoma progression [26]; however, the relatively small patient numbers in the various subgroups is a limitation of our study. Our finding that high APE1 mRNA is associated with poor survival in melanoma is supported by previous studies [42] and provides additional evidence for targeting APE1 in melanoma.
The success of PARP inhibitors in BRCA-deficient breast and ovarian tumours [13, 14] implies that a synthetic lethality approach may be an attractive strategy in melanomas with DSB repair deficiency. PTEN-deficient melanoma cells with defective DSB repair may be reliant upon BER as a frontline defence to prevent damage accumulation, replication fork collapse, DSB formation and consequent cell death. Thus, blockade of BER through APE1 inhibition could lead to synthetic lethality in PTEN-deficient cells. To test this hypothesis, we first profiled PTEN-deficient and PTEN-proficient melanoma cells for DNA repair factor expression, finding that expression of several genes involved in HR was impaired at protein and mRNA level in PTEN-deficient cells. In addition we observed downregulation of genes involved in NHEJ, NER, MMR and MGMT. Given the role of PTEN in the regulation of HR expression as well as in HR recruitment to sites of DNA damage [19-21], it is perhaps not surprising that genomic instability in PTEN-null cells may, over a period of time, eventually lead to acquisition of new defects in other DNA repair pathways. Recent studies that positively link PTEN with NER [43], MMR [44], and MGMT expression [45] suggests that such a mechanism may be operating in cells, but detailed mechanistic studies are required to confirm this hypothesis. An interesting observation across PTEN-deficient cell lines was the loss of BRCA1 protein expression, consistent with previous evidence in UACC62 melanoma cells (http://cancer.sanger.ac.uk/cancergenome/
projects/cosmic/). This observation is also consistent with a recent study in breast cancer, wherein high frequency of PTEN loss was observed in BRCA1-associated breast tumours [46]. In a separate study, PTEN loss was found to be a predictor of BRCA1 germ-line mutations in women with early onset breast cancer [47]. Taken together, this evidence suggests a possible functional link between PTEN and BRCA1 in melanomas. These observations, however, require detailed mechanistic studies to confirm functional interactions between PTEN and BRCA1.
PTEN-deficient melanoma cells were shown in the current study to be sensitive to APE1 inhibitors. Similar hypersensitivity observed following exposure to the PARP inhibitor NU1025 is consistent with previous observations using PTEN-deficient colorectal cells [24]. We have concluded that the interrelationship between BER and PTEN may be a valid therapeutic target in melanoma for the following reasons: a) PTEN-deficient cells are sensitive to APE1 repair domain inhibitors; b) PTEN-deficient cells are sensitive to methoxyamine, an indirect inhibitor of APE1; c) PTEN-deficient cells are sensitive to the PARP inhibitor NU1025; and d) upon APE1 inhibitor treatment, PTEN-deficient cells accumulate DNA DSBs, resulting in the induction of apoptosis. It should be noted that the observed level of apoptosis in PTEN-deficient cells following inhibitor exposure is lower than may be predicted from survival analyses using clonogenic assays. Although apoptotic cell accumulation may occur over a period of time following chronic exposure to APE1 inhibitor, another possible explanation is ‘synthetic sickness’, a synthetic lethal-type relationship wherein the overall outcome is reduced fitness rather than cellular lethality [48]. Accumulation of DNA damage, including specific DSB accumulation, has been associated with the induction of cellular senescence [49]. More recently, PTEN loss has also been shown to induce a senescent phenotype [50]. Therefore we speculate that DSB accumulation in PTEN-deficient cells following APE1 inhibition, besides induction of apoptosis, may also drive the cell population into senescence, contributing to reduced clonogenicity. Although further study is required to assess for definitive evidence of senescence induction in this setting, our data raise the intriguing possibility of a future avenue for pro-senescence therapy.
In conclusion, our study provides the first evidence that blockage of BER by APE1 inhibition has attractive potential for a novel therapeutic approach in PTEN-deficient melanomas. This strategy could have significant translational applications for personalised therapy in melanoma patients.
METHODS
Clinical study
Gene expression analysis: Whole genome gene expression (~30,000 probes) was measured using Illumina DASL approach in 240 formalin fixed primary melanoma tumours from Leeds Melanoma Cohort [31], including 29 duplicates to serve in QC checks. After QC, the cohort contained 191 patients including 106 who had relapsed and 101 who had died. Histology data were derived from clinical histopathology reports. For a subset of tumors diagnostic H+E slides were reviewed by a dermatopathologist (Dr Andy Boon (Cohort study) to standardize reporting across the specimens. BRAF and NRAS mutation status was also available for 182 of these tumours, derived using pyrosequencing as previously described [32]. The study was approved by the National Research ethics committee (UK).
