
INTRODUCTION
The non-spore-forming, anaerobic Gram-negative bacteria, Fusobacterium is part of the normal flora in the human mouth and gut mucosa. Although Fusobacterium species are part of the gut microbiome in human, their invasive [1,2], adherent [3,4], and pro-inflammatory [5–7] features have been noted. Fusobacterium have been associated with inflammatory disorders such as periodontitis [8], cerebral abscesses [9], acute appendicitis [10] and inflammatory bowel diseases [1,11,12]. Moreover, emerging evidence suggest a possible link between Fusobacterium infection and colorectal carcinogenesis through altering the host immune responses [13–16]. Enrichment of Fusobacterium have been especially associated with colorectal cancers and adenomas with methylation phenotypes, suggesting the potential role of this bacteria in DNA methylation related colorectal tumorigenesis [17, 18].
Since Fusobacterium has a reported association with inflammatory bowel diseases (IBD), including both ulcerative colitis (UC) and Crohn’s diseases [1, 11, 12], and IBD is one of the highest risk factors for colorectal cancer [19]. In particular, UC associated colorectal cancers are characterized as high degree of methylation accumulation through continuous colonic inflammation [20, 21]. The aim of this study was to investigate a potential link between Fusobacterium enrichment and DNA methylation accumulation in the inflammatory colonic mucosa in UC.
RESULTS
Detection of fusobacterium in the inflammatory colonic mucosa in UC patients and its association with methylation status of candidate genes
Among 86 inflammatory colonic mucosa from UC patients, pan-fusobacterium was heavily enriched in ten (11.6%) cases using the same cut off value of our recent study [22]. Supplementary Figure 1 illustrates the results of an unsupervised clustering analysis based on the methylation status of a panel of 24 candidate genes. This analysis showed that samples with Fusobacterium high (FB-high) distributed as moderately methylated samples but were not clustered clearly each other. Since the analyzed genes can be divided into type A (for age-related: N33, MYOD1, ER1, HPP1, and SFRP1), type C (for cancer-specific: MINT1, 2, 12, and 31, RASSF1A, P16, NEOUROG1, TERT, and MGMT) and other colorectal cancer related (GARA2, IGF2, DPYS, NKX2-5, DOK5, RARB2, SLC16A12, CDH13 and SPOCK2) genes [23–26]. We then divided analyzed genes into these groups in relation to the Fusobacterium status. This analysis demonstrated that the mean methylation Z score of type C genes was significantly higher among FB-high group compared to Fusobacterium low and negative (FB-low/neg) group (P<0.01). On the other hand, no significant associations between mean methylation Z score of all, type A and other genes and Fusobacterium status were observed (Figure 1). Analysis of individual panel showed significantly higher methylation of known type C genes in colorectal cancer such as (MINT2 and 31, P16 and NEUROG1) in FB high samples [23, 24], while subset of type A and other colorectal cancer related genes (SFRP1, DOK5, GARA2) [24, 25, 26] also presented significantly higher methylation in the FB-high samples (Figure 2). On the other hand, we did not find genes hypermethylated in FB-low/neg samples among these candidate panels (data not shown). We also evaluated the methylation status of the LINE1 repetitive element, which is an indicator for genome wide hypomethylation [27]. However, we did not observe significant association between LINE1 methylation status and Fusobacterium status (Supplementary Figure 2).
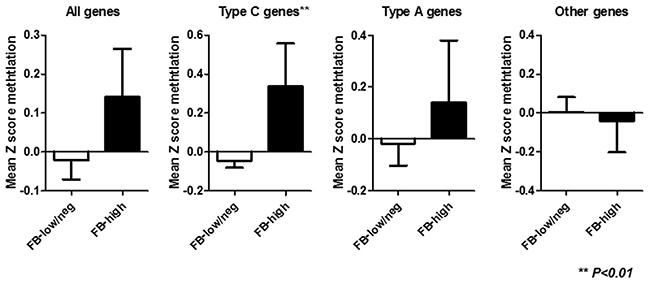
Figure 1: Mean Z score methylation of all, type C, type A and other genes in relation to the Fusobacterium status. FB-low/neg, Fusobacterium low and negative samples; FB-high, Fusobacterium high samples; Statistical analysis was performed using Student’s t-test.
