
INTRODUCTION
The sensitivity of the mammalian myocardium to pathological alterations is related to its highly limited regenerative potential [1, 2]. Interestingly, in a variety of pathophysiological conditions including ischemia, hypertrophy, and atrophy, a common response of the heart is the induction of fetal gene reprogramming associated with complex changes in gene expression profiles, often suggested to support cell adaptation and survival [3]. These include modulation of the expression of genes coding for myosin heavy chains (MHCs). The expression of the α isoform of MHC (MHCα; encoded by the Myh6 gene) decreases, while the expression of the β isoform (MHCβ; encoded by Myh7 gene) increases. Physiologically, the ratio of MHC isoform expressions changes in the opposite manner after birth under the control of thyroid hormone [4–6]. The critical pathological event responsible for such regulation is not yet well understood; however, it has been suggested that it is most likely related to alterations in the myocardium microenvironment [7].
The acute switch from an aerobic to an anaerobic metabolism occurring in cardiac cells appears to be a necessary prerequisite for immediate cell survival during hypoxia, the oxygen deprivation of tissues and cells [7, 8]. The modulation of the energetic metabolism is suggested by various authors as an important factor in the induction of the evolutionarily conserved fetal gene program [7, 8]. Interestingly, a positive correlation between the glycolytic metabolism and MHCβ isoform expression has also been described in primary rat cardiomyocytes cultured in vitro [9].
The key regulatory factor involved in cell response to a decreased oxygen level is hypoxia-inducible factor (HIF) [10]. Transcriptionally active HIF is composed of two subunits; one of the three oxygen-dependent HIF-α subunits (HIF-1α, -2α, or -3α) forms dimers with HIF-1β [11, 12]. The most intensively studied subunit, HIF-1α, is upregulated by oxygen at two levels – protein stability and transcriptional activity – via prolyl hydroxylase domain enzymes (PHD1–3) and factor inhibiting HIF, respectively [13, 14]. Murine HIF-1α is expressed at high levels in the ventricular wall, but decreases throughout embryonal development [15–17]. There is no detectable level of HIF-1α protein in healthy adult ventricles [18]. Interestingly, a number of cardiac pathophysiologies are associated with increased HIF-1α [18]. An increase in HIF-1α mRNA levels is one of the earliest responses to myocardial ischemia [19]. Interestingly, hypoxia was suggested to decrease MHCα and to increase MHCβ transcript levels in rat in vivo and in vitro models [20]. Moreover, the pharmacological stabilization of HIF-1α, mediated by PHD inhibitor dimethyloxalylglycine (DMOG), was shown to be cardioprotective [21, 22].
Besides HIF-directed hypoxic responses, there are also several other hypoxia-related pathways which ensure ATP conservation by limiting energy-consuming processes in favor of processes that are critical for the maintenance of cell viability [23]. The mammalian (also mechanistic) target of rapamycin (mTOR) is a serine/threonine protein kinase which regulates protein translation, cellular metabolism, and the organization of actinin in response to nutrient, growth factors, and O2 availability [23, 24]. mTOR is inhibited in the majority of cells in response to hypoxia [23].
Notably, the expression of MHCs is currently understood to be repressed by antisense RNA molecules transcribed in the healthy myocardium [25–27]. Antisense Myh7 RNA was shown to inhibit the processing of sense pre-mRNA into mature mRNA, thus turning off Myh7 gene expression [26, 28]. In addition, the long noncoding RNA (lncRNA) Mhrt transcribed in the Myh7 locus was recently reported to protect the heart from pathological hypertrophy [29].
Considering the association between changes in oxygen supply and the modulation of Myh6 and Myh7 gene expression, we suggest here that the primary trigger crucial for activation of the fetal gene program is the limited amount of oxygen. Uncovering the possible role of hypoxia, particularly reflected by the role of HIF-1α, in the regulation of MHC gene expression is therefore the main goal of this work.
RESULTS
Hypoxic conditions and the pharmacological stabilization of HIF induce MHCβ expression in cardiomyocytes derived from mESC
We engaged a model system represented by preselected (pMHCα-Neo-Hyg vector) cardiomyocytes differentiated in vitro from mESC (HG8) to observe the impact of hypoxic exposure. Hypoxia (1% O2) or the presence of DMOG, inducing the pharmacological stabilization of HIF-α subunits by the inhibition of PHDs [30], upregulated the gene expression of Myh7 (Figure 1A, 1C) as well as total MHCα and β protein levels (Figure 1B) in comparison with normoxic cultivation. Aware of the fact that in the HG8 model the MHCα expression profile might be biased by cardiomyocyte pre-selection based on Myh6 promoter activity, we also tested the expression of both cardiac MHC isoforms upon hypoxic exposure in the parental R1 cell line and again observed the upregulation of Myh7 mRNA expression (Figure 1D) and total MHCα and β protein levels (Figure 1E).
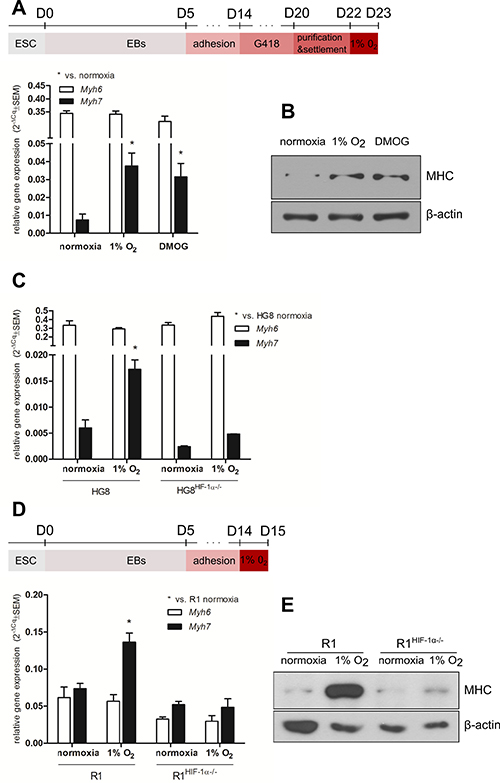
Figure 1: Effect of hypoxia and the pharmacological stabilization of HIF on MHC expression. Time-scheme of cardiomyocyte derivation from A top) HG8 and D top) R1 and R1HIF-1α−/− mESC. Myh6 and Myh7 relative mRNA levels in (A) cardiomyocytes derived from HG8 mESC (n = 3), (C) cardiomyocytes derived from parental HG8 and HG8HIF-1α−/− mESC (n = 3), and (D) cardiomyocytes derived from R1 and R1HIF-1α−/− mESC determined by qRT-PCR (n = 4 (R1), n = 3 (R1HIF-1α−/−)). Data are presented as 2−ΔCq ± SEM (groups were compared using one-way ANOVA with the Tukey (HSD) post hoc test *p < 0.05). Total MHCα and β protein levels (B) in cardiomyocytes derived from HG8 mESC (n = 2) and (E) in cardiomyocytes derived from R1 and R1HIF-1α−/− mESC (cropped representative western blots from total n = 2 are shown).