Statistical analyses
Data was normalised using background correction and robust spline smoothing with Lumi R package. The associations between PTEN and APE1 mRNA expressions and histological factors were assessed using linear and logistic regression of log2 transformed gene expression data. Separate and joint effects of PTEN and APE1 mRNA expressions on relapse-free and overall survival was tested. Expression data were used as continuous after log2 transformation and after dichotomisation using expression cut-offs determined using X-Tile software [33]. In stratified analysis, Cox proportional hazard model was applied on dichotomized PTEN expression stratifying on presence of BRAF mutations, NRAS mutations and no mutations.
Cell lines
Clinically relevant previously well-characterised melanoma cell lines were chosen for the pre-clinical study. MeWo is a BRAF wildtype and NRAS wildtype melanoma cell line. SkMel28, HT144 and UACC62 are BRAF V600 mutant and NRAS wildtype melanoma cells lines (http://cancer.sanger.ac.uk/cancergenome/
projects/cosmic/).
Generation of doxycycline inducible PTEN knockdown MeWo cell line by lentiviral shRNA transduction
TRIPZ shRNA plasmid against PTEN was purchased from Thermo Scientific (Clone ID: V3THS_312158: 5’-GGAAAGAATCAAGGAGG-3’, Loughborough, UK). Lentiviral production was performed per the supplied protocol using the Lenti-X high-titer lentiviral packaging system in Lenti-X 293T cells (Clontech, Mountain View, USA). After 48 hours incubation, lentivirus-containing supernatant was harvested and applied to MeWo cells at an MOI of 1 or 3. Once stable in culture, puromycin selection was initiated at a concentration of 1μg/ml. To induce shRNA expression, doxycycline was added to culture medium at a concentration of 0.5-1μg/ml and replaced every 72 hours. Confirmation of reduced transcript production by qRT-PCR was performed on an Applied Biosystems 7500 FAST cycler using Qiagen PCR primers against PTEN [Qiagen Quantitect primers (HS_PTEN_4_SG and HS_GAPDH_1_SG)]. Absence of PTEN protein expression was confirmed on Western blot.
qRT-PCR analysis of DNA repair gene expression in melanoma cell lines
RNA was extracted from melanoma cell lines using the RNeasy Mini Kit (Qiagen) and quantified using a microvolume spectrophotometer. cDNA synthesis was performed using the RT2 First Strand Kit (Qiagen). Real time PCR was carried out on an Applied Biosystems 75000 FAST cycler in a commercially available 96-well plate format, allowing assessment of 88 DNA repair genes simultaneously (RT2 Profiler DNA Repair PCR Array). Threshold cycle was calculated for each well and exported to online software for further analysis (www.SABiosciences.com/pcrarraydataanalysis.php). All experiments were performed in triplicate.
Western blot analysis
Primary antibody details are summarised in Supplementary Table S4. Protein expression was examined by infrared dye-labelled secondary antibody (Li-Cor, 1:15000 dilution) detected by Li-Cor Odyssey Scanner. All experiments were performed in triplicate.
