
INTRODUCTION
Gliomas account for approximately 80% of all malignant primary central nervous system tumors [1, 2]. Stage I and II gliomas are lower-risk tumors and have a better prognosis, whereas stage III and IV gliomas, including anaplastic astrocytomas and glioblastomas, are high-grade malignant tumors [3, 4]. Although progress has been made in treatment modalities such as surgery, chemotherapy, and radiotherapy, recurrence after standard therapies is inevitable, and the median survival of patients with high-grade gliomas is no more than 14 months [5, 6]. The 5-year survival rate of patients with glioblastoma is less than 3% [4]. Thus, finding susceptible cells and molecules on which to apply new therapeutic options has been the focus of diagnosis and treatment of gliomas.
Studies confirm the presence of cancer stem cells (CSCs) in various cancer tissues [7, 8]. CSCs are undifferentiated cells within tumors that possess high tumorigenic activity and the capacity to self-renew and undergo multilineage differentiation [9–11]. CSCs possess biological characteristics such as rapid repair of damaged DNA [12, 13], metabolic reprogramming [14], adaption to hyperinflammatory microenvironments [10], and resistance to oxidative stress and anticancer drugs [10, 15]. Evidence indicates that malignant glioma CSCs induce cancer and promote cancer development [16, 17]. Moreover, glioma CSCs promote cancer resistance to treatment and recurrence, leading to high mortality [18]. Although opinions differ, most reports define CD133-positivity (CD133+) as a marker of glioma CSCs [19–21].
Evidence indicates that inflammation is a factor in the initiation, proliferation, progression, and dissemination of certain cancers [22–26]. Inflammation is induced by direct reaction with pathogens [27]. Toll-like receptors (TLRs) are type I transmembrane molecules that react with foreign pathogens such as bacteria and viruses, and trigger immune responses [28, 29]. TLRs are correlated with development and progression of various tumors [30]. Toll-like receptor 4 (TLR4) is expressed on the cell surface and reacts with the membrane components of foreign pathogens [29, 31]. TLR4 reacts with lipopolysaccharide (LPS) in the cell wall of Gram-negative bacteria [32]. TLR4 is expressed in various cancers and functions in cancer biology [33, 34]. In colorectal cancer (CRC), increased TLR4 levels are correlated with cancer stage, histological grade, metastasis, progression, and prognosis of CRC patients [34–36]. LPS enhances the expression of VEGF-C, which promotes cell motility and metastasis by triggering TLR4 signaling, and TLR4-deficient mice are protected from CRC oncogenesis [34, 37]. Activation of the TLR4 signaling pathway results in cancer cell proliferation and angiogenesis in pancreatic cancer [38, 39]. LPS-triggered TLR4 signaling enhances cancer cell proliferation and leads to drug resistance in hepatoblastoma cells [40]. LPS promotes the stemness of CD133+ CSCs in hepatoma [32]. However, little is known about the function of LPS-induced TLR4 signaling in glioma CD133+ CSCs. Therefore, we investigated the function of TLR4 in glioma CD133+ CSCs. We found that LPS promotes the proliferation and drug resistance of glioma CD133+ CSCs and leads to immune evasion of CD133+ CSCs from cytotoxic T lymphocyte-induced cytolysis.
RESULTS
Identification of glioma CSCs
We generated CSCs from glioma cell lines or patient samples and identified CSCs by flow cytometry. The expression of CSC markers, including CD133, Nanog, SSEA-1, Msil and Nestin in glioma CSCs generated from one glioma patient are shown in Figure 1.
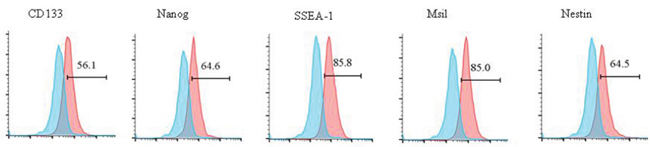
Figure 1: Identification of glioma CSCs via flow cytometry. Surface proteins of CD133, Nanog, SSEA-1, Msil, and Nestin are detected by flow cytometry in CSCs generated from one glioma patient.
TLR4 is expressed in human glioma CD133+ CSCs and human glioma tissues
We investigated the expression of TLR4 in human glioma CD133+ CSCs isolated from six CSCs derived from two glioma cell lines, SF295 and U251, and four fresh human surgical glioma tissues, patient (pT) 1 to pT4. Flow cytometry (Figure 2a) and Western blot analysis (Figure 2b) demonstrated that TLR4 protein was expressed in six human CD133+ glioma CSCs. Magnetic resonance imaging (MRI) was performed to identify intracranial gliomas of two patients (Figure 2c). TLR4 expression was analyzed in two glioma patients by immunohistochemistry (IHC). We identified expression of TLR4 protein in glioma tissues (Figure 2c).
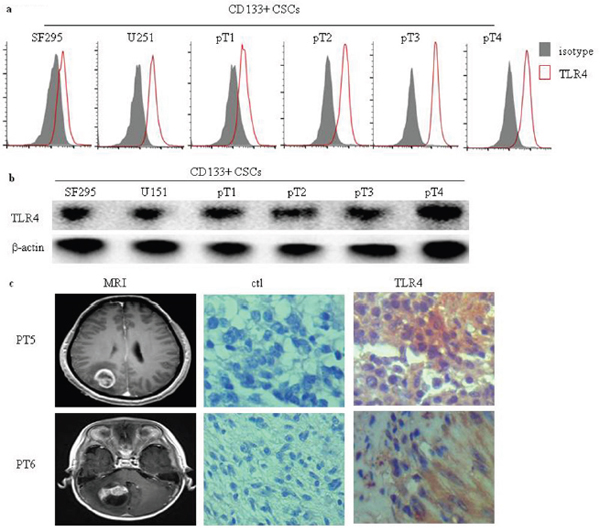
Figure 2: TLR4 expression in human glioma CD133+ CSCs and glioma tissues. Surface (a) and total (b) TLR4 protein expression in six human glioma CD133+ CSCs isolated from CSCs derived from SF295 and U251 and four fresh human surgical glioma tissue samples (pT1 to pT4) was detected by flow cytometry and Western blot analysis. (c) Representative MRI images and immunochemistry staining of TLR4 expression in two glioma patients. An isotype control antibody was used for negative control staining. Original magnification ×400.
