
INTRODUCTION
The CUL4A E3 ubiquitin ligase belongs to the family of cullin proteins and is a component of the cullin-RING ligase 4A complex [1]. This complex plays a crucial role in the regulation of both stability and degradation of a huge spectrum of cellular proteins through the ubiquitin-proteasome system [1, 2]. CUL4A confers substrate specificity and it is involved in the control of cell cycle, DNA replication and DNA repair depending on the nature of ubiquitinated proteins [3-5]. CUL4A has been found amplified and/or overexpressed in different types of carcinomas and a putative oncogenic role has been proposed for this gene [6-9]. Breast cancer patients with strong expression of CUL4 show shorter overall and disease-free survival [6, 10] which has led to the proposal that CUL4A might confer an aggressive behavior in this malignancy. The functional in vitro and in vivo characterization of the putative oncogenic role of CUL4A has been previously studied in prostate cancer and mesothelioma [7, 8]. In breast cancer, a recent functional analysis of CUL4A has revealed a pivotal role of the gene in regulating the metastatic behavior of breast cancer cells [11].
In previous studies we showed that the overexpression of CUL4A was associated with markers of proliferation and tumor aggressiveness in familiar and sporadic primary breast tumors [12]. In addition we found that, although not exclusively, the 13q34 amplification would be one of the mechanisms triggering CUL4A overexpression in breast cancer. Importantly, we observed that the 13q34 amplification presents increased frequency in BRCA1-defective tumors and seems to be associated with primary breast tumors of basal-like phenotype [12, 13]. Since basal-like breast cancers are characterized by lack of effective targeted therapy and poor clinical outcome [14], the elucidation of the specific role of CUL4A in the carcinogenic process of this tumor subtype is of major interest.
In this study we have evaluated in vitro and in vivo the functional significance of CUL4A down-regulation and up-regulation in basal-like breast cancer models that exhibit features resembling those found in primary breast tumors. Our results support the role of CUL4A as a driver oncogene at the 13q34 amplification in basal-like breast cancer. Also, suggest an implication of CUL4A overexpression in cancer aggressiveness and progression rather than a major function as a transformation-initiating event. In addition, our proteomics-derived data provide evidence for novel molecules and pathways that might be involved in mediating the tumorigenic process orchestrated by CUL4A.
RESULTS
CUL4A is overexpressed in breast cancer cell lines
We examined the expression of CUL4A in 11 human breast cancer cell lines and two non-transformed human mammary epithelial cell lines by quantitative PCR (qPCR) and Western blot (WB). In both analysis the non-transformed cells (HBL100 and 184B5) were within the lower relative expression levels indicating that CUL4A is overall up-regulated in breast cancer cell lines compared with normal mammary epithelial cells (Figure 1A and B). CUL4A expression varied across the breast cancer cell lines. MDAMB157 with extra DNA copies at 13q34 was found among those cell lines exhibiting the highest CUL4A relative expression values. Also, HCC1937 and MDAMB436 cells, with BRCA1-impairment, were found in the upper limit of the expression range. With few exceptions cell lines ordered according to their CUL4A relative expression by qPCR matched the order obtained by WB (Figure 1A and B), with both data set exhibiting a high correlation (ρ=0.714, P=0.004). Since the 13q34 amplification and/or CUL4A overexpression is more frequent in BRCA1-associated and basal-like primary breast tumors [12] we selected the BRCA1-null HCC1937 and the 13q34 amplified MDAMB157 cell lines to further characterize CUL4A function in breast cancer progression. Both cell lines are of basal-like subtype [15, 16] and as shown by our experiments express high levels of CUL4A. In addition, we included the basal-type and low CUL4A-expressing 184B5 mammary epithelial cells as a non-tumorigenic model to assess the implication of CUL4A overexpression in the initial steps of the transformation process.
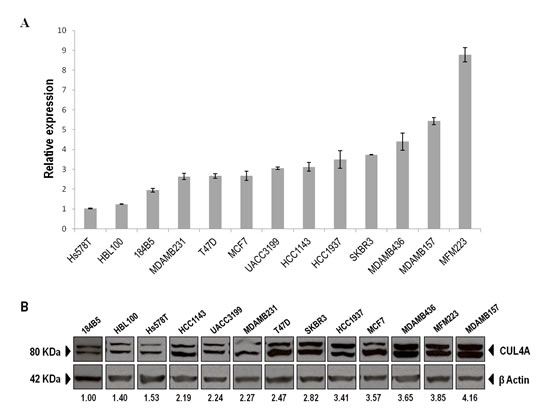
Figure 1: CUL4A expression in the panel of human breast epithelial cell lines. (A) CUL4A mRNA expression was evaluated by qRT-PCR in the panel of breast cell lines and normalized to β-actin expression. Cell lines are shown from low to high CUL4A expression. Bars represent median and standard deviation (SD) of three technical replicates. (B) CUL4A protein levels were assessed by Western blot. Image of WB was cropped to organize cell lines from low to high CUL4A protein levels. The anti-CUL4A antibody recognizes a doublet of CUL4A. The upper band corresponds to the neddylated form of the protein. Numbers under the image correspond to intensity of CUL4A bands (neddylated and non-neddylated forms) for each cell line normalized to β-ACTIN intensity value and relative to the lowest CUL4A-expressing cells (184B5).
CUL4A down-regulation inhibits breast cancer tumorigenesis in vitro and in vivo
To directly assess the contribution of endogenous CUL4A overexpression on the transformed phenotype of human breast cancer cells, we examined the effects of knocking down CUL4A in the HCC1937 and MDAMB157 breast cancer cell lines where CUL4A is amplified and/or overexpressed. Cells transduced with a CUL4A-specific shRNA construct (SH4) (see Methods) presented reduced CUL4A protein levels by at least 90% compared with control cells (transduced with scrambled vector) (Figure 2A). The CUL4A-knocked-down cells showed decreased proliferation compared with control cells in viability assays (Figure 2B). In agreement, a reduced S-phase population was observed in both CUL4A knocked-down cells compared with controls (Figure 2C). In addition, HCC1937- and MDAMB157-CUL4A-silenced cells showed reduced ability to form colonies in anchorage-dependent conditions relative to controls (Figure 2D). Proliferation was also inhibited in CUL4A-knocked-down HCC1937 cells when grown in anchorage-independent conditions (Figure 2E). Modifications in anchorage-independent growth could not be evaluated in the MDAMB157 model as these cells do not grow in soft-agar. The 184B5-CUL4A-silenced cells (Figure 3A) did not show significant changes in proliferation (Figure 3B) and colony formation abilities (Figure 3C). This would be consistent with a specific dependence of the CUL4A function acquired in CUL4A-overexpressing cancer cells. In agreement with our in vitro results, orthotopical inoculation of CUL4A-silenced cells in immunodeficient mice generated tumors with significantly slower growth rate than that of Scramble-derived tumors (Figure 4A). At the end point of these in vivo experiments the average volume of tumors induced by CUL4A-silenced cells was half the average volume exhibited by tumors produced by control cells (Figure 4B).