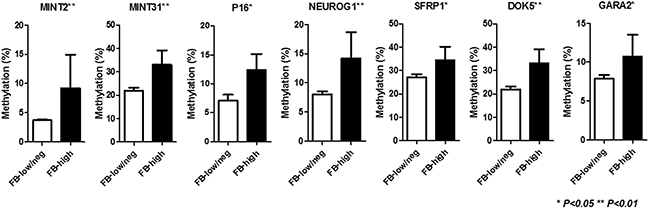
Figure 2:Methylation status of specific genes in relation to the Fusobacterium status. FB-low/neg, Fusobacterium low and negative samples; FB-high, Fusobacterium high samples; The statistical analysis was performed using Student’s t-test.
Since methylation status of colonic mucosa in UC patients would be influenced by the clinicopathological factors [28]. We then performed multivariate analysis assessing the factors related to the hypermethylation of type C genes in the colonic mucosa in UC patients. Age, gender, duration, extension of disease, clinical course, number of hospitalization, presence of refractory or steroid dependency were included for this analysis with the Fusobacterium status. The mean Z score methylation of type C genes in the colonic mucosa in UC patients presented an approximately Gaussian distribution, with over representation of hypermethylated cases, we set cut-off value of 0.18 (mean Z score methylation) for the definition samples with hypermethylated (methylation high) cases. The result demonstrated that FB-high status held an increased likelihood for methylation high as an independent factor (odds ratio: 16.18, 95% confidence interval: 1.94-135.2, P=0.01), while other clinicopethological factors were not significantly associated with methylation high cases (Table 1).
Table 1: Multivariate analysis assessing the factors related to the methylation-high* in type C genes
Variables |
Odds ratio (95% confidence interval) |
P value |
|---|---|---|
Age (40y =<) |
1.68 (0.39-7.17) |
0.49 |
Gender (female) |
5.39 (0.94-30.91) |
0.06 |
Duration (10y=<) |
0.58 (0.10-3.23) |
0.53 |
Extension (total colitis) |
1.49 (0.31-7.18) |
0.62 |
Clinical course (flare-up) |
0.42 (0.04-4.36) |
0.47 |
Number of hospitalization (2=<) |
0.91 (0.16-5.14) |
0.91 |
Refractory |
1.35 (0.17-10.59) |
0.78 |
Steroid dependency |
1.84 (0.16-21.05) |
0.62 |
FB-high |
16.18 (1.94-135.2) |
0.01 |
*Methylation-high, mean Z score of methylation>0.18;
The genome-wide methylation status of CpG islands distinguishes FB-high cases
To determine the methylation changes occurred in the colonic inflammatory mucosa of the UC patients with FB-high cases, we used an Infinium HumanMethylation450 BeadChip array, which allowed us to query methylation status of >450,000 CpG sites within the genome. The Infinium HumanMethylation450 BeadChip array data were available for ten patients [28], all these were considered to be FB-low/neg cases. The methylation levels of FB low/neg cases were considerably different among the samples (Supplementary Figure 1). Among the ten samples of these the Infinium HumanMethylation450 BeadChip array data were available, 6 and 4 belonged to hypermethylated (methylation-high) and hypomethylated cluster (methylation-low) based on the unsupervised clustering analysis of 24 candidate panels (Suppelementary Figure 1). We then included additional four genomic samples from FB-high cases for the analysis. Based on GRCh37/hg19, we first checked the methylation status of 473,864 CpG sites and divided the sites into CpG islands (CGI: n=145,842) and non-CpG islands (NCGI: n=328,022). We found that accelerated methylation among the FB-low/neg methylation-high samples compared to the FB-low/neg methylation-low samples especially at the CGI sites rather than the NCGI sites. Similar result was also observed for the comparison of FB-high samples and FB-low/neg methylation-low samples (Figure 3). When a gain in methylation was defined as a methylation level ≥20% (β-value≥0.2), the numbers of methylated sites in the CGI was significantly greater in both FB-low/neg methylation-high and FB-high samples compared to the FB-low/neg methylation-low samples (Both P<0.0001), while the numbers of methylated sites was not significantly different among the FB-low/neg methylation-high and FB-high samples (P>0.1) (Figure 3). On the other hand, we did not observe any significant association between the numbers of methylated sites among three groups in the NCGI (all P>0.1) (Figure 3).
