
INTRODUCTION
The evolution of cancer therapy is diverse, and continues to be expanded. Tumor reductive therapies include classic therapies (e.g. surgery, chemotherapy, and radiation therapy), modern local and systemic treatment modalities, (e.g. radiation frequency ablation (RFA), local transarterial chemoembolization (TACE), high intensity focused ultrasound (HIFU)), and drugs that target specific molecules in the cancer cells). All cancer reductive therapies have only one objective: to reduce tumor burden through direct killing of tumor cells. A new category of cancer treatment, immunotherapy, emerged in the last few years and they are categorized separately from cytotoxic treatments (chemo and radiation therapy). The most prominent examples include cellular therapies, such as lymphokine-activated killer (LAK) and chimeric antigen receptor T cells CAR-T) and immune checkpoint inhibitors, such as anti-program cell death (PD)-1 and cytotoxic T lymphocyte antigen (CTLA-4). Other modes of tumor destruction have come into clinical use with time. Examples range from older modalities such as radiation frequency ablation (RFA) to novel techniques such as irreversible electroporation (i.e. NanoKnife). With each new piece of oncology knowledge gained, a multitude of questions follow.
TUMOR REDUCTIVE THERAPY AND ITS CONSEQUENCES
The role of surgery in tumor reductive therapies
Our initial hypotheses of how tumor reductive therapies work were based solely on their physical or chemical mechanisms; however, with time, these hypotheses became increasingly challenged. For example, surgery is the oldest and most promising mode of tumor reduction that achieves clinical cure in many “early stage” patients. But our recent understanding of tumor mobility confounds this statement. While it was assumed that surgery could eradicate early stage cancer because the primary tumor has not yet metastasized, recent studies with more sensitive analysis show that tumor cell dissemination is a very early event, taking place soon after a tumor becomes vascularized [1, 2]. In most cases, tumors cannot grow over 1-2mm in size without an independent blood supply [3, 4]. By that, one can assume that all primary tumors detectable by imaging and fit for surgery should have already spread to distant sites. This indicates that cancer becomes a systemic disease in a very early stage. How is it that a local therapy such as surgery is able to cure this systemic condition? Indeed, assays have detected metastasized tumor cells in bone marrows of patients with various early stage solid tumors [5–9]. Furthermore, cases of organ transplant recipients, transferred from donors whose solid tumors were cured years before, developing cancer, indicate that tumor bearing is a lifetime event even when patients are clinically “cured” [10–12].
The relationship between primary tumor and its metastases
Often, tumor metastases do not appear before the removal of the primary tumor. For example, hepatocellular carcinoma rarely presents with extra-hepatic metastases at diagnosis regardless of how large the primary tumor may be (and they are often quite large). Yet extra-hepatic metastases can develop well after removing the primary tumor. This inhibition of metastasis by primary tumor is well known, and the reason might be due to the production of anti-angiogenesis factors such as angiostatin and endostatin, leading to the inhibitory effect of primary tumor on its metastasis [13, 14]. Yet there is no clear evidence for such factors in patients presenting with primary tumors without metastasis in liver cancer [15], and often a negative correlation was seen in other cancers [16]. Furthermore, clinical applications of these protein molecules do not prevent or eliminate metastases [17].
Cancer chemotherapy and multi-drug resistance (MDR)
While it is understandable that inherent drug resistance is likely correlated with decreased clinical response, the opposite (i.e. sensitivity to chemotherapy drug) is not always true [18]. If chemotherapy is purely direct toxicity on tumor cells, then one would expect that the higher the drug dose, the better response (regardless of patient status); this is simply not always the case in clinical observations and individual patient response to chemotherapy is often unpredictable [19, 20]. A given drug’s clinical efficacy varies greatly among patients bearing similar tumors (e.g. adenocarcinoma of the lung). A patient who fails responding to one drug may respond to another completely unrelated drug. But once the tumor acquires drug resistance, its response to all other drugs decrease significantly. Development of drug resistance has been explained by molecular mechanisms such as proteins of the multiple drug resistance (MDR) gene family, yet there is no clear evidence from clinical samples to verify the overwhelming population of chemotherapy -resistant tumor cells. In fact, studies comparing tumor samples from pre- and post-development of so-called chemotherapy resistance consistently find little change of cellular sensitivity to in vitro drug testing [21–24]. In addition, means of reversing multiple drug resistance have been developing for years but have not made any significant clinical progress [25, 26], challenging whether this explanation is the true mechanism of acquired chemotherapy resistance.
On the other hand, some of the local treatment modalities seem to have systemic effects as well. Two such examples are the abscopal effect of radiation therapy and the RFA down-staging strategy for the treatment of liver cancers before liver transplant. In the first example, radiation treatment of tumor in one location could cause regression of another distant tumor [27–31]. In the second example, treatment of tumor nodules in a diseased liver by RFA followed by removal and replacement with a non-diseased liver prevented tumor recurrence post-transplant [32]. This practice principle cannot be explained solely by tumor burden reduction to the “allowable” tumor size by pre-transplant criteria – regardless of size, the entire tumor in the diseased liver is removed completely during the liver transplant. In fact, research models indicate that RFA may actually promote residual and distant tumor progression due to generation of local wound-healing factors [33]. The contrasting findings indicate that local therapies may not be as local as initially assumed; rather their mechanisms must be further elucidated to optimize treatment.
