
Introduction
Albeit intensive efforts that have focused on therapeutics development, cancer is still a leading health problem worldwide [1]. In past few decades, the concept of chemoprevention has emerged and become a new strategy in fighting against cancers [2-4]. World Health Organization (WHO) indicates that at least 30% of all cancer deaths are preventable [5]. Since diverse phytochemicals were reported to interfere with a specific stage of the carcinogenic process, numerous efforts have been devoted to identifying phytochemicals and phytochemical-derived agents with cancer preventive properties [4, 6-7].
Isorhapontigenin (ISO) is a new derivative of stilbene isolated from the Chinese herb Gnetum Cleistostachyum [8]. ISO was also recently identified from wine grapes that are the main dietary source of stilbene [9]. Despite several investigations on biological properties of ISO such as its antioxidant effect [10-11], the anti-cancer activity of this compound has not been evaluated until quite recently, and it has been found that ISO triggers apoptosis in multiple human cancer cell lines [12-13]. Mechanistically, ISO treatment is shown to downregulate XIAP and cyclin D1 expression by promoting transcription factor Sp1 protein degradation [12-13]. However, ISO chemopreventive effects have not been explored thus far. In the current study, therefore, we using TPA/EGF-induced mouse Cl41 cell transformation model sought to investigate the potential chemopreventive activity of ISO and molecular mechanisms underlying its activity. We found that ISO was capable of inhibiting TPA/EGF-induced cell transformation with induction of G0/G1 cell-cycle arrest by downregulating cyclin D1 transcription via both upregulating MKP-1 expression and deactivating MKK7/JNK cascade.
Results
ISO inhibited cell transformation and induced G0/G1 cell-cycle arrest with no redundant cytotoxic effects on non-transformed cells
To investigate the potential chemopreventive activity of ISO, TPA/EGF-induced Cl41 cell transformation model was employed. Given that ISO could reduce cell viability in T24T bladder cancer cells with an approximate IC50 of 55 μM [12], we thus treated mouse epidermal Cl41 cells with ISO in concentrations of 30, 40, and 50 μM with exposure to TPA/EGF. As shown in Figs. 1A and 1B, co-incubation of cells with ISO for 3 weeks significantly inhibited TPA/EGF-induced anchorage-independent colony formation in a dose-dependent manner in Cl41 cells, indicating that ISO is a potential preventive agent. To further explore whether the inhibitory effect of ISO on cell transformation is due to its induction of apoptosis and/or cell cycle arrest, high-resolution flow cytometry analysis of PI-stained nuclei was performed. The data revealed that treatment of cells with ISO at the same concentrations for 48 hours was capable of significantly reversing TPA/EGF-induced G1/S phase progression in a dose-dependent manner, whereas almost no apoptosis was triggered under the same experimental condition (Figs. 1C and 1D). Considering that an ideal chemopreventive agent should be able to impart apoptotic/anti proliferative effects specifically in carcinogen/tumor promoter-treated cells without affecting normal cells [6], we thus evaluate the cytotoxic effect of ISO on normal non-transformed Cl41 cells using ATPase assay. The data showed that ISO did not exert any notable growth inhibition at the concentration range 30-50 μM at 48 hours after the treatment (Fig. 1E). These results demonstrated that ISO could remarkably inhibit the growth of transformed Cl41 cells via arresting G1/S progression without redundant cytotoxic effects on non-transformed cells.
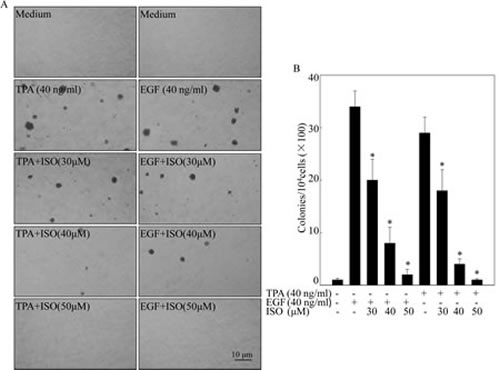
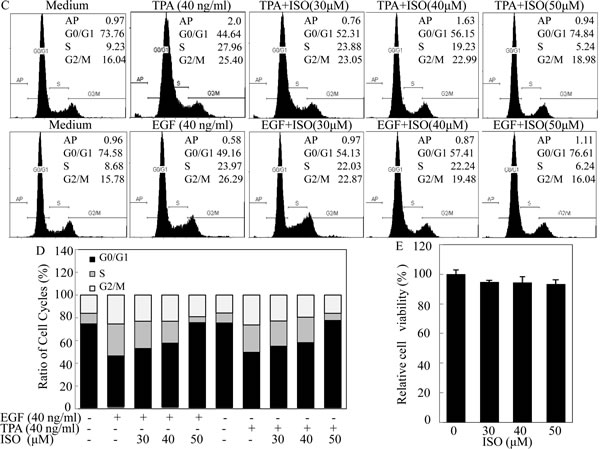
Figure 1: ISO inhibited cell transformation and induced G0/G1 cell-cycle arrest with no redundant cytotoxic effects on non-transformed Cl41 cells. (A) Representative images of colonies of Cl41 cells in soft agar assay. Cells were co-treated with TPA/EGF (40 ng /ml) and various concentrations of ISO as indicated. (B) The number of colonies was counted under microscopy in soft agar after 3 weeks and the results were presented as colonies per 10,000 cells from three independent experiments. The asterisk (*) indicates a significant difference in Cl41 cells treated with different doses of ISO compared with cell treated with TPA or EGF alone respectively (P<0.05). (C and D) Flow cytometric analysis of cell cycle distribution. Cl41 cells were pretreated with various concentrations of ISO for 30 min and then co-incubated with ISO and TPA/EGF (40 ng /ml) as described in “Materials and Methods”. Data was represent one of three different experiments. (E) Cl41 cells were treated with various concentrations of ISO (30, 40 or 50 μM) for 48 hours. Percentage of cell viability was expressed as relative to medium control in triplicate.
ISO downregulation of cyclin D1 expression was responsible for its induction of G0/G1 phase arrest and inhibition of cell transformation
To delineate the molecular basis of ISO-induced inhibition of cell transformation and reversion of G1/S phase transition due to TPA or EGF treatment, we performed Western blot analysis to probe the molecular targets that could be potentially regulated by this phytochemical. Cells were pretreated with ISO in different concentrations for 30 min and then co-incubated with TPA/EGF for 24 hours. The data showed that cyclin D1, a pivotal regulator of G1/S phase progression [14], was significantly downregulated by ISO in a dose-dependent manner (Fig. 2A). Furthermore, time course analysis by Western blot revealed that induction of cyclin D1 expression by TPA or EGF was profoundly reduced by 50 μM of ISO at 24 hours in comparison to those in the cells treated with TPA/EGF alone (Figs. 2B and 2C). In contrast, the expression of anti-apoptotic protein XIAP and transcription factor Sp1, which has shown to be suppressed by ISO in human cancer cells [12], was not affected upon ISO treatment in Cl41 cells (Fig. 2A). These data suggested a distinct molecular mechanism of the ISO chemopreventive activity in non-transformed Cl41 cells.