LPS stimulation induces proliferation of CD133+ CSCs and protects CD133+ CSCs from Adriamycin-induced apoptosis
To study the activity of TLR4 in human glioma CD133+ CSCs, we incubated CD133+ CSCs isolated from CSCs derived from SF295 and U251 glioma cell lines with LPS at different concentrations and time points. The proliferation of CD133+ CSCs was upregulated on a dose-dependent (Figure 3a and 3b; P < 0.05, compared to CD133+ CSCs without LPS stimulation) and time-dependent (Figure 3c and 3d; P < 0.05, compared to CD133+ CSCs without LPS stimulation) basis. After stimulation with LPS, CD133+ CSCs isolated from four CSC samples derived from four fresh human surgical glioma tissues, pT1 to pT4, also demonstrated enhanced proliferation (Figure 3e; P < 0.01, compared to CD133+ CSCs without LPS stimulation), whereas PBMCs did not (Figure 3e). The number of CD133+ CSCs increased after LPS stimulation compared with cells without LPS stimulation (Figure 3f; P < 0.05). We used Pam3C to activate TLR2 signaling and found that TLR2 activation did not promote the proliferation of CD133+ CSCs (data not shown). To verify the function of TLR4 in the proliferation of CD133+ CSCs, TLR4 expression in CD133+ CSCs was knocked down by use of TLR4-specific short-hairpin RNA (shRNA) lentiviral particles. In the present study, CD133+ CSCs were classified as wild-type (wt), control-shRNA transfected (shRNA-ctl), and TLR4-shRNA transfected (shRNA-TLR4) cells. Western blot (Figure 4a) and flow cytometry analysis (Figure 4b) revealed that shRNA-TLR4 downregulated the expression of TLR4. Compared with wt or shRNA-ctl CD133+ CSCs, TLR4 knockdown resulted in a reduced response of CD133+ CSCs to LPS exposure (Figure 4c; P < 0.01).
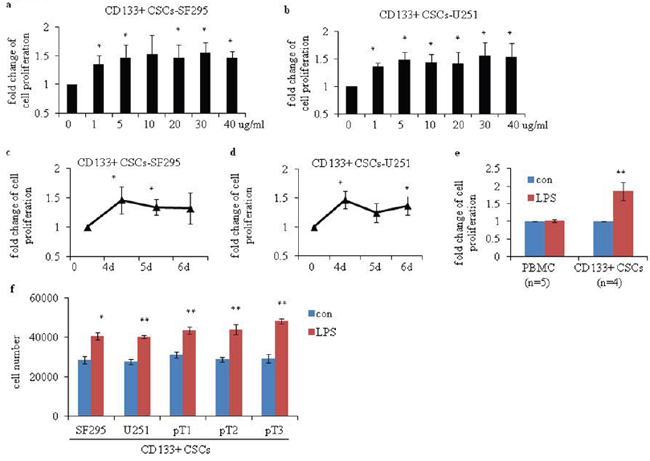
Figure 3: Proliferation of human glioma CD133+ CSCs after stimulation with LPS. CCK-8 assays were performed to determine the proliferation of CD133+ CSCs isolated from CSCs derived from (a) SF295 (CD133+ CSCs-SF295) and (b) U251 (CD133+ CSCs-U251) after stimulation with 0 to 40 μg/mL LPS for 4 days. Proliferation of (c) CD133+ CSCs-SF295 and (d) CD133+ CSCs-U251 after stimulation with 5 μg/mL LPS for 4, 5, and 6 days. (e) Proliferation of five PBMC samples (isolated from five healthy donors) and four CD133+ CSCs derived from four fresh human surgical glioma tissues (pT1 to pT4) after stimulation with 1 μg/mL LPS for 4 days. (f) Cell number was counted to determine the proliferation of CD133+ CSCs isolated from CSCs derived from SF295, U251, pT1, pT2 and pT3 after stimulation with 1 μg/mL LPS for 6 days. *P < 0.05, **P < 0.01, compared with the control (con) cells.
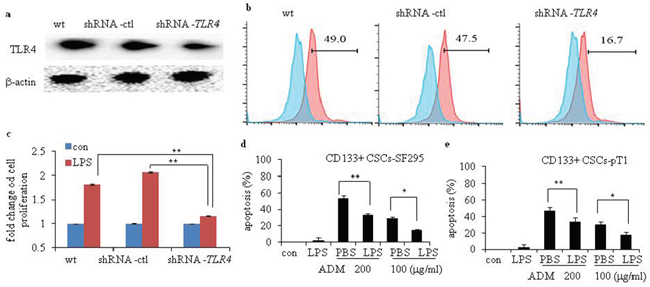
Figure 4: Efficiency of TLR4 knockdown and chemotherapy-induced apoptosis of human glioma CD133+ CSCs after stimulation with LPS. Efficiency of TLR4 knockdown in wild-type (wt) CD133+ CSCs and cells transfected with control shRNA (shRNA-ctl) or TLR4-specific (shRNA-TLR4) lentiviral particles were evaluated by Western blot analysis (a) and flow cytometry (b). (c) Proliferation of wt, shRNA-ctl, and shRNA-TLR4 CD133+ CSCs. (d) LPS protected CD133+ CSCs-SF295 from ADM-induced apoptosis. (e) LPS protected CD133+ CSCs-pT1 from ADM -induced apoptosis. *P < 0.05, **P < 0.01, compared with the control (con) cells.
We next examined the function of LPS on chemotherapy-induced apoptosis of CD133+ CSCs. We determined that Adriamycin (ADM, 200 μg/mL or 100 μg/mL) is an effective chemotherapy drug for CD133+ CSCs. Preincubation of CD133+ CSCs derived from SF295 and one fresh human surgical glioma tissue sample, pT1, with LPS partially inhibited ADM-induced apoptosis (Figure 4d and 4e; P < 0.01).
LPS exposure promoted the expression of gene markers in CD133+ CSCs
To investigate the function of LPS in the regulatory expression of surface markers in CD133+ CSCs isolated from CSCs generated from U251, the mRNA expression of CD133, Nanog, SSEA-1, Msil, and Nestin were examined. After LPS exposure, the mRNA levels of CD133 (Figure 5a; P < 0.01), Nanog (Figure 5b; P < 0.01), and Nestin (Figure 5c; P < 0.01) increased, compared to CD133+ CSCs without LPS stimulation. We detected the surface protein expression of CD133, Nanog, SSEA-1, Msil, and Nestin by flow cytometry. The surface expression of CD133 (Figure 5d), Nanog (Figure 5e), and Nestin (Figure 5f) increased after LPS stimulation. No changes of mRNA and protein levels of SSEA-1 and Msil were observed (data not shown).
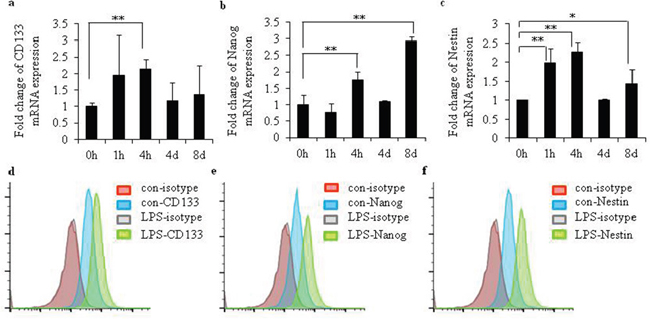
Figure 5: LPS increased expression levels of gene markers in glioma CD133+ CSCs. mRNA levels of (a) CD133, (b) Nanog, and (c) Nestin after stimulation with LPS (25 μg/mL) for 0 hours, 1 hour, 4 hours, 4 days, and 8 days in glioma CD133+CSCs isolated from CSCs generated from U251. Surface protein expression of (d) CD133, (e) Nanog, and (f) Nestin after stimulation with LPS (25 μg/mL) for 24 hours in glioma CD133+CSCs isolated from CSCs generated from U251. *P < 0.05, **P < 0.01, compared with the control groups.