The hypoxia-induced upregulation of MHCβ expression is dependent on the presence of HIF-1α
To confirm the significance of HIF-1α in the described phenomenon we employed HG8HIF-1α−/− and R1HIF-1α−/− cells deficient in HIF-1α. Interestingly, neither the induction of Myh7 mRNA (Figure 1C, 1D, Supplementary Figure 1) nor the increase in total MHCα and β protein levels (Figure 1E) was observed in cardiomyocytes differentiated from these HIF-1α-deficient cells. The missing inducibility of Myh7/ total MHCα and β protein in the cell cultures derived from HG8HIF-1α−/− and R1HIF-1α−/− argues for the dependence of the observed effect on the presence of HIF-1α. Indeed, HIF-1α protein was stabilized in R1-derived cardiomyocytes exposed to hypoxia, but was missing in R1HIF-1α−/−-derived cells, as expected (Figure 2A). Accordingly, in HG8-derived cardiomyocytes, hypoxia- or DMOG-induced Myh7/total MHCα and β protein upregulation goes hand-in-hand with both HIF-1α and HIF-2α protein stabilization under hypoxic conditions, with even higher levels of both proteins observed in the case of DMOG (Figure 2B). The transcriptional activity of stabilized HIFs is documented by the increased expression of genes known to be under the control of HIF-1α, particularly Glut1 and VEGF (Figure 2C, 2D).
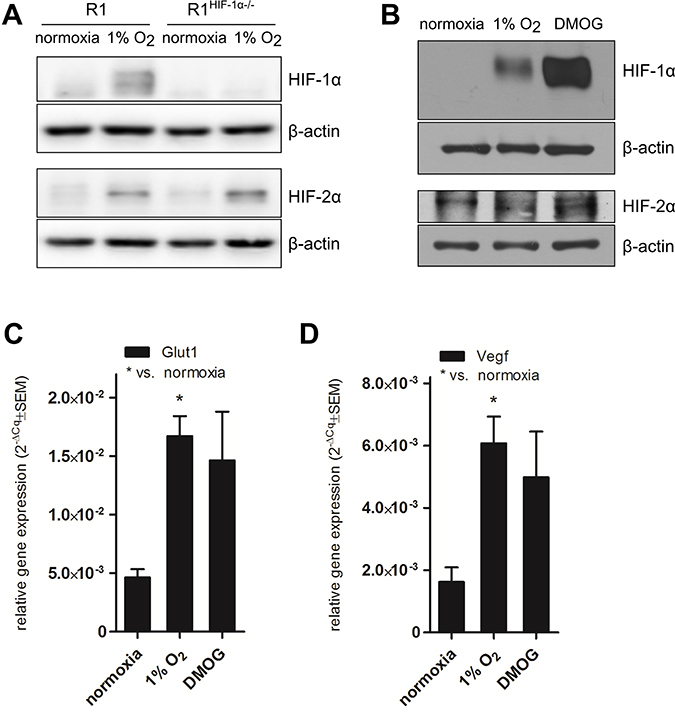
Figure 2: HIF protein stabilization and HIF-1α transcriptional activity. Representative western blot showing protein levels of HIF-1α and HIF-2α (n = 3) in (A) cardiomyocytes derived from R1 and R1HIF-1α−/− mESC exposed to 1% O2 and (B) cardiomyocytes derived from HG8 mESC exposed to 1% O2 and DMOG. Cropped representative western blots are shown from total of n = 3. mRNA levels of (C) Glut1 and (D) VEGF in cardiomyocytes derived from HG8 mESC exposed to 1% O2 or DMOG determined by qRT-PCR (n = 3). Data are presented as 2−ΔCq ± SEM (groups were compared using one-way ANOVA with the Tukey (HSD) post hoc test *p < 0.05).
Analysis of HIF-1α binding to the DNA locus encoding MHCα and MHCβ
The observed correlation between HIF-1α protein stabilization and Myh7/total MHCα and β protein expression induced by hypoxia and DMOG (Figures 1, 2) encouraged us to test the hypothesis that HIF-1α might control either Myh7 or Myh6 expression via its direct binding to the putative HREs predicted in the corresponding DNA locus. We therefore performed in silico analysis of the ~100 kb-long DNA region encompassing mouse Myh7 and Myh6 genes and their adjacent UTR sequences. One putative HIF binding site in intron 25–26 of the Myh7 gene and another 28 potential clusters of HIF binding sites were predicted by different web-based algorithms (Figure 3A; only clusters within the 10kb surroundings of the coding sequences are depicted; see details in section Materials and Methods). For the ChIP analysis, we selected clusters 9 and 11 (Myh7 upstream), clusters 13a and 13b (Myh7 encoding region), cluster 19 (intergenic region) and the putative HIF-binding site located in Myh7 intron 25–26, i.e. HRE-rich regions identified by the majority of predictive algorithms. ChIP analysis of samples from 10-day-differentiated R1 mESCs exposed to 1% O2 hypoxia for 24 h, in which both HIF stabilization and MHC expression were observed (Figure 3B and Supplementary Figure 2), did not confirm the hypothesized direct binding of HIF-1α.
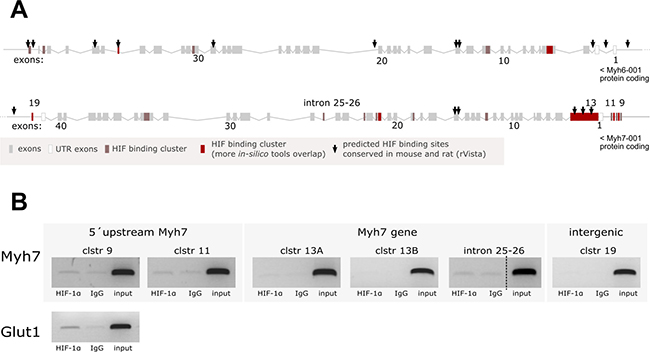
Figure 3: Analysis of HIF-1α binding to the DNA locus encoding MHCα and MHCβ. (A) Graphical representation of predicted HIF binding sites in the locus encoding for mouse MHCα and MHCβ; sites conserved in mouse and rat are marked with arrows. (B) ChIP analysis of HIF-1α binding to predicted binding sites performed on samples from 10 day-differentiated R1 mESCs exposed to 1% O2 hypoxia for 24 h. HIF-1α binding to its target gene Glut1 served as ChIP assay positive control (cropped representative gels are shown from n = 4 (intron 25–26, Glut1) or n = 3 (other clusters)).
Hypoxia and the pharmacological stabilization of HIF slow down the induction of Myh6 in a mouse embryonal heart model
To monitor the effect of hypoxia on the changes in the tested MHC expressions in heart tissue, we established a model derived from explanted mouse embryonal hearts (Figure 4A). Fetal mouse hearts predominantly express the MHCβ isoform, which is gradually displaced by the adult MHCα isoform after birth. Considering this and the previously described phenomenon that the expression of both cardiac MHC isoforms is regulated by thyroid hormone [4–6], we cultured excised hearts from 13.5-dpc-old mouse embryos in vitro in the presence of FBS containing thyroid hormone.