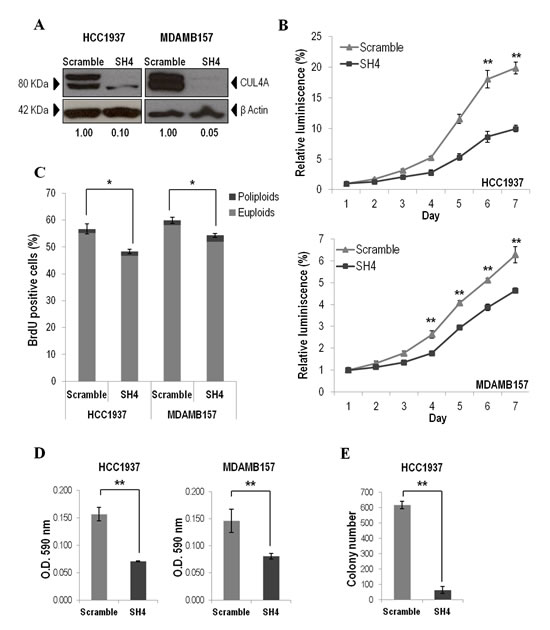
Figure 2: CUL4A down-regulation in HCC1937 and MDAMB157 breast cancer cell lines. (A) Western blot analysis of CUL4A expression in cells transduced with CUL4A specific shRNA (SH4) and control shRNA (Scramble). Numbers under the image correspond to intensity of CUL4A bands (neddylated and non-neddylated forms) for each cell line normalized to β-ACTIN intensity value. (B) Viability of CUL4A-silenced (SH4) and Scrambled cells. Cells were seeded in 96-well plates at 1500 cells/well and incubated for indicated time. The quantity of viable cells was determined by CellTiter-Glow assay. Each point represents mean and standard deviation (SD) of 6 replicates. Experiments were carried out 3 times and showed similar results. (C) Percentage of BrdU positive cells in CUL4A-knocked down (SH4) and Scrambled cells. Bars represent the means of triplicate experiments and error bars indicate the SD. Light and dark grey illustrates the partial contribution of euploids and poliploids cells, respectively, to the total counting. (D) CUL4A-depleted breast cancer cell lines (SH4) and control cells (Scramble) were grown in anchorage-dependent conditions. Cells were fixed and stained with crystal violet. Crystal violet was solubilized and readouts (optical density at 590 nm) were obtained. Representative example of three-independent experiments is shown. Data are expressed as mean ± SD of three replicates. (E) CUL4A-knocked down (SH4) HCC1937 and control cells (Scramble) were grown in soft agar to evaluate anchorage-independent growth ability. Graphics represents the average number of colonies ± SD. **P<0.01, *P<0.05.
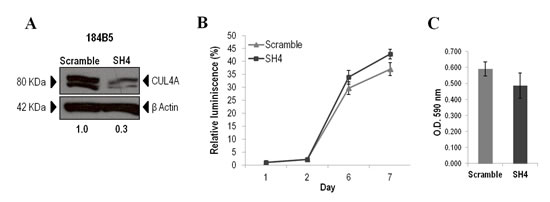
Figure 3: CUL4A down-regulation in the non-transformed 184B5 mammary epithelial cells. (A) Western blot analysis of CUL4A expression in 184B5 cells transduced with CUL4A specific shRNA (SH4) and control shRNA (Scramble). Numbers under the image correspond to intensity of CUL4A bands (neddylated and non-neddylated forms) for each cell line normalized to β-ACTIN intensity value. (B) Viability of CUL4A-silenced (SH4) and Scrambled cells (Scramble). Cells were seeded in 96-well plates at 1500 cells/well and incubated for indicated time. The quantity of viable cells was determined by CellTiter-Glow assay. Each point represents mean and standard deviation (SD) of 6 replicates. Experiments were carried out 3 times and showed similar results. (C) CUL4A-depleted 184B5 cells (SH4) and control cells (Scramble) were grown in anchorage-dependent conditions. Cells were fixed and stained with crystal violet. Then crystal violet was solubilized and readouts (optical density at 590 nm) were obtained. Representative example of three-independent experiments is shown. Data are expressed as mean ± SD of three replicates.
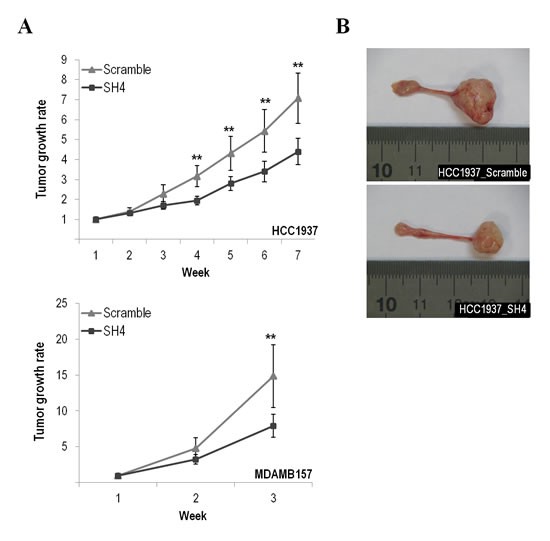
Figure 4: CUL4A down-regulation effect in the tumor growth abilities of the breast cancer cell lines. (A) CUL4A-silenced (SH4) and control cells (Scramble) were orthotopically injected into the mammary glands of nude mice and the tumor onset and growth were evaluated at the indicated time points. Error bars represent 95% confidence intervals. (B) Representative images showing the tumor size formed by HCC1937-silenced and HCC1937-scramble cells. **P<0.01.
CUL4A overexpression promotes in vitro transformation of human mammary epithelial cells
To elucidate the role played by CUL4A in the initial steps of the carcinogenic process the 184B5 immortalized non-transformed human mammary breast cells were transduced with a CUL4A expression vector. Cells with exogenous CUL4A expression showed effective increase in CUL4A protein levels compared with cells infected with empty vector (Figure 5A) and showed increased viability with respect to control cells in cell-based luminescence assays (Figure 5B). Although CUL4A overexpression also induced an S-phase population increase relative to control (Figure 5C) differences did not reach statistically significance. However, when we specifically compared the subpopulation of polyploid cells we found significantly higher BrdU incorporation in the CUL4A-overexpressing cells compared to control cells (3-fold increase, P<0.05). These results suggest that CUL4A overexpression might enhance proliferation through promotion of S-phase entry. In particular, significant increase of poliploid cells in S-phase suggests that CUL4A expression might contribute to tetraploid checkpoint failure mostly leading to propagation of poliploid cells. Consistently, cell cycle analysis revealed that 184B5 cells transduced with the CUL4A vector exhibited a significant increase of polyploid cells compared to those transduced with the empty vector (Figure 5D). We next performed colony formation assays and observed a significant enhancement of the capability of the CUL4A up-regulated cells to form colonies both, in anchorage-dependent (Figure 5E) and anchorage-independent conditions (Figure 5F). Altogether these results support the CUL4A contribution to malignant transformation of normal breast epithelial cells. Despite these in vitro evidences overexpression of CUL4A did not generate 184B5-derived tumors in orthotopically xenografted mice.