LPS enhances the production of cytokines in glioma CD133+ CSCs
We examined LPS-induced secretion of cytokines in glioma CD133+ CSCs isolated from CSCs derived from SF295 and U251 and three fresh human surgical glioma tissue samples, pT1 to pT3. We incubated CD133+ CSCs with LPS for 96 hours and collected the supernatants for enzyme-linked immunosorbent assay (ELISA). Compared to CD133+ CSCs without LPS stimulation, levels of monocyte chemotactic protein 1 (MCP-1) (Figure 6a; P < 0.01), macrophage inflammatory protein-1 alpha (MIP-1α) (Figure 6b; P < 0.01), tumor necrosis factor-alpha (TNF-α) (Figure 6c; P < 0.01), interleukin-1 beta (IL-1β) (Figure 6d; P < 0.01), interleukin-6 (IL-6) (Figure 6e; P < 0.01), and interleukin-10 (IL-10) (Figure 6f; P < 0.01) were elevated after LPS stimulation.
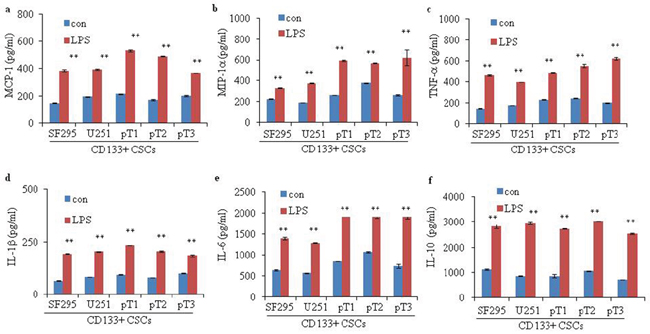
Figure 6: Cytokine secretion of glioma CD133+ CSCs upon LPS stimulation. Secretion of (a) MCP-1, (b) MIP-1α, (c) TNF-α, (d) IL-1β, (e) IL-6, and (f) IL-10 in CD133+ CSCs derived from two glioma cell lines, SF295 and U251, and three fresh human surgical glioma tissue samples, pT1 to pT3, without or with pretreatment with LPS (1 μg/mL) for 4 days. **P < 0.01, compared with the control (con) groups.
Proteins and signaling pathways associated with TLR4 activation in glioma CD133+ CSCs
LPS stimulation-activated signaling pathways in glioma CD133+ CSCs isolated from CSCs generated from SF295 were further investigated. First, we found that LPS activated cyclin and cyclin-dependent kinase (CDK) complexes, including CDK4, CDK6, and cyclin E in CD133+ CSCs (Figure 7). Second, we analyzed the expression of apoptosis-related molecules such as bcl-2, survivin, and BAX in CD133+ CSCs. The protein expression of bcl-2 increased (Figure 7), whereas there was no obvious change of survivin and BAX (data not shown) after LPS stimulation. Third, we demonstrated that LPS exposure increased the levels of nuclear transcription factor-κB (NF-κB), phosphorylated p38 (p-p38), phosphorylated c-Jun N-terminal kinase (p-JNK), phosphorylated extracellular signal-regulated kinase (p-ERK), and phosphorylated protein kinase B (p-Akt) in CD133+ CSCs (Figure 7). The total protein of p38, JNK, ERK, and Akt did not increase after LPS stimulation (Figure 7).
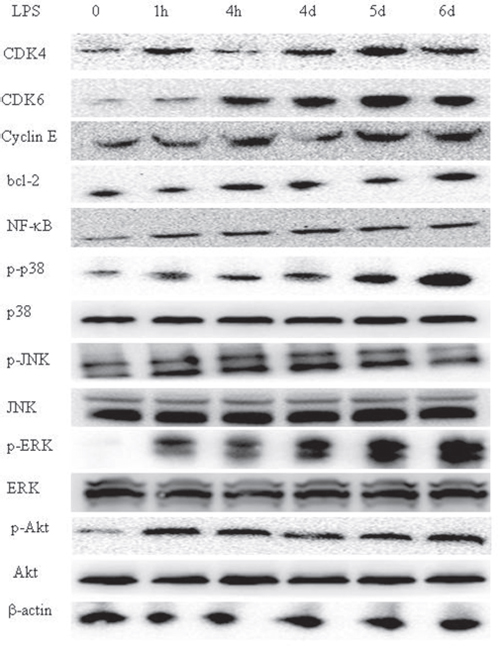
Figure 7: LPS triggers the TLR4 signaling pathway in glioma CD133+ CSCs. Expression of CDK4, CDK6, cyclin E, bcl-2, NF-κB, phosphorylated p38, p38, phosphorylated JNK, JNK, phosphorylated ERK, ERK, phosphorylated Akt, Akt, and β-actin in glioma CD133+ CSCs isolated from CSCs generated from SF295 after stimulation with LPS (1 μg/mL) at different time points (0 hours to 6 days).
TLR4 signaling facilitates immune evasion of glioma CD133+ CSCs
We investigated the function of TLR4 signaling in cytotoxic T lymphocyte (CTL)-induced cytolysis to glioma CD133+ CSCs. CD133+ CSC-reactive CTL1 to CTL3 were established via co-culture of CD3+ T cells with three types of irradiated CD133+ CSCs derived from SF295 and U251 and one fresh human surgical glioma tissue sample, pT1, that were pretreated with or without LPS. The sensitivity of CTL generated against target LPS-pretreated CD133+ CSCs (CTL-LPS) was significantly reduced compared with CTL generated against target control CD133+ CSCs (CTL-con) (Figure 8a, 8b and 8c; P < 0.01). Flow cytometry analysis showed the proportion of CD8+ T cells expressing interferon- γ (IFN-γ) was lower in the CTL-LPS than in the CTL-con (Figure 8d). Moreover, pretreatment of CD133+ CSCs cells with LPS before cytotoxic assays inhibited the killing capacity of CTL compared with pretreatment with medium only (Figure 8e; P < 0.01). However, when pretreated with LPS, CD133+ CSCs transfected with shRNA-TLR4 were more sensitive than control or wt cells to CTL-induced killing (Figure 8f; P < 0.01). These results suggest that TLR4 signaling is involved in immune evasion by glioma CD133+ CSCs. We also determined whether the CTL kill normal cells and K562 cells. Purified normal peripheral blood mononuclear cells (PBMC) from glioma patients and K562 cells were used in the test and it demonstrated that less killing occurred in normal PBMC or K562 cells, whereas CTL kill glioma CD133+ CSCs (Figure 8g).