To further prove that ISO downregulation of cyclin D1 protein expression was responsible for the induction of G0/G1 phase arrest and inhibition of cell transformation, Cl41 cells stably transfected with the plasmid encoding FLAG-cyclin D1 T286A mutant, which is refractory to phosphorylation by GSK-3β and thus this point mutation impairs the cyclin D1 proteasomal degradation [15],were established and utilized to test the contribution of cyclin D1 downregulation to the inhibition of cell transformation and induction of G0/G1 phase arrest due to ISO treatment. Overexpression of FLAG-cyclin D1 T286A mutant in the stable transfectant of Cl41 cells was verified as indicated in Fig. 2D. Although ISO treatment did not reduce expression level of FLAG-cyclin D1 T286A mutant, It markedly blocked endogenous cyclin D1 expression (Fig. 2E). As expected, ectopic expression of FLAG-cyclin D1 T286A mutant substantially assisted to bypass ISO-induced G0/G1 phase arrest and attenuated the ISO inhibition of cell transformation induced by TPA or EGF (Fig. 2F-2I), thereby buttressing that ISO-downregulated cyclin D1 expression accounted for its induction of G0/G1 phase arrest and inhibition of cell transformation in Cl41 cells.
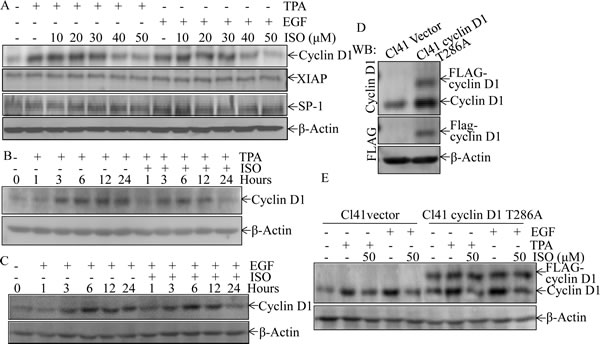
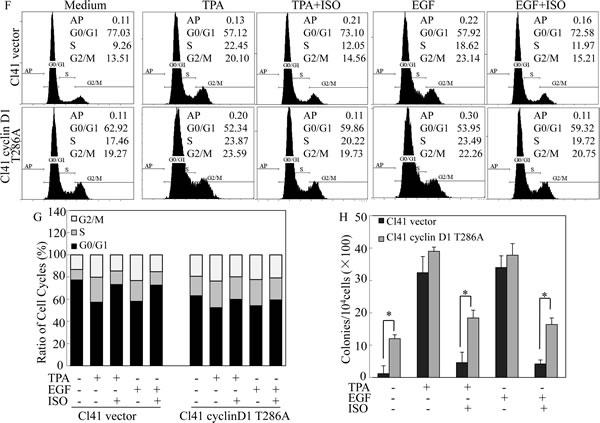
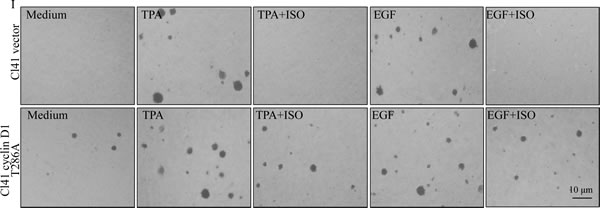
Figure 2: Downregulation of cyclin D1 expression mediated the inhibition of colony formation and induction of G0/G1 phase arrest following ISO treatment. (A-C) Cl41 cells were pretreated with indicated doses of ISO for 30 min and then co-treated with ISO and TPA/EGF (40 ng /ml) for 24 hours (A) or pretreated with 50 μM ISO for 30 min and then co-incubated with ISO and TPA/EGF for the indicated time periods (B and C). The protein expression levels were determined by Western blot, and β-actin levels were used to as protein loading control. Data was representative one of three independent experiments. (D and E) The FLAG-cyclin D1 expression levels were determined by Western blot in Cl41 cells transfected with plasmid encoding FLAG-tagged cyclin D1 T286A mutant and its vector control. Antibodies specifically against cyclin D1 and FLAG were used, respectively, to detect the protein as indicated in the figure (D). Transfectants were treated with TPA/EGF (40 ng /ml) alone, or pretreated with 50 μM ISO for 30 min and then co-incubated with ISO and TPA/EGF for 24 hours. Antibody specifically against cyclin D1 was used to determine cyclin D1 expression (E). (F and G) Cl41 cells were treated with TPA/EGF (40 ng /ml) alone, or pretreated with 50 μM ISO for 30 min and then co-incubated with ISO and TPA/EGF for 24 hours. Cell cycle distribution was determined by flow cytometric analysis. Data represent one of three different experiments. (H and I) Cells stably transfected with vector control or FLAG-cyclinD1 T286A were treated with medium containing either DMSO or TPA/EGF( 40 ng /ml) with or without exposure to ISO (50 μM) for 3 weeks. Representative images of colonies of transfectants in soft agar assay were presented (I). Colonies were counted after 3 weeks and the results were presented as colonies per 10,000 cells from three independent experiments (H). The asterisk (*) indicates a significant difference of colony number between Cl41 FLAG-cyclinD1 T286A cells and Cl41 vector cells (P<0.05).
ISO inhibited cyclin D1 transcription via abolishing C-Jun/AP-1 activation
To clarify the underlying mechanism of cyclin D1 downregulation by ISO, we examined cyclin d1 mRNA level and the transcriptional activity of cyclin D1 promoter after cells were co-treated with ISO and TPA/EGF. RT-PCR analysis and luciferase reporter assay demonstrated that ISO treatment resulted in the reduction of TPA- or EGF-induced both cyclin d1 mRNA level and its promoter-dependent transcriptional activity in a dose-dependent manner (Figs. 3A and 3B), suggesting that ISO was capable of suppressing cyclin D1 transcription in Cl41 cells.
To identify the transcription factor responsible for ISO downregulation of cyclin D1 transcription, we evaluated the changes in the nuclear translocation of related transcription factors upon ISO treatment for 9 hours. Compared to cells treated with TPA/EGF alone, cells pretreated with 50 μM of ISO displayed the substantial reduction of c-Jun phosphorylation at Ser63 and Ser73 in the nuclear protein extract, whereas c-Jun total protein expression was not affected by ISO (Fig. 3C). Meanwhile, no significant suppression of other transcription factors, including Sp1, NF-kB p65 or p50, NFAT1, CREB, Jun-D, Jun-B, and C-fos was observed following ISO treatment (Fig. 3C). These data suggested that ISO was able to specifically inhibit c-Jun phosphorylation at Ser63 and Ser73. To further substantiate this finding, the inhibitory effect of ISO on c-Jun phosphorylation was examined by means of the time course studies. In comparison to cells treated with TPA/EGF at the corresponding time point, cells with 50 μM ISO pretreatment presented the decreased phosphorylation of c-Jun at Ser63 and Ser73 at approximately 12-24 hours (Figs. 3D and 3E). It was noted that ISO had no impact on c-Jun total expression, although TPA or EGF treatment led to increase in total protein expression (Figs. 3D and 3E).These results together demonstrated that ISO was indeed capable of suppressing c-Jun phosphorylation and activation.
C-Jun is a major component of transcription factor AP-1. We next examined ISO’s effect on AP-1 transactivation in Cl41 cells stably transfected with AP-1-luciferase reporter containing seven tandem AP-1 binding sites[16]. As anticipated, ISO treatment significantly blocked AP-1 transactivation in a dose-dependent manner (Fig. 3F). To determine whether ISO-suppressed cyclin D1 transcription was mediated by c-Jun/AP-1, we tested the effect of ISO on cyclin D1 transcription in Cl41 cells stably transfected with luciferase reporter containing cyclin D1 promoters with mutated AP-1 binding sites in compared to that with WT cyclin D1 promoter luciferase reporter. As indicated in Fig. 3G, treatment of cells with either TPA or EGF led to 4 fold induction of cyclin D1 promoter activity in WT cyclin D1 promoter reporter transfectant, and the induction was crippled in the transfectants of AP-1 binding site-mutated cyclin D1 promoter-luciferase reporter. Moreover, co-incubation of cells with ISO exhibited a marked inhibition of cyclin D1 promoter activity in WT cyclin D1 promoter reporter transfectant, whereas it did not show significant effect in AP-1 binding site-mutated cyclin D1 promoter-luciferase reporter (Fig. 3G).Thus, our results strongly implicated that ISO treatment resulted in the suppression of TPA/EGF-induced c-Jun/AP-1 activation, which would in turn inhibit cyclin D1 transcription in Cl41 cells.