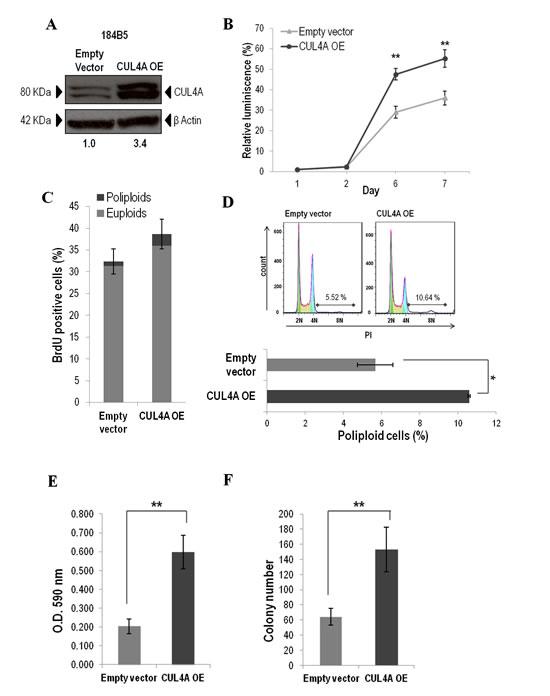
Figure 5: CUL4A overexpression in the 184B5 human mammary epithelial cells. (A) Western blot analysis of CUL4A expression in CUL4A-overexpressing cells (OE) and cells infected with the empty vector. Numbers under the image correspond to intensity of CUL4A bands (neddylated and non-neddylated forms) for each cell line normalized to β-ACTIN intensity value. (B) Viability of CUL4A-overexpressing and control cells. Cells at a concentration of 1500 cells were seeded in a 96-well plate and incubated for indicated time. The quantity of viable cells was determined by CellTiter-Glow assay. Each point represents mean and standard deviation (SD) of 6 replicates. Experiments were carried out 3 times and showed similar results. (C) Percentage of BrdU positive cells in CUL4A-overexpressing and control cells. Bars represent the mean of triplicate experiments and error bars indicate the SD. Light and dark grey illustrates the partial contribution of euploids and poliploids cells, respectively, to the total counting. (D) Cell cycle distribution of CUL4A-overexpressing and control cells. Upper, representative graphs. (E) CUL4A-overexpressing and control cells were grown in anchorage-dependent conditions. Cells were fixed and stained with crystal violet. Then crystal violet was solubilized and readouts (optical density at 590 nm) were obtained. Representative example of three-independent experiments is shown. Data are expressed as mean ± SD of three replicates. (F) CUL4A-overexpressing and control cells were grown in soft agar to evaluate anchorage-independent growth ability Graphics represents the average number of colonies ± SD. **P<0.01, *P<0.05.
CUL4A overexpression collaborates with H-Ras in the transformation of 184B5 cells
In order to continue deciphering CUL4A role in the transformation of human mammary epithelial cells we evaluated CUL4A cooperation with known oncogenes. To this end by using a retroviral system we further modified the CUL4A-overexpressing and control (transduced with the empty vector) 184B5 cells to stably express c-Myc or the mutant active H-Ras (H-Ras-V12).
Overexpression of c-Myc or H-Ras-V12 was confirmed by western blot (Figure 6A) and transforming capacities were evaluated by growing cells in soft agar. Cells with exogenous expression of H-Ras-V12 that concomitantly overexpressed CUL4A presented a statistically significant increase in the number of colonies compared with control cells (transduced with empty vector and H-Ras-V12) (Figure 6B). Cells with ectopic expression of c-Myc that in addition overexpressed CUL4A also presented higher number of colonies than control cells (cells transduced with empty vector and c-Myc) however, differences did not reach significance (Figure 6B). The magnitude of the relative increment in colony number in 184B5 cells that co-expressed H-Ras-V12 and CUL4A was significantly larger (4 fold versus control) than the effect induced in cells that overexpressed CUL4A alone (2 fold versus control) (Figure 6B).
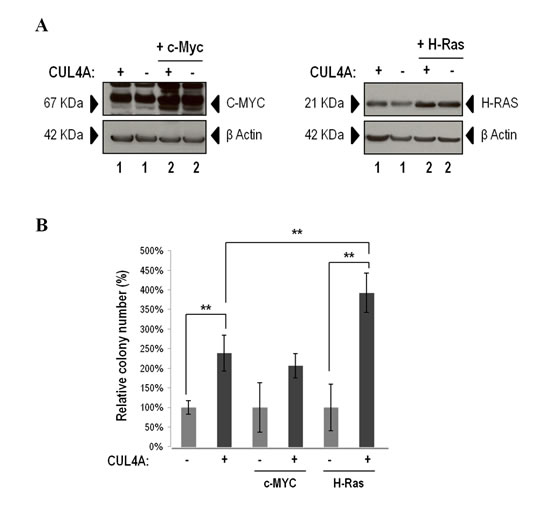
Figure 6: CUL4A cooperation experiment. (A) Western blot analysis of C-MYC (left) and H-RAS (right) ectopic expression in CUL4A-overexpressing and control cells. Numbers under the image correspond to intensity of C-MYC or H-RAS bands for each cell line normalized to β-ACTIN intensity. (B) CUL4A-overexpresing and control cells transduced with c-Myc and H-Ras-V12 were seeded in soft agar, stained with MTT and counted. Values correspond to mean number of colonies ± standard deviation (SD) of three independent replicates. Data show percentage of colonies relative to control. **P<0.01. (+) CUL4A overexpression; (-) Empty vector.
Proteome expression profiling analysis reveals several mediators of CUL4A activity
To better understand the molecular consequences associated with the modulation of CUL4A expression, we examined changes in protein expression of our CUL4A-modified cellular models. To this end, CUL4A-silenced and overexpressed cells and their corresponding controls (cells transduced with scrambled shRNA and empty vector, respectively) were analyzed using a quantitative proteomic approach. We confidently identified and quantified 5,087 proteins (Supplementary Table S4) from which 764 showed differential expression in at least one of the three experiments conducted (Supplementary Figure S1) (see Materials and Methods). As expected, and technically validating our approach, CUL4A was one of the top-ranking differentially expressed proteins being down-regulated in silenced cells and up-regulated in overexpressed cells (Figure 7A). We then defined a subset of 64 differentially expressed proteins that showed opposite behavior between the CUL4A-overexpressing cells and at least one of the CUL4A knocked down models. Proteins that did not show regulation or with no available data in the CUL4A-overexpressing cells but that showed same behavior in both CUL4A silenced cells were also included in the list. Among the 64 differentially expressed proteins we found a large variety of protein classes (Figure 7B) including nucleic acid binding proteins (e.g. HMGA1, PDCD4, MAX), hydrolases (e.g. DDX60, SETX, MX1) and transporters (e.g. TAP1, ABCF3, SLC39A9).