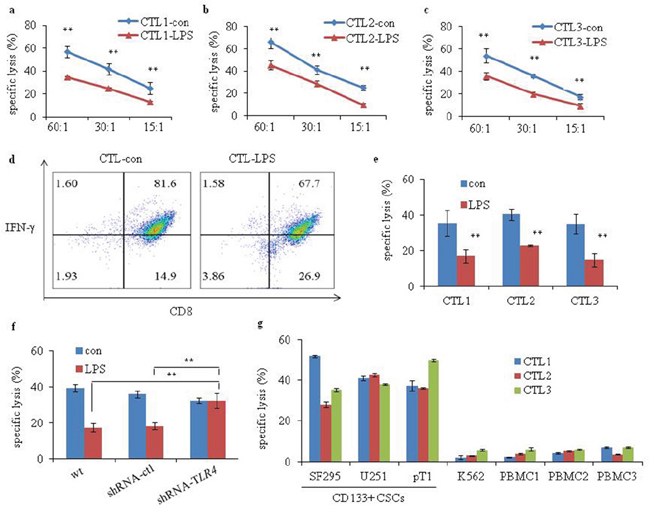
Figure 8: Cytotoxicity of glioma CD133+ CSC-reactive CTL. Cytotoxicity of (a) CTL1-con and CTL1-LPS (generated without or with irradiated LPS-stimulated glioma CD133+ CSCs isolated from CSCs generated from SF295), (b) CTL2-con and CTL2-LPS (generated without or with irradiated LPS-stimulated glioma CD133+ CSCs isolated from CSCs generated from U251) and (c) CTL3-con and CTL3-LPS (generated without or with irradiated LPS-stimulated glioma CD133+ CSCs isolated from CSCs generated from one fresh human surgical glioma tissue, pT1) against target cells. The ratios of effector cells: target cells were 60:1, 30:1, and 15:1, respectively. A ratio of 30:1 was used for the following function assay. (d) Flow cytometry analysis showing the percentages of IFN-γ-expressing CD8+ T cells in the CTL-con and CTL-LPS. (e) CTL1 to CTL3 against specific CD133+ CSCs pretreated without or with LPS (1 μg/mL) for 4 days. (f) CTL against CD133+ CSCs (wt, shRNA-ctl, and shRNA-TLR4) pretreated without or with LPS (1 μg/mL) for 4 days. (g) Cytotoxicity of CTL1 to CTL3 against target cells, including CD133+ CSCs isolated from CSCs derived from U251, SF295 and pT1, K562, and PBMC (PBMC1-PBMC3). K562 was used as a control for NK-cell activity. **P < 0.01, compared with the control (con) groups.
Glioma CD133+ CSC-reactive CTL is memory effector T cell
We detected the surface expression of CD28, CD45RA, CD45RO, CCR7, and CD44 in CTL and found CD28, CD45RO, and CD44 were expressed in glioma CD133+ CSC-reactive CTL, whereas CD45RA and CCR7 were rarely expressed (Figure 9). Thus, memory effector T cells function in LPS-induced immune evasion.
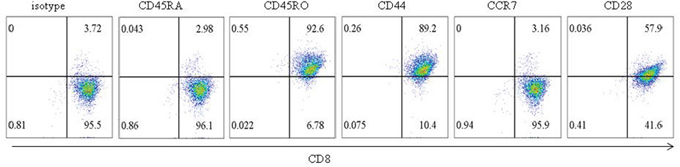
Figure 9: Phenotype of glioma CD133+ CSC-reactive CTL. The surface expression of CD45RA, CD45RO, CD44, CCR7, and CD28 in glioma CD133+ CSC-reactive CTL was examined by flow cytometry.
DISCUSSION
Previous studies reported TLR4 to be absent on glioblastoma cells, and LPS-induced antitumoral effects might depend on microglia and inflammatory cells [41]. No TLR4 expression was observed in astrocytes and oligodendroglial cells [42]. However, another study demonstrates that TLR4 is expressed on human astrocytes and glioma cell lines GL261, U373MG, U118, and U87 [43–46]. Immunohistochemical detection revealed that TLR4 protein is overexpressed in tissues from glioblastoma patients [47]. LPS-activated TLR4 signaling promotes the proliferation of glioma cell lines [43, 46]. LPS also induces chemotactic migration of glioma cells [48]. Although opinions differ, it is accepted that TLR4 is expressed in glioma. However, the expression and function of TLR4 in glioma CD133+ CSCs are unknown. In the present study, we generated CSCs from glioma cell lines and patient samples. We identified CSCs by detection of surface expression of CD133, Nanog, SSEA-1, Msil, and Nestin via flow cytometry. Glioma CD133+ CSCs were isolated by use of the MACS CD133 kit for further study. We demonstrated that surface and total TLR4 protein is expressed in glioma CD133+ CSCs. IHC analysis demonstrated that TLR4 is expressed in the tissue of glioma patients. And LPS was used to investigate the function of the TLR4 signaling pathway in glioma CD133+ CSCs. LPS-activated TLR4 signaling promotes the proliferation and chemoresistance of glioma CD133+ CSCs and triggers the release of various cytokines, resulting in the resistance of glioma CD133+ CSCs to CTL-induced lysis. Knockdown of TLR4 by shRNA in glioma CD133+ CSCs inhibited LPS-stimulated CSC proliferation and reversed the inhibitory effect of LPS-induced glioma CD133+ CSCs on the cytotoxicity of CTLs. These effects were mediated by memory effector T cells. Thus, TLR4 expression in glioma CD133+ CSCs may lead to cancer progression and immune evasion.
A previous study reported that LPS maintains the stemness of CD133+ CSCs by facilitating the expression of CD133 [32]. Because CD133, Nanog, and Nestin are considered markers of glioma CSCs [49], we examined the gene and protein levels of CD133, Nanog, and Nestin in glioma CD133+ CSCs. After LPS stimulation, the mRNA and protein levels of CD133, Nanog, and Nestin increased in glioma CD133+ CSCs. Thus, LPS helped preserve the stemness of glioma CD133+ CSCs. Whether LPS helped the colony formation, migration, and invasion of glioma CD133+ CSCs remains to be investigated.
Glioma cells produce various inflammatory mediators, such as IL-1β, IL-6, IL-8, IL-10, MCP-1, and MIP [22, 50]. Increased expression of IL-1β, IL-6, and IL-8 was also observed in glioma patient samples and was correlated with cancer invasiveness and survival of patients [50–52]. IL-1β activates signaling pathways and induces the expression of genes that promote carcinogenesis and cancer progression [22]. IL-6 signaling is essential for the proliferation, differentiation, aggressiveness, and migration of glioma cells [22, 53, 54]. IL-6 also enhances neurosphere formation capacity and promotes the stemness of glioma CSCs [55]. Elevated IL-8 expression is associated with enhanced growth, angiogenesis, and invasion of glioma [56–59]. In our present study, activation of the TLR4 signaling pathway promoted the production of IL-1β, IL-6, IL-10, MCP-1, MIP-1α, and TNF-α in glioma CD133+ CSCs. Consistent with our findings, LPS exposure induces release of IL-1β and IL-6 in U373-MG human astrocytoma cells [46]. LPS promotes the release of TNF-α and MIP-1α from monocytes [60, 61], and TNF-α induces the release of IL-8 [62, 63]. In lung cancer cells, LPS-triggered TLR4 activation enhances the secretion of IL-10 [64]. Thus, LPS-activated interaction between cytokines and glioma CD133+ CSCs might establish a complicated microenvironment that facilitates cancer progression and immune evasion.
Cell proliferation is activated by CDKs and cyclin proteins [65]. CDK4/6 and cyclin E are necessary to promote transition of cells from G1 to S phase of the cell cycle and are factors in cell adhesion [66, 67]. We found that LPS induced the expression of CDK4/6 and cyclin E in glioma CD133+ CSCs, which might promote increased proliferation of these cells. LPS also increased the protein expression of bcl-2.