Altogether, the above phenomena increasingly suggest that the classic tumor reductive therapies (i.e., by reducing tumor burden) may not work as we previously thought. There are some other factors at play that we do not see. At least one of them is antitumor immunity.
THE OLD AND NEW CANCER IMMUNOTHERAPIES
The history of tumor immunotherapy extends beyond all other classic tumor reductive therapies except for surgery. The dream of treating cancer by activating one’s own immune system has continued to linger, but it was not accepted into standard cancer care until most recently. Immunotherapy, as it is called in modern term, was always relevant to never-ending reports of spontaneous tumor regression, albeit rare [35, 36]. It is these observations that encourage the curious minds to try to duplicate the miracles [37].
Immunotherapy using viruses and bacteria
Many biological substances ranging from infectious viruses and bacteria to their cellular components have been tested in cancer patients [38–40], some with striking results. The best known (but not necessarily the earliest) example of immunotherapy is that of Coley’s Toxin in the late 1890’s [34]. Since the identification of lipopolysaccharides (LPS or endotoxin) as the true active ingredient of Coley’s toxin in the 1940’s, scientists have tried to pinpoint its mechanism. The subsequent description of a LPS-induced blood factor that can cause tumor necrosis [41, 42] fanned great enthusiasm in clinical application. It drove immunology into its modern age via the molecular cloning technique initially intended to produce tumor necrosis factor (TNF), interferon-gamma, and IL-2. Cytokines were discovered and created in mass quantity and tested in clinical trials against cancer, hoping to duplicate the miracles of Coley’s Toxin. But when pure TNF was made available for clinical use, we did not have a wonder drug; instead, more mysteries ensued [43]. For example, cachectin, a well-known factor that was associated with cachexia, was found to be the same molecule as TNF [44] as confirmed by clinical experiment [45]. How could a cytokine that is highly toxic and associated with the most deleterious effect of late stage cancer death be the factor that accounts for Coley’s Toxin effect? If not, what are the alternative explanations of the antitumor activity of endotoxins? Furthermore, effects of most of the so-called Biological Response Modifiers (substances that activate host antitumor immunity) have been observed in patients, but vary greatly [46]. This variation was observed in those early Coley’s trials using his toxin [40]. This elicits further questions as to what factor(s) predispose a response to immune stimulation, and whether non-response was due to failed immune activation.
Immunotherapy using vaccines and T cells
Alternative perspectives of immunotherapy arise from other therapeutic strategies, such as tumor vaccines and T cell modulation. With sensitive assays and the precise knowledge of antigens, we saw that modern tumor vaccines did activate specific immune responses in patients [47, 48]. However, the lack of significant overall clinical efficacy triggered tumor immunologists to question this approach [49, 50]. Yet, the belief that the immune system has the power to eradicate cancer was sustained due to occasional outliers of extreme efficacy. For example, though the number of patients was low, recombinant human IL-2 was able to produce dramatic antitumor response in a few patients [51]. The in vitro expansion of tumor-infiltrating T cells from cancer patients followed by re-infusion has resulted in clear clinical responses in some patients [52–55]. But the manipulation of an individual patient’s immune system does not yield the same results as that of another. This has been the enigma all throughout the history of tumor immunology. Today, no one argues against the potential of antitumor immunity, but the reliability of it. The various efforts to activate one’s own immune system to fight cancer, ranging from the amateur approach of Coley’s toxin to highly sophisticated cancer vaccine and tumor-specific T cells, have not yet granted us the key to perfect immunotherapy.
Immunotherapy using immune checkpoints
The recent fanfare for the immune checkpoint therapy, signified by anti-PD-1/PD-L1 antibodies, represents another wave of enthusiasm we have repeatedly seen for cancer immunotherapy. We have added another ammunition in our fight against cancer, one with a novel mechanism that is effective even in patients who have failed all previous therapies. Clinical trials showed that the responses to the new therapies were much broader and lasted longer than past immunotherapies in patients [56–61]. Similar to previous immunotherapy trials, there are several miracles of complete tumor eradication even after the therapy had long stopped [56]. It is this kind of observations that keeps the idea of immunotherapy from dying completely. However, the same question is posed on the variable range of patient responses. The indubitable activation of antitumor immunity in this way (without assistance from any other tumor reductive therapies), tumor regression during the clinical response, followed by gradual loss of efficacy and subsequent tumor relapse in many patients are all events that continue to be observed as a natural course of disease treatment despite persistent therapy [61]. How does the tumor overcome the drug suppression of tumor immunity and halt regression? Furthermore, while it is simply known that the therapy works by PD-L1 inhibition, there are still mysteries shrouding the complete picture of this mechanism. This class of drugs works by removing the blockade of antitumor immunity through PD-L1 expression, similar to removing the brakes on a downward moving truck. That means the truck was already gaining momentum on the slope before it was stopped; antitumor immunity was coexistent with the tumor in order for immune checkpoint therapy to work. If so, why didn’t we see it before? How did this antitumor immunity emerge initially and what possible nurturing effect could it have had on the tumor? While the current state of cancer immunotherapy is no panacea, answers to these questions may help us get slightly closer to just that.