To get further insights into the functional roles of such proteomic changes, we performed enrichment analysis of the 64 proteins by using Ingenuity Pathway Analysis (IPA) which showed a significant over-representation of functions such as “cell cycle”, “cellular growth and proliferation”, “cellular movement” or “antigen presentation” and of canonical pathways such as “interferon signaling” (Benjamini-Hochberg test, P<0.05). Interestingly, several known or putative oncogenes showed up-regulation upon CUL4A overexpression in the 184B5 cells and exhibited down-regulation in the CUL4A-silenced models (Figure 7A). Among them, SSRP1 and SEPT2 that participate in cell cycle progression [17, 18] or MYCBP, HMGA1 and MT2A involved in cell growth and proliferation and previously linked to tumor progression [19-21]. Also, replication-dependent histones such as HIST1H4A and HIST2H3A were up-regulated in CUL4A-overexpressing cells and down-regulated in at least one of the silencing models. Conversely, a number of putative or known tumor suppressors were down-regulated in CUL4A-overexpressing cells and/or overexpressed in CUL4A knocked down cells (Figure 7A). Among them, the Programmed cell death protein 4 (PDCD4) that blocks cell cycle progression and cell proliferation and the anti-tumoral activity-related protein CRABP2 that is involved in cellular growth inhibition [22, 23]. In addition a set of 15 proteins that participate in the antitumor immune response such as the tumor suppressors PSMB9, ERAP1 and TAP1 or the interferon-stimulated genes IFIT1 and MX1 were found down-regulated in CUL4A-overexpresing cells and up-regulated in CUL4A-silencing models (Figure 7A). Given the novelty of a possible relation between the CUL4A E3 ubiquitin ligase and immune response modulation in breast cancer we interrogated IPA to specifically evaluate the potential link between the ubiquitin degradation system and the differentially expressed immune system-related proteins from our list. Remarkably, IPA generated a protein network showing multiple connections between this set of proteins and ubiquitin, which would be consistent with a putative role of CUL4A as modulator of immune system processes (Figure 7C).
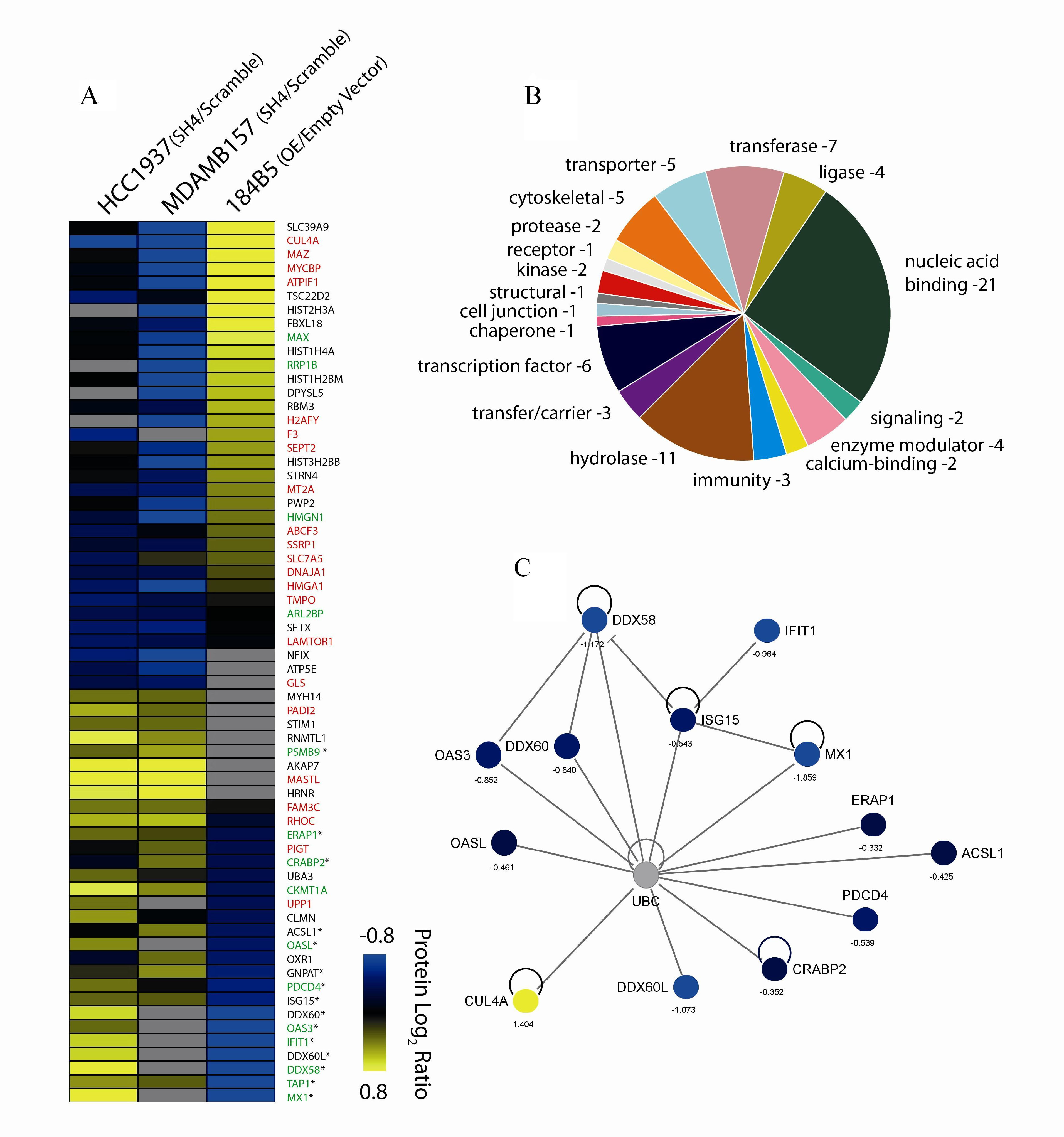
Figure 7: Differentially regulated proteins. (A) Candidate proteins modulated by CUL4A expression identified by quantitative proteomics in the silencing (HCC1937, MDAMB157) and overexpression (184B5) cell line models. Protein log2 ratios are represented by colors, where “blue” means differentially down-regulated protein and “yellow”, differentially up-regulated protein. Grey and black colors refer to not found or not modified proteins, respectively. Protein names in red indicate collaboration with the oncogenic process, green involvement in tumor suppressor activities, and (*) implication in the immune system, as described in the literature. Detailed information such as accession number, gene symbol, averaged ratio, and full protein names for these proteins are listed in Supplementary Table S4. (B) Classification of candidate proteins based on their molecular functional annotations using gene ontology (GO). (C) Network generated by IPA including the immune system-related proteins and CUL4A. Color code for differential regulation as described in (A).
DISCUSSION
The overexpression of the CUL4A E3 ubiquitin ligase has been related to tumor aggressiveness and poor clinical outcome in breast cancer [6, 10, 24]. One of the mechanisms triggering CUL4A overexpression is the 13q34 amplification and this genomic aberration has been shown to be associated with breast tumors characterized by exhibiting BRCA1 impairment or a basal-like phenotype [12, 13]. With these antecedents we were interested in studying the role of CUL4A in the carcinogenic process of the basal-like breast tumor subtype, which is aggressive in nature and lacks effective targeted therapies. Our results show that CUL4A would contribute to the tumorigenicity of basal-like breast cancers through the modulation of cell growth and anti-tumor immune response.