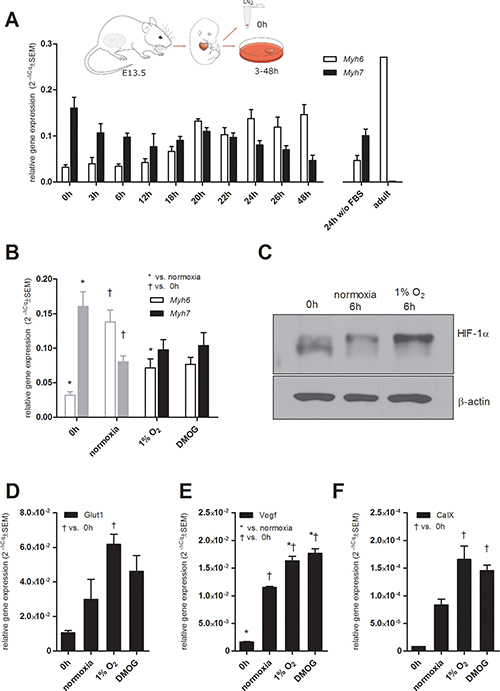
Figure 4: Mouse fetal heart model. (A) Scheme showing the derivation of fetal hearts and kinetics of changes in Myh6 and Myh7 mRNA levels in mouse explanted fetal hearts cultured in vitro determined by qRT-PCR; for comparison, Myh6 and Myh7 mRNA levels in an adult heart sample are shown. Data are expressed as mean 2−ΔCq ± SEM Myh6 and Myh7 (n = 10 (48 h), n = 7 (0 h, 24 h), n = 4 (3 h, 22 h, 2 h wo FBS), n = 3 (6 h, 12 h, 18 h, 20 h, 26 h)), n = 1 (adult heart). Effect of hypoxia and the pharmacological stabilization of HIF on (B) Myh6 and Myh7 relative mRNA levels determined by qRT-PCR (n = 9 (0 h), n = 7 (normoxia), n = 8 (1% O2), n = 5 (DMOG); same data depicted in Figure 4A are displayed in grey for comparison); (C) stabilization of HIF-1α protein after 6 h of in vitro culture in normoxia and 1% O2 (cropped representative western blots are shown, n = 3), mRNA levels of (D) Glut1, (E) Vegf, and (F) CaIX after 24 h determined by qRT-PCR (n = 3), data are presented as 2−ΔCq ± SEM (groups were compared using one-way ANOVA with the Tukey (HSD) post hoc test *p < 0.05; †p < 0.05).
The kinetics of changes in Myh6 and Myh7 expression profiles was analyzed over time and clearly confirmed the induction of Myh6 by FBS and the overall suppression of Myh7 expression (Figure 4A). For comparison, levels of Myh6 and Myh7 expression profiles in hearts incubated in the absence of FBS and in an adult heart sample are shown (Figure 4A).
In order to further analyze hypoxia/HIF-mediated changes in the expression patterns of the tested MHCs, we exposed whole mouse fetal hearts to hypoxic conditions (24 h). Indeed, hypoxic treatment significantly delayed the increase in Myh6 mRNA observed in fetal hearts cultivated in medium with FBS under normoxic conditions (Figure 4B). Furthermore, the pharmacological stabilization of HIF-α subunits by DMOG resulted in the same effect (Figure 4B).
In an effort to elucidate the role of HIF-1α and HIF-2α in this observation, we analyzed the protein levels of both transcription factors and the expression patterns of their target genes, Glut1, CaIX and Vegf. The stabilization of HIF-1α protein in hypoxia-exposed fetal hearts after 6 h (Figure 4C) was followed by the subsequent upregulation of mRNA levels of all three tested HIF targets after 24 h (Figure 4D–4F). Interestingly, analysis of the stabilization of HIF-1α protein after 24 h revealed lower levels of HIF-1α protein in samples incubated in hypoxia or with DMOG compared to control samples and also to samples from hearts without any incubation (Supplementary Figure 3). These data suggest that the observed regulatory effects of HIF-1α on MHC gene expression are driven by its transcriptional activity during the earlier phases of its stabilization by hypoxia or DMOG. Further, we were unable to credibly detect HIF-2α protein in fetal heart samples (data not shown).
This model of explanted hearts was also tested for HIF-1α binding to the DNA locus encoding MHCα and MHCβ. In agreement with the R1-derived model, no binding of HIF-1α in any of the six selected HREs-predicted clusters was detected by ChIP in fetal hearts incubated for 24 h in 1% O2 (n = 3, data not shown).
mTOR inhibition increases Myh7 expression in HG8-derived cardiomyocytes
Another possible mechanism previously shown to be engaged during hypoxia is mTOR signaling, which is known to be inhibited under hypoxic conditions. To test its role in our model, we employed treatment with rapamycin, an inhibitor of mTOR, in order to simulate hypoxia-related mTOR inhibition, and then we analyzed the expression of both Myhs in HG8-derived cardiomyocytes. Interestingly, we did not observe the downregulation of phosphorylation of its downstream target p70s6 kinase (Thr 389) in hypoxic conditions (Figure 5A). Rapamycin-induced mTOR inhibition was validated by the absence of phosphorylated p70s6k (Figure 5A). This resulted in the upregulation of Myh7 expression, even in cardiomyocytes cultivated under normoxic conditions; however, this effect does not reach statistical significance (Figure 5B). Interestingly, the combination of rapamycin and hypoxic conditions induced the expression of Myh7 even more potently than hypoxia itself (Figure 5B). As expected, HIF levels were stabilized after incubation under hypoxic conditions (Figure 5A).
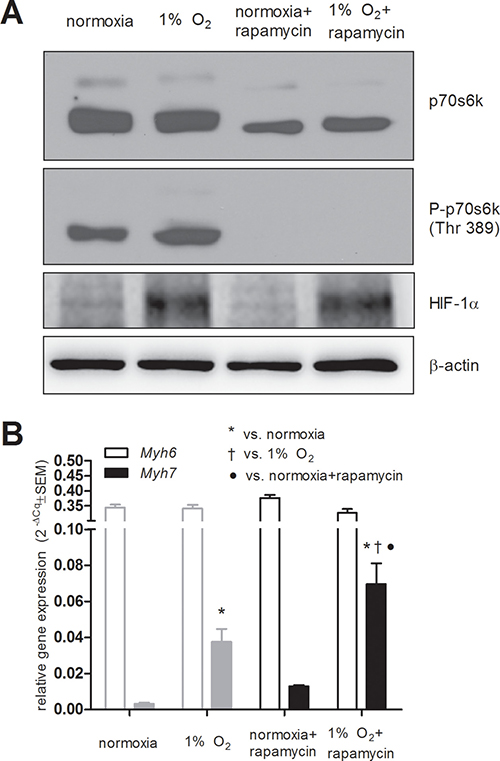
Figure 5: Effects of mTOR inhibition using rapamycin (10 ng/ml). (A) mTOR inhibition using rapamycin validated by western blot detection of its downstream target p70 s6 kinase (n = 6) and the stabilization of HIF-1α protein by hypoxia (n = 4) (B) Myh6 and Myh7 relative mRNA levels in normoxia and in 1 % O2 hypoxia determined by qRT-PCR. Data are presented as 2−ΔCq ± SEM (n = 5 (normoxia), n = 4 (1% O2), n = 3 (rapamycin treatment); in grey same data depicted in Figure 1A are displayed for comparison; groups were compared using one-way ANOVA with the Fisher’s LSD post hoc test *p < 0.05; †p < 0.05).