CONCOMITANT ANTITUMOR IMMUNITY: AN INVISIBLE ASPECT OF CANCER MANAGEMENT
Existence of concomitant antitumor immunity
Unless the antigen is known, specific antitumor immunity is not directly measurable, but that does not disprove its existence. If there was no pre-existing antitumor immunity, how could removing its inhibition have worked as a successful therapy? In this case, the presence of a concomitant antitumor immunity is inferred rather than directly detected. Animal tumor models have long demonstrated this in as early as the 1950s. Many experiments have established that the presence of one tumor, when inoculated into a distant physiological site, prevented the growth of the tumor in the remote site [62–66]. Since this phenomenon was found to depend on host T cells and was tumor-specific [67], it was deduced that the induction of host antitumor immunity prevented the second tumor from grafting. Indeed, subsequent studies have shown that immune cells from tumor-bearing mice could be transferred into another naive mouse, and the recipient was able to reject grafting of the same tumor. By carrying out this transfer at different time points during tumor progression, the initiation and decline of antitumor immunity in the tumor-bearing host was reported [68]. The antitumor immunity subsides in the tumor-bearing host, never fully eliminated. It becomes activated when certain treatments are performed on the tumor-bearing mice. For example, antitumor immunity is “restored” when the tumor is removed by surgery; in several studies, the tumor-excised mice resisted when re-challenged with the same tumor [69–71]. Otherwise, antitumor immunity often stays dormant, co-existing with the progressing tumor.
Other studies have also demonstrated that this concomitant antitumor immunity needs to be in place for certain therapies to work. For example, after the identification of TNF, scientists thought that this factor was the explanation of Coley’s toxin because it was the single component that was responsible for the antitumor activity in animal models [72, 73]. However, TNF alone could not duplicate the antitumor activity of LPS despite the fact that it is able to produce tumor necrosis. In order for the tumor to regress completely after administering LPS, the tumor-bearing mice needed to have concomitant antitumor immunity [74]. The same was true when these mice were cured by a combination of chemotherapy and LPS [75].
The facts about concomitant antitumor immunity
Despite these findings from animal studies, concomitant antitumor immunity has not been accepted into considerations for designing clinical treatments in cancer patients. There are a few reasons for this. First, there is a general belief that human cancers are not immunogenic, thus do not carry concomitant antitumor immunity. This concept was derived from an animal study in which “spontaneous” tumors (i.e., tumors arising naturally rather than induced by chemical carcinogens) tend to be less immunogenic or immuno-stimulatory [76]. Since all human tumors arise spontaneously, it was argued that human cancers are not immunogenic. As such, animal models in which antitumor immunity is a critical component are often considered unrealistic or not closely related to human cancers [77].
The second reason may be that this concomitant antitumor immunity in patients is immeasurable by current assay standards. In animal models, simply removing T cells in the host may reveal the effects of antitumor immunity. The tumor tends to progress more rapidly than the controls [78]. It may also be measured by taking T cells from the host to test their reactivity in vitro such as by direct tumor cell killing using cytotoxic T lymphocyte (CTL) assay or measuring cytokine release [79]. Finally, concomitant antitumor immunity is also induced by challenging a tumor-bearing host with the same tumor, or transferring the tumor-bearing host spleen cells to a naive host, followed by tumor challenge and protection assays [80]. But these assays are impractical in human cases. In vitro reactivity of tumor-infiltrating T cells (TIL) has been demonstrated [79], supporting the presence of concomitant antitumor immunity in human patients. But these assays are technologically challenging and cannot be performed on every patient.
Another way that supports concomitant antitumor immunity is the existence of T cell infiltration in tumors from surgery. It has been repeatedly reported that significant T cell presence in solid tumors from surgery correlates with better post-surgery prognosis [81–90]. What this correlation means is that concomitant antitumor immunity is able to protect against post-surgery cancer metastases. But despite repeated publications of similar results, this conclusion has largely been overlooked by the medical society. One of the reasons is the difficulty or subjectivity of interpreting the findings. T cells are often found in tumor samples, but sometimes are associated with poorer prognosis [91, 92]. In addition, the type of T cells may vary greatly ranging from antitumor T cells [93] to suppressive Treg cells [94–96]. Thus, simply estimating the number of intratumor T cells does not predict post-surgery prognosis in one specific patient; however, the overall trend towards a better prognosis is found for multiple tumors with increased T cell presence [97]. On the other hand, since tumors are where antitumor T cells exert their effects, another way to look for the presence of true antitumor T cells in a given tumor would be to quantify them with the negative correlation between tumor infiltrating T cells and tumor growth in T cell-heavy areas [98]. Despite these findings, no current treatment design is based on this analysis. For example, decision for surgery is made on tumor resectability, not whether the patient will likely have post-surgical recurrence and metastases. Even post-surgery treatments are not based on this hypothesis. Therefore, the situation is that on one hand, we see clear presence of concomitant antitumor immunity in cancer patients; on the other hand we do not know how to use this concomitant antitumor immunity for the benefits of the patients. Figure 1 is a diagram summarizing the initiation, establishment, and function of antitumor immunity in cancer patients.