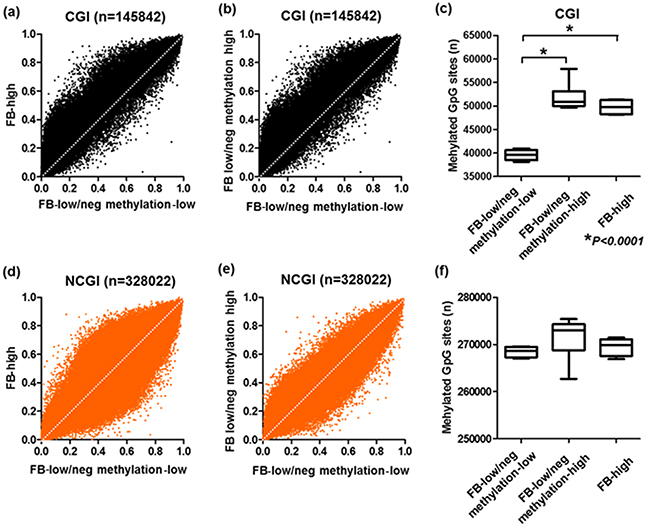
Figure 3: Genome scale analysis using HumanMethylation450 BeadChip array. Comparison of mean methylationβ-value of Fusobacterium high (FB-high) samples, Fusobacterium low and negative (FB-low/neg) with methylation low samples and Fusobacterium low and negative (FB-low/neg) with methylation-high samples (A)-(D). Mean number of methylated sites among FB-high, FB-low/neg methylation-low and FB-low/neg methylation-high samples (E) (F). All sites were divided into CpG islands (CGI, upper) and non-CpG islands (NCGI, lower). The statistical analysis was performed Student’s t-Test.
We next aimed to investigate whether the methylated sites in FB-high group would be exclusive in this group compared to the FB-low/neg groups. Unsupervised clustering analysis using the 10% of the probes with the greatest variation very clearly distinguished the samples from the FB-high cases in both the CGI and NCGI. FB-high cases were clustered together as the moderately methylated samples in the CGI and also clustered together as the hypermethylated samples in the NCGI (Figure 4). The tight cluster of the FB-high cases as the moderately methylated samples was also confirmed when dividing the CGI into the promoter CGI (PCGI: n=1684) and outside the promoter CGI (NPCGI: n=15611) (Supplementary Figure 3). On the other hand, dividing NCGI into the promoter NCGI (PNCGI: n=3231) and outside the promoter NCGI (NPNCGI: n=42270) showed that FB high cases were clustered as hypomethylated samples in the PNCGI, while the same samples were clustered as hypermethylated samples in the NPNCGI (Supplementary Figure 3).
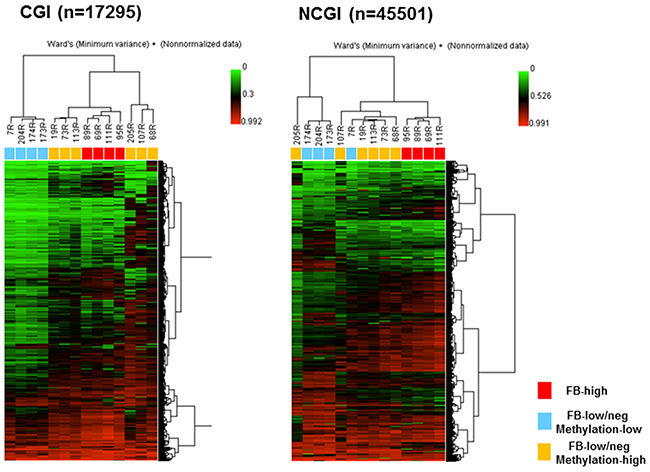
Figure 4:Unsupervised hierarchical clustering analysis of CpG islands (CGI, left) and non-CpG islands (NCGI, right) using 10% most variant probes among fourteen UC patients. Red boxes, Fusobacterium high (FB-high) samples; Blue boxes, Fusobacterium low and negative (FB-low/neg) with methylation low samples; Yellow boxes, Fusobacterium low and negative (FB-low/neg) with methylation high samples; Samples ID number were listed above the boxes.
Identification of the hypermethylated promoter CpG islands in the severe phenotypes
We next explored genes that were exclusively hypermethylated in FB-high cases. For this analysis, we focused on the PCGIs because of their influence on gene expression. When a gain in methylation was defined as a methylation level ≥20% (β-value≥0.2), we identified 344 genes that were exclusively hypermethylated in the FB-high group. Gene ontology analysis using DAVID revealed that these genes are significantly codified genes related to the catalytic activity (P=0.039: Figure 5 and Supplementary Table 2 and 3). Gene ontology analysis using methylated genes in the common elements in all three groups (FB-high, FB-low/neg methylation-low and high groups) showed that these genes are significantly codified genes related to the binding (P=0.016: Figure 5 and Supplementary Tables 2 and 3). On the other hand, methylated genes among other groups or elements did not show any significant functional enrichment using the same analysis.