Hypoxia induces a decrease in antisense Myh7 RNA transcript and Mhrt lncRNA level
In the light of recent discoveries concerning the regulation of MHC gene expression by naturally present antisense transcripts and lncRNAs, we tested the effect of hypoxia on the level of antisense Myh7 transcript and Mhrt lncRNA. In both tested models (explanted fetal hearts and HG8-derived cardiomyocytes), hypoxia downregulated the levels of antisense Myh7 transcript (Figure 6A) as well as RNA levels of Mhrt lncRNA (Figure 6B, 6C).
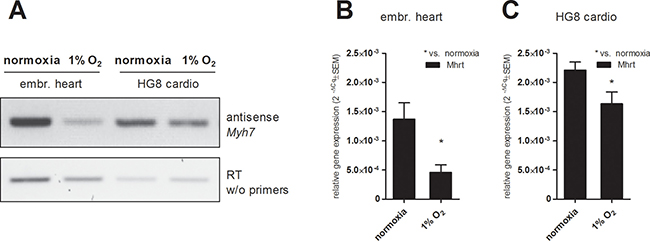
Figure 6: Effect of hypoxic exposure on the expression levels of (A) antisense Myh7 transcript in fetal hearts and HG8-derived cardiomyocytes analyzed by antisense PCR and subsequent agarose electrophoresis (a template from the reverse-transcription without primers was used as a negative control (cropped representative gels are shown, (n = 3)) and of Mhrt transcript in (B) fetal hearts and (C) HG8 derived-cardiomyocytes determined by qRT-PCR. Data are presented as 2−ΔCq ± SEM (n = 7 (embr. hearts 1% O2, HG8 normoxia), n = 4 (HG8 1% O2), n = 5 (embr. hearts normoxia); groups were compared using one-way ANOVA with the Tukey (HSD) post hoc test *p < 0.05).
DISCUSSION
Herein, we reveal the hypoxic regulation of MHC expression in cardiomyocytes, specifically the expression of the MHCβ isoform. We show for the first time that the effect of hypoxia on the cardiac MHC expression profile is HIF-1α-dependent, as no significant changes in MHC expression profiles were observed in HIF-1α-deficient cells exposed to hypoxia, even in the presence of HIF-2α. In all three presented models, HIF-1α protein was stabilized under hypoxic conditions and also transcriptionally active, according to HIF-target gene expression data measured in both the in vitro and the ex vivo model.
In line with this observation, hypoxia was indeed previously shown to increase MHCβ transcript levels in rat models in vivo (in rats exposed to 24 and 48 h of hypobaric hypoxia (11 % oxygen)) and in vitro (in neonatal rat cardiomyocytes incubated for up to 48 h in a hypoxic chamber) [20]. Further, justifying our hypothesis, hypoxia was previously shown to stimulate the expression of muscle slow-oxidative type I myosin in an HIF-1α dependent manner [31]. Although Razeghi et al. also showed a hypoxia-induced decrease in Myh6 transcript levels in the rat models specified above, we did not observe any significant effect of hypoxia or DMOG on the Myh6 expression profile in our mESC cell-derived models of cardiomyocytes [20]. This putative discrepancy could be explained by biased Myh6 expression levels in HG8-derived cardiomyocytes purified on the basis of Myh6 promoter activity and by the obvious partial variation of mESC-derived in vitro models and the in vivo situation. We therefore decided to study the negative effect of hypoxia (and DMOG) on Myh6 transcript expression rather in the context of real cardiac tissue, and we, indeed, observed the delayed onset of FBS-induced Myh6 upregulation in explanted mouse fetal hearts exposed to hypoxia and also DMOG. Notably, the Myh7 expression pattern in hypoxia- and DMOG-treated explanted hearts tended to be slightly enhanced; however, this effect was only mild and did not pass the criteria of significance of the employed statistical method. Interestingly, lower levels of HIF-1α were detected in samples incubated in hypoxia or with DMOG compared to control samples after 24 h suggesting the observed regulatory effects of HIF-1α on MHC gene expression to be driven by HIF-1α transcriptional activity during the earlier phases of HIF-1α stabilization. We can speculate that increased levels of HIF-1α in samples incubated in normoxia for 24 h can reflect stress response in explanted hearts.
Since hypoxia is a condition common in fetal heart development and the failing adult heart, HIF-1α is a good candidate for being the regulator responsible for the postnatal switch and the return to the fetal gene program. Considering this, we decided to address the possibility of the Myh6 and Myh7 genes being direct gene targets of HIF-1α. However, we did not observe the direct binding of HIF-1α protein to either of the tested binding sites predicted in the corresponding DNA locus. Several considerations, however, must be kept in mind. First, the existence of a distant regulatory motif sensitive to the presence of HIF-1α cannot be excluded. Second, no reliable genome-wide study dedicated to the mapping of HIF-1α binding sites in cardiomyocytes exposed to hypoxic stress has been published so far. Nevertheless, the proposed indirect effect of HIF-1α on the induction of Myh7 mRNA is strongly supported by the observation of no significant changes in Myh7 gene expression during the short-term exposure of HG8-derived cardiomyocytes to hypoxia or DMOG (6 h) (data not shown). Interestingly, although we observed statistically significant hypoxia- or DMOG-mediated slowdown of FBS-induced Myh6 upregulation in the model of mouse explanted hearts, a published suggestion based on another model that HIF-dependent gene suppression is almost entirely indirect [32] discouraged us from testing direct HIF-1α binding in genomic DNA regions flanking and corresponding to the Myh6 gene. Thus, the involvement of other mechanisms triggered by hypoxia and/or HIF-1α protein stabilization has to be considered, these acting as potential regulators of MHC gene expression.
mTOR complex 1 (mTORC1) was previously shown to be necessary for embryonic cardiovascular development and for the postnatal maintenance of cardiac structure and function as well as for adaptation to pressure overload and the development of compensatory hypertrophy [33]. It is suggested that mTOR inhibition decreases the translation of the HIF-α subunit in mild hypoxia or even normoxia [34, 35] and HIF-1α is suggested to be responsible for glycolytic mTORC1 action [36]. However, hypoxic mTORC1 regulation is suggested to be HIF-1α-independent [37], and, notably, the HIF-2α-dependent regulation of mTORC1 activity has been proposed [38]. Interestingly, rapamycin was shown to have significant benefits for cardiovascular function [39] similarly to DMOG [21, 22]. Herein, we revealed that the inhibition of mTOR by rapamycin induces an increase in Myh7 mRNA level similarly to the presence of hypoxia or DMOG. However, under our conditions, mTOR signaling was probably not responsible for the increase in Myh7 mRNA level observed after the exposure of HG8-derived cardiomyocytes to hypoxia, since no change in the phosphorylated/active status of p70 s6 kinase, its downstream target, was simultaneously detected. The lack of inhibition of p70s6k phosphorylation by exposure to hypoxia in our model can be related to different kinetics and to a lower level of intensity of the inhibition of p70s6k phosphorylation in response to the employed level of hypoxia than in response to rapamycin. We can speculate that prolonged and more severe hypoxia would have a more profound inhibitory effect on p70s6k phosphorylation. The limited nature of the effect of the employed hypoxic conditions in our model is further supported by the observation that the combination of rapamycin and hypoxic conditions induced the expression of Myh7 more potently than hypoxia itself. Interestingly, Knaup et al. found that under severe hypoxia, no influence of mTOR inhibitors was observed; thus, the stimulation of HIF1α by mTOR may only be relevant under mild hypoxia or even normoxia [34].