CUL4A silencing in CUL4A-overexpressing basal-like breast cancer cells induced a reduction of cell proliferation and colony formation in short-term assays and long-term soft agar assays and decreased the tumor growth rate of cells injected into mammary glands of nude mice. Importantly, we specifically demonstrated this effect in 13q34 amplification-bearing cells, which is biologically relevant given the association found between this amplification and primary breast tumors of basal-like phenotype [12, 13]. We previously found an increased frequency of the 13q34 amplification in BRCA1 tumors [12] and in this study we also demonstrate the growth inhibitory effect of CUL4A silencing in BRCA1-deficient cells. Altogether our results support the role of CUL4A as target gene of the 13q34 amplification. We provide evidence that, rather than being a passenger alteration derived from the increased genomic instability shown by basal-like and BRCA1 primary breast tumors [14, 25], CUL4A overexpression would confer selective advantage to tumor cells.
This dependence or ´addiction´ to CUL4A for maintenance of the malignant phenotype and cell survival of CUL4A-overexpressing cells would be similar to that observed in breast tumor cells that overexpress ERBB2 or C-MYC [26, 27]. Hence, it may offer the opportunity to take advantage of CUL4A up-regulation in order to develop targeted therapies. In this regard our data suggest that the 13q34 amplification could be a robust biomarker for identification of up to 20% basal-like breast cancer patients [12, 13] that might eventually benefit from CUL4A-targeted therapy. Interestingly, in previous studies we observed increased sensitivity to trabectedin in breast cancer cell lines exhibiting high levels of CUL4A in combination with a “BRCAness” status [28]. The potential therapeutic option of CUL4A- targeting in certain subtypes of breast cancer has also been discussed in a recent study reporting a fundamental role of CUL4A in regulating the metastatic behavior of breast cancer cells [11]. However, the authors did not provide data on the effect of CULA4 silencing in normal mammary cells. We showed that CUL4A silencing would not present detrimental consequences on the non-tumorigenic 184B5 mammary epithelial cells. These observations would be consistent with lack of CUL4A dependence in cells that do not overexpress the gene and might suggest low toxicity of eventual CUL4A-targeted inhibitory therapeutic approaches.
Although exogenous overexpression of CUL4A was sufficient to promote proliferation of 184B5 cells, CUL4A up-regulation alone was not enough to generate 184B5-derived tumors in xenografted mice. This suggests that CUL4A overexpression, rather than an initiating event, would constitute a secondary oncogenic hit acquired and selected during tumoral evolution. Our results support a potential synergistic effect between CUL4A overexpression and the constitutive activation of the RAS mitogenic signal in the transformation of human mammary epithelial cells, which is consistent with the high frequency of RAS pathway activation in basal-like breast tumors [29, 30].
To our knowledge, this is the first study that aims to define mediators of CUL4A transforming capacities in breast cancer using a proteomics approach. By using this methodology we propose novel molecules and pathways that might modulate the oncogenic effect of CUL4A in basal-like tumors. This can shed light on the understanding of the molecular events driven by CUL4A that may be explored in order to develop new therapeutic strategies.
Among the proteins that showed up-regulation upon induction of CUL4A exogenous expression were HMGA1, SSRP1 and MT2A. Interestingly, HMGA1, SSRP1 and MT2A overexpression has been reported in breast cancer [19, 20, 31] and HMGA1 was previously described as a master regulator of the tumor progression in triple-negative breast cancer cells [32]. These molecules may be good candidates to develop targeted therapies in CUL4A-overexpressing tumors. In particular, SSRP1 is reported to be a prospective target for anti-cancer small molecules [31, 33]. On the other hand, PDCD4 and CRABP2 protein levels were reduced with CUL4A overexpression. Consistently, low levels of PDCD4 in breast tumors have been associated with poor prognosis [34] and high CRABP2 levels have been shown to be indicative of longer overall survival in breast cancer patients [22].
For the first time, we report a putative role of CUL4A in bypassing the immune system in breast cancer through the down-regulation of several molecules involved in the anti-tumor immune surveillance. The overexpression of CUL4A induced a reduction of TAP1, IFIT1 and MX1 protein levels. Low or defective TAP1 in breast tumors predicts higher risks for developing metastasis [35] and down-regulation of the interferon-stimulated genes IFIT1 and MX1 has been linked to immune evasion mechanisms and tumor progression [36-39]. Interestingly, E3 ligases are involved in the efficient regulation of the immune system [40-42] and alterations in the ubiquitination machinery/pathway are associated with immune deficiencies and cancer [43].
In summary, we propose CUL4A as an important contributor to the development and progression of basal-like breast cancers. Our results indicate that CUL4A might play a central role in the oncogenic process by modulating the expression of a number of oncogenes and tumor suppressors. Interestingly, there is a growing number of E3 ubiquitin ligases being involved in cancer. For example, MDM2 and IAP are highly expressed in several human cancers [44, 45] and inhibitors for these enzymes are being evaluated in clinical trials [43]. Thereby CUL4A would constitute a promising target for therapeutic intervention and our data reinforce its clinical value in basal-like breast cancers.
MATERIALS AND METHODS
Cell lines and cell culture
A panel of 13 human breast epithelial cell lines was included in this study (Supplementary Table S1). Eight corresponded to sporadic breast cancer tumors (MDAMB157, MFM223, HCC1143, SKBR3, Hs578T, MCF7, MDAMB231 and T47D). Two derived from breast tumors of BRCA1 mutation carrier patients (HCC1937 and MDAMB436) and one derived from a breast tumor with somatic inactivation of the BRCA1 gene (UACC3199). Two human mammary breast cell lines derived from normal breast tissue (184B5, HBL100). SKBR3, T47D, HBL100, MCF7, MDAMB231, Hs578T, and UACC3199 were obtained from the Cancer Epigenetics Group at the Bellvitge Institute for Biomedical Research (Barcelona, Spain); MDAMB157, HCC1143 and HCC1937 were kindly provided by Dr. P. Edwards (Department of Pathology, University of Cambridge, Cambridge, UK); MDAMB436 was provided by Dr. K.S. Massey- Brown (Department of Pharmacology and Toxicology, University of Arizona, Tucson, AZ); MFM223 cells were purchased from DSMZ (Braunschweig, Germany); 184B5 was acquired from the ATCC (Manassas, VA, USA). Authentication was not carried out. MDAMB157 presented amplification at the 13q34 region [12, http://www.sanger.ac.uk/] and were grown in Dulbecco’s Modified Eagle’s Medium (Gibco, Life Technologies, Grand Island, NY). 184B5 was cultured in a 1:1 mixture of Dulbecco’s modified Eagle’s medium and F12 medium (DMEM-F12, Sigma-Aldrich, St. Louis, MO) supplemented with 5% horse serum (Invitrogen, Life Technologies, Carlsbad, CA), insulin (10 µg/ml), hydrocortisone (0.5 µg/ml), cholera toxin (100 ng/ml), and epidermal growth factor (20 ng/ml) (Sigma-Aldrich). The remaining cell lines were grown in RPMI-1640 (Sigma-Aldrich) or DMEM (Gibco, Life Technologies). All mediums were completed with 10% FBS, 1% penicillin/streptomycin, and 0.5% fungizone (Gibco, Life Technologies). Cells were maintained in an atmosphere of 5% CO2 in air at 37ºC.