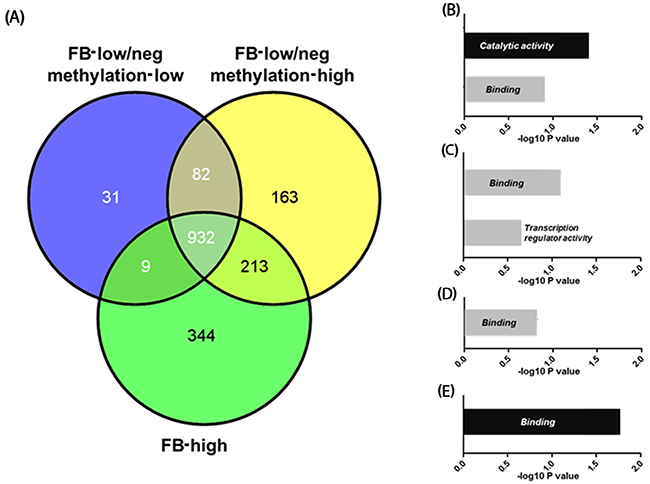
Figure 5:Venn-diagram representing the number of genes methylated in Fusobacterium high (FB-high), Fusobacterium low and negative (FB-low/neg) with methylation low and Fusobacterium low and negative (FB-low/neg) with methylation high samples (A). Results of the gene ontology analysis of the genes methylated exclusively in the specific groups or common elements (B)-(E). Genes methylated exclusively in FB-high groups (B), common elements in FB-low/neg methylation high and FB-high groups (C), common elements in FB-low/neg methylation low and FB-low/neg methylation high groups (D), and common elements in all three groups (E). Categories with significant enrichment by the Benjamini method are shown by black boxes, while no significant enrichment are shown by grey boxes.
DISCUSSION
An unsupervised clustering analysis based on the methylation status of a panel of 24 candidate genes showed that FB-high cases were distributed as moderately methylated samples but were not clustered clearly. However, the FB-high cases presented hypermethylation especially in the type C genes, including MINT2 and 31, P16 and NEUROG1, these methylation have been reported in the colorectal cancers with methylation phenotype [23, 24]. Multivariate analysis with adjustment of clinicopathological factors demonstrated that FB-high held an increased likelihood for hypermethylation of the type C genes.
Enrichment of Fusobacterium has been reported in the colorectal cancer and adenoma tissues especially in cases with methylation phenotype [17, 18]. The methylation phenotype of colorectal cancers are characterized as accumulation of cancer specific methylation of type C genes [23, 24]. The current potential association between Fusobacterium enrichment and methylation of type C genes in inflamed mucosa of UC might provide evidence, supporting the potential link between Fusobacterium and methylation related carcinogenesis in UC. Fusobacterium have been associated with several inflammatory diseases such as such as periodontitis [8], cerebral abscesses [9], acute appendicitis [10] and inflammatory bowel diseases [1, 11, 12]. It is interesting to note that the colorectal cancer with methylation phenotype that is associated with Fusobacterium have a distinct immune response with abundant tumor infiltrating lymphocytes. This inflammatory reaction has been thought to be a host immune response to the tumor cells [29, 30]. It has been also shown that the infection of Fusobacterium accelerates the pro-inflammatory cytokine expressions in adenoma tissues in the mice model [16]. Since chronic inflammation induced by pro-inflammatory cytokines have roles in the methylation induction [21]. It is reasonable to expect that Fusobacterium alters inflammatory immune response and influence the methylation status in UC patients.
On the other hand, UC associated colorectal cancer and its background mucosa have been characterized as hypermethylation in type A genes [20]. It has also been reported that methylation phenotypes are rather rare in UC associated colorectal cancers [31–32]. Whether hypermethylation of type C genes in the inflamed mucosa in UC linked to the future risk of colorectal cancer is currently unknown. The role of Fusobacterium enrichment in UC associated colorectal cancer is not clearly demonstrated, while our previous study demonstrated that Japanese UC patients with Fusobacterium enrichment was associated with chronic continuous inflammation in the colonic mucosa [22], which may link to accelerated DNA methylation and colorectal cancer. Potential role hypermethylation of type C genes in UC with FB high cases need to be further evaluated in relation to their biological and clinical significances.