Further, HIF-1α was previously shown to maintain the expression of the sarcomeric protein titin, but not that of the sarcomeric proteins EH-myomesin and α-actinin, during cardiac development [17]. Under conditions promoting changes in the tested MHC expression profiles we did not observe changes in sarcomeric α-actinin mRNA levels when compared to other reference gene Rpl13a (data not shown).
Significantly, and for the first time, we show here the hypoxia induces a decrease in Myh7 antisense transcript. Antisense Myh7 RNA was also found to be significantly decreased in 12-day pressure-overloaded hearts [26, 40]. Moreover, the relative level of MHC-associated RNA transcript (Mhrt), previously identified as the lncRNA molecule transcribed from Myh7 loci and proposed as a cardioprotective molecule [29], was shown here to decrease in response to hypoxia. Interestingly, alterations in miRNA expression in heart failure were also reported to display a pattern similar to that observed in the fetal heart [41]. Thus, the role of other regulatory RNAs in MHC gene expression control remains to be elucidated.
Importantly, we also have to consider the difference between the regulation of MHC gene expression in humans and rodents. In humans, with slower heart rates than mice or rats, MHCβ is the predominant isoform throughout their whole life [42]. However, the ratio of MHCα to MHCβ isoforms is reduced in failing human ventricles, similarly as in the mouse heart during pathological processes [3, 43–46].
In conclusion, since ChIP analysis did not confirm direct interaction between HIF-1α and the proposed HREs located in the MHCα and MHCβ encoding DNA regions, we suggest that other hypoxia-associated and HIF-1α-dependent mechanisms are involved in the regulation of Myh7 gene expression; in particular, the role of antisense RNA molecules remains to be elucidated. Another interesting finding is that the presence of HIF-2α itself is not sufficient to rescue the hypoxia-mediated induction of Myh7, as shown in the R1 and R1HIF-1α−/− models. However, as discussed above, several other mechanisms which seem to regulate MHC isoform expression in heart have previously been described [29, 47]. The question is whether all these mechanisms compete or play a collective role, whether they are independent or synchronized by one upstream master regulator, and whether the overall mechanism works through the induction of repressors or by a decrease in stimulation. This knowledge could further help to elucidate the mechanism, when the adult heart returns to the fetal gene program under pathophysiological conditions, and possibly help to identify novel therapeutic targets for the treatment of heart failure.
MATERIALS AND METHODS
Mouse embryonic stem cell (mESC) culture
Feeder-free adapted maternal line R1, R1HIF-1α−/− (derived from R1, the kind gift of Dr. P. Carmeliet [48]) and HG8 (derived from the R1 line transfected with pMHC-Neo-Hyg vector, the kind gift of Dr. Loren J. Field) mESC cell lines were propagated in an undifferentiated state as described earlier [49]. Briefly, mESC were cultured on gelatinized tissue culture plastic in Dulbecco’s modified Eagle’s medium (DMEM) containing 15% fetal calf serum (ESC tested; both from Invitrogen, USA), 1 × non-essential amino acid, 0.05 mM β-mercaptoethanol, 100 IU/ml of penicillin, and 0.1 mg/ml of streptomycin (all from PAA Laboratories, Austria), supplemented with 1 000 U/ml of leukemia inhibitory factor (LIF; Chemicon, USA), here referred to as the ES medium [30, 50].
To prepare the HG8 cell line deficient in HIF-1α, HG8 cells grown to confluency in ES medium in a 60mm cell culture dish were transfected with 2ug of HIF-1α Double Nickase Plasmid (sc-420856-NIC, Santa Cruz Biotechnology, USA), by means of Lipofectamine 2000 (Invitrogen, USA). After 5 h, the medium was changed for fresh ES medium supplemented by 5 μg/ml puromycine. After 24 h of selection, the medium was exchanged again for fresh ES medium. After 4 days, single clones were picked, trypsinized, and propagated. The successful generation of HG8 HIF-1α−/− lines was confirmed by western blot analysis of cells under conditions stabilizing HIF-1α.
For the induction of differentiation into cardiac-like cells, cells were transferred onto agar-coated cell culture dishes for 5 days in ES medium without the addition of LIF to form embryoid bodies (EBs) [49]. On day 5, EBs were suspended in ITS medium (DMEM/F12, Insulin, Trasferine, Selenium (ITS), Invitrogen, USA; Penicilin/Streptomycin) and transferred onto a gelatin-coated culture surface inducing adhesion. R1 and R1HIF-1α−/− cells were exposed to hypoxia on day 14 of differentiation for 24 hours. Purified cardiomyocytes (the selection cassette coding neomycin phosphotransferase enabling G418 resistance was driven by Myh6 promoter) were prepared from differentiated HG8 cells as described earlier [49, 51]. Briefly, the cells were treated with G418 (0.5 mg/ml) on days 14 and 17. On day 20, cells were treated with 0.15 % collagenase and allowed to sediment several times to remove dead cell debris. The resulting pellet was then suspended in ES:ITS medium (1:1) seeded onto a gelatinized cell culture surface for 48 hours. After 48 hours, the medium was exchanged and the cells were treated with DMOG (0.5 mM) or cultivated in 1% O2 for 24 hours. In the case of rapamycin (Selleckchem, USA) pretreatment, the medium was exchanged for medium containing rapamycin (10 ng/ml) for the first 8 hours, and then exchanged again for fresh medium without rapamycin. Samples were collected after a total of 24 hours of treatment (the time-line of the cultivation is depicted in Figure 1A (HG8) and 1B (R1)).
Animals and explanted mouse fetal hearts
C57BL/6 mice were obtained from the Laboratory Animal Breeding and Experimental Facility of the Faculty of Medicine, Masaryk University, Brno, Czech Republic. The mice were kept under controlled conditions; a standardized pelleted diet and HCl or UV light-treated tap water were available ad libitum. Experiments were performed in accordance with national and international guidelines on laboratory animal care and with the approval of the Institute’s Ethical Committee conforming to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
Pregnant mice were euthanized 13.5 days post coitum (dpc) by gradually filling the chamber with CO2. Unless otherwise stated, mouse fetal hearts were extracted from mouse embryos, washed in PBS, and either immediately frozen (0 h samples) or incubated on 0.1% bovine skin gelatin-coated cell culture plastic in high glucose Dulbecco’s modified Eagle’s medium (PAA Laboratories, Austria) supplemented with 100 IU/ml of penicillin, 0.1 mg/ml of streptomycin (Sigma-Aldrich, USA), and 10% FBS (PAA Laboratories, Austria) under various conditions and for various time periods (Figure 1A). For particular experiments DMOG (Calbiochem, USA) at a final concentration of 0.5 mM was added to cultivation media.