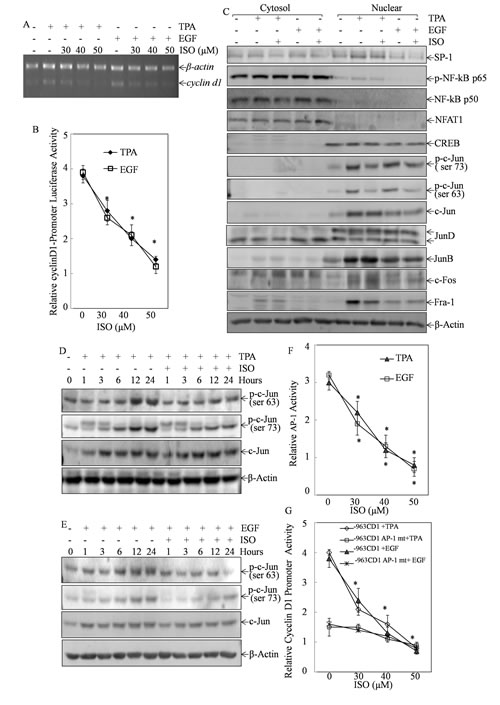
Figure 3: The inhibition of c-Jun/AP-1 by ISO mediated the suppression of cyclin D1 transcription. (A) Cl41 cells were pretreated with ISO at the indicated dose for 30 min and then co-incubated with ISO and TPA/EGF (40 ng /ml) for 12 hours. RT-PCR was performed to determine cyclin d1 mRNA levels. β-actin was used as loading control. Data are representative of three independent experiments. (B) Cl41 cells stably transfected with cyclin D1-promoter luciferase reporter were pretreated with ISO in the indicated concentration for 30 min followed by co-incubation with ISO and TPA/EGF (40 ng /ml) for 24 hours. The luciferase activity was measured as described in “Materials and methods”. The results were presented as relative cyclin D1 promoter activity. The symbol (*) indicates a significant decrease as compared with Cl41 cells treated with TPA/EGF alone (P<0.05). (C) Cytoplasmic and nuclear extracts from Cl41 cells (treated with TPA/EGF (40 ng /ml) alone, or pretreated with ISO (50 μM) for 30 min and then co-incubated with ISO and TPA/EGF for 9 hours were prepared as described in “Materials and methods”, and then subjected to Western blot analysis with specific antibodies against Sp-1, c-Fos, p-c-Jun (Ser73), p-c-Jun (Ser63), c-Jun, Jun D, Jun B, p-NF-kB p65, NF-kB p50, NFAT1, and CREB, respectively. (D and E) Cl41 cells were treated with TPA/EGF (40 ng/ml), or pretreated with 50 μM ISO for 30 min and then co-incubated with ISO and TPA/EGF for indicated time periods. The p-c-Jun (Ser73), p-c-Jun (Ser63) and c-Jun expression levels were determined by Western blot analysis. (F) AP-1-luciferase reporter containing seven tandem AP-1 binding sites was stably transfected into Cl41 cells. The transfectants were treated with TPA/EGF (40 ng /ml) alone, or pretreated with ISO in the indicated concentrations for 30 min followed by co-incubation with ISO and TPA/EGF for 24 hours. The results were presented as relative AP-1 activity compared to that treated with TPA/EGF alone. The symbol (*) indicates a significant decrease as compared with the indicated cells treated with TPA/EGF alone (P<0.05). (G) Cyclin D1 promoter-driven Luciferase reporter (-963CD1) or Cyclin D1 promoter-driven Luciferase reporter with AP-1 binding site mutation (-963CD1 mt) were stably transfected into Cl41 cells. The transfectants were pretreated with ISO in the indicated concentrations for 30 min followed by co-incubation with ISO and TPA/EGF (40 ng /ml) for 24 hours. The luciferase activity was measured as described in “Materials and methods”. The results were presented as relative cyclin D1 promoter activity compared to that treated with TPA/EGF alone. The symbol (*) indicates a significant decrease as compared with the indicated cells treated with TPA/EGF alone (P<0.05).
ISO treatment upregulated MKP-1 expression and deactivated MKK7/JNK cascade
The mitogen-activated protein kinases (MAPKs), including ERK, p38, and JNK, have been proven as activators of c-Jun/AP-1 in many cellular settings [17-20]. We thus tested whether TPA/EGF-induced MAPKs activation could be deactivated by ISO in Cl41 cells. The results showed that ISO treatment downregulated the TPA/EGF-induced phosphorylation of JNK at 6-24 hours, but did not affect JNK total expression. By contrast, the induction of phosphorylation of ERK and p38 by TPA/EGF was not impacted by ISO treatment (Figs. 4A and 4B). These data indicated that ISO could selectively deactivate TPA/EGF-induced JNK activation. Given that MKK4 and MKK7 are the two upstream kinases required for JNK full activation [21-23], we next tested whether ISO treatment could likewise attenuate TPA/EGF-induced phosphorylation of MKK4 and MKK7 along the time course. As a consequence, only the obvious reduction of MKK7 phosphorylation was observed when cells were treated with ISO, while MKK7 total expression did not change (Figs. 4A and 4B), thereby suggesting that MKK7 might be a kinase involved in the deactivation of JNK by ISO.
In addition to kinases, phosphatases might be also involved in JNK deactivation upon ISO treatment. Thus, time course analysis was performed to test whether potential phosphatases are induced by ISO treatment. As shown in Figs. 5A and 5B, ISO treatment upregulated MKP-1 expression, but did not affect the expression of other phosphatases, including PHLPP1, PP2A-A, PP2A-B, and PTEN. Given that MKP-1 is a phosphatase that could directly regulate JNK phosphorylation [24-25], our results indicated that ISO treatment resulted in MKP1 induction, which might subsequently deactivate JNK phosphorylation induced by TPA/EGF
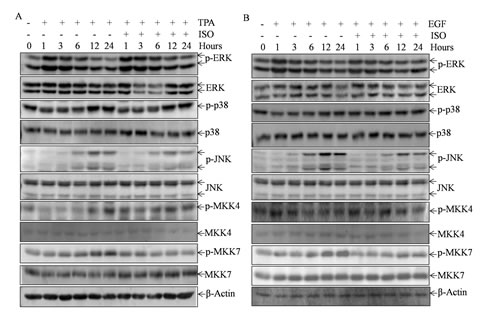
Figure 4: ISO treatment deactivated MKK7/JNK cascade.(A and B) Cl41 cells were pretreated with ISO (50 μM) for 30 min and then co-incubated with ISO and TPA/ EGF (40 ng /ml) for indicated time periods. The total cell extracts were subjected to Western blot analysis with specific antibodies against p-ERK, ERK, p-p38, p38, p-JNK, JNK, p-MKK4, MKK4, p-MKK7, and MKK7. β-Actin protein was used as protein loading control. Data are representative of three independent experiments.