A unique methylome signature of UC with FB-high cases were also highlighted through the genome-wide methylation analysis. Compared to the FB-low/neg methylation-low cases, methylation accumulation shown in FB-high cases seemed to be more striking at CGI, rather than the NCGI, which appeared to be similar to that of FB-low/neg methylation high samples. On the other hand, FB-high cases were very tightly clustered as unique samples including PCGI, NPCGI, PNCGI and NPCGI by the clustering analysis of the 10% of the probes with the greatest variation. We also showed that group of PCGIs that were exclusively hypermethylated in FB-high cases significantly codified the genes related to the catalytic activity. The enrichment of genes related to the catalytic activity was not observed in other groups of samples nor their common elements, suggesting that Fusobacterium accelerates DNA methylation in specific groups of genes. Although the association between genes related to the catalytic activity and UC associated carcinogenesis remain unknown, methylated genes in FB-high cases included several cancer related genes, for example, DAG1 and RBM7. Deletion of DAG1 is associated to the worse outcome of breast cancer patients [33]. Lacking of RBM7 has been also reported to be associated with DNA damage hypersensitivity that can linked to cancer predisposition [34]. Enrichment of Fusobacterium has been associated with colorectal and pancreatic cancers [13, 14, 35]. The discovery of the specific methylation induction in Fusobacterium emphasizes the importance of an improved understanding of pathway-specific molecular changes in UC associated carcinogenesis and raises the possibility that specific epigenetic therapies that target alterations in proteins related to catalytic activity could be useful in the treatment and chemoprevention of UC associated colorectal cancer. In the same time, our findings also deserve to be tested in animal models, where one could specifically explore the possibility of therapeutic intervention modulating DNA methylation with antibiotics, anti-inflammatory or demethylation agents in the prevention or treatment of UC associated colorectal cancer.
MATERIALS AND METHODS
Tissue samples
We used genomic DNA samples of 86 UC patients who underwent colonoscopy at the Fujita Health University Hospital (Toyoake, Japan). All of the samples were extracted from fresh frozen endoscopic biopsies taken from inflamed mucosae of the rectum. These patients included 48 males and 38 females. The median age and clinical duration were 35 and 4.5 years, respectively. Regarding their clinical course, 9 patients presented only one attack and the remaining cases showed at least one time flare-up of disease. Five and two cases eventually underwent surgery due to toxic megacolon and UC-associated colorectal cancer, respectively. All cases were clinically in remission at the time of endoscopy. The histopathological examinations revealed mild or moderate inflammation but no evidence of dysplasia or neoplasia at any of the sites from which the biopsies were taken. Based on the appearances during endoscopy, 19 patients exhibited inflammatory mucosae only in the rectum, and 25 patients exhibited extensions of the inflammatory mucosae into the left side of the colon (sigmoid and descending colons). The remaining 41 patients exhibited extensions of the inflammatory mucosae into proximal sites (the transverse and ascending colons and the cecum). This cohort was recruited from our previous studies that investigated the associations of DNA methylation with clinical phenotypes, host genetic factors and telomere lengths [28, 36–38]. This study was approved by the Human Research Ethics Committee of the Fujita Health University School of Medicine. Each participant provided written informed consent for the use of his or her clinical and laboratory data for publication and research purposes. The study was conducted according to the principles outlined in the Declaration of Helsinki.
Quantitative PCR analysis for fusobacterium
Quantitative real-time PCR analysis for Fusobacterium was performed using the Universal PCR Master Mix (Bio-Rad) and StepOnePlus™ Real-Time PCR System (Applied Biosystems). The pan-fusobacterium TaqMan primer/probe set used in this study were described previously [13, 39]. The cycle threshold (Ct) values for pan-fusobacterium were normalized to the amount of human DNA in each reaction by using a primer/probe set for the reference gene, prostaglandin transporter (PGT), as described previously [40]. All assays were done in duplicate and we averaged the results. We have reported that subset of UC cases show heavy enrichment of Fusobacterium in the inflamed colonic mucosa [22]. In this cohort, we identified ten cases (11.6%) of UC with enrichment of Fusobacterium using the same cut off value [22]. We then defined these cases as Fusobacterium high (FB-high) cases. Since the amount of Fusobacterium in detectable cases except the FB-high cases was much lower than that of FB-high cases and had no relevance to the clinic-pathological features of patients [22], we then attached these cases with Fusobacterium undetectable cases and defined as Fusobacterium low and negative (FB-low/neg) cases.