RNA isolation and qRT-PCR
ES cells were washed in PBS and lysed in lysis buffer (UltraClean® Tissue & Cells RNA Isolation Kit; MO BIO Laboratories, USA). Hearts were snap-frozen in liquid nitrogen either immediately after extraction or after in vitro incubation. The hearts were then disrupted in 200 μl lysis buffer (High Pure RNA Kit, Roche, Germany). Total RNA was isolated according to the kit manufacturer’s recommendations. The concentration and purity of isolated RNA was assessed using a Nanodrop 1000 instrument (Thermo Scientific, USA) and only samples with an A280/A260 absorbance ratio greater than 1.9 were used for further investigation. 45 ng of total RNA was reverse transcribed in a 20 μl reaction mixture using Sensiscript Reverse Transcription kit (Qiagen, Germany), Anchored-Oligo (dt)18 Primer (1 μM final concentration), Random Hexamer Primer (10 μM final concentration), and Protector RNase Inhibitor (10 units/20 μl reaction) (all from Roche, Germany). Relative mRNA levels of genes encoding MHCα (Myh6), MHCβ (Myh7), Vascular endothelial growth factor (Vegf), Glucose transporter 1 (Glut1), Carbonic anhydrase (CaIX), sarcomeric α actinin (Actn2), and lncRNA Mhrt were analyzed, with Actn2 serving as reference genes. A reaction using the Universal Probe Library System and a LightCycler Probe Master was performed in a total volume of 10 μl on a LightCycler 480 Instrument (Roche, Germany). Specific combinations of primers and probes were designed using the Assay Design Center on the manufacturer’s web pages; primers and probes were used at final concentrations of 10 μM and 0.5 μM, respectively. For the specific combinations of primers and UPL probes, see Table 1. The cycling program was set as follows: 95°C for 10 min followed by 45 cycles of 95°C for 10 s, 60°C for 30 s, and 72°C for 1 s, with data acquisition. Results are expressed as the fold difference between the target gene and the reference gene, determined by the relative quantification 2−ΔCq method. Data are presented as 2-(Cq(target)-Cq(reference)) ± SEM. The mRNA expression profiles of Myh6 and Myh7 genes were also verified using TaqManGene Expression Assay (Applied Biosytems, USA).
Table 1: Specific combinations of primers and UPL probes
Target |
Left primer 5′→3′ |
Right primer 5′→3′ |
UPL probe |
|---|---|---|---|
Actn2 |
CCGGATTCTGGCTTCTGAT |
GAGGCAGCTCTCGACGAA |
53 |
Myh6 |
CGCATCAAGGAGCTCACC |
CCTGCAGCCGCATTAAGT |
6 |
Myh7 |
CGCATCAAGGAGCTCACC |
CTGCAGCCGCAGTAGGTT |
6 |
Vegf |
CAGGCTGCTGTAACGATGAA |
GCTTTGGTGAGGTTTGATCC |
9 |
Glut 1 |
GGACCCTGCACCTCATTG |
GCCACGATGCTCAGATAGG |
20 |
CaIX |
ATTCCTGCTTCACTGCTGGT |
CTTTGGTCCCACTTCTGTGC |
16 |
Mhrt |
ACACAGATGGACGCTCTGG |
GGATGAGGCGGAGGAGAG |
95 |
Antisense Myh7 transcript analysis
The expression of mouse antisense Myh7 transcript was analyzed using forward 5′-AGCCAGGCCTGCTACCAAAGACACC-3′ and reverse 5′-CCCTAGAACACCACAGAGCATAAA-3′ primers designed according to Haddad [28]. Briefly, total RNA was isolated as described above and equal amounts of total RNA were reverse-transcribed by means of Sensiscript Reverse Transcription kit (Qiagen, Germany) using only forward primer in the reaction mixture. 3 μl of 4 × diluted resulting specific cDNA was subsequently amplified in 30 cycles of PCR using Hot Start Titanium Taq Polymerase kit (Clontech, USA) employing forward and reverse primers [28]. The PCR conditions were set as follows according to the polymerase kit manufacturer’s recommendations: 95°C for 5 min followed by 30 cycles of 95°C for 30 s and 68°C for 20 s, ending the reaction with 68°C for 1 min. The PCR product was visualized on 2% agarose gel supplemented with ethidium bromide. As a negative control, a sample resulting from reverse-transcription with no forward primer was used as a template in the amplification step [40].
Western blot analysis
Cell sample harvesting and immunoblot analysis were performed as presented previously [50]. To detect proteins, the primary antibodies β-actin (sc-47778, Santa Cruz Biotechnology, USA), vinculin (Sigma-Aldrich), HIF-1α (GTX127309, Genetex, USA), HIF-2α (NB100–122, Novus Biologicals, USA or GTX30114, Genetex, USA), p70 s6 kinase and phospho-p70 s6 kinase (Thr 389) (9202 and 9205 respectively, both from Cell Signaling, USA), and MF20 (a hybridoma producing both antibody detecting α and β MHC isoforms; Developmental Studies Hybridoma Bank, Iowa City, IA, USA) were used in 5% non-fat milk/TBS-T at 4°C overnight. Corresponding secondary HRP-conjugated anti-rabbit or anti-mouse antibodies (in 5% non-fat milk/TBS-T, 1 h, RT; Cell Signaling, USA) were employed. Immunoreactive bands were detected using ECL detection reagent kit (Pierce, USA) and exposed to radiographic film (AGFA, Belgium). The adjustment of brightness and contrast was applied to the whole image.
In silico analysis of the DNA locus harbouring Myh6/7 genes
The coding DNA sequences of mouse Myh6 and Myh7 genes, the intergenic regions, and the flanking regions located 30 kb upstream and downstream of both 5′ and 3′ ends were analyzed for the presence of potential HIF binding sites (HREs); for further analysis, only 10 kb flanking regions were considered. The genomic sequence was sourced by the Ensembl genome browser and screened for HIF-1, HIF-1 ancillary sequence, and HIF1beta/ARNT by means of the following online tools using different algorithms for the prediction of the presence of transcription factor binding sites: MatInspector (genomatix.de) [52], Match, Patch, P-Match [53, 54], and AliBaba2 (gene-regulation.com) [55]. For the analysis of evolutionarily conserved putative HREs, the rVista 2.0 tool (rvista.dcode.org) was employed as described previously [56]. Predicted HREs containing the [A/G]CGT core motif were further screened and scored by the position-specific matrix of frequencies according to recommendations described earlier [57]. The calculated score (Table 2.) reflects the similarity of the analyzed sequence to known functional hypoxia-responsive sequences.