MKP-1 knockdown attenuated ISO inhibition of c-Jun/JNK, suppression of cyclin D1 expression, G0/G1 growth arrest and inhibition of cell transformation in Cl41 cells
To further confirm that MKP-1 acted as the precursor regulating the JNK/c-Jun pathway responsible for cyclin D1 downregulation and growth inhibition following ISO treatment, MKP-1 was knocked down in Cl41 cells with a short hairpin RNAi construct that specifically targeted mkp-1. Knockdown of MKP1 in two stable mass transfectants was verified in Fig. 5C. As a result, cyclin D1 downregulation and the reduction of phosphorylated c-Jun and JNK by ISO treatment were abrogated in MKP-1 knockdown cells in comparison to that from nonsense transfectant (Fig. 5D). Furthermore, MKP-1 knockdown also reversed ISO-caused G0/G1 cell cycle arrest and inhibition of cell transformation in Cl41 cells (Fig. 5E-5H). Taken together, our results demonstrated a critical role of MKP-1 in the inhibition of the JNK/c-Jun cascade and in turn downmodulates cyclin D1 expression, as well as induction G0/G1 arrest and inhibition of cell transformation following ISO treatment.
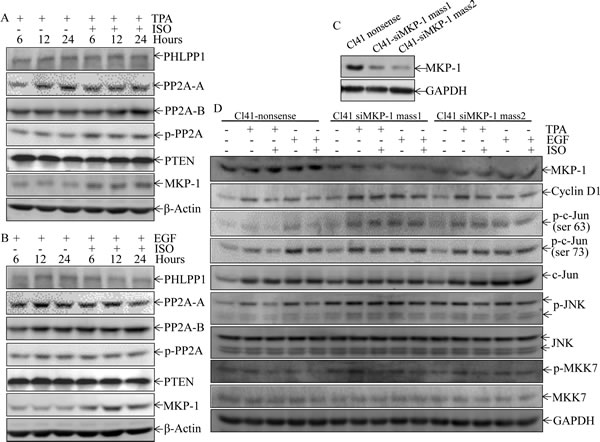
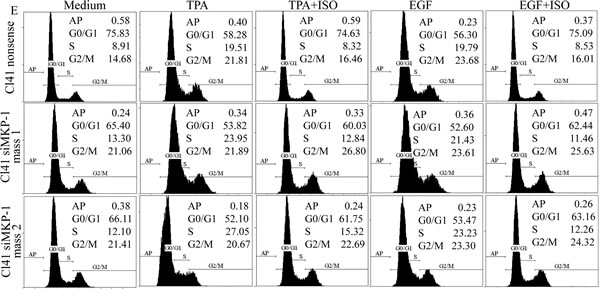
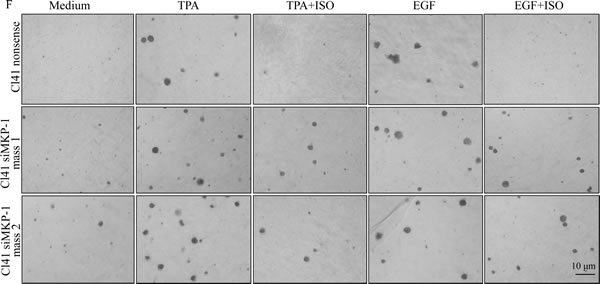
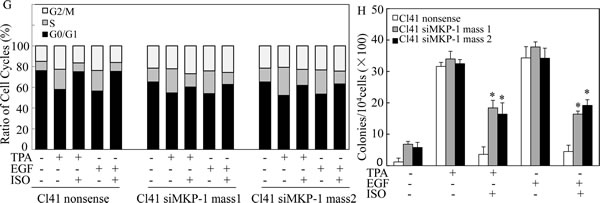
Figure 5: ISO treatment upregulated MKP-1 expression. (A and B) Cl41 cells were treated with TPA/EGF (40 ng /ml) alone, or pretreated with ISO (50 μM) for 30 min and then co-incubated with ISO and TPA/EGF for indicated time periods. The cell extracts were subjected to Western blot analysis with specific antibodies against PHLPP1, PP2A-A, PP2A-B, p-PP2A, PTEN, and MKP-1. (C and D) Cl41 cells were transfected with the siMKP-1 or nonsense constructs. Stable transfectants were established and cell extracts were subjected to Western blot analysis with specific antibody against MKP-1(C). The indicated transfectants were treated with medium or TPA/EGF (40 ng /ml) alone, or pretreated with ISO (50 μM) for 30 min and then co-incubated with ISO and TPA/EGF for 24 hours. The cell extracts were subjected to Western blot analysis with specific antibodies against MKP-1, cyclin D1, p-c-JunSer63, p-c-JunSer73, c-Jun, p-JNK, JNK, p-MKK7, and MKK7 (D). (E and G) Cl41 cells were treated with medium or TPA/EGF (40 ng /ml) alone, or pretreated with 50 μM of ISO for 30 min and then co-incubated with ISO and TPA/EGF for 48 hours as described in “Materials and Methods”. Data represent one of three different experiments as indicated in Flow cytometric analysis of cell cycle distribution (E) and percentage of cell-cycle phase (G). (F and H) The cell transformation was determined using the indicated Cl41 stable transfectants in the presence of TPA/EGF (40 ng /ml)with or without ISO (50 μM). Representative images of colonies of transformed cells in soft agar assay were presented (F), and the colonies were counted under microscopy and presented as colonies per 10,000 cells from three independent experiments (H). The symbol (*) indicates a significant increase as compared with that in nonsense transfectants treated with TPA/EGF alone (P<0.05).
ISO treatment increased mkp-1 mRNA stability
To elucidate mechanism underlying ISO upregulation of MKP-1 expression, we determined mkp-1 mRNA level following co-incubation of ISO with either TPA or EGF. The results revealed that mkp-1 mRNA level was inhibited in Cl41 cells following either TAP or EGF treatment (Fig. 6A). However, this inhibition by TPA or EGF was completely reversed to the level that is even higher than basal level by co-incubation of cells with ISO, suggesting that ISO upregulated MKP-1 expression at either transcription or mRNA stability. To address this question, the MKP-1 promoter-driven luciferase reporter was transfected into Cl41 cells and the MKP-1 promoter activity in the transfectant was determined in the cells treated with ISO followed TPA or EGF. As shown in Fig. 6B, ISO treatment significantly downregulated MKP-1 promoter activity, suggesting ISO treatment might upregulate MKP-1 expression via increase mkp-1 mRNA stability. This notion was greatly supported by the results showing that pretreatment of cells with ISO for 6 hours followed by the treatment of actinomycin D for 20 min increased mkp-1 mRNA half-life (T1/2 increased to 42 min) in comparison to that observed in cells treated with actinomycin D alone (T1/2=14 min) (Figs. 6C& 6D). Thus, our data proved that ISO treatment stabilized mkp-1 mRNA stability.
Several RNA-binding proteins, e.g. HNRPD, VHL, Hur, and Nucleolin, have been reported to bind their target mRNA and increased mRNA stability [26-28]. We hence tested whether any of these proteins was involved in ISO upregulation of mkp-1 mRNA stability. Western blot analysis showed that ISO treatment could clearly increase the Nucleolin expression, and moreover Nucleolin protein upregulation was associated with the MKP-1 protein induction along the time course studies (Fig. 6E), suggesting that Nucleolin might participated in the increased mkp-1 mRNA stability following ISO treatment.