CpG methylation analysis of candidate panels for colorectal cancer
For this cohort, we have characterized the methylation status of candidate 45 CpG islands, in relation to their clinicopethological features [28]. Among the 45 genes, we selected a panel of 24 genes that were reported to be associated with colorectal carcinogenesis [23–26]. In colorectal cancer, there appears to be two types of methylation that are associated with cancer progression: type A (for age-related) methylation, and type C (for cancer-specific) methylation [41]. Based on this, we selected 9 and 5 genes that are reported to be associated with type A and type C methylation, respectively [23–26]. The type A genes included N33, MYOD1, ER1, HPP1, and SFRP1 [23, 24]. The type C genes included MINT1, 2, 12, and 31, RASSF1A, P16, NEOUROG1, TERT, and MGMT [23, 24]. We also included other 9 genes (GARA2, IGF2, DPYS, NKX2-5, DOK5, RARB2, SLC16A12, CDH13 and SPOCK2) that have been associated with colorectal tumorigenesis in human or mice [25, 26]. We also evaluated the methylation status of the LINE1 repetitive element. All the methylation analysis was performed by the bisulfite pyrosequencing. The bisulfite treatment of the DNA was performed with an EpiTect bisulfite kit (Qiagen, Tokyo, Japan) according to the manufacturer’s protocol. Pyrosequencing was performed using a PSQ24 system with a Pyro-Gold reagent kit (QIAGEN, Tokyo, Japan), and the results were analyzed using PyroMark Q24 software (QIAGEN). List of the genes and the primers used for pyrosequencing are listed in Supplementary Table 1.
Genome-wide methylation analysis
We performed array-based DNA methylation analyses using the Infinium HumanMethylation450 BeadChip array, which allowed us to query the methylation status of >450,000 CpG sites within the genome and to cover 99% of the RefSeq genes. Genomic DNA samples from the inflamed rectal mucosae of fourteen UC patients were used for this analysis. Among them, the data were available for ten patients [28]. All these ten cases were considered to be Fusobacterium low and negative (FB-low/neg) case. We then used additional four genomic samples from Fusobacterium high (FB-high) cases for the Infinium HumanMethylation450 BeadChip array experiment. Bisulfite modification of the genomic DNA was performed using an EZ DNA Methylation Kit (Zymo Research). The bisulfite conversion efficiency was determined based on sample-dependent controls on the chip and was displayed in the quality control panel in the software. All samples passed the quality control measurements. The samples were run on an Infinium HumanMethylation450K BeadChip (Illumina) and scanned on an Illumina iScan instrument according to the manufacturer’s instructions. The methylation values for the individual CpG sites in each sample were obtained as β-values. The β-value generated for each CpG locus reflected a measure of the intensities of the methylated (β = 1) and unmethylated probes (β = 0). The β-values are continuous variable that are calculated by dividing the intensity of the methylated beads by the combined intensity, and the resultant values range from 0 to 1. The genomic regions were defined according to National Center for Biotechnology Information coordinates, which were downloaded from the University of California, Santa Cruz website in February 2009 (GRCh37/hg19). We removed probes that were targeted for an annotated SNP (dbSNP137) and for either the X or Y chromosome. Information about the CpG islands and promoters (surrounding gene transcription start sites) was also obtained based on the GRCh37/hg19.
Clustering analysis
Unsupervised clustering analysis using ArrayTrack™ (http://www.fda.gov/ScienceResearch/BioinformaticsTools/Arraytrack/) was performed to identify distinct subgroups based on the methylation status.
Gene ontology analysis
Functional enrichment of the methylated genes was determined by gene ontology analysis using DAVID Bioinformatics Resources 6.7 (http://david.abcc.ncifcrf.gov/). P-values were corrected for multiple hypotheses testing using the Benjamini method.
Statistical analysis
The continuous variables were compared between two and more groups using Student’s t-test. P values <0.05 were considered statistically significant.
Abbreviations
UC, Ulcerative colitis; IBD, inflammatory bowel diseases; FB-high, Fusobacterium high; FB-low/neg, Fusobacterium low and negative;
Author contributions
TT designed the study, performed experiment, analyzed the data and wrote the manuscript. ST analyzed the data and performed pathological assessment. TT, MU, MK performed pathological assessment. NN, NH, TK, MO, HY, DY, TO, KM, NK, HI, YJ, TK, MN and YN collected the samples and assessed clinical information.
IH, TT, MU, TS, MK, and NO designed and supervised the study.
CONFLICTS OF INTEREST
We declare that we have no conflicts of interest.
FUNDING
This study was funded by the Uehara Memorial Foundation.
REFERENCES
1. Strauss J, Kaplan GG, Beck PL, Rioux K, Panaccione R, Devinney R, Lynch T, Allen-Vercoe E. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm Bowel Dis. 2011;17:1971–1978.
2. Han YW1 Shi W, Huang GT, Kinder Haake S, Park NH, Kuramitsu H, Genco RJ. Interactions between periodontal bacteria and human oral epithelial cells: Fusobacterium nucleatum adheres to and invades epithelial cells. Infect Immun. 2000;68:3140–3146.