RNA isolation and real-time quantitative PCR (qPCR)
Total RNA was extracted from three independent plates for each cell line (RNAeasy Kit, Quiagen, Valencia, CA) and converted to cDNA by using 500ng as template (High Capacity cDNA RT kit, Applied Biosystems, Foster City, CA). qPCR assays were designed for target gene (CUL4A) and endogenous control (β-ACTIN ) using the Roche Universal Probe Library Assay Design Centre web site (Roche Applied Science, Indianapolis, IN) (Supplementary Table S2). Reactions were performed in triplicate using the ABI Prism 7900HT Sequence Detection System according to the manufacturer’s protocol (Applied Biosystems, Foster City, CA). Relative expression was determined using the qBase software that allows for PCR efficiency correction and implements normalization by endogenous genes [46].
Protein extraction and immunoblot analysis
Cells were harvested and lysed in RIPA buffer [50 mmol/L Tris-HCl, pH 8.0, with 150 mmol/L NaCl, 1.0% Igepal CA-630 (NP-40), 0.5% sodium deoxycholate, and 0.1% SDS; Sigma-Aldrich, St. Louis, MO] and protease inhibitor cocktail (Roche Applied Science, Foster City, CA). Protein concentration of samples was determined by detergent-compatible colorimetric assay (Bio-Rad Laboratories, Hercules, CA). Ninety micrograms of total protein were subjected to electrophoresis on NUPAGE 4% to 12% Bis–Tris gels and MOPS running buffer (Novex, Carlsbad, CA) followed by blotting to nitrocellulose membranes. The blots were analized using polyclonal rabbit anti-CUL4A (1:100; #2699; Cell Signaling Technology, Danvers, MA), polyclonal rabbit anti-Myc (1:1000; #06-340; Millipore, Billerica, MA), mouse anti-Pan-Ras (1:1000; #OP40; Millipore, Billerica, MA) and monoclonal mouse anti-β-actin (1:12,000, clone AC-15, #A5441, Sigma-Aldrich, St. Louis, MO) antibodies. The immunoblot signals were quantified using the ImageJ 1.43u software (Wayne Rasband, National Institutes of Health). CUL4A bands intensity were normalized to β-actin bands.
Lentiviral constructs
Cell line models were modified for the expression of CUL4A. For gene down-regulation short hairpin RNAs (shRNA) for human CUL4A (NM_003589) were obtained from Sigma-Aldrich (MISSION shRNA Bacterial Glycerol Stock). Five different shRNA constructs were transduced, and the one providing better knockdown efficiency and non-citotoxic effects was selected (SH4, TRCN0000006530; Supplementary Table S3) to silence CUL4A expression in the HCC1937 and MDAMB157 cell lines. A MISSION Non-target shRNA vector (Scramble SHC002) was used as negative control. In order to up-regulate the expression of CUL4A a human cDNA of the gene (ID CNIO: hAE5834) containing an HA tag was cloned into FG12-CMV vector (kindly provided by Dr. M. Soengas at CNIO) that expresses green fluorescent protein (GFP). FG12-CMV empty vector was used as negative control.
CUL4A-overexpressing cell models and controls were modified for the expression of H-Ras-V12 and c-MYC by using a retroviral system. The retroviral vectors pBabe-Neo-H-Ras-V12 and pBabe-Puro-c-Myc were kindly provided by Dr. Víctor Javier Sánchez-Arévalo at CNIO.
Generation of stable cell lines
Lentiviral vectors were cotransfected with lentiviral packaging plasmids into 293FT cells (Invitrogen, Carlsbad, CA) in the presence of Fugene 6 (Promega, Madison, WI). The virus-containing supernatants were collected after 36 hours of transfection and filtered. Cells were infected and the stably transduced cells were selected either by adding medium containing 1 mg/mL of puromycin (CUL4A down-regulation) or by GFP sorting by flow cytometry (FACS Calibur; BD Biosciences, San Jose, CA) (CUL4A up-regulation). Modification of CUL4A levels was confirmed by q-PCR and Western blot. Retroviral vectors (pBabe-Neo-H-Ras-V12 or pBabe-Puro-c-Myc) were cotransfected with pCL-Ampho retroviral packaging plasmid into 293FT cells. Infection was carried out as explained above and the selection was performed by using neomycin or puromycin, respectively.
Cell viability assay
Viability was measured by using the CellTiter-Glo Luminescent Cell Viability Assay from Promega, according to the manufacturer’s recommendations. Cells were seeded on 96-well plates at densities of 1500 or 2500 cells per well. Luminescence was recorded using a plate reader (Wallac 1420 VICTOR2™, PerkinElmer, Waltham, MA).
BrdU incorporation assay
Cells were incubated with 10 µM bromodeoxyuridine (BrdU) for 30 minutes, fixed, permeabilized and processed using anti-BrdU antibody (BD Bioscence, San Jose CA) and fluorescein isothiocianate (FITC) secondary antibody (Dako Cytomation, Glostrup, Denmark). At least 10,000 single events were assembled. Staining was analyzed by flow cytometry (FACS Calibur; BD Biosciences). All data were analyzed using FlowJo software (TreeStar, Oregon).
Cell-cycle analysis
Cells were fixed in ethanol and incubated with 50 µg/ml propidium iodide and 10 mg/ml RNAse A (Sigma-Aldrich, St. Louis, MO) followed by analysis on a FACS Calibur flow cytometer (BD Biosciences). At least 10,000 single events were collected. All data were analyzed using FlowJo software (TreeStar, Oregon).
Colony formation assay
Cells were seeded at densities of 1,500 or 2500 cells per well in 6-well plates and incubated for 9 to 10 days. Medium was removed and cells were washed, fixed, and stained with 0.1% crystal violet. Cell density was assessed by solubilization of crystal violet with 15% acetic acid and absorbance of the solution was measured at 590 nm (Wallac 1420 VICTOR2™, PerkinElmer, Waltham, MA).
Soft agar assay
30,000 cells were embedded in 0.3% agar over a 0.6% base agar layer. Soft agar cultures were fed with growth media once a week and incubated during 3 weeks. To visualize colonies, cultures were stained with MTT and counted manually.
Orthotopic xenografts
Primary tumorigeneis in vivo was evaluated by orthotopicaly inoculating CUL4A-modified breast cell lines into the mammary glands of 6-month-old female adult athymic nude mice (Hsd:Athymic Nude-Foxn1nu). One million HCC1937 cells and two million MDAMB157 cells infected with the CUL4A-shRNA vector (or with the Scramble-shRNA) were inoculated. Tumor growth was monitored weekly with a caliper and tumor volumes were calculated using the formula volume= [(length x width2)/2]. The experiment was stopped, mice were sacrificed, and tumors were harvested when tumors in control animals reached approximately 1 cm3. Mean volume of tumors per group of animals (injected with CUL4A-silenced and control cells) was measured each week. Animal experimentation at the CNIO, Madrid, was performed according to protocols approved by the CNIO-ISCIII Ethics Committee for Research and Animal Welfare (CEIyBA).