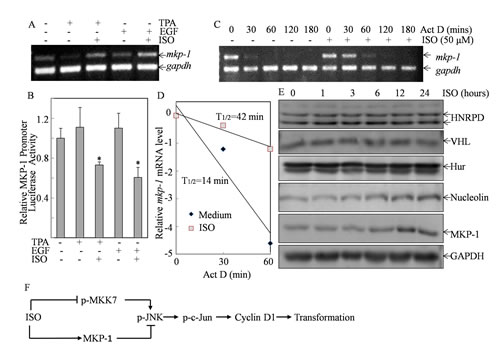
Figure 6: ISO treatment increased mkp-1 mRNA stability. (A) Cl41 cells were treated with medium or TPA/EGF (40 ng /ml) alone, or pretreated with 50μM of ISO for 30 min and then co-incubated with ISO and TPA/EGF for 12 hours. RT-PCR was performed to determine mkp-1 mRNA levels. The gapdh mRNA levels were used as loading control. (B) Cl41 cells stably transfected with MKP-1 promoter luciferase reporter were treated with medium or TPA/EGF (40 ng /ml) alone, or pretreated with 50 μM of ISO for 30 min followed by co-incubation with ISO and TPA/EGF for 24 hours. The luciferase activity was measured as described in “Materials and Methods”. The results were presented as relative MKP-1 promoter activity compared with that of the cells treated with TPA/EGF. The symbol (*) indicates a significant decrease as compared with cells treated with TPA/EGF alone (P<0.05). (C) Cl41 cells were pretreated with ISO (50 μM) for 6 hours and then co-incubated with ISO and actinomycin D (20 μM) for indicated time periods. Total RNA was isolated and RT-PCR was then performed to determine mkp-1 mRNA levels. The result was a representative one from three independent experiments. (D) The relative mRNA level of mkp-1 was determined by ImageQuant 5.2 (GE Healthcare). Natural logarithm of ratio [mkp-1]t/[mkp-1]0 was plotted against the time and the half life of mkp-1 mRNA was calculated via linear regression. (E) Cl41 cells were treated with ISO (50 μM) for indicated time periods. The total cell extracts were subjected to Western blot analysis with specific antibodies against HNRPD, VHL, Hur, Nucleolin, and MKP-1. (F) The diagram indicates mechanisms responsible for ISO inhibition of cell transformation.
Discussion and Conclusions
The naturally occurring compound ISO [8] has been proved to possess anti-cancer activities against multiple human cancer cell lines [12]. Mechanistic studies revealed that ISO downregulation of XIAP and cyclin D1 protein expression accounted for its anti-cancer effects in human cancer cells [12-13]. In the current study, we investigated potential cancer chemopreventive activity of this phytochemical by exploring its potential inhibitory effect on cell transformation in Cl41 cells. Our results demonstrated that ISO treatment was capable of inhibiting TPA/EGF-induced colony formation via upregulating MKP-1 expression and arresting G1/S cell-cycle progression, whereas it did not produce redundant cytotoxic effects on normal non-transformed cells, thereby suggesting the cancer chemopreventive activity of ISO. Further study indicated that ISO-upregulated MKP-1 expression mediated the inhibition of JNK/C-Jun/AP-1 activation and in turn led to cyclin D1 downregulation, G1/S cell-cycle arrest, and cell transformation inhibition. Therefore, we identified a novel function of ISO as a cancer chemopreventive agent that is mediated through a molecular mechanism distinct from that responsible for ISO cancer therapeutic effect.
Abnormal expression of cyclin D1 has been reported in many human cancers [29]. Particularly, cyclin d1 mRNA amplification has been observed in approximately 45% breast carcinomas [30]. It is also reported that more than 50% patients presented cyclin D1 overexpression in a total of 307 patients with non-melanocytic skin cancer [31]. Cyclin D1 has been shown to play a crucial role in cancer development, especially skin carcinogenesis [32-33]. Transgenic mice with conditional cyclin D1 overexpression are much easier to develop skin tumor when exposed to dimethylbenz[a]anthracene [33]. Also, cyclin D1 knockdown was capable of attenuating arsenite-induced cell transformation in mouse Cl41 cells and human HaCat cells [34-35]. Hence, cyclin D1 has been regarded as an important player in cancer development as well as a target for cancer prevention and therapy [36]. Our previous study, for example, has reported that suberoylanilide hydroxamic acid (SAHA) is able to inhibit EGF-induced cell transformation through reduction of cyclin d1 mRNA stability [37]. In the present study, we found that ISO inhibition of cell transformation and cell cycle progression in Cl41 cells was mediated through downregulation of cyclin D1 protein expression. Further investigation proved that ISO regulated cyclin D1 through inhibiting its transcription, and this process was mediated through the suppression of its transcription factor c-Jun/AP-1 activation via a novel mechanism, i.e. upregulating MKP-1 expression.
MKK7/JNK/AP-1 cascade is an important oncogenic signaling pathway [1, 38]. Murine genetic loss-of-function studies have demonstrated that targets of JNK signaling cascade, particularly c-Jun and c-fos, may contribute to chemical-induced murine epidermal neoplasia [39-41]. JNK/AP-1 has been reported to play a role in inducing aberrant cell proliferation associated with human skin carcinogenesis [38-43]. Moreover, previous studies have also reported that TNFR1/MKK7/JNK/AP-1 cascade promoted human neoplasia and inhibition of this pathway represented a potential therapeutic approach for squamous cell carcinoma (SCC) [38]. Therefore, the discovery in the current investigation that ISO could inhibit the activation of MKK7/JNK/c-Jun/AP-1 cascade triggered by TPA/EGF in Cl41 mouse epidermal cells reveals the application of this phytochemical for the cancer prevention.
In addition to MKK7, phosphatase MKP-1 was also shown to be upregulated and mediate JNK deactivation following ISO treatment. Further results indicated that ISO treatment could augment mkp-1 mRNA stability. Yet, although our data suggested that Nucleolin might be involved in the stabilization of mkp-1 mRNA, which has never been reported before, the detailed mechanism underlying this process remains elusive and thus is currently underway of investigation in our laboratory. On the other hand, it is intriguing that albeit with profoundly decreased promoter activity, mkp-1 mRNA level was upregulated following ISO treatment. This seemingly paradox might be due to fact that MKP-1 is regulated by c-Jun, which has been reported in sympathetic neurons [44] and thus c-Jun deactivation might result in the downregulation of MKP-1 promoter transcriptional activity following ISO treatment. But the detailed mechanism responsible for ISO downregulation of MKP-1 transcription in Cl41 cells is still under investigation.
In summary, our studies demonstrated that ISO exerted an inhibitory effect of TPA/EGF-induced cell transformation with induction G0/G1 cell-cycle arrest in Cl41 cells. Mechanistic insight into cell-cycle arrest induction suggested that this inhibitory effect is through MKP-1 upregulation and MKK7 deactivation, both of which would concertedly deactivate JNK/c-Jun cascade, thereby leading to cyclin D1 downregulation (Fig. 6F). These findings implicate the potential utilization of ISO as a cancer chemopreventive agent.
Material and Methods
Cell Culture and Reagents
Mouse epidermal Cl41 cells and their transfectants were cultured in 5% Fetal bovine Serum (FBS) MEM containing with 1% penicillin/streptomycin and 2 mM L-glutamine (Life Technologies). Cells were maintained in a humidified incubator at 37°C with 5% CO2 atmosphere[45]. The antibodies specific against phospho-c-Jun, c-Jun, phospho-JNKs, JNKs, phospho-ERKs, ERKs, phospho-p38, p38, phospho-MKK7, MKK7, phospho-MKK4, MKK4, PP2A A subunit, XIAP, SP1, β-Actin and GAPDH were purchased from Cell Signaling Technology Inc (Beverly, MA). The antibodies specific against cyclin D1, c-Fos, CREB, Fra1, p-NF-kB p65, NF-kB p50, JunD, JunB, PTEN and MKP-1 were bought from Santa Cruz Biotechnology (Santa Cruz, CA). The antibody against FLAG was obtained from Covance Inc. (Princeton, NJ). The antibodies against phospho-PP2A and PP2A B subunit were purchased from Epitomics (Burlingame, CA). Actinomycin D was purchased from Calbiochem (Billerica, MA). ISO with over 98% purity was purchased by Higher Biotech (Shanghai, China). The preparation of ISO was performed as described in our previous studies [12].