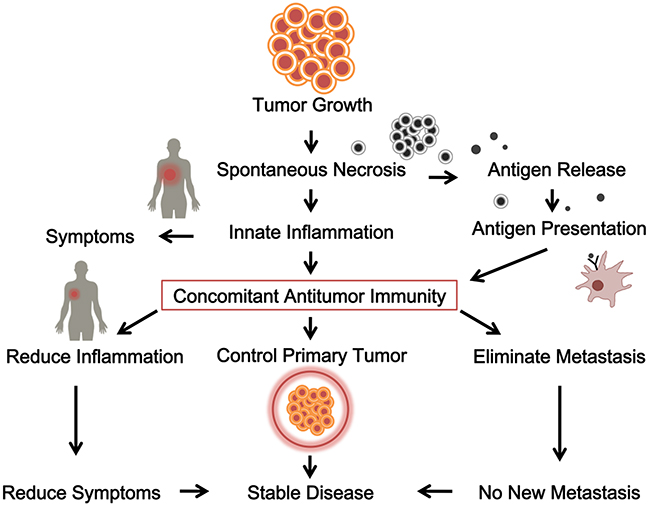
Figure 1: The initiation, establishment and function of a concomitant antitumor immunity in cancer patients. Tumor growth releases antigen and induces innate inflammation, stimulates concomitant antitumor immunity, which contributes to control the primary tumor and eliminates metastasis.
IMMUNOLOGICAL VIEWS OF CANCER SURGERY
Cancer surgery and antitumor immunity
Cancer surgery, once thought to be a simple resection of offending tumors, is intricately related to immunology. The known traditional effects of cancer surgery are as follows: 1) reduction of tumor burden to alleviate symptoms associated with it; 2) prevention of tumor dissemination by removing the source; 3) stimulation of new metastases through wound-healing process. The first two functions are obvious and are the reasons behind many rushed surgeries immediately after diagnosis. The third effect is well-established [99, 100] and is likely the basis for recommendations against surgery on metastatic disease. Early scientists, pressured by the fear of metastasis, assumed that early removal of the primary tumor would prevent cancer cell dissemination. This belief, however, is not supported by facts. Cancer cell dissemination is an early and continuous process that takes place soon after a tumor forms independent blood supply through angiogenesis (2). This complex process involves multiple enzymes and growth factors to facilitate individual cancer cells to move out of the circulation and settle down in remote tissues. This is only half of the metastatic process established; the other half requires the cells to produce sufficient factors to attract blood supply through angiogenesis (3). As such, it is a highly variable process among different tumors, even among disseminated tumor cells from the same primary tumor.
However, the tumors found at the point of clinical diagnosis, especially those that already induce symptoms, have most likely spread into circulation (blood and lymphatics) and established dormant or active micro-metastases at distant organs. Several sensitive assays have found circulating tumor cells in almost all patients at diagnosis of solid tumors [6]. This is also consistent with the observation of tumor-containing organs from clinically cured cancer patients who donated their organs many years after cancer surgery [11]. These findings shape the argument that a clinical cure by surgery is not only due to the tumor being contained in one area, nor is it due to the tumor’s lack of ability to metastasize. How does surgery, a local therapy, cure cancer, which is systemic by nature? The clue comes from the potential role of concomitant antitumor immunity. Animal studies have shown that the presence of antitumor immunity is able to prevent establishment of tumor metastasis [78]. The positive correlation between T cell infiltration and post-surgical disease-free survival suggests that this also takes place in human cancer. Perhaps cancer survivors lived without recurrence not due to the complete surgical resection of the tumor, but rather due to a residual tumor preventing further cancer metastasis with its corresponding antitumor immunity.
Cancer surgery and immunological memory
Each individual patient’s immunity has evolved alongside its target antigens. While the immunity is enhanced with greater antigen levels, it also contracts according to the reduction of its target antigen [101]. The critical aspect is what happens when the antigen is cleared. The formation of immune memory requires the antigen clearance [102]. During a course of infection, successful clearance of the antigen leads to the establishment of immunological memory for that specific antigen. This is the basis for immunization with vaccines. Low-level antigen persistence prevents memory formation and promotes immune exhaustion or tolerance [103, 104]. When these rules are applied to immunity to tumor antigen, we can explain why complete removal of all visible tumor burden (excluding dormant tumor deposits) is critical [66, 105]. Surgery, compared to other forms of tumor reduction therapy, is more suitable for achieving this goal in cases where no established metastases are present. Incomplete tumor resection would create a situation of antigen reduction but not clearance, thus inducing the antitumor immunity to shrink without being able to form a memory mechanism. As a result, the antitumor immunity wanes and becomes ineffective in preventing future metastases [64, 105]. This explains why incomplete cancer surgery is often deleterious than beneficial and underlines the need for complete tumor resection as indicated by cancer surgery guidelines [106]. On the other hand, surgery as a means to reduce tumor burden benefits a patient’s antitumor immunity under certain conditions. For example, under the balance of small antitumor immunity against a large tumor burden, the immunity will likely become exhausted simply because of the overwhelming antigen load [107]. In such situations, surgery can help to significantly tilt the balance toward better disease control due to the pre-surgery concomitant antitumor immunity that needs to be activated [108–110]; otherwise, the antitumor immunity will likely contract with antigen reduction and the patient will lose protection against recurrence and metastases [64]. This explains some of the cases where known incomplete surgery still resulted in disease-free survival. In such situations, surgeons apply electrocauterization in situ that burns and destroys tumor deposits, similar to a microwave or radiofrequency-induced ablation causing in situ release of tumor antigen, coupled with inflammation to present the tumor antigens to further activate antitumor immunity. It is through these immunological impacts that make surgery a means to cure a systemic disease.
As such, the most critical prerequisite for surgery to be effective is the presence of concomitant antitumor immunity. Without it, surgery would result only in local but not systemic tumor control. In fact, without the help of concomitant antitumor immunity, surgery alone may be tumor stimulatory as the research models demonstrate [64, 100], explaining many immediate post-surgical appearances of metastases that were not seen before surgery. In this regard, since tumor resection impacts antitumor immunity by reducing or clearing antigens and achieves curative efficacy through it, we can view classic cancer surgery as a form of immunotherapy. Figure 2 depicts the interaction of various effects of cancer surgery and the possible outcome.