APE1 inhibitors and other compounds
We have previously identified a number of APE1 inhibitors. [34, 35] Cell biology experiments performed here utilised structural analogues of N-(4-fluorophenyl)-2-[4-phenylsulfonyl-2-(p-tolyl)oxazol-5-yl]sulfanyl-acetamide (previously characterised in [12]: N-(4-fluorophenyl) -2- (2-phenyl-4- phenylsulfonyl-1,3-oxazol-5-yl]sulfanyl-acetamide (Inhibitor-1) and N-(4-fluorophenyl)-2-(2-phenyl-4-phenylsulfonyl-oxazol-5-yl)sulfanyl-acetamide (Inhibitor-2), purchased from ChemDiv. Additional inhibitors previously identified by Rai et al. [35] were synthesised by the National Institutes of Health (NIH) Chemical Genomics Center (NCGC): N-(3-(benzo[d]-thiazol-2-yl)-6-isopropyl-4,5,6,7-tetrahydrothieno[2,3-c]-pyridin-2-yl)acetamide (Inhibitor-3) and its analogue N-(3-(benzo[d]thiazol-2-yl)-5,6-dihydro-4H-thieno[2,3-c]pyrrol-2-yl)acetamide (Inhibitor-4). APE1 inhibitors investigated here are highly potent and specific for APE1. They do not bind to DNA and have no activity against E. coli endonuclease IV (a functional homolog with no sequence or structural homology to APE1). IC50 for APE1 endonuclease activity inhibiton in purified protein fluoresence based biochemical assays are as follows; inhibitor 1= 0.2µM, inhibitor 2= 0.1µM , inhibitor 3= 2µM and inhibitor 4=3.3µM . The compounds also block AP site cleavage activity in HeLa whole cell extract assays, and potentiate the cytotoxicity of alkylating agents in cancer cell lines [34, 35]. Molecular modelling studies indicate that these APE1 inhibitors dock onto the active site of APE1 [8, 34]. Methoxyamine, a non-specific indirect inhibitor of APE1, binds irreversibly to AP sites in DNA [36] and prevents APE1 (and endonuclease IV) from processing the adducted AP lesion. Methoxyamine (indirect APE1 inhibitor) was purchased from Sigma. NU1025 (PARP inhibitor) was purchased from Tocris Bioscience.
Clonogenic survival assay
Cell lines were plated into 6-well plates at a density of 200-400 cells per well. Cells were allowed to adhere for 4 hours, after which inhibitory compound was added at varying concentrations. Cells were incubated for 14 days under normal incubator conditions. After this time, media was discarded and cells were stained using 10% crystal violet in 70% aqueous ethanol to allow quantification of colony number. All experiments were performed in triplicate.
Aldehyde Reactive Probe (ARP) assay
Cell-based ARP assay was performed according to the manufacturer’s instructions (Abcam). Cells were plated at high density in a 96-well plate, allowed to grow for 24 hours, then treated with inhibitor. Results were presented as percent increase in fluorescence as a surrogate for AP site accumulation.
Neutral COMET assay
Cell lines were plated into 6cm petridishes at a density of 105 cells per plate. Cells were allowed to adhere for 24 hours, after which time inhibitory compound was added at a single concentration. Cells were harvested at 0, 2, 4, 8, 24 and 48 hour time points, and neutral COMET assay was performed as described previously [12]. All experiments were performed in triplicate.
γH2AX immunocytochemistry
Cell lines were seeded onto sterile coverslips in 6-well plates at a density of 105 cells per well. Cells were allowed to adhere for 24 hours, after which time inhibitory compound was added. After 24 hours exposure, γH2AX immunocytochemistry was performed [12]. The number of γH2AX foci per nucleus was determined in 100 cells per slide. Nuclei containing more than 6 γH2AX foci were considered positive. All experiments were performed in triplicate.
Apoptosis detection by FITC-annexin V flow cytometric analysis
Cell lines were plated into 6-well plates at a density of 5 x 105 cells per well. Cells were allowed to adhere for 24 hours, after which time inhibitory compound was added. After 24 hours exposure, cells were gently trypsinised, washed twice in ice-cold PBS, and resuspended in Annexin V binding buffer (FITC Annexin V Apoptosis Detection Kit I, BD Pharmingen). Cells were incubated for 15 minutes in the dark with FITC-Annexin V and propidium iodide (PI), then analysed by flow cytometry using a BC Accuri C6 Flow Cytometer. The percentage of induced apoptosis (FITC-Annexin V positive, PI negative and FITC-Annexin V positive, PI positive) was determined by comparison to a control population of untreated cells. All experiments were performed in triplicate [12].
Conflict of interest
The authors disclose no potential conflicts of interest
Funding
This work was supported in part by an NIH grant to DMWIII (1 R03 MH086444-01), the Intramural Research Program of the National Institute on Aging, NIH, the Molecular Libraries Common Fund Program of the NIH, the National Center for Advancing Translational Sciences (USA), University of Nottingham and Nottingham University Hospitals Charity (UK). In Leeds, the work was supported by Cancer Research UK (project grants C8216/A6129 and C8216/A8168), and program grants C588/A4994 and C588/A10589), and by the NIH (R01 CA83115). RJ was in receipt of a Bramall Fellowship and a Medical Research Council Clinical Research Training Fellowship (G0802123). RA was in receipt of a Medical Research Council Research Training Fellowship (G1000252).