Plasmids and Cell Transfection
The -963cyclin D1 promoter-driven luciferase reporter and its AP-1 binding site mutant were kindly provided by Dr. Richard G Pestell (Thomas Jefferson University Jefferson Medical College) [46-47]. The cyclin D1 promoter region containing -963 to +14 was subcloned into the pA3LUC and named as the -963 cyclin D1 promoter-driven Luciferase reporter (-963CD1). The -963 CD1AP-1 mt was derived from -963CD1 with two nucleotide mutation within AP-1 binding site of the cyclin D1 promoter. The MKP-1 promoter-driven luciferase reporter was a gift from Dr. Yusen Liu (National Institutes of Health) [48]. Cyclin D1 promoter-driven luciferase reporter was described in our previously study [49]. The AP-1-driven luciferase reporter containing seven tandem AP-1 binding sites (TGACTAA) was purchased from Stratagene (La Jolla, CA). FLAG–cyclin D1 T286A/pCMV5 expression vector was a gift of Dr. Udit N. Verma (University of Texas Southwestern Medical Center at Dallas) [50]. The specific small-interference RNA (siRNA) vector targeted mouse mkp1 (dusp1) was purchased from Sigma-Aldrich Chemical (St. Louis, MO). Stable co-transfections were performed with specific cDNA constructs together with pSUPER-puro vector using PolyJetTM DNA In Vitro Transfection Reagent (SignaGen Laboratories, Gaithersburg, MD) according to the manufacturer’s instructions. For stable transfection selection, cultures were subjected to puromycin selection for 4–6 weeks, and surviving cells were pooled as stable mass transfectants. These stable transfectants were cultured in the selected antibiotic-free medium for at least two passages before utilization for experiments[51-53].
Anchorage-independent Growth Assay
Soft agar assay was performed as described previously [54]. Briefly, 2.5 ml of 0.5% agar in basal modified Eagle’s medium (BMEM) supplemented with 10% FBS with or without ISO, EGF, TPA, EGF+ISO or TPA+ISO respectively was layered onto each well of 6-well tissue culture plates. 1×104 Cl41 cells or their transfectants were mixed with 1 ml of 0.5% agar BMEM supplemented with 10% FBS with or without EGF/TPA and ISO, and then layered on top of the 0.5% agar layer. The plates were incubated at 37 °C in 5% CO2 for 3 weeks. The colonies with more than 32 cells were scored and the results were presented as colonies/104 cells[45, 47, 51].
Flow Cytometry Assay
Cells (1×105) were cultured in each well of 6-well plates till 60-70% confluence with normal culture medium. Cells were synchronized by the replacing the medium containing 0.1% FBS for 24 hours, and then replacing medium containing 1% FBS with or without EGF/TPA and ISO, and cultured for another 24 hours as indicated in figure legends. The cells were harvested and fixed with 3 ml of ice-cold 80% ethanol overnight. The fixed cells were washed twice with PBS, and then suspended in Propidium Iodide staining solution (Propidium Iodide 50 mg /ml, RNAse A 10 mg /ml, and 0.1% Triton X-100) (Sigma-Aldrich Chemical, St. Louis, MO) for at least 1 hour at 4°C. The DNA content was determined by means of Flow Cytometric Analysis with the Epics XL FACS (Beckman Coulter, Miami, FL) and EXPO 32 software as described previously [45, 51, 55].
Cell Proliferation Assay
Confluent monolayers of Cl41 cells were trypsinized and 2×103 viable cells suspended in 100 μl MEM supplemented with 5% FBS were added to each well of 96-well plates. After adherent, cells were synchronized by replacing the medium containing 0.1% FBS for 24 hours, and then replacing culture medium containing 1% FBS with or without ISO, and cells treated with ISO at indicated doses were cultured for 48 hours. The viability of the cells was determined using CellTiter-Glo® Luminescent Cell Viability Assay kit (Promega, Madison, WI) with a luminometer (Wallac 1420 Victor 2 multipliable counter system). The results were presented as proliferation index (relative luminescence signal to medium control)[56-57].
Western Blotting
Cell extracts were prepared with cell lysis buffer (10 mM Tris-HCl, pH 7.4, 1% SDS, and 1 mM Na3VO4) and protein concentrations were determined by NanoDrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE). 30~80 μg of protein sample from the cell extracts were separated on SDS-polyacrylamide gels (SDS-PAGE), transferred, and probed with indicated antibody. The protein band that was specifically bound to the primary antibody was detected using an alkaline phosphatase-linked secondary antibody and an ECF Western blotting system (Amersham Biosciences, Piscataway, NJ)[47, 58-59].
RT-PCR
Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA), and cDNAs were synthesized with the ThermoScript RT-PCR system (Invitrogen, Carlsbad, CA). The mouse cyclin d1 cDNA fragments were amplified by primers 5’-TCCCTTGACTGCCGAGAAG-3’ and 5’-AGACCAGCCTCTTCCTCCAC-3’. The primers for mouse mkp-1 are 5’-GAA GCG TTT TCG GCT TCC TG-3’ and 5’- AGG TAA GCA AGG CAG ATG GTG-3’. The primers for mouse gapdh are 5’-GGA GGT TGT CAT CCC TCAGA-3’ and 5’-TCC TCC TCA GCC ACA CTC TT-3’. The primers for mouse β-actin are 5’-ATA TCG CTG CGC TGG TCG TC-3’ and 5’-AGG ATG GCG TGA GGG AGA GC-3’. The PCR products were separated onto 3% agarose gels, stained with EB, and scanned the images from a UV light as described previously[45, 52, 60-61].
Luciferase Assay
Cl41 cells were transfected with the indicated luciferase reporter construct in combination with the pRL-TK vector (Promega, Madison, WI) as an internal control. The transfectants were seeded into 96-well plates. After the cell density reached 70-80%, cells were treated as indicated in the figure legends, and were then extracted with luciferase assay lysis buffer (Promega, Madison, WI). The luciferase activity was determined by the microplate luminometer LB 96V (Berthold GmbH & Co. KG, Bad Wildbad, Germany) using the luciferase assay system (Promega Corp., Madison, WI) as described [53, 61-62].
Nuclear Extract Preparation
Preparation of nuclear extracts was assessed as previously described [62]. Cl41 cells were plated into 10-cm culture dishes at 80% confluence, treated with EGF/TPA alone or pre-treated with 50 μM ISO for 30 mins and then co-treated with EGF or TPA for 9 hours. The nuclear proteins were extracted according to the protocol of the Nuclear/Cytosol Fractionation Kit (BioVison Technologies, Mountain View, CA). Equal protein concentrations were determined using a protein quantification assay kit (Bio-Rad, Hercules, CA). Nuclear extracts were stored at -80 °C until they were used.
Statistical Analysis
The student’s t-test was used to determine the significance between treated and untreated group. The results are expressed as mean ± SD from at least three independent experiments. P<0.05 was considered as a significant difference between compared groups.
Acknowledgements
We thank Dr. Richard G Pestell from Thomas Jefferson University Jefferson Medical College for providing the -963 cyclin D1 promoter-driven luciferase reporter and its AP-1 binding site mutant; Dr. Yusen Liu from National Institutes of Health for the gift of MKP-1 promoter-driven luciferase reporter; Dr. Udit N. Verma from University of Texas Southwestern Medical Center at Dallas for the gift of FLAG–cyclin D1 T286A/pCMV5 expression vector.