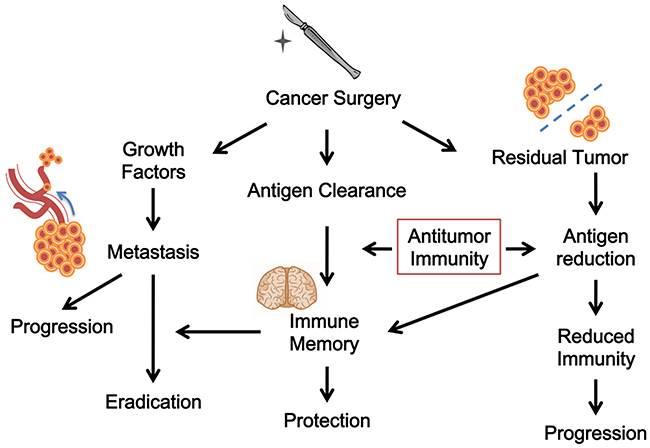
Figure 2: The immunological views of cancer surgery. Surgery may cause antigen clearance and antitumor immunity. It could also be tumor stimulatory. Surgery may have different outcomes because of the interaction of various effects.
IMMUNOLOGICAL VIEWS OF OTHER TUMOR REDUCTIVE THERAPY
Chemotherapy and antitumor immunity
Chemotherapy is another major tumor reductive therapy. Its mechanism is thought to be through cellular toxicity. While it is true that chemotherapy drugs are cytotoxic to cancer cells, the antitumor efficacy, however, may not be attributed entirely to this direct cytotoxicity. In animal models where the difference of efficacy in the presence and absence of host antitumor immunity were compared, chemotherapy was found to be more effective in the presence of immunity [111–113]. More recent studies have confirmed these early findings [114–116] while also detailing the molecular parts of chemotherapy-induced immune activation [117]. When chemotherapy drugs eradicate tumor cells, the cells release antigens via specific tumor death (necrosis or apoptosis) that are detected by antitumor immunity. Because every tumor and host HLA combination is unique, not every tumor death by a given drug will lead to the same antigen release, even with the same drug, same tumor type, and same tumor death mechanism. This variation has already been reported in different animal models arguing whether it is better to induce a necrotic cell death or apoptosis [118, 119]. In most of these studies, the requirement of a pre-existing antitumor immunity (concomitant immunity) has been ignored, presuming that as long as tumor cells die an “immunogenic death”, antitumor immunity will be activated. But this is unlikely true in that de novo activation of antitumor immunity may not be possible and a pre-existing antitumor immunity is necessary [115].
Since not every patient possesses the same conco-mitant antitumor immunity, there is an unpredictability of activation of antitumor immunity. In patients who do not carry concomitant antitumor immunity regardless of the way in which the tumor cells die and release antigens, there would be no activation of immunity due to the lack of responders. Since antitumor immunity may contribute significantly to the overall chemotherapeutic efficacy [113, 115], its presence or absence and the diverse ways of antigen release will likely cause significant variation among patients with the same tumor treated by the same drugs. This in turn may explain the observed variability and unpredictability during cancer chemotherapy. By the same principle, this explanation also postulates that there would not be a single drug that will give consistent responses as long as participation of antitumor immunity is involved. This hypothesis is supported by the observations made with classic chemotherapeutic drugs thus far.
Targeted therapy and antitumor immunity
However, recent drugs of targeted therapy, such as small molecular drugs inhibiting certain tumor proliferation-associated receptor kinases (e.g., EGFR) are the exceptions to this rule. Patients who have certain EGFR mutations respond well to targeted inhibitor drugs [120]. This would argue that such a drug-induced response is not dependent on antitumor immunity. Indeed, in most cases, there are no obvious signs of contribution by antitumor immunity. One pattern of response by this immunity is durability, a continued response long after therapy cessation [121]; this is not the case in most patients taking EGFR inhibitors. Symptomatic response and tumor relapse are often immediate upon initiating and terminating these drugs, respectively. The quick response to targeted therapy is not tumor-burden size dependent, but is likely a result of immediate suppression of tumor-induced local inflammation. This is consistent with the known mechanism of apoptosis-induced killing of tumor cells by these drugs [122, 123]. It is likely that continued tumor apoptosis suppresses rather than stimulates adaptive immunity [124]. But this is not absolute truth; in rare cases with large tumor burdens, even these targeted drugs may induce antitumor immunity (our own observation). The differences in response patterns between classic cytotoxic drugs and modern targeted therapy seem to support the participation of antitumor immunity as a major factor behind chemotherapy, thus arguing that this therapy is, in essence, immunotherapy. Figure 3 is a diagram of this view.