LPS-triggered TLR4 activation in turn triggers the mitogen-activated protein kinase signaling pathway [68], which promotes tumor cell proliferation and facilitates inflammation [69, 70]. The phosphatidylinositol 3 kinase (PI3K)/Akt pathway is a factor in the cell cycle and apoptosis [65]. LPS stimulation increases the expression of p-p38, p-JNK, p-ERK and p-Akt in glioma CD133+ CSCs.
We generated glioma CD133+ CSC-reactive CTL by co-culturing isolated CD3+ T cells from healthy donors with different glioma CD133+ CSCs as antigen-presenting cells. We investigated CD133+ CSC-reactive CTL-induced killing of glioma CD133+ CSCs with CD8+ T cells sorted from the generated CD133+ CSC-reactive CTL. Compared with the control cells, CTL primed with LPS-stimulated CD133+ CSCs had less capacity to kill glioma CD133+ CSCs. Moreover, preincubation of glioma CD133+ CSCs with LPS inhibited CD133+ CSC-reactive CTL lysis of target CD133+ CSCs. This effect was reversed by shRNA-TLR4 in glioma CD133+ CSCs. CD133+ CSC-reactive CTL did not kill normal PBMC or K562 cells. We proved that memory effector T cells function in LPS-induced immune evasion. A previous study reported that TLR4 ligand LPS might be a factor in cancer immune evasion. LPS-activated TLR4 signaling renders tumor cells resistant to CTL-induced killing, which might be attributed to increased expression of B7-H1, B7-H2, and CD40 and decreased expression of Fas [68]. TLR4 signaling in human mantle cell lymphoma cells inhibited T cell proliferation and CTL-induced cytolysis, and this effect was partially restored by neutralization of IL-10 and/or VEGF [71]. LPS-pretreated multiple myeloma cells suppressed the generation of CTL by inducing expression of PD-L1 [72].
Our study demonstrated the importance of TLR4-induced effects on glioma CD133+ CSCs. Further investigations are needed to elucidate TLR4-induced signaling pathways in the pathogenesis and immune evasion of glioma CD133+ CSCs. TLR4 might provide novel therapeutic approaches for patients with glioma.
MATERIALS AND METHODS
Cell culture and isolation of glioma CD133+ CSCs
Glioma cell line U251 and SF295 (Cell Bank of the Chinese Academy of Sciences, Shanghai, China) were cultured in Dulbecco Modified Eagle’s medium (DMEM) (Hyclone,USA) supplemented with 10% fetal bovine serum (Gibco, USA) and penicillin/streptomycin (1×; Gibco, USA) at 37° C in a 5% CO2 incubator. The medium was changed every 2 to 3 days. Fresh glioma tissues were collected from patients undergoing craniotomy at the Department of Neurosurgery, Linyi People’s Hospital (Linyi, Shandong Province, China). All patients gave written consent. To obtain CSCs from U251 or SF295 cells (CSCs-U251 or CSCs-SF295), cells were digested with collagenase D (Roche, Switzerland) and DNase I (Sigma-Aldrich, USA) when they had reached logarithmic growth phase, and then resuspended in serum-free medium (SFM) consisting of DMEM/F12 medium (Hyclone), B27 Supplement (1×; Gibco, USA), human EGF (20 ng/mL; PeproTech, USA), human FGF-basic (20 ng/mL; PeproTech, USA), leukemia inhibitory factor (10 ng/mL; PeproTech, USA), L-glutamine (2 mM; Gibco, USA), and penicillin/streptomycin (1×; Gibco, USA). SFM was renewed every 3 to 4 days and cells grew as spheres. CSCs derived from glioma patients were obtained according to the protocols described previously [49]. When cells had reached a high density at the third passage, CD133+ CSCs were isolated by use of the MACS CD133 kit (Miltenyi Biotec, Germany).
RNA extraction, cDNA synthesis and real-time quantitative PCR
Details of RNA extraction and cDNA synthesis were described previously [71]. Real-time quantitative PCR (qPCR) was performed in triplicate for each sample on an Applied Biosystems QuantStudio 5 PCR system (Waltham, USA) with SYBR Green Real-time PCR Master Mix (TOYOBO, Osaka, Japan). Primers are listed in Table 1. The qPCR conditions were pre-denaturation at 95 °C for 1 minute, followed by 40 cycles at 95 °C for 15 seconds, 60 °C for 15 seconds, and 72 °C for 60 seconds. Data were collected and analyzed by ABI QuantStudio Design & Analysis Software Version 1.3.1. The mRNA levels of specific genes in each sample were normalized to GAPDH mRNA levels by use of mean values of triplicates and expressed as relative expression compared with GAPDH.
Table 1: Primer sequences for qPCR
Gene |
Forward (5′→3′) |
Reverse (5′→3′) |
|---|---|---|
CD133 |
CCCGTGGATGCAGAACTTGA |
CCTGAATAGGAAGACGCTGAGT |
Nanog |
CCCAGCTGTGTGTACTCAAT |
CAGGCATCCCTGGTGGTAG |
Nestin |
CTGAAAAGTTCCAGCTGGCTGT |
GCTGAGGGACATCTTGAGGTG |
GAPDH |
CGGAGTCAACGGATTTGGTCGTAT |
AGCCTTCTCCATGGTGGTGAAGAC |
Flow cytometry
Antibodies against human TLR4, CD133, Nestin, SSEA-1, Msil, CD28, CD45RA, CD45RO, CCR7, CD44, IFN-γ and isotype control were purchased from eBioscience (San Diego, USA). Antibody against human Nanog was purchased from BD Biosciences (USA). Data were obtained with a flow cytometer (FACS Canto; BD Biosciences, USA).
Immunohistochemistry
IHC was performed according to previous literature [50]. The primary antibody against TLR4 and the isotype-negative control antibody (both Abcam, UK) were used at a dilution of 1:200.
Cell proliferation detection
CD133+ CSC proliferation was detected by use of a Cell Counting Kit-8 (CCK-8) or cell number counting. CD133+ CSCs (2 × 104/200 μL per well) were cultured in 96-well culture plates (Corning Inc., Corning, USA) and treated with LPS (Sigma, USA) at different time points or concentrations. At the end of each experiment, CCK-8 was added to the culture system for 3 hours and detected, by use of a microplate reader (Bio-Rad, USA).
Western blot
Details of the Western blot analysis were described previously [43]. Phosphorylated p38, phosphorylated JNK, phosphorylated ERK, p38, JNK, ERK, and β-actin monoclonal antibodies were used (all Abcam, UK). Phosphorylated Akt, Akt, bcl-2, survivin, BAX, CDK4, CDK6, and cyclin E monoclonal antibodies were purchased from Cell Signaling Technology (Beverly, USA).
ELISA
ELISA kits (all from eBioscience, USA) were used to detect the levels of IL-1β, IL-6, IL-10, MCP-1, MIP-1α, and TNF-α in cell supernatants collected from glioma CD133+ CSCs according to the manufacturer’s instructions.
TLR4 shRNA transfection of glioma CD133+ CSCs by lentivirus
TLR4-specific or control shRNA lentiviral particles were obtained from Santa Cruz Biotechnology Inc. (Dallas, USA), and glioma CD133+ CSCs were transfected according to the manufacturer’s protocol.