This work was partially supported by NIH/NCI CA177665, CA165980 and CA112557, and NSFC81229002 and NSFC30900639.
Conflicts of interest
None.
References
1. Lee KW, Bode AM and Dong Z. Molecular targets of phytochemicals for cancer prevention. Nature reviews Cancer. 2011; 11(3):211-218.
2. Sporn MB. Approaches to prevention of epithelial cancer during the preneoplastic period. Cancer research. 1976; 36(7 PT 2):2699-2702.
3. Wattenberg LW. Chemoprevention of cancer. Cancer research. 1985; 45(1):1-8.
4. Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nature reviews Cancer. 2003; 3(10):768-780.
5. Bode AM and Dong Z. Cancer prevention research - then and now. Nature reviews Cancer. 2009; 9(7):508-516.
6. She QB, Ma WY, Wang M, Kaji A, Ho CT and Dong Z. Inhibition of cell transformation by resveratrol and its derivatives: differential effects and mechanisms involved. Oncogene. 2003; 22(14):2143-2150.
7. Szekeres T, Fritzer-Szekeres M, Saiko P and Jager W. Resveratrol and resveratrol analogues--structure-activity relationship. Pharmaceutical research. 2010; 27(6):1042-1048.
8. Huang KS, Zhou S, Lin M and Wang YH. An isorhapontigenin tetramer and a novel stilbene dimer from Gnetum hainanense. Planta medica. 2002; 68(10):916-920.
9. Fernandez-Marin MI, Guerrero RF, Garcia-Parrilla MC, Puertas B, Richard T, Rodriguez-Werner MA, Winterhalter P, Monti JP and Cantos-Villar E. Isorhapontigenin: a novel bioactive stilbene from wine grapes. Food chemistry. 2012; 135(3):1353-1359.
10. Liu Y and Liu G. Isorhapontigenin and resveratrol suppress oxLDL-induced proliferation and activation of ERK1/2 mitogen-activated protein kinases of bovine aortic smooth muscle cells. Biochemical pharmacology. 2004; 67(4):777-785.
11. Li HL, Wang AB, Huang Y, Liu DP, Wei C, Williams GM, Zhang CN, Liu G, Liu YQ, Hao DL, Hui RT, Lin M and Liang CC. Isorhapontigenin, a new resveratrol analog, attenuates cardiac hypertrophy via blocking signaling transduction pathways. Free radical biology & medicine. 2005; 38(2):243-257.
12. Fang Y, Yu Y, Hou Q, Zheng X, Zhang M, Zhang D, Li J, Wu XR and Huang C. The Chinese herb isolate isorhapontigenin induces apoptosis in human cancer cells by down-regulating overexpression of antiapoptotic protein XIAP. The Journal of biological chemistry. 2012; 287(42):35234-35243.
13. Fang Y, Cao Z, Hou Q, Ma C, Yao C, Li J, Wu XR and Huang C. Cyclin D1 Downregulation Contributes to Anti-Cancer Effect of Isorhapontigenin (ISO) on Human Bladder Cancer Cells. Mol Cancer Ther. 2013.
14. Baldin V, Lukas J, Marcote MJ, Pagano M and Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes & development. 1993; 7(5):812-821.
15. Diehl JA, Cheng M, Roussel MF and Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes & development. 1998; 12(22):3499-3511.
16. Li J, Chen H, Tang MS, Shi X, Amin S, Desai D, Costa M and Huang C. PI-3K and Akt are mediators of AP-1 induction by 5-MCDE in mouse epidermal Cl41 cells. J Cell Biol. 2004; 165(1):77-86.
17. Leppa S, Saffrich R, Ansorge W and Bohmann D. Differential regulation of c-Jun by ERK and JNK during PC12 cell differentiation. The EMBO journal. 1998; 17(15):4404-4413.
18. Brandt B, Abou-Eladab EF, Tiedge M and Walzel H. Role of the JNK/c-Jun/AP-1 signaling pathway in galectin-1-induced T-cell death. Cell death & disease. 2010; 1:e23.
19. Leppa S and Bohmann D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene. 1999; 18(45):6158-6162.
20. Eriksson M, Taskinen M and Leppa S. Mitogen activated protein kinase-dependent activation of c-Jun and c-Fos is required for neuronal differentiation but not for growth and stress response in PC12 cells. Journal of cellular physiology. 2007; 210(2):538-548.
21. Song L, Li J, Zhang D, Liu ZG, Ye J, Zhan Q, Shen HM, Whiteman M and Huang C. IKKbeta programs to turn on the GADD45alpha-MKK4-JNK apoptotic cascade specifically via p50 NF-kappaB in arsenite response. The Journal of cell biology. 2006; 175(4):607-617.
22. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000; 103(2):239-252.
23. Weston CR and Davis RJ. The JNK signal transduction pathway. Current opinion in genetics & development. 2002; 12(1):14-21.
24. Sun H, Charles CH, Lau LF and Tonks NK. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell. 1993; 75(3):487-493.
25. Keyse SM. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Current opinion in cell biology. 2000; 12(2):186-192.
26. Abdelmohsen K and Gorospe M. Posttranscriptional regulation of cancer traits by HuR. Wiley interdisciplinary reviews RNA. 2010; 1(2):214-229.
27. Audic Y and Hartley RS. Post-transcriptional regulation in cancer. Biology of the cell / under the auspices of the European Cell Biology Organization. 2004; 96(7):479-498.
28. Ginisty H, Sicard H, Roger B and Bouvet P. Structure and functions of nucleolin. Journal of cell science. 1999; 112 ( Pt 6):761-772.
29. Knudsen KE, Diehl JA, Haiman CA and Knudsen ES. Cyclin D1: polymorphism, aberrant splicing and cancer risk. Oncogene. 2006; 25(11):1620-1628.
30. Buckley MF, Sweeney KJ, Hamilton JA, Sini RL, Manning DL, Nicholson RI, deFazio A, Watts CK, Musgrove EA and Sutherland RL. Expression and amplification of cyclin genes in human breast cancer. Oncogene. 1993; 8(8):2127-2133.
31. Liang SB, Furihata M, Takeuchi T, Iwata J, Chen BK, Sonobe H and Ohtsuki Y. Overexpression of cyclin D1 in nonmelanocytic skin cancer. Virchows Archiv : an international journal of pathology. 2000; 436(4):370-376.
32. Bianchi AB, Fischer SM, Robles AI, Rinchik EM and Conti CJ. Overexpression of cyclin D1 in mouse skin carcinogenesis. Oncogene. 1993; 8(5):1127-1133.
33. Yamamoto H, Ochiya T, Takeshita F, Toriyama-Baba H, Hirai K, Sasaki H, Sakamoto H, Yoshida T, Saito I and Terada M. Enhanced skin carcinogenesis in cyclin D1-conditional transgenic mice: cyclin D1 alters keratinocyte response to calcium-induced terminal differentiation. Cancer research. 2002; 62(6):1641-1647.
34. Ding J, Ning B, Huang Y, Zhang D, Li J, Chen CY and Huang C. PI3K/Akt/JNK/c-Jun signaling pathway is a mediator for arsenite-induced cyclin D1 expression and cell growth in human bronchial epithelial cells. Curr Cancer Drug Targets. 2009; 9(4):500-509.
35. Ouyang W, Luo W, Zhang D, Jian J, Ma Q, Li J, Shi X, Chen J, Gao J and Huang C. PI-3K/Akt pathway-dependent cyclin D1 expression is responsible for arsenite-induced human keratinocyte transformation. Environmental health perspectives. 2008; 116(1):1-6.