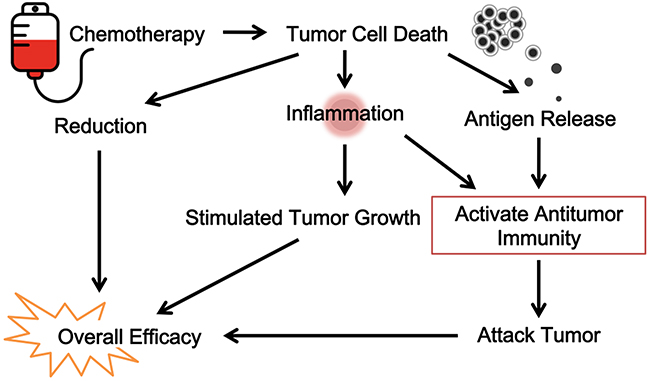
Figure 3: The immunological view of chemotherapy. Classic cytotoxic drugs and modern targeted therapy contributes to antitumor immunity. Chemotherapy is, in essence, immunotherapy.
Radiation therapy and antitumor immunity
The same may be true for classic radiation therapy. The abscopal effect of radiation is a historically consistent clinical observation where treatment of one tumor site induces responses in other distant sites [28, 30, 31, 125]. This effect is shown to be the result of activation of antitumor immunity in animal model and in clinical trials [126, 127]. Since killing tumor cells may result in local inflammation and antigen presentation, it is expected that other local treatments may also activate antitumor immunity. One such example is radiofrequency ablation (RFA). In animal models, RFA has been shown to activate antitumor immunity [128–130]. This provides a good explanation for its application in liver cancer treatment via transplant [131]. Guidelines for liver transplant require that the primary tumor be limited in size because larger tumor nodules correlate with high cancer recurrence following liver transplant. In cases where stable reduction of primary tumor size using RFA could be achieved, subsequent liver transplants had significantly reduced recurrence rate [132]. This long-term post-transplant effect cannot be explained solely by tumor nodule reduction perse, since the diseased liver is removed entirely during transplant. Disease relapse is due to re-establishment of tumor nodules by previously disseminated tumor cells outside the diseased liver; thus prevention or elimination of these newly established tumor metastases is the mechanism of disease control. From an immunotherapy point of view, RFA treatment may activate pre-existing antitumor immunity through inflammation and antigen release. This is the factor behind the control of metastases after a cancerous liver is severed. Following the RFA, there is a stable reduction of tumor burden [133].
Animal experiments show that RFA may also actually stimulate intra-hepatic metastases [33], yet this is not always observed in clinical practice. There is a bigger factor at play in the human physiology. The stable reduction of tumor burden by RFA suggests that antitumor immunity is activated, controlling tumor progression. Indeed, only those patients with stable tumor reduction achieved satisfactory prognosis following liver transplant [134]. RFA is therefore also a test to detect the success of immunity activation. It acts as a form of tumor immunotherapy by concurrently enhancing patient’s own immunity and subsequently controlling tumor metastases post-transplant.
CONCLUSIONS
Classic cancer therapies have been manipulated into various regimens to achieve superior efficacy over nearly a century; but the quiet, incremental breakthroughs in chemotherapy do not result in a glorious outcry by the media every time, or by the patients who benefit from them. We have come to expect instant gratification and consistent remedy from our modern medicine. Yet these therapies cure as many times as they fail. This is the major reason that they are much less appreciated than some of the newer developments (immune checkpoint therapy, for example) although it is highly debatable whether the novel treatments can entirely replace the older ones. These classic therapies all work through activation or preservation of antitumor immunity. The variation seen in patient responses are often due to each patient’s underlying immune status rather than the direct impact of the therapies themselves. In essence, these are immunotherapies.
The status of antitumor immunity in a given patient is determined by many factors and is likely to be unique to each patient. This predisposes each cancer patient to a unique pattern of responses to a commonly applied therapy. For example, a patient with decent concomitant antitumor immunity before surgery should be able to achieve good prognosis with post-surgical, immunity-mediated protection against future metastasis. With tumors such as breast cancer, a total mastectomy may not require post-surgical chemotherapy. On the other hand, patients without sufficient antitumor immunity before surgery should receive chemotherapy to prevent surgery-induced metastases. In these cases, it is vital to determine the status of host antitumor immunity. Current state of clinical testing does not allow measurement of specific antitumor immunity. Therefore, future developments are needed to derive biomarkers that accurately determine a patient’s antitumor immunity status.
Presently, physicians could rely on specific clinical clues to make judgments. For example, a patient with inflammation-induced symptoms that subside naturally under general care is an indication of innate immunity activation, with possible establishment of adaptive antitumor immunity. This could be supported by tests that range from the changes of tumor markers to tumor site metabolism imaging with PET-CT. The appearance of enlarged lymph nodes without metabolic activity under PET-CT may indicate a history of metastases and subsequent control (eradication or suppression) by concomitant antitumor immunity. Activation of this immunity by chemotherapy or radiation followed by surgery may provide a clinical cure. Conversely, a patient presenting with a highly active single primary tumor discovered during regular check-up without any sign of symptoms (inflammation) has likely not established concomitant antitumor immunity. Although the patient may be a good candidate for surgery, the lack of antitumor immunity will not be able to provide post-surgery protection against future metastases. Therefore, when such metastases later arise, the patient will have a bleak outlook for all other therapies.
The abundant animal studies and cases discussed in this review are not only theoretical examples; the clinical evidence is presented to us everyday. Antitumor immunity is intricately woven into every cancer treatment modality in many ways that are still unknown. Future research should focus on solving this puzzle to truly enhance patient’s immunity for cancer cure.
ACKNOWLEDGMENTS
The authors would like to thank Dr. Yangmin Chen of MediMedia Managed Markets (an ICON plc company) for editorial assistance.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, Leach SD, Stanger BZ. EMT and dissemination precede pancreatic tumor formation. Cell. 2012; 148: 349-61.