Generation of CD133+ CSC-reactive CTL and function assays
CD3+ T cells were co-cultured with irradiated CD133+ CSCs to generate glioma CD133+ CSC-reactive CTL, which were expanded for function assays, as described previously [73]. CTL1 was generated against irradiated CD133+ CSCs isolated from CSCs derived from SF295, CTL2 was generated against irradiated CD133+ CSCs isolated from CSCs derived from U251, and CTL3 was generated against irradiated CD133+ CSCs isolated from CSCs derived from one fresh human surgical glioma tissue, pT1. CTL-con was generated against untreated glioma CD133+ CSCs and CTL-LPS were generated against glioma CD133+ CSCs stimulated with LPS. For cytotoxic assays, CD8+ T cells were sorted by use of the MACS CD8 kit (Miltenyi Biotec, Germany). A cytotoxicity assay kit (Promega Corporation, USA) was used to measure the killing capacity of CTL against target glioma CD133+ CSCs.
Statistical analysis
Student’s t-test was applied to compare groups and P values < 0.05 were considered statistically significant. Unless otherwise indicated, data are presented as the mean ± standard deviation.
Author contributions
FYC, XYH, YFD and LJW designed the project. FYC, JWY, YCQ, XLY and LJW performed experiments and analyzed data. FYC, YFD and LJW wrote the manuscript.
CONFLICTS OF INTEREST
We have no conflicts of interest to declare.
FUNDING
This work was supported by the Key Research Project program of Shandong Province (grant no. 2016GSF201056), the Natural Science Foundation of Shandong Province (grant no. ZR2014HM077), the Medical Health Science and Technology Development Plan of Shandong Province (grant no. 2014WS0287), the China Postdoctoral Science Foundation (grant no. 2015M580594), and the Postdoctoral Innovation Foundation of Shandong Province (grant no. 201502008).
REFERENCES
1. Wang L, Li G, Liu N, Wang Z, Xu X, Qi J, Ren D, Zhang P, Zhang Y, Tu Y. Association of genetic variants in the CART gene with glioma susceptibility in a Chinese population. Oncotarget. 2016. doi: 10.18632/oncotarget.11763. [Epub ahead of print].
2. Chen B, Liang T, Yang P, Wang H, Liu Y, Yang F, You G. Classifying lower grade glioma cases according to whole genome gene expression. Oncotarget. 2016; 7:74031–42. doi: 10.18632/oncotarget.12188.
3. Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006; 2:494-503; quiz 1 p following 16. doi: 10.1038/ncpneuro0289.
4. Qi L, Ding L, Wang S, Zhong Y, Zhao D, Gao L, Wang W, Lv P, Xu Y, Wang S. A network meta-analysis: the overall and progression-free survival of glioma patients treated by different chemotherapeutic interventions combined with radiation therapy (RT). Oncotarget. 2016; 7:69002–13. doi: 10.18632/oncotarget.10763.
5. Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010; 60:166-93. doi: 10.3322/caac.20069.
6. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352:987-96. doi: 10.1056/NEJMoa043330.
7. Balasubramaniyan V, Vaillant B, Wang S, Gumin J, Butalid ME, Sai K, Mukheef F, Kim SH, Boddeke HW, Lang F, Aldape K, Sulman EP, Bhat KP, et al. Aberrant mesenchymal differentiation of glioma stem-like cells: implications for therapeutic targeting. Oncotarget. 2015; 6:31007-17. doi: 10.18632/oncotarget.5219.
8. Pan Z, Shi Z, Wei H, Sun F, Song J, Huang Y, Liu T, Mao Y. Magnetofection based on superparamagnetic iron oxide nanoparticles weakens glioma stem cell proliferation and invasion by mediating high expression of microRNA-374a. J Cancer. 2016; 7:1487-96. doi: 10.7150/jca.15515.
9. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414:105-11. doi: 10.1038/35102167.
10. Ning X, Shu J, Du Y, Ben Q, Li Z. Therapeutic strategies targeting cancer stem cells. Cancer Biol Ther. 2013; 14:295-303. doi: 10.4161/cbt.23622.
11. O'Brien CA, Kreso A, Jamieson CH. Cancer stem cells and self-renewal. Clin Cancer Res. 2010; 16:3113-20. doi: 10.1158/1078-0432.CCR-09-2824.
12. Maugeri-Sacca M, Bartucci M, De Maria R. DNA damage repair pathways in cancer stem cells. Mol Cancer Ther. 2012; 11:1627-36. doi: 10.1158/1535-7163.MCT-11-1040.
13. Skvortsov S, Debbage P, Lukas P, Skvortsova I. Crosstalk between DNA repair and cancer stem cell (CSC) associated intracellular pathways. Semin Cancer Biol. 2015; 31:36-42. doi: 10.1016/j.semcancer.2014.06.002.
14. Shen YA, Wang CY, Hsieh YT, Chen YJ, Wei YH. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle. 2015; 14:86-98. doi: 10.4161/15384101.2014.974419.
15. Dean M. ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia. 2009; 14:3-9. doi: 10.1007/s10911-009-9109-9.
16. Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006; 5:67. doi: 10.1186/1476-4598-5-67.
17. García-Romero N, González-Tejedo C, Carrión-Navarro J, Esteban-Rubio S, Rackov G, Rodríguez-Fanjul V, Oliver-De La Cruz J, Prat-Acín R, Peris-Celda M, Blesa D, Ramírez-Jiménez L, Sánchez-Gómez P, Perona R. Cancer stem cells from human glioblastoma resemble but do not mimic original tumors after in vitro passaging in serum-free media. Oncotarget. 2016; 7:65888-901. doi: 10.18632/oncotarget.11676.
18. Kenig S, Faoro V, Bourkoula E, Podergajs N, Ius T, Vindigni M, Skrap M, Lah T, Cesselli D, Storici P, Vindigni A. Topoisomerase IIbeta mediates the resistance of glioblastoma stem cells to replication stress-inducing drugs. Cancer Cell Int. 2016; 16:58. doi: 10.1186/s12935-016-0339-9.
19. Wilson TJ, Zamler DB, Doherty R, Castro MG, Lowenstein PR. Reversibility of glioma stem cells' phenotypes explains their complex in vitro and in vivo behavior. Discovery of a novel neurosphere-specific enzyme, cGMP-dependent protein kinase 1, using the genomic landscape of human glioma stem cells. Oncotarget. 2016; 7:63020–41. doi: 10.18632/oncotarget.11589.
20. Lenkiewicz M, Li N, Singh SK. Culture and isolation of brain tumor initiating cells. Curr Protoc Stem Cell Biol. 2009; Chapter 3:Unit3. doi: 10.1002/9780470151808.sc0303s11.
21. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003; 63:5821-8.
22. Yeung YT, McDonald KL, Grewal T, Munoz L. Interleukins in glioblastoma pathophysiology: implications for therapy. Br J Pharmacol. 2013; 168:591-606. doi: 10.1111/bph.12008.
23. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008; 454:436-44. doi: 10.1038/nature07205.
24. Kwon OJ, Zhang L, Ittmann MM, Xin L. Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. Proc Natl Acad Sci U S A. 2014; 111:E592-600. doi: 10.1073/pnas.1318157111.
25. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004; 431:461-6. doi: 10.1038/nature02924.