36. Musgrove EA, Caldon CE, Barraclough J, Stone A and Sutherland RL. Cyclin D as a therapeutic target in cancer. Nature reviews Cancer. 2011; 11(8):558-572.
37. Zhang J, Ouyang W, Li J, Zhang D, Yu Y, Wang Y, Li X and Huang C. Suberoylanilide hydroxamic acid (SAHA) inhibits EGF-induced cell transformation via reduction of cyclin D1 mRNA stability. Toxicology and applied pharmacology. 2012; 263(2):218-224.
38. Zhang JY, Adams AE, Ridky TW, Tao S and Khavari PA. Tumor necrosis factor receptor 1/c-Jun-NH2-kinase signaling promotes human neoplasia. Cancer research. 2007; 67(8):3827-3834.
39. Tournier C, Dong C, Turner TK, Jones SN, Flavell RA and Davis RJ. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes & development. 2001; 15(11):1419-1426.
40. Cooper SJ, MacGowan J, Ranger-Moore J, Young MR, Colburn NH and Bowden GT. Expression of dominant negative c-jun inhibits ultraviolet B-induced squamous cell carcinoma number and size in an SKH-1 hairless mouse model. Molecular cancer research : MCR. 2003; 1(11):848-854.
41. Wang XJ, Greenhalgh DA, Donehower LA and Roop DR. Cooperation between Ha-ras and fos or transforming growth factor alpha overcomes a paradoxic tumor-inhibitory effect of p53 loss in transgenic mouse epidermis. Molecular carcinogenesis. 2000; 29(2):67-75.
42. Zhang JY, Green CL, Tao S and Khavari PA. NF-kappaB RelA opposes epidermal proliferation driven by TNFR1 and JNK. Genes & development. 2004; 18(1):17-22.
43. Zhang JY, Tao S, Kimmel R and Khavari PA. CDK4 regulation by TNFR1 and JNK is required for NF-kappaB-mediated epidermal growth control. The Journal of cell biology. 2005; 168(4):561-566.
44. Kristiansen M, Hughes R, Patel P, Jacques TS, Clark AR and Ham J. Mkp1 is a c-Jun target gene that antagonizes JNK-dependent apoptosis in sympathetic neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010; 30(32):10820-10832.
45. Zhang J, Gao G, Chen L, Deng X, Li J, Yu Y, Zhang D, Li F, Zhang M, Zhao Q and Huang C. Cheliensisin A inhibits EGF-induced cell transformation with stabilization of p53 protein via a hydrogen peroxide/Chk1-dependent axis. Cancer Prev Res (Phila). 2013; 6(9):949-958.
46. Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A and Pestell RG. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. The Journal of biological chemistry. 1995; 270(40):23589-23597.
47. Cao Z, Zhang R, Li J, Huang H, Zhang D, Zhang J, Gao J, Chen J and Huang C. XIAP regulation of cyclin D1 expression and cancer cell anchorage-independent growth via Its E3 ligase-mediated protein phosphatase 2A/C-Jun axis. J Biol Chem. 2013.
48. Li J, Gorospe M, Hutter D, Barnes J, Keyse SM and Liu Y. Transcriptional induction of MKP-1 in response to stress is associated with histone H3 phosphorylation-acetylation. Molecular and cellular biology. 2001; 21(23):8213-8224.
49. Ouyang W, Li J, Ma Q and Huang C. Essential roles of PI-3K/Akt/IKKbeta/NFkappaB pathway in cyclin D1 induction by arsenite in JB6 Cl41 cells. Carcinogenesis. 2006; 27(4):864-873.
50. Kwak YT, Li R, Becerra CR, Tripathy D, Frenkel EP and Verma UN. IkappaB kinase alpha regulates subcellular distribution and turnover of cyclin D1 by phosphorylation. The Journal of biological chemistry. 2005; 280(40):33945-33952.
51. Zhang R, Wang Y, Li J, Jin H, Song S and Huang C. The Chinese herb isolate YHL-14 induces G2/M arrest in human cancer cells by up-regulating p21 expression through P53-independent cascade. J Biol Chem. 2014.
52. Huang H, Ma L, Li J, Yu Y, Zhang D, Wei J, Jin H, Xu D, Gao J and Huang C. NF-κB1 inhibits c-Myc protein degradation through suppression of FBW7 expression. oncotarget. 2014; 5(2):493-505.
53. Cao Z, Zhang R, Li J, Huang H, Zhang D, Zhang J, Gao J, Chen J and Huang C. X-linked inhibitor of apoptosis protein (XIAP) regulation of cyclin D1 protein expression and cancer cell anchorage-independent growth via its E3 ligase-mediated protein phosphatase 2A/c-Jun axis. J Biol Chem. 2013; 288(28):20238-20247.
54. Zhang D, Li J, Costa M, Gao J and Huang C. JNK1 mediates degradation HIF-1alpha by a VHL-independent mechanism that involves the chaperones Hsp90/Hsp70. Cancer research. 2010; 70(2):813-823.
55. Luo W, Liu J, Li J, Zhang D, Liu M, Addo JK, Patil S, Zhang L, Yu J, Buolamwini JK, Chen J and Huang C. Anti-cancer effects of JKA97 are associated with its induction of cell apoptosis via a Bax-dependent and p53-independent pathway. The Journal of biological chemistry. 2008; 283(13):8624-8633.
56. Fang Y, Yu Y, Hou Q, Zheng X, Zhang M, Zhang D, Li J, Wu XR and Huang C. The Chinese herb isolate isorhapontigenin induces apoptosis in human cancer cells by down-regulating overexpression of antiapoptotic protein XIAP. J Biol Chem. 2012; 287(42):35234-35243.
57. Zhang J, Ouyang W, Li J, Zhang D, Yu Y, Wang Y, Li X and Huang C. Suberoylanilide hydroxamic acid (SAHA) inhibits EGF-induced cell transformation via reduction of cyclin D1 mRNA stability. Toxicol Appl Pharmacol. 2012; 263(2):218-224.
58. Zhang D, Li J, Zhang M, Gao G, Zuo Z, Yu Y, Zhu L, Gao J and Huang C. The requirement of c-Jun N-terminal kinase 2 in regulation of hypoxia-inducing factor-1alpha mRNA stability. J Biol Chem. 2012; 287(41):34361-34371.
59. Yu J, Zhang D, Liu J, Li J, Yu Y, Wu XR and Huang C. RhoGDI SUMOylation at Lys-138 increases its binding activity to Rho GTPase and its inhibiting cancer cell motility. J Biol Chem. 2012; 287(17):13752-13760.
60. Zhang J, Gao G, Chen L, Li J, Deng X, Zhao Q and Huang C. Hydrogen Peroxide/ATR-Chk2 Activation Mediates p53 Protein Stabilization and Anti-Cancer Activity of Cheliensisin A in Human Cancer Cells. Oncotarget. 2014.
61. Liu J, Zhang D, Mi X, Xia Q, Yu Y, Zuo Z, Guo W, Zhao X, Cao J, Yang Q, Zhu A, Yang W, Shi X, Li J and Huang C. p27 suppresses arsenite-induced Hsp27/Hsp70 expression through inhibiting JNK2/c-Jun- and HSF-1-dependent pathways. The Journal of biological chemistry. 2010; 285(34):26058-26065.
62. Cai T, Li X, Ding J, Luo W, Li J and Huang C. A cross-talk between NFAT and NF-kappaB pathways is crucial for nickel-induced COX-2 expression in Beas-2B cells. Current cancer drug targets. 2011; 11(5):548-559.