Author contributions
RA designed the work, acquired data, played an important role in interpreting the results, drafted the manuscript and approved the final version; RJ acquired human melanoma data, played an important role in interpreting the results, drafted the manuscript and approved the final version; JN acquired human melanoma data, played an important role in interpreting the results, drafted the manuscript and approved the final version; DJM synthesised APE1 inhibitors, acquired data, played an important role in interpreting the results, revised the manuscript and approved the final version; AS synthesised APE1 inhibitors, acquired data, played an important role in interpreting the results, revised the manuscript and approved the final version, CS played an important role in interpreting the results, drafted the manuscript and approved the final version; FE acquired human melanoma data, played an important role in interpreting the results drafted the manuscript and approved the final version , JL acquired human melanoma data, played an important role in interpreting the results, drafted the manuscript and approved the final version, AJ synthesised APE1 inhibitors, acquired data, played an important role in interpreting the results, revised the manuscript and approved the final version; CW acquired human melanoma data, played an important role in interpreting the results drafted the manuscript and approved the final version , AG played an important role in interpreting the results, revised the manuscript and approved the final version, GB played an important role in interpreting the results, drafted the manuscript and approved the final version; PMP played an important role in interpreting the results, drafted the manuscript and approved the final version , JNB acquired human melanoma data, played an important role in interpreting the results, drafted the manuscript and approved the final version, DMWIII played an important role in interpreting the results drafted the manuscript and approved the final version; SM designed the work, played an important role in interpreting the results drafted the manuscript, revised the manuscript and approved the final version
REFERENCES
1. Abbotts, R and Madhusudan S. Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treat Rev. 2010; 36(5):425-435.
2. Dianov, GL, Sleeth KM, Dianova, II and Allinson SL. Repair of abasic sites in DNA. Mutat Res. 2003; 531(1-2):157-163.
3. Li, M and Wilson DM, 3rd. Human Apurinic/Apyrimidinic Endonuclease 1. Antioxid Redox Signal. 2014;20(4): 678-707.
4. Evans, AR, Limp-Foster M and Kelley MR. Going APE over ref-1. Mutat Res. 2000; 461(2):83-108.
5. Wang, D, Xiang DB, Yang XQ, Chen LS, Li MX, Zhong ZY and Zhang YS. APE1 overexpression is associated with cisplatin resistance in non-small cell lung cancer and targeted inhibition of APE1 enhances the activity of cisplatin in A549 cells. Lung Cancer. 2009; 66(3):298-304.
6. Madhusudan, S, Smart F, Shrimpton P, Parsons JL, Gardiner L, Houlbrook S, Talbot DC, Hammonds T, Freemont PA, Sternberg MJ, Dianov GL and Hickson ID. Isolation of a small molecule inhibitor of DNA base excision repair. Nucleic Acids Res. 2005; 33(15):4711-4724.
7. Mohammed, MZ, Vyjayanti VN, Laughton CA, Dekker LV, Fischer PM, Wilson DM, 3rd, Abbotts R, Shah S, Patel PM, Hickson ID and Madhusudan S. Development and evaluation of human AP endonuclease inhibitors in melanoma and glioma cell lines. Br J Cancer. 104(4):653-663.
8. Simeonov, A, Kulkarni A, Dorjsuren D, Jadhav A, Shen M, McNeill DR, Austin CP and Wilson DM, 3rd. Identification and characterization of inhibitors of human apurinic/apyrimidinic endonuclease APE1. PLoS One. 2009; 4(6):e5740.
9. Dorjsuren, D, Kim D, Vyjayanti VN, Maloney DJ, Jadhav A, Wilson DM, 3rd and Simeonov A. Diverse small molecule inhibitors of human apurinic/apyrimidinic endonuclease APE1 identified from a screen of a large public collection. PLoS One. 7(10):e47974.
10. Rai, G, Vyjayanti VN, Dorjsuren D, Simeonov A, Jadhav A, Wilson DM, 3rd and Maloney DJ. Synthesis, biological evaluation, and structure-activity relationships of a novel class of apurinic/apyrimidinic endonuclease 1 inhibitors. J Med Chem. 55(7):3101-3112.
11. Wilson, DM, 3rd and Simeonov A. Small molecule inhibitors of DNA repair nuclease activities of APE1. Cell Mol Life Sci. 2012; 67(21):3621-3631.