Table 2: Primers used for the amplification of immunoprecipitated DNA and the similarity of individual predicted binding sites to functional HRE (Score)
Target |
Left primer 5′→3′ |
Right primer 5′→3′ |
Score |
|---|---|---|---|
Cluster 9 |
AAAGCATCCCAGGAAGAAAGCAGCA |
AGTCGAGACCCACAGGCTGAGAACC |
7.15 |
Cluster 11 |
AATCTGACACTACCGGCCACCTGCT |
ACCCCAGCACTCCTGTGGCAAAATA |
7.72/7.58 |
Cluster 13a |
CGTAGGAATGTGGGAGCCCAAGTGT |
AGCCTCATGATTCCAACAGCCCTCT |
6.34/7.84 |
Cluster 13b |
TCAGAGGGCTGTTGGAATCATGAGG |
AGAAAGGAACCTGCCAGGCTTTGG |
8.85 |
Cluster 19 |
TATCCACAGTGTCCCTTGCCCCTTC |
TGGTCAGAACACAGAGGGCTGTTGA |
8.33 |
Intron 25–26 |
ACTCCCGTGCCATTCTGCCCTAGTAA |
GACCAGCCATGAACCACAAGGTAGGA |
7.34 |
Glut1 |
TCAGCAAAGCCTTTTCAAGCCCTACC |
CCCTGGAGGTGACTTCAGCCCTTAAA |
7.79 |
Chromatin immunoprecipitation (ChIP)
ChIP assay was performed on 10-day differentiated R1 exposed to 1% O2 for 24 h using 2 ug of HIF-1α specific antibody (GTX127309 Genetex, USA) according to a previously described protocol [58]. An alternative protocol using ExactaChIP Human/Mouse HIF-1α Chromatin Immunoprecipitation Kit (R&D Systems USA) was applied according to the manufacturer’s recommendations in embryonal hearts exposed to 1% O2 for 24 h (data not shown). Immunoprecipitated DNA was purified using QIAquick PCR Purification Kit (Qiagen, Germany) and subsequently amplified in a PCR step using Titanium TaqPCR Kit (Clontech Laboratories, Inc., USA) with primer sets corresponding to putative HIF-binding DNA clusters predicted in the Myh7/Myh6 encoding locus (Table 2). The products were separated and visualized by means of EtBr-based agarose electrophoresis. As a positive control, the presence of Glut1 enhancer sequence 5′-CACAGGCGTGCCGTCTGACACGCA-3′ [59] in the HIF-1α-immunoprecipitated DNA sample was analyzed (primers listed in Table 2).
Statistical analysis
Unless otherwise stated, data are presented as the average of at least 3 independent measurements ± SEM. Groups were compared using one-way ANOVA with the Tukey (HSD) post hoc test or with the Fisher’s LSD post hoc test. Values of p equal to, or less than 0.05 were considered significant and are marked with asterisks or a cross symbol.
Abbreviations
DMEM: Dulbecco’s modified Eagle’s medium; DMOG: dimethyloxalylglycine; HIF: hypoxia inducible factor; HRE: HIF response element; ChIP: Chromatin immunoprecipitation; lncRNA: long noncoding RNA; mESC: mouse embryonic stem cell; MHC: myosin heavy chain; mTOR: mammalian (mechanistic) target of rapamycin; mTORC1: mTOR complex 1; PHD: prolyl hydroxylase domain enzyme.
Authors’ contributions
LB – mice studies, qRT-PCR, western blots, ChIP, statistical analysis, the writing of the manuscript; KAR HG-8-derived samples, the preparation of HG8HIF-1α−/− cell lines; JPr – the writing of the manuscript and design of the study; JKuč – R1-derived samples and a portion of the western blots; JKud – part of data on R1 cardiomyogenesis; JPa R1 – derived samples and design of the study; LK – design and supervision of the study, the writing of the manuscript.
ACKNOWLEDGMENTS
We are grateful to Dr. Loren J. Field of Wells Pediatric Center for Research, IUPUI, School of Medicine, R4–376, Indianapolis, IN 46202 for providing the pMHC-Neo-Hyg vector and to prof. Peter Carmeliet, Vesalius Research Center, Leuven, Belgium for providing R1HIF-1α−/− mESCs clones. Further, we would like to thank Dr. Karel Soucek, Dr. Stjepan Uldrijan, and Dr. Jirina Medalova for kind material support. We would also like to thank Vladimir Cicmanec for compiling an algorithm for the calculation of the score according to a position-specific matrix of frequencies. This work was supported by grants from the Ministry of Education, Youth and Sport of the Czech Republic Cost CZ LD11015 and COST Action TD0901. L. K., J. Kuč., and J. Kud. were supported by project no. LQ1605 from the National Program of Sustainability II (MEYS CR).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
1. Mahmoud AI, Porrello ER. Turning back the cardiac regenerative clock: lessons from the neonate. Trends Cardiovasc Med. 2012; 22:128–133.
2. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011; 331:1078–1080.
3. Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation. 2001; 104:2923–2931.
4. Wright CE, Haddad F, Qin AX, Bodell PW, Baldwin KM. In vivo regulation of beta-MHC gene in rodent heart: role of T3 and evidence for an upstream enhancer. Am J Physiol. 1999; 276:C883–891.
5. Gustafson TA, Markham BE, Morkin E. Effects of thyroid hormone on alpha-actin and myosin heavy chain gene expression in cardiac and skeletal muscles of the rat: measurement of mRNA content using synthetic oligonucleotide probes. Circ Res. 1986; 59:194–201.
6. Edwards JG, Bahl JJ, Flink IL, Cheng SY, Morkin E. Thyroid hormone influences beta myosin heavy chain (beta MHC) expression. Biochem Biophys Res Commun. 1994; 199:1482–1488.
7. Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program A suggested metabolic link to gene expression in the heart. Ann N Y Acad Sci. 2010; 1188:191–198.
8. Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Failure Reviews. 2007; 12:331–343.
9. Kakinuma Y, Miyauchi T, Suzuki T, Yuki K, Murakoshi N, Goto K, Yamaguchi I. Enhancement of glycolysis in cardiomyocytes elevates endothelin-1 expression through the transcriptional factor hypoxia-inducible factor-1 alpha. Clin Sci (Lond). 2002; 103:210S–214S.
10. Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992; 12:5447–5454.
11. Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995; 270:1230–1237.
12. Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem. 1996; 271:32253–32259.
13. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001; 107:43–54.
14. Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001; 294:1337–1340.
15. Jain S, Maltepe E, Lu MM, Simon C, Bradfield CA. Expression of ARNT, ARNT2, HIF1 alpha, HIF2 alpha and Ah receptor mRNAs in the developing mouse. Mech Dev. 1998; 73:117–123.
16. Nau PN, Van Natta T, Ralphe JC, Teneyck CJ, Bedell KA, Caldarone CA, Segar JL, Scholz TD. Metabolic adaptation of the fetal and postnatal ovine heart: regulatory role of hypoxia-inducible factors and nuclear respiratory factor-1. Pediatr Res. 2002; 52:269–278.
17. Krishnan J, Ahuja P, Bodenmann S, Knapik D, Perriard E, Krek W, Perriard JC. Essential Role of Developmentally Activated Hypoxia-Inducible Factor 1 alpha for Cardiac Morphogenesis and Function. Circ Res. 2008; 103:1139–1146.
18. Lee SH, Wolf PL, Escudero R, Deutsch R, Jamieson SW, Thistlethwaite PA. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N Engl J Med. 2000; 342:626–633.
19. Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol. 2011; 76:347–353.
20. Razeghi P, Essop MF, Huss JM, Abbasi S, Manga N, Taegtmeyer H. Hypoxia-induced switches of myosin heavy chain iso-gene expression in rat heart. Biochem Biophys Res Commun. 2003; 303:1024–1027.
21. Natarajan R, Salloum FN, Fisher BJ, Kukreja RC, Fowler AA 3rd. Hypoxia inducible factor-1 activation by prolyl 4-hydroxylase-2 gene silencing attenuates myocardial ischemia reperfusion injury. Circ Res. 2006; 98:133–140.