3. Bachrach G, Ianculovici C, Naor R, Weiss EI. Fluorescence based measurements of Fusobacterium nucleatum coaggregation and of fusobacterial attachment to mammalian cells. FEMS Microbiol Lett. 2005;248:235–240.
4. Uitto VJ, Baillie D, Wu Q, Gendron R, Grenier D, Putnins EE, Kanervo A, Firth JD. Fusobacterium nucleatum increases collagenase 3 production and migration of epithelial cells. Infect Immun. 2005;73:1171–1179.
5. Krisanaprakornkit S, Kimball JR, Weinberg A, Darveau RP, Bainbridge BW, Dale BA. Inducible expression of human beta-defensin 2 by Fusobacterium nucleatum in oral epithelial cells: multiple signaling pathways and role of commensal bacteria in innate immunity and the epithelial barrier. Infect Immun. 2000;68:2907–2915.
6. Peyret-Lacombe A, Brunel G, Watts M, Charveron M, Duplan H. TLR2 sensing of F. nucleatum and S. sanguinis distinctly triggered gingival innate response. Cytokine. 2009;46:201–210.
7. Moore RA, Warren RL, Freeman JD, Gustavsen JA, Chénard C, Friedman JM, Suttle CA, Zhao Y, Holt RA. The sensitivity of massively parallel sequencing for detecting candidate infectious agents associated with human tissue. PLoS One. 2011;6:e19838.
8. Signat B, Roques C, Poulet P, Duffaut D. Fusobacterium nucleatum in periodontal health and disease. Curr Issues Mol Biol. 2011;13:25–36.
9. Kai A, Cooke F, Antoun N, Siddharthan C, Sule O. A rare presentation of ventriculitis and brain abscess caused by Fusobacterium nucleatum. J Med Microbiol. 2008;57:668–671.
10. Swidsinski A, Dörffel Y, Loening-Baucke V, Theissig F, Rückert JC, Ismail M, Rau WA, Gaschler D, Weizenegger M, Kühn S, Schilling J, Dörffel WV. Acute appendicitis is characterised by local invasion with Fusobacterium nucleatum/necrophorum. Gut. 2011;60:34–40.
11. Neut C, Bulois P, Desreumaux P, Membré JM, Lederman E, Gambiez L, Cortot A, Quandalle P, van Kruiningen H, Colombel JF. Changes in the bacterial flora of the neoterminal ileum after ileocolonic resection for Crohn’s disease. Am J Gastroenterol. 2002;97:939–946.
12. Ohkusa T, Sato N, Ogihara T, Morita K, Ogawa M, Okayasu I. Fusobacterium varium localized in the colonic mucosa of patients with ulcerative colitis stimulates species-specific antibody. J Gastroenterol Hepatol. 2002;17:849–853.
13. Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, Holt RA. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306.
14. Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, Baselga J, Liu C, Shivdasani RA, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292–298.
15. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14:195–206.
16. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, El-Omar EM, Brenner D, Fuchs CS, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207–215.
17. Tahara T, Yamamoto E, Suzuki H, Maruyama R, Chung W, Garriga J, Jelinek J, Yamano HO, Sugai T, An B, Shureiqi I, Toyota M, Kondo Y, et al. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. 2014;74:1311–1318.
18. Ito M, Kanno S, Nosho K, Sukawa Y, Mitsuhashi K, Kurihara H, Igarashi H, Takahashi T, Tachibana M, Takahashi H, Yoshii S, Takenouchi T, Hasegawa T, et al. Association of Fusobacterium nucleatum with clinical and molecular features in colorectal serrated pathway. Int J Cancer. 2015;137:1258–1268
19. Herrinton LJ, Liu L, Levin TR, Allison JE, Lewis JD, Velayos F. Incidence and mortality of colorectal adenocarcinoma in persons with inflammatory bowel disease from 1998 to 2010. Gastroenterology. 2012;143:382–389.
20. Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001;61:3573–3577.
21. Tahara T, Arisawa T. Potential usefulness of DNA methylation as a risk marker for digestive cancer associated with inflammation. Expert Rev Mol Diagn. 2012;12:489–497.
22. Tahara T, Shibata T, Kawamura T, Okubo M, Ichikawa Y, Sumi K, Miyata M, Ishizuka T, Nakamura M, Nagasaka M, Nakagawa Y, Ohmiya N, Arisawa T, Hirata I. Fusobacterium detected in colonic biopsy and clinicopathological features of ulcerative colitis in Japan. Dig Dis Sci. 2015;60:205–210.
23. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–8686.
24. Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, Hernandez NS, Chen X, Ahmed S, Konishi K, Hamilton SR, Issa JP. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci U S A. 2007;104:18654–18659.
25. Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, Zhang N, Liang S, Donehower LA, Issa JP. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010;20:332–340.
26. Chung W, Kwabi-Addo B, Ittmann M, Jelinek J, Shen L, Yu Y, Issa JP. Identification of novel tumor markers in prostate, colon and breast cancer by unbiased methylation profiling. PLoS One. 2008;3:e2079.
27. Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–196.
28. Tahara T, Hirata I, Nakano N, Nagasaka M, Nakagawa Y, Shibata T, Ohmiya N. Comprehensive DNA methylation profiling of inflammatory mucosa in ulcerative colitis . Inflamm Bowel Dis. 2017;23:165–173.
29. Ogino S, Nosho K, Irahara N, Meyerhardt JA, Baba Y, Shima K, Glickman JN, Ferrone CR, Mino-Kenudson M, Tanaka N, Dranoff G, Giovannucci EL, Fuchs CS. Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clin Cancer Res. 2009;15:6412–6420.
30. Ogino S, Odze RD, Kawasaki T, Brahmandam M, Kirkner GJ, Laird PW, Loda M, Fuchs CS. Correlation of pathologic features with CpG island methylator phenotype (CIMP) by quantitative DNA methylation analysis in colorectal carcinoma. Am J Surg Pathol. 2006;30:1175–1183.
31. Tahara T, Arisawa T. Potential usefulness of DNA methylation as a risk marker for digestive cancer associated with inflammation. Expert Rev Mol Diagn. 2012;12:489–497.
32. Konishi K, Shen L, Wang S, Meltzer SJ, Harpaz N, Issa JP. Rare CpG island methylator phenotype in ulcerative colitis-associated neoplasias. Gastroenterology. 2007;132:1254–1260.
33. Alvarez C, Aravena A, Tapia T, Rozenblum E, Solís L, Corvalán A, Camus M, Alvarez M, Munroe D, Maass A, Carvallo P. Different Array CGH profiles within hereditary breast cancer tumors associated to BRCA1 expression and overall survival. BMC Cancer. 2016;16:219.
34. Blasius M, Wagner SA, Choudhary C, Bartek J, Jackson SP. A quantitative 14-3-3 interaction screen connects the nuclear exosome targeting complex to the DNA damage response. Genes Dev. 2014;28:1977–1982.
35. Mitsuhashi K, Nosho K, Sukawa Y, Matsunaga Y, Ito M, Kurihara H, Kanno S, Igarashi H, Naito T, Adachi Y, Tachibana M, Tanuma T, Maguchi H, et al. Association of Fusobacterium species in pancreatic cancer tissues with molecular features and prognosis. Oncotarget. 2015;6:7209–7220. doi: 10.18632/oncotarget.3109.
36. Tahara T, Shibata T, Okubo M, Ishizuka T, Nakamura M, Nagasaka M, Nakagawa Y, Ohmiya N, Arisawa T, Hirata I. DNA methylation status of epithelial-mesenchymal transition (EMT)--related genes is associated with severe clinical phenotypes in ulcerative colitis (UC). PLoS One. 2014;9:e107947.
37. Okubo M, Tahara T, Shibata T, Yamashita H, Nakamura M, Yoshioka D, Yonemura J, Kamiya Y, Ishizuka T, Nakagawa Y, Nagasaka M, Iwata M, Yamada H, et al. Association study of common genetic variants in pre-microRNAs in patients with ulcerative colitis. J Clin Immunol. 2011;31:69–73.
38. Tahara T, Shibata T, Okubo M, Kawamura T, Sumi K, Ishizuka T, Nakamura M, Nagasaka M, Nakagawa Y, Ohmiya N, Arisawa T, Hirata I. Telomere length in non-neoplastic colonic mucosa in ulcerative colitis (UC) and its relationship to the severe clinical phenotypes. Clin Exp Med. 2015;15:327–332.
39. Boutaga K, van Winkelhoff AJ, Vandenbroucke-Grauls CM, Savelkoul PH. Periodontal pathogens: a quantitative comparison of anaerobic culture and real-time PCR. FEMS Immunol Med Microbiol. 2005;45:191–199.
40. Wilson GM, Flibotte S, Chopra V, Melnyk BL, Honer WG, Holt RA. DNA copy-number analysis in bipolar disorder and schizophrenia reveals aberrations in genes involved in glutamate signaling. Hum Mol Genet. 2006;15:743–749.
41. Toyota M, Issa JP. CpG island methylator phenotypes in aging and cancer. Semin Cancer Biol. 1999;9:349–357.