Proteomic analysis
An iTRAQ proteomics approach was performed to identify proteins and pathways associated with CUL4A modulation. To this end we looked for differentially expressed proteins between each of the CUL4A-overexpressing cell line models (HCC1937 and MDAMB157 transduced with CUL4A specific shRNA, SH4) and their corresponding control cells (transduced with scrambled shRNA). We also looked for differentially expressed proteins between the CUL4A-overexpresing 184B5 cells (transduced with CUL4A cDNA) and control cells (transduced with empty vector). Cell pellets were extracted and proteins were digested using the FASP protocol [47]. Peptides were labeled with iTRAQ reagents and samples were pooled. Then, the complex mixture was subjected to IEF fractionation. The resulting fractions were separated by on-line nano-LC and analyzed by electrospray MS/MS using a LTQ Orbitrap Velos mass spectrometer (Thermo Scientific, Bremen, Germany). Raw files were searched against SwissProt human database (release date: March 21, 2012; 20329 entries) using MASCOT [48] as a search engine through the Proteome Discoverer (Thermo Scientific) software. Peptides were filtered at 1% FDR using a concatenated target-decoy database. Proteins were considered differentially regulated using a cutoff > 1.25-fold. A more detailed description of methodology is provided in the Supplementary Information.
Statistical analyses
We used SPSS version 13 software to conduct the statistical analysis of our data. Two-tailed Student t tests were used for comparisons between different groups. Statistical analysis of the mean tumor volumes was done by U test of Mann-Whitney analysis. P< 0.05 and P< 0.01 were considered to be significant. Unless otherwise stated, the error bars represent standard deviation of the biologic triplicates.
ACKNOWLEDGMENTS
We thank the excellent technical support from the CNIO Flow Cytometry Core Unit and the Animal Facility. LPSC is funded by the “Ministerio Español de Sanidad y Consumo, PFIS FI08/00155”. MJG is recipient of a research contract from “Instituto de Salud Carlos III” of the “Ministerio Español de Sanidad y Consumo” (Miguel Servet Tipo II Program, CPII 13/00047). PXE is a recipient of the Fondo de Investigaciones Sanitarias (FIS) grant (CA10/01231). This work has been funded in part by MEC grants PI12/00070 and PI09/1120. The CNIO Proteomics Unit is member of Proteored, PRB2-ISCIII, supported by grant PT13/0001.
REFERENCES
1. Lee J and Zhou P. Pathogenic Role of the CRL4 Ubiquitin Ligase in Human Disease. Front Oncol. 2012; 2:21.
2. Sugasawa K. The CUL4 enigma: culling DNA repair factors. Mol Cell. 2009; 34(4):403-404.
3. Liu L, Lee S, Zhang J, Peters SB, Hannah J, Zhang Y, Yin Y, Koff A, Ma L and Zhou P. CUL4A abrogation augments DNA damage response and protection against skin carcinogenesis. Mol Cell. 2009; 34(4):451-460.
4. Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T and Tsurimoto T. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J Biol Chem. 2008; 283(43):29045-29052.
5. Nag A, Bagchi S and Raychaudhuri P. Cul4A physically associates with MDM2 and participates in the proteolysis of p53. Cancer Res. 2004; 64(22):8152-8155.
6. Chen LC, Manjeshwar S, Lu Y, Moore D, Ljung BM, Kuo WL, Dairkee SH, Wernick M, Collins C and Smith HS. The human homologue for the Caenorhabditis elegans cul-4 gene is amplified and overexpressed in primary breast cancers. Cancer Res. 1998; 58(16):3677-3683.
7. Hung MS, Mao JH, Xu Z, Yang CT, Yu JS, Harvard C, Lin YC, Bravo DT, Jablons DM and You L. Cul4A is an oncogene in malignant pleural mesothelioma. J Cell Mol Med. 2009; 15(2):350-358.
8. Ren S, Xu C, Cui Z, Yu Y, Xu W, Wang F, Lu J, Wei M, Lu X, Gao X, Liang Y, Mao JH and Sun Y. Oncogenic CUL4A determines the response to thalidomide treatment in prostate cancer. J Mol Med (Berl). 2012; 90(10):1121-1132.
9. Yasui K, Arii S, Zhao C, Imoto I, Ueda M, Nagai H, Emi M and Inazawa J. TFDP1, CUL4A, and CDC16 identified as targets for amplification at 13q34 in hepatocellular carcinomas. Hepatology. 2002; 35(6):1476-1484.
10. Schindl M, Gnant M, Schoppmann SF, Horvat R and Birner P. Overexpression of the human homologue for Caenorhabditis elegans cul-4 gene is associated with poor outcome in node-negative breast cancer. Anticancer Res. 2007; 27(2):949-952.
11. Wang Y, Wen M, Kwon Y, Xu Y, Liu Y, Zhang P, He X, Wang Q, Huang Y, Jen KY, Labarge MA, You L, Kogan SC, Gray JW, Mao JH and Wei G. CUL4A Induces Epithelial-Mesenchymal Transition and Promotes Cancer Metastasis by Regulating ZEB1 Expression. Cancer Res. 2013; 74(2):520-531.
12. Melchor L, Saucedo-Cuevas LP, Munoz-Repeto I, Rodriguez-Pinilla SM, Honrado E, Campoverde A, Palacios J, Nathanson KL, Garcia MJ and Benitez J. Comprehensive characterization of the DNA amplification at 13q34 in human breast cancer reveals TFDP1 and CUL4A as likely candidate target genes. Breast Cancer Res. 2009; 11(6):R86.
13. Melchor L, Honrado E, Garcia MJ, Alvarez S, Palacios J, Osorio A, Nathanson KL and Benitez J. Distinct genomic aberration patterns are found in familial breast cancer associated with different immunohistochemical subtypes. Oncogene. 2008; 27(22):3165-3175.
14. Foulkes WD, Smith IE and Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2012; 363(20):1938-1948.
15. Kao J, Salari K, Bocanegra M, Choi YL, Girard L, Gandhi J, Kwei KA, Hernandez-Boussard T, Wang P, Gazdar AF, Minna JD and Pollack JR. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One. 2009; 4(7):e6146.
16. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006; 10(6):515-527.
17. Tan BC, Chien CT, Hirose S and Lee SC. Functional cooperation between FACT and MCM helicase facilitates initiation of chromatin DNA replication. EMBO J. 2006; 25(17):3975-3985.
18. Yu W, Ding X, Chen F, Liu M, Shen S, Gu X and Yu L. The phosphorylation of SEPT2 on Ser218 by casein kinase 2 is important to hepatoma carcinoma cell proliferation. Mol Cell Biochem. 2009; 325(1-2):61-67.