12. Sultana, R, McNeill DR, Abbotts R, Mohammed MZ, Zdzienicka MZ, Qutob H, Seedhouse C, Laughton CA, Fischer PM, Patel PM, Wilson DM, 3rd and Madhusudan S. Synthetic lethal targeting of DNA double-strand break repair deficient cells by human apurinic/apyrimidinic endonuclease inhibitors. Int J Cancer. 2012;131(10):2433-44.
13. Farmer, H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC and Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434(7035):917-921.
14. Bryant, HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ and Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434(7035):913-917.
15. Song, MS, Salmena L and Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 13(5):283-296.
16. Conde-Perez, A and Larue L. PTEN and melanomagenesis. Future Oncol. 8(9):1109-1120.
17. Aguissa-Toure, AH and Li G. Genetic alterations of PTEN in human melanoma. Cell Mol Life Sci. 2012; 69(9):1475-1491.
18. Zhou, XP, Gimm O, Hampel H, Niemann T, Walker MJ and Eng C. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. Am J Pathol. 2000; 157(4):1123-1128.
19. Gupta, A, Yang Q, Pandita RK, Hunt CR, Xiang T, Misri S, Zeng S, Pagan J, Jeffery J, Puc J, Kumar R, Feng Z, Powell SN, Bhat A, Yaguchi T, Wadhwa R, et al. Cell cycle checkpoint defects contribute to genomic instability in PTEN deficient cells independent of DNA DSB repair. Cell Cycle. 2009; 8(14):2198-2210.
20. Puc, J, Keniry M, Li HS, Pandita TK, Choudhury AD, Memeo L, Mansukhani M, Murty VV, Gaciong Z, Meek SE, Piwnica-Worms H, Hibshoosh H and Parsons R. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell. 2005; 7(2):193-204.
21. Shen, WH, Balajee AS, Wang JL, Wu H, Eng C, Pandolfi PP and Yin YX. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007; 128(1):157-170.
22. Fraser, M, Zhao H, Luoto KR, Lundin C, Coackley C, Chan N, Joshua AM, Bismar TA, Evans A, Helleday T and Bristow RG. PTEN deletion in prostate cancer cells does not associate with loss of RAD51 function: implications for radiotherapy and chemotherapy. Clin Cancer Res. 2012; 18(4):1015-1027.
23. Bassi, C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B and Stambolic V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science. 341(6144):395-399.
24. Mendes-Pereira, AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, Waldman T, Lord CJ and Ashworth A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. Embo Molecular Medicine. 2009; 1(6-7):315-322.
25. Dedes, KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, Vatcheva R, Savage K, Mackay A, Lord CJ, Ashworth A and Reis-Filho JS. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2(53):53ra75.
26. Stahl, JM, Cheung M, Sharma A, Trivedi NR, Shanmugam S and Robertson GP. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003; 63(11):2881-2890.
27. Dankort, D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr., You MJ, DePinho RA, McMahon M and Bosenberg M. BRAF(V600E) cooperates with PTEN loss to induce metastatic melanoma. Nat Genet. 2009; 41(5):544-552.
28. Colombino, M, Capone M, Lissia A, Cossu A, Rubino C, De Giorgi V, Massi D, Fonsatti E, Staibano S, Nappi O, Pagani E, Casula M, Manca A, Sini M, Franco R, Botti G, et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J Clin Oncol. 30(20):2522-2529.
29. Chapman, PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011; 364(26):2507-2516.
30. Paraiso, KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson AR, Ribas A, Palma MD, Nathanson KL, Koomen JM, Messina JL and Smalley KS. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011; 71(7):2750-2760.
31. Newton-Bishop, JA, Beswick S, Randerson-Moor J, Chang YM, Affleck P, Elliott F, Chan M, Leake S, Karpavicius B, Haynes S, Kukalizch K, Whitaker L, Jackson S, Gerry E, Nolan C, Bertram C, et al. Serum 25-hydroxyvitamin D3 levels are associated with breslow thickness at presentation and survival from melanoma. J Clin Oncol. 2009; 27(32):5439-5444.
32. Jewell, R, Chambers P, Harland M, Laye J, Conway C, Mitra A, Elliott F, Cook MG, Boon A and Newton-Bishop J. Clinicopathologic features of V600E and V600K melanoma--letter. Clin Cancer Res. 2012; 18(24):6792; author’s reply p 6793.