26. Li G, Wang Z, Ye J, Zhang X, Wu H, Peng J, Song W, Chen C, Cai S, He Y, Xu J. Uncontrolled inflammation induced by AEG-1 promotes gastric cancer and poor prognosis. Cancer Res. 2014; 74:5541-52. doi: 10.1158/0008-5472.CAN-14-0968.
27. Munn LL. Cancer and inflammation. Wiley Interdiscip Rev Syst Biol Med. 2016; 9. doi: 10.1002/wsbm.1370.
28. Deng S, Zhu S, Qiao Y, Liu YJ, Chen W, Zhao G, Chen J. Recent advances in the role of toll-like receptors and TLR agonists in immunotherapy for human glioma. Protein Cell. 2014; 5:899-911. doi: 10.1007/s13238-014-0112-6.
29. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011; 34:637-50. doi: 10.1016/j.immuni.2011.05.006.
30. Alvarado AG, Lathia JD. Taking a toll on self-renewal: TLR-mediated innate immune signaling in stem cells. Trends Neurosci. 2016; 39:463-71. doi: 10.1016/j.tins.2016.04.005.
31. Beutler BA. TLRs and innate immunity. Blood. 2009; 113:1399-407. doi: 10.1182/blood-2008-07-019307.
32. Lai FB, Liu WT, Jing YY, Yu GF, Han ZP, Yang X, Zeng JX, Zhang HJ, Shi RY, Li XY, Pan XR, Li R, Zhao QD, et al. Lipopolysaccharide supports maintaining the stemness of CD133(+) hepatoma cells through activation of the NF-κB/HIF-1α pathway. Cancer Lett. 2016; 378:131-41. doi: 10.1016/j.canlet.2016.05.014.
33. Jouhi L, Renkonen S, Atula T, Makitie A, Haglund C, Hagstrom J. Different toll-like receptor expression patterns in progression toward cancer. Front Immunol. 2014; 5:638. doi: 10.3389/fimmu.2014.00638.
34. Li TT, Ogino S, Qian ZR. Toll-like receptor signaling in colorectal cancer: carcinogenesis to cancer therapy. World J Gastroenterol. 2014; 20:17699-708. doi: 10.3748/wjg.v20.i47.17699.
35. Eiro N, Gonzalez L, Gonzalez LO, Fernandez-Garcia B, Andicoechea A, Barbon E, Garcia-Muniz JL, Vizoso FJ. Toll-like receptor-4 expression by stromal fibroblasts is associated with poor prognosis in colorectal cancer. J Immunother. 2013; 36:342-9. doi: 10.1097/CJI.0b013e31829d85e6.
36. Eyking A, Ey B, Runzi M, Roig AI, Reis H, Schmid KW, Gerken G, Podolsky DK, Cario E. Toll-like receptor 4 variant D299G induces features of neoplastic progression in Caco-2 intestinal cells and is associated with advanced human colon cancer. Gastroenterology. 2011; 141:2154-65. doi: 10.1053/j.gastro.2011.08.043.
37. Zhu G, Huang Q, Huang Y, Zheng W, Hua J, Yang S, Zhuang J, Wang J, Ye J. Lipopolysaccharide increases the release of VEGF-C that enhances cell motility and promotes lymphangiogenesis and lymphatic metastasis through the TLR4- NF-κB/JNK pathways in colorectal cancer. Oncotarget. 2016; 7:73711-24. doi: 10.18632/oncotarget.12449.
38. Grimmig T, Moench R, Kreckel J, Haack S, Rueckert F, Rehder R, Tripathi S, Ribas C, Chandraker A, Germer CT, Gasser M, Waaga-Gasser AM. Toll like receptor 2, 4, and 9 signaling promotes autoregulative tumor cell growth and VEGF/PDGF expression in human pancreatic cancer. Int J Mol Sci. 2016; 17:2060.
39. Sun Y, Wu C, Ma J, Yang Y, Man X, Wu H, Li S. Toll-like receptor 4 promotes angiogenesis in pancreatic cancer via PI3K/AKT signaling. Exp Cell Res. 2016; 347:274-82. doi: 10.1016/j.yexcr.2016.07.009.
40. Hsiao CC, Chen PH, Cheng CI, Tsai MS, Chang CY, Lu SC, Hsieh MC, Lin YC, Lee PH, Kao YH. Toll-like receptor-4 is a target for suppression of proliferation and chemoresistance in HepG2 hepatoblastoma cells. Cancer Lett. 2015; 368:144-52. doi: 10.1016/j.canlet.2015.08.004.
41. Chicoine MR, Zahner M, Won EK, Kalra RR, Kitamura T, Perry A, Higashikubo R. The in vivo antitumoral effects of lipopolysaccharide against glioblastoma multiforme are mediated in part by Toll-like receptor 4. Neurosurgery. 2007; 60:372-80; discussion 81. doi: 10.1227/01.NEU.0000249280.61761.2E.
42. Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002; 22:2478-86. doi: 20026268.
43. Sarrazy V, Vedrenne N, Billet F, Bordeau N, Lepreux S, Vital A, Jauberteau MO, Desmouliere A. TLR4 signal transduction pathways neutralize the effect of Fas signals on glioblastoma cell proliferation and migration. Cancer Lett. 2011; 311:195-202. doi: 10.1016/j.canlet.2011.07.018.
44. Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002; 61:1013-21.
45. Grauer OM, Molling JW, Bennink E, Toonen LW, Sutmuller RP, Nierkens S, Adema GJ. TLR ligands in the local treatment of established intracerebral murine gliomas. J Immunol. 2008; 181:6720-9.
46. Niranjan R, Nagarajan R, Hanif K, Nath C, Shukla R. LPS induces mediators of neuroinflammation, cell proliferation, and GFAP expression in human astrocytoma cells U373MG: the anti-inflammatory and anti-proliferative effect of guggulipid. Neurol Sci. 2014; 35:409-14. doi: 10.1007/s10072-013-1530-6.
47. Tewari R, Choudhury SR, Ghosh S, Mehta VS, Sen E. Involvement of TNFα-induced TLR4-NF-κB and TLR4-HIF-1α feed-forward loops in the regulation of inflammatory responses in glioma. J Mol Med (Berl). 2012; 90:67-80. doi: 10.1007/s00109-011-0807-6.
48. Lee SY, Lee HY, Kim SD, Jo SH, Shim JW, Lee HJ, Yun J, Bae YS. Lysophosphatidylserine stimulates chemotactic migration in U87 human glioma cells. Biochem Biophys Res Commun. 2008; 374:147-51. doi: 10.1016/j.bbrc.2008.06.117.
49. Kim SS, Pirollo KF, Chang EH. Isolation and culturing of glioma cancer stem cells. Curr Protoc Cell Biol. 2015; 67: 23.10.1-10. doi: 10.1002/0471143030.cb2310s67.
50. Yao Y, Ye H, Qi Z, Mo L, Yue Q, Baral A, Hoon DS, Vera JC, Heiss JD, Chen CC, Hua W, Zhang J, Jin K, et al. B7-H4(B7x)-mediated cross-talk between glioma-initiating cells and macrophages via the IL6/JAK/STAT3 pathway lead to poor prognosis in glioma patients. Clin Cancer Res. 2016; 22:2778-90. doi: 10.1158/1078-0432.CCR-15-0858.