19. Cherian MG, Jayasurya A and Bay BH. Metallothioneins in human tumors and potential roles in carcinogenesis. Mutat Res. 2003; 533(1-2):201-209.
20. Di Cello F, Shin J, Harbom K and Brayton C. Knockdown of HMGA1 inhibits human breast cancer cell growth and metastasis in immunodeficient mice. Biochem Biophys Res Commun. 2013; 434(1):70-74.
21. Xiong J, Du Q and Liang Z. Tumor-suppressive microRNA-22 inhibits the transcription of E-box-containing c-Myc target genes by silencing c-Myc binding protein. Oncogene. 2010; 29(35):4980-4988.
22. Liu RZ, Graham K, Glubrecht DD, Germain DR, Mackey JR and Godbout R. Association of FABP5 expression with poor survival in triple-negative breast cancer: implication for retinoic acid therapy. Am J Pathol. 2011; 178(3):997-1008.
23. Wei N, Liu SS, Chan KK and Ngan HY. Tumour suppressive function and modulation of programmed cell death 4 (PDCD4) in ovarian cancer. PLoS One. 2012; 7(1):e30311.
24. Abba MC, Fabris VT, Hu Y, Kittrell FS, Cai WW, Donehower LA, Sahin A, Medina D and Aldaz CM. Identification of novel amplification gene targets in mouse and human breast cancer at a syntenic cluster mapping to mouse ch8A1 and human ch13q34. Cancer Res. 2007; 67(9):4104-4112.
25. Popova T, Manie E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, Delattre O, Sigal-Zafrani B, Bollet M, Longy M, Houdayer C, Sastre-Garau X, Vincent-Salomon A, Stoppa-Lyonnet D and Stern MH. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012; 72(21):5454-5462.
26. Carey LA. Breast cancer: HER2--a good addiction. Nat Rev Clin Oncol. 2012; 9(4):196-197.
27. Kuehl WM and Bergsagel PL. MYC addiction: a potential therapeutic target in MM. Blood. 2012; 120(12):2351-2352.
28. Garcia MJ, Saucedo-Cuevas LP, Munoz-Repeto I, Fernandez V, Robles MJ, Domingo S, Palacios J, Aracil M, Nieto A, Tercero JC and Benitez J. Analysis of DNA repair-related genes in breast cancer reveals CUL4A ubiquitin ligase as a novel biomarker of trabectedin response. Mol Cancer Ther. 2013; 12(4):530-541.
29. Hoeflich KP, O’Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, Januario T, Savage H, Punnoose E, Truong T, Zhou W, Berry L, Murray L, Amler L, Belvin M, Friedman LS, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009; 15(14):4649-4664.
30. Network TCGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490(7418):61-70.
31. Koman IE, Commane M, Paszkiewicz G, Hoonjan B, Pal S, Safina A, Toshkov I, Purmal AA, Wang D, Liu S, Morrison C, Gudkov AV and Gurova KV. Targeting FACT complex suppresses mammary tumorigenesis in Her2/neu transgenic mice. Cancer Prev Res (Phila). 2012; 5(8):1025-1035.
32. Shah SN, Cope L, Poh W, Belton A, Roy S, Talbot CC, Jr., Sukumar S, Huso DL and Resar LM. HMGA1: a master regulator of tumor progression in triple-negative breast cancer cells. PLoS One. 2013; 8(5):e63419.
33. Gasparian AV, Burkhart CA, Purmal AA, Brodsky L, Pal M, Saranadasa M, Bosykh DA, Commane M, Guryanova OA, Pal S, Safina A, Sviridov S, Koman IE, Veith J, Komar AA, Gudkov AV, et al. Curaxins: anticancer compounds that simultaneously suppress NF-kappaB and activate p53 by targeting FACT. Sci Transl Med. 2012; 3(95):95ra74.
34. Meric-Bernstam F, Chen H, Akcakanat A, Do KA, Lluch A, Hennessy BT, Hortobagyi GN, Mills GB and Gonzalez-Angulo AM. Aberrations in translational regulation are associated with poor prognosis in hormone receptor-positive breast cancer. Breast Cancer Res. 2012; 14(5):R138.
35. Liu Y, Komohara Y, Domenick N, Ohno M, Ikeura M, Hamilton RL, Horbinski C, Wang X, Ferrone S and Okada H. Expression of antigen processing and presenting molecules in brain metastasis of breast cancer. Cancer Immunol Immunother. 2012; 61(6):789-801.
36. Calmon MF, Rodrigues RV, Kaneto CM, Moura RP, Silva SD, Mota LD, Pinheiro DG, Torres C, de Carvalho AF, Cury PM, Nunes FD, Nishimoto IN, Soares FA, da Silva AM, Kowalski LP, Brentani H, et al. Epigenetic silencing of CRABP2 and MX1 in head and neck tumors. Neoplasia. 2009; 11(12):1329-1339.
37. Critchley-Thorne RJ, Simons DL, Yan N, Miyahira AK, Dirbas FM, Johnson DL, Swetter SM, Carlson RW, Fisher GA, Koong A, Holmes S and Lee PP. Impaired interferon signaling is a common immune defect in human cancer. Proc Natl Acad Sci U S A. 2009; 106(22):9010-9015.
38. Mushinski JF, Nguyen P, Stevens LM, Khanna C, Lee S, Chung EJ, Lee MJ, Kim YS, Linehan WM, Horisberger MA and Trepel JB. Inhibition of tumor cell motility by the interferon-inducible GTPase MxA. J Biol Chem. 2009; 284(22):15206-15214.
39. Reiser J, Hurst J, Voges M, Krauss P, Munch P, Iftner T and Stubenrauch F. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J Virol. 2011; 85(21):11372-11380.
40. Kumar KG, Tang W, Ravindranath AK, Clark WA, Croze E and Fuchs SY. SCF(HOS) ubiquitin ligase mediates the ligand-induced down-regulation of the interferon-alpha receptor. EMBO J. 2003; 22(20):5480-5490.
41. Lee YR, Yuan WC, Ho HC, Chen CH, Shih HM and Chen RH. The Cullin 3 substrate adaptor KLHL20 mediates DAPK ubiquitination to control interferon responses. EMBO J. 2010; 29(10):1748-1761.
42. Yuan C, Qi J, Zhao X and Gao C. Smurf1 protein negatively regulates interferon-gamma signaling through promoting STAT1 protein ubiquitination and degradation. J Biol Chem. 2012; 287(21):17006-17015.
43. Fulda S, Rajalingam K and Dikic I. Ubiquitylation in immune disorders and cancer: from molecular mechanisms to therapeutic implications. EMBO Mol Med. 2012; 4(7):545-556.
44. Fulda S and Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012; 11(2):109-124.
45. Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, Onel K, Yip L, Hwang SJ, Strong LC, Lozano G and Levine AJ. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004; 119(5):591-602.
46. Hellemans J, Mortier G, De Paepe A, Speleman F and Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007; 8(2):R19.
47. Wisniewski JR, Zougman A, Nagaraj N and Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009; 6(5):359-362.
48. Perkins DN, Pappin DJ, Creasy DM and Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999; 20(18):3551-3567.