33. Camp, RL, Dolled-Filhart M and Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res. 2004; 10(21):7252-7259.
34. Mohammed, MZ, Vyjayanti VN, Laughton CA, Dekker LV, Fischer PM, Wilson DM, 3rd, Abbotts R, Shah S, Patel PM, Hickson ID and Madhusudan S. Development and evaluation of human AP endonuclease inhibitors in melanoma and glioma cell lines. Br J Cancer. 2011; 104(4):653-663.
35. Rai, G, Vyjayanti VN, Dorjsuren D, Simeonov A, Jadhav A, Wilson DM, 3rd and Maloney DJ. Synthesis, biological evaluation, and structure-activity relationships of a novel class of apurinic/apyrimidinic endonuclease 1 inhibitors. Journal of medicinal chemistry. 2012; 55(7):3101-3112.
36. Liu, L and Gerson SL. Therapeutic impact of methoxyamine: blocking repair of abasic sites in the base excision repair pathway. Curr Opin Investig Drugs. 2004; 5(6):623-627.
37. Dedes, KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, Vatcheva R, Savage K, Mackay A, Lord CJ, Ashworth A and Reis-Filho JS. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010; 2(53):53ra75.
38. McEllin, B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, Bachoo RM and Burma S. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 2010; 70(13):5457-5464.
39. Bowman, KJ, Newell DR, Calvert AH and Curtin NJ. Differential effects of the poly (ADP-ribose) polymerase (PARP) inhibitor NU1025 on topoisomerase I and II inhibitor cytotoxicity in L1210 cells in vitro. Br J Cancer. 2001; 84(1):106-112.
40. Bassi, C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B and Stambolic V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science. 2013; 341(6144):395-399.
41. Zhang, C, Kang C, Wang P, Cao Y, Lv Z, Yu S, Wang G, Zhang A, Jia Z, Han L, Yang C, Ishiyama H, Teh BS, Xu B and Pu P. MicroRNA-221 and -222 regulate radiation sensitivity by targeting the PTEN pathway. Int J Radiat Oncol Biol Phys. 2011; 80(1):240-248.
42. Yang, S and Meyskens FL. Apurinic/apyrimidinic endonuclease/redox effector factor-1(APE/Ref-1): a unique target for the prevention and treatment of human melanoma. Antioxid Redox Signal. 2009; 11(3):639-650.
43. Ming, M, Feng L, Shea CR, Soltani K, Zhao B, Han W, Smart RC, Trempus CS and He YY. PTEN positively regulates UVB-induced DNA damage repair. Cancer Res. 2011; 71(15):5287-5295.
44. Zhou, XP, Kuismanen S, Nystrom-Lahti M, Peltomaki P and Eng C. Distinct PTEN mutational spectra in hereditary non-polyposis colon cancer syndrome-related endometrial carcinomas compared to sporadic microsatellite unstable tumors. Hum Mol Genet. 2002; 11(4):445-450.
45. Neto, JC, Ikoma MM, Carvalho KC, Vassallo J, De Brot M, Gobbi H, Soares FA and Rocha RM. MGMT and PTEN as potential prognostic markers in breast cancer. Exp Mol Pathol. 2012; 92(1):20-26.
46. Martins, FC, De S, Almendro V, Gonen M, Park SY, Blum JL, Herlihy W, Ethington G, Schnitt SJ, Tung N, Garber JE, Fetten K, Michor F and Polyak K. Evolutionary pathways in BRCA1-associated breast tumors. Cancer Discov. 2012; 2(6):503-511.
47. Phuah, SY, Looi LM, Hassan N, Rhodes A, Dean S, Taib NA, Yip CH and Teo SH. Triple-negative breast cancer and PTEN (phosphatase and tensin homologue)loss are predictors of BRCA1 germline mutations in women with early-onset and familial breast cancer, but not in women with isolated late-onset breast cancer. Breast Cancer Res. 2011; 14(6):R142.
48. Ferrari, E, Lucca C and Foiani M. A lethal combination for cancer cells: Synthetic lethalityscreenings for drug discovery. E J Cancer. 2010; 46:2889–2895.
49. Parrinello, S, E. Samper, A. Krtolica, J. Goldstein, S. Melov, J. Campisi. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. . Nat Cell Biol. 2003; 5:741–747.
50. Alimonti, A, Carracedo, A., Clohessy, J.G., Trotman, L.C., Nardella, C., Egia. A., Salmena, L., Sampieri K, Haveman WJ, Brogi E, Richardson AL, Zhang J, Pandolfi PP. Subtle variations in PTEN dose determine cancer susceptibility. Nat Genet. 2010; 42:454-458.