22. Philipp S, Jurgensen JS, Fielitz J, Bernhardt WM, Weidemann A, Schiche A, Pilz B, Dietz R, Regitz-Zagrosek V, Eckardt KU, Willenbrock R. Stabilization of hypoxia inducible factor rather than modulation of collagen metabolism improves cardiac function after acute myocardial infarction in rats. Eur J Heart Fail. 2006; 8:347–354.
23. Simon MC, Liu L, Barnhart BC, Young RM. Hypoxia-induced signaling in the cardiovascular system. Annu Rev Physiol. 2008; 70:51–71.
24. Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003; 278:29655–29660.
25. Boheler KR, Chassagne C, Martin X, Wisnewsky C, Schwartz K. Cardiac expressions of alpha- and beta-myosin heavy chains and sarcomeric alpha-actins are regulated through transcriptional mechanisms. Results from nuclear run-on assays in isolated rat cardiac nuclei. J Biol Chem. 1992; 267:12979–12985.
26. Haddad F, Bodell PW, Qin AX, Giger JM, Baldwin KM. Role of antisense RNA in coordinating cardiac myosin heavy chain gene switching. J Biol Chem. 2003; 278:37132–37138.
27. Luther HP, Morwinski R, Wallukat G, Haase H, Morano I. Expression of sense and naturally occurring antisense mRNA of myosin heavy chain in rat heart tissue and cultivated cardiomyocytes. J Mol Cell Cardiol. 1997; 29:27–35.
28. Haddad F, Qin AX, Bodell PW, Jiang W, Giger JM, Baldwin KM. Intergenic transcription and developmental regulation of cardiac myosin heavy chain genes. Am J Physiol Heart Circ Physiol. 2008; 294:H29–40.
29. Han P, Li W, Lin CH, Yang J, Shang C, Nurnberg ST, Jin KK, Xu W, Lin CY, Lin CJ, Xiong Y, Chien HC, Zhou B, et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014; 514:102–106.
30. Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998; 394:485–490.
31. Radaszkiewicz KA, Sýkorová D, Karas P, Kudová J, Kohút L, Binó L, Večeřa J, Víteček J, Kubala L, Pacherník J. Simple non-invasive analysis of embryonic stem cell-derived cardiomyocytes beating in vitro. Review of Scientific Instruments. 2016; 87:024301.
32. Kucera J, Bino L, Stefkova K, Jaros J, Vasicek O, Vecera J, Kubala L, Pachernik J. Apocynin and Diphenyleneiodonium Induce Oxidative Stress and Modulate PI3K/Akt and MAPK/Erk Activity in Mouse Embryonic Stem Cells. Oxid Med Cell Longev. 2016; 2016:7409196.
33. Bino L, Kucera J, Stefkova K, Svihalkova Sindlerova L, Lanova M, Kudova J, Kubala L, Pachernik J. The stabilization of hypoxia inducible factor modulates differentiation status and inhibits the proliferation of mouse embryonic stem cells. Chem Biol Interact. 2016; 244:204–214.
34. Klug MG, Soonpaa MH, Koh GY, Field LJ. Genetically selected cardiomyocytes from differentiating embronic stem cells form stable intracardiac grafts. J Clin Invest. 1996; 98:216–224.
35. Haddad F, Qin AX, Bodell PW, Zhang LY, Guo H, Giger JM, Baldwin KM. Regulation of antisense RNA expression during cardiac MHC gene switching in response to pressure overload. Am J Physiol Heart Circ Physiol. 2006; 290:H2351–2361.
36. Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005; 21:2933–2942.
37. Chekmenev DS, Haid C, Kel AE. P-Match: transcription factor binding site search by combining patterns and weight matrices. Nucleic Acids Res. 2005; 33:W432–437.
38. Kel AE, Gossling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003; 31:3576–3579.
39. Grabe N. AliBaba2: context specific identification of transcription factor binding sites. In Silico Biol. 2002; 2:S1–15.
40. Loots GG, Ovcharenko I. rVISTA 2.0: evolutionary analysis of transcription factor binding sites. Nucleic Acids Res. 2004; 32:W217–221.
41. Pescador N, Cuevas Y, Naranjo S, Alcaide M, Villar D, Landazuri MO, Del Peso L. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem J. 2005; 390:189–197.
42. Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H, Cao Y, Berkenstam A, Poellinger L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature. 2001; 414:550–554.
43. Ebert BL, Firth JD, Ratcliffe PJ. Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct Cis-acting sequences. J Biol Chem. 1995; 270:29083–29089.
44. Slot IG, Schols AM, Vosse BA, Kelders MC, Gosker HR. Hypoxia differentially regulates muscle oxidative fiber type and metabolism in a HIF-1alpha-dependent manner. Cell Signal. 2014; 26:1837–1845.
45. Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, Ratcliffe PJ. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009; 284:16767–16775.
46. Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014; 114:549–564.
47. Knaup KX, Jozefowski K, Schmidt R, Bernhardt WM, Weidemann A, Juergensen JS, Warnecke C, Eckardt KU, Wiesener MS. Mutual regulation of hypoxia-inducible factor and mammalian target of rapamycin as a function of oxygen availability. Mol Cancer Res. 2009; 7:88–98.
48. Land SC, Tee AR. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007; 282:20534–20543.
49. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010; 39:171–183.
50. Tan CY, Hagen T. Post-translational regulation of mTOR complex 1 in hypoxia and reoxygenation. Cell Signal. 2013; 25:1235–1244.
51. Elorza A, Soro-Arnaiz I, Melendez-Rodriguez F, Rodriguez-Vaello V, Marsboom G, de Carcer G, Acosta-Iborra B, Albacete-Albacete L, Ordonez A, Serrano-Oviedo L, Gimenez-Bachs JM, Vara-Vega A, Salinas A, et al. HIF2alpha acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol Cell. 2012; 48:681–691.
52. Flynn JM, O’Leary MN, Zambataro CA, Academia EC, Presley MP, Garrett BJ, Zykovich A, Mooney SD, Strong R, Rosen CJ, Kapahi P, Nelson MD, Kennedy BK, et al. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell. 2013; 12:851–862.
53. Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007; 116:258–267.
54. England J, Loughna S. Heavy and light roles: myosin in the morphogenesis of the heart. Cell Mol Life Sci. 2013; 70:1221–1239.
55. Lowes BD, Minobe W, Abraham WT, Rizeq MN, Bohlmeyer TJ, Quaife RA, Roden RL, Dutcher DL, Robertson AD, Voelkel NF, Badesch DB, Groves BM, Gilbert EM, et al. Changes in gene expression in the intact human heart. Downregulation of alpha-myosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest. 1997; 100:2315–2324.
56. Nakao K, Minobe W, Roden R, Bristow MR, Leinwand LA. Myosin heavy chain gene expression in human heart failure. J Clin Invest. 1997; 100:2362–2370.
57. Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000; 86:386–390.
58. Lowes BD, Zolty R, Shakar SF, Brieke A, Gray N, Reed M, Calalb M, Minobe W, Lindenfeld J, Wolfel EE, Geraci M, Bristow MR, Cleveland J Jr. Assist devices fail to reverse patterns of fetal gene expression despite beta-blockers. J Heart Lung Transplant. 2007; 26:1170–1176.
59. Morkin E. Control of cardiac myosin heavy chain gene expression. Microsc Res Tech. 2000; 50:522–531.