2. Kang Y, Pantel K. Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell. 2013; 23: 573-81.
3. Holmgren L, O’Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995; 1: 149-53.
4. Demicheli R. Tumour dormancy: findings and hypotheses from clinical research on breast cancer. Semin Cancer Biol. 2001; 11: 297-306.
5. Janni W, Rjosk D, Braun S. Clinical relevance of occult metastatic cells in the bone marrow of patients with different stages of breast cancer. Clin Breast Cancer. 2000; 1: 217-25.
6. Joosse SA, Gorges TM, Pantel K. Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol Med. 2015; 7: 1-11.
7. Riethdorf S, Wikman H, Pantel K. Review: biological relevance of disseminated tumor cells in cancer patients. Int J Cancer. 2008; 123: 1991-2006.
8. Hinz S, Bockhorst J, Roder C, Egberts JH, Schafmayer C, Kuchler T, Becker T, Kalthoff H. Disseminated tumor cells in the bone marrow negatively influence survival after resection of colorectal liver metastases. Ann Surg Oncol. 2012; 19: 2539-46.
9. Shiozawa Y, Taichman RS. Cancer stem cells and the bone marrow microenvironment. Bonekey Rep. 2012; 1: 48-55.
10. Baehner R, Magrane G, Balassanian R, Chang C, Millward C, Wakil AE, Osorio RW, Waldman FM. Donor origin of neuroendocrine carcinoma in 2 transplant patients determined by molecular cytogenetics. Hum Pathol. 2000; 31: 1425-9.
11. MacKie RM, Reid R, Junor B. Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. N Engl J Med. 2003; 348: 567-8.
12. Kim JK, Carmody IC, Cohen AJ, Loss GE. Donor transmission of malignant melanoma to a liver graft recipient: case report and literature review. Clin Transplant. 2009; 23: 571-4.
13. O’Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994; 79: 315-28.
14. O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997; 88: 277-85.
15. Poon RT, Ho JW, Tong CS, Lau C, Ng IO, Fan ST. Prognostic significance of serum vascular endothelial growth factor and endostatin in patients with hepatocellular carcinoma. Br J Surg. 2004; 91: 1354-60.
16. Mo HY, Luo DH, Qiu HZ, Liu H, Chen QY, Tang LQ, Zhong ZL, Huang PY, Zhao ZJ, Zhang CQ, Zhang Y, Mai HQ. Elevated serum endostatin levels are associated with poor survival in patients with advanced-stage nasopharyngeal carcinoma. Clin Oncol (R Coll Radiol). 2013; 25: 308-17.
17. Schellens JH, Ratain MJ. Endostatin: are the 2 years up yet? J Clin Oncol. 2002; 20: 3758-60.
18. Perez-Tomas R. Multidrug resistance: retrospect and prospects in anti-cancer drug treatment. Curr Med Chem. 2006; 13: 1859-76.
19. Lloyd KL, Cree IA, Savage RS. Prediction of resistance to chemotherapy in ovarian cancer: a systematic review. BMC Cancer. 2015; 15: 117.
20. Nieboer P, de Vries EG, Mulder NH, van der Graaf WT. Relevance of high-dose chemotherapy in solid tumours. Cancer Treat Rev. 2005; 31: 210-25.
21. Zwaan CM, Kaspers GJ, Pieters R, Hahlen K, Huismans DR, Zimmermann M, Harbott J, Slater RM, Creutzig U, Veerman AJ. Cellular drug resistance in childhood acute myeloid leukemia is related to chromosomal abnormalities. Blood. 2002; 100: 3352-60.
22. Candoni A, Michelutti A, Simeone E, Damiani D, Baccarani M, Fanin R. Efficacy of liposomal daunorubicin and cytarabine as reinduction chemotherapy in relapsed acute lymphoblastic leukaemia despite expression of multidrug resistance-related proteins. Eur J Haematol. 2006; 77: 293-9.
23. Styczynski J, Wysocki M. Ex vivo drug resistance in childhood acute myeloid leukemia on relapse is not higher than at first diagnosis. Pediatr Blood Cancer. 2004; 42: 195-9.
24. Arts HJ, Katsaros D, de Vries EG, Massobrio M, Genta F, Danese S, Arisio R, Scheper RJ, Kool M, Scheffer GL, Willemse PH, van der Zee AG, Suurmeijer AJ. Drug resistance-associated markers P-glycoprotein, multidrug resistance-associated protein 1, multidrug resistance-associated protein 2, and lung resistance protein as prognostic factors in ovarian carcinoma. Clin Cancer Res. 1999; 5: 2798-805.
25. Modok S, Mellor HR, Callaghan R. Modulation of multidrug resistance efflux pump activity to overcome chemoresistance in cancer. Curr Opin Pharmacol. 2006; 6: 350-4.
26. Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006; 5: 219-34.
27. Mole RH. Whole body irradiation; radiobiology or medicine? Br J Radiol. 1953; 26: 234-41.
28. Nobler MP. The abscopal effect in malignant lymphoma and its relationship to lymphocyte circulation. Radiology. 1969; 93: 410-2.
29. Kingsley DP. An interesting case of possible abscopal effect in malignant melanoma. Br J Radiol. 1975; 48: 863-6.
30. Antoniades J, Brady LW, Lightfoot DA. Lymphangiographic demonstration of the abscopal effect in patients with malignant lymphomas. Int J Radiat Oncol Biol Phys. 1977; 2: 141-7.
31. Ohba K, Omagari K, Nakamura T, Ikuno N, Saeki S, Matsuo I, Kinoshita H, Masuda J, Hazama H, Sakamoto I, Kohno S. Abscopal regression of hepatocellular carcinoma after radiotherapy for bone metastasis. Gut. 1998; 43: 575-7.
32. Hanje AJ, Yao FY. Current approach to down-staging of hepatocellular carcinoma prior to liver transplantation. Curr Opin Organ Transplant. 2008; 13: 234-40.
33. Rozenblum N, Zeira E, Scaiewicz V, Bulvik B, Gourevitch S, Yotvat H, Galun E, Goldberg SN. Oncogenesis: an “off-target” effect of radiofrequency ablation. Radiology. 2015; 276: 426-32.
34. Nauts HC, Swift WE, Coley BL. The treatment of malignant tumors by bacterial toxins as developed by the late William B. Coley, M.D., reviewed in the light of modern research. Cancer Res. 1946; 6: 205-16.
35. Dunphy JE. Some observations on the natural behavior of cancer in man. N Engl J Med. 1950; 242: 167-72.
36. Pectasides E, Miksad R, Pyatibrat S, Srivastava A, Bullock A. Spontaneous regression of hepatocellular carcinoma with multiple lung metastases: a case report and review of the literature. Dig Dis Sci. 2016.
37. Cole WH. Efforts to explain spontaneous regression of cancer. J Surg Oncol. 1981; 17: 201-9.
38. Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007; 15: 651-9.
39. Linnebacher M, Maletzki C, Klier U, Klar E. Bacterial immunotherapy of gastrointestinal tumors. Langenbecks Arch Surg. 2012; 397: 557-68.
40. Wiemann B, Starnes CO. Coley’s toxins, tumor necrosis factor and cancer research: a historical perspective. Pharmacol Ther. 1994; 64: 529-64.
41. Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975; 72: 3666-70.
42. Old LJ. Tumor necrosis factor. Sci Am. 1988; 258: 59-60, 9-75.
43. Gamm H, Lindemann A, Mertelsmann R, Herrmann F. Phase I trial of recombinant human tumour necrosis factor alpha in patients with advanced malignancy. Eur J Cancer. 1991; 27: 856-63.
44. Tracey KJ, Lowry SF, Cerami A. Cachectin: a hormone that triggers acute shock and chronic cachexia. J Infect Dis. 1988; 157: 413-20.
45. Selby P, Hobbs S, Viner C, Jackson E, Jones A, Newell D, Calvert AH, McElwain T, Fearon K, Humphreys J, Shiga T. Tumour necrosis factor in man: clinical and biological observations. Br J Cancer. 1987; 56: 803-8.
46. Bisht M, Bist SS, Dhasmana DC. Biological response modifiers: current use and future prospects in cancer therapy. Indian J Cancer. 2010; 47: 443-51.
47. Butterfield LH. Cancer vaccines. BMJ. 2015; 350: h988.
48. Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001; 411: 380-4.
49. Prehn RT. On the nature of cancer and why anticancer vaccines don’t work. Cancer Cell Int. 2005; 5: 25.
50. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004; 10: 909-15.
51. Lotze MT, Rosenberg SA. Results of clinical trials with the administration of interleukin 2 and adoptive immunotherapy with activated cells in patients with cancer. Immunobiology. 1986; 172: 420-37.
52. Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986; 233: 1318-21.
53. Aebersold P, Hyatt C, Johnson S, Hines K, Korcak L, Sanders M, Lotze M, Topalian S, Yang J, Rosenberg SA. Lysis of autologous melanoma cells by tumor-infiltrating lymphocytes: association with clinical response. J Natl Cancer Inst. 1991; 83: 932-7.
54. Arienti F, Belli F, Rivoltini L, Gambacorti-Passerini C, Furlan L, Mascheroni L, Prada A, Rizzi M, Marchesi E, Vaglini M, Parmiani G, Cascinelli N. Adoptive immunotherapy of advanced melanoma patients with interleukin-2 (IL-2) and tumor-infiltrating lymphocytes selected in vitro with low doses of IL-2. Cancer Immunol Immunother. 1993; 36: 315-22.
55. Goedegebuure PS, Douville LM, Li H, Richmond GC, Schoof DD, Scavone M, Eberlein TJ. Adoptive immunotherapy with tumor-infiltrating lymphocytes and interleukin-2 in patients with metastatic malignant melanoma and renal cell carcinoma: a pilot study. J Clin Oncol. 1995; 13: 1939-49.
56. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, Leming PD, Lipson EJ, Puzanov I, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014; 32: 1020-30.
57. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, Minor D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015; 372: 2006-17.
58. Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr, Lao CD, Linette GP, Thomas L, Lorigan P, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015; 16: 375-84.
59. Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, Horn L, Lena H, Minenza E, Mennecier B, Otterson GA, Campos LT, Gandara DR, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 2015; 16: 257-65.
60. Sundar R, Cho BC, Brahmer JR, Soo RA. Nivolumab in NSCLC: latest evidence and clinical potential. Ther Adv Med Oncol. 2015; 7: 85-96.
61. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, Barlesi F, Kohlhaufl M, Arrieta O, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015; 373: 1627-39.
62. Foley EJ. Antigenic properties of methylcholanthrene