51. Tchirkov A, Khalil T, Chautard E, Mokhtari K, Veronese L, Irthum B, Vago P, Kemeny JL, Verrelle P. Interleukin-6 gene amplification and shortened survival in glioblastoma patients. Br J Cancer. 2007; 96:474-6. doi: 10.1038/sj.bjc.6603586.
52. Yeung YT, Bryce NS, Adams S, Braidy N, Konayagi M, McDonald KL, Teo C, Guillemin GJ, Grewal T, Munoz L. p38 MAPK inhibitors attenuate pro-inflammatory cytokine production and the invasiveness of human U251 glioblastoma cells. J Neurooncol. 2012; 109:35-44. doi: 10.1007/s11060-012-0875-7.
53. Liu Q, Li G, Li R, Shen J, He Q, Deng L, Zhang C, Zhang J. IL-6 promotion of glioblastoma cell invasion and angiogenesis in U251 and T98G cell lines. J Neurooncol. 2010; 100:165-76. doi: 10.1007/s11060-010-0158-0.
54. Zhang J, Sarkar S, Cua R, Zhou Y, Hader W, Yong VW. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis. 2012; 33:312-9. doi: 10.1093/carcin/bgr289.
55. Jin X, Kim SH, Jeon HM, Beck S, Sohn YW, Yin J, Kim JK, Lim YC, Lee JH, Kim SH, Kang SH, Pian X, Song MS, et al. Interferon regulatory factor 7 regulates glioma stem cells via interleukin-6 and Notch signalling. Brain. 2012; 135:1055-69. doi: 10.1093/brain/aws028.
56. de la Iglesia N, Konopka G, Lim KL, Nutt CL, Bromberg JF, Frank DA, Mischel PS, Louis DN, Bonni A. Deregulation of a STAT3-interleukin 8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J Neurosci. 2008; 28:5870-8. doi: 10.1523/JNEUROSCI.5385-07.2008.
57. Bonavia R, Inda MM, Vandenberg S, Cheng SY, Nagane M, Hadwiger P, Tan P, Sah DW, Cavenee WK, Furnari FB. EGFRvIII promotes glioma angiogenesis and growth through the NF-κB, interleukin-8 pathway. Oncogene. 2012; 31:4054-66. doi: 10.1038/onc.2011.563.
58. Raychaudhuri B, Vogelbaum MA. IL-8 is a mediator of NF-κB induced invasion by gliomas. J Neurooncol. 2011; 101:227-35. doi: 10.1007/s11060-010-0261-2.
59. Brat DJ, Bellail AC, Van Meir EG. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol. 2005; 7:122-33. doi: 10.1215/S1152851704001061.
60. Wensink AC, Kemp V, Fermie J, Garcia Laorden MI, van der Poll T, Hack CE, Bovenschen N. Granzyme K synergistically potentiates LPS-induced cytokine responses in human monocytes. Proc Natl Acad Sci U S A. 2014; 111:5974-9. doi: 10.1073/pnas.1317347111.
61. Guzzo C, Ayer A, Basta S, Banfield BW, Gee K. IL-27 enhances LPS-induced proinflammatory cytokine production via upregulation of TLR4 expression and signaling in human monocytes. J Immunol. 2012; 188:864-73. doi: 10.4049/jimmunol.1101912.
62. Honda K, Wada H, Nakamura M, Nakamoto K, Inui T, Sada M, Koide T, Takata S, Yokoyama T, Saraya T, Kurai D, Ishii H, Goto H, et al. IL-17A synergistically stimulates TNF α-induced IL-8 production in human airway epithelial cells: a potential role in amplifying airway inflammation. Exp Lung Res. 2016; 42:205-16. doi: 10.1080/01902148.2016.1190796.
63. O'Dwyer AM, Lajczak NK, Keyes JA, Ward JB, Greene CM, Keely SJ. Ursodeoxycholic acid inhibits TNF α-induced IL-8 release from monocytes. Am J Physiol Gastrointest Liver Physiol. 2016; 311:G334-41. doi: 10.1152/ajpgi.00406.2015.
64. Li C, Li H, Jiang K, Li J, Gai X. TLR4 signaling pathway in mouse Lewis lung cancer cells promotes the expression of TGF-beta1 and IL-10 and tumor cells migration. Biomed Mater Eng. 2014; 24:869-75. doi: 10.3233/BME-130879.
65. Park GB, Song H, Kim YS, Sung M, Ryu JW, Lee HK, Cho DH, Kim D, Lee WJ, Hur DY. Cell cycle arrest induced by engagement of B7-H4 on Epstein-Barr virus-positive B-cell lymphoma cell lines. Immunology. 2009; 128:360-8. doi: 10.1111/j.1365-2567.2009.03111.x.
66. Hennessy EJ, Sheedy FJ, Santamaria D, Barbacid M, O'Neill LA. Toll-like receptor-4 (TLR4) down-regulates microRNA-107, increasing macrophage adhesion via cyclin-dependent kinase 6. J Biol Chem. 2011; 286:25531-9. doi: 10.1074/jbc.M111.256206.
67. Darzynkiewicz Z, Zhao H, Zhang S, Lee MY, Lee EY, Zhang Z. Initiation and termination of DNA replication during S phase in relation to cyclins D1, E and A, p21WAF1, Cdt1 and the p12 subunit of DNA polymerase delta revealed in individual cells by cytometry. Oncotarget. 2015; 6:11735-50. doi: 10.18632/oncotarget.4149.
68. Sun Z, Luo Q, Ye D, Chen W, Chen F. Role of toll-like receptor 4 on the immune escape of human oral squamous cell carcinoma and resistance of cisplatin-induced apoptosis. Mol Cancer. 2012; 11:33. doi: 10.1186/1476-4598-11-33.
69. Liao T, Wen D, Ma B, Hu JQ, Qu N, Shi RL, Liu L, Guan Q, Li DS, Ji QH. Yes-associated protein 1 promotes papillary thyroid cancer cell proliferation by activating the ERK/MAPK signaling pathway. Oncotarget. 2016; 8:11719–28. doi: 10.18632/oncotarget.14319.
70. Bhatia HS, Baron J, Hagl S, Eckert GP, Fiebich BL. Rice bran derivatives alleviate microglia activation: possible involvement of MAPK pathway. J Neuroinflammation. 2016; 13:148. doi: 10.1186/s12974-016-0615-6.
71. Wang L, Zhao Y, Qian J, Sun L, Lu Y, Li H, Li Y, Yang J, Cai Z, Yi Q. Toll-like receptor-4 signaling in mantle cell lymphoma: effects on tumor growth and immune evasion. Cancer. 2013; 119:782-91. doi: 10.1002/cncr.27792.
72. Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D, Saudemont A, Quesnel B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. 2007; 110:296-304. doi: 10.1182/blood-2006-10-051482.
73. Wang L, Qian J, Lu Y, Li H, Bao H, He D, Liu Z, Zheng Y, He J, Li Y, Neelapu S, Yang J, Kwak LW, et al. Immune evasion of mantle cell lymphoma: expression of B7-H1 leads to inhibited T-cell response to and killing of tumor cells. Haematologica. 2013; 98:1458-66. doi: 10.3324/haematol.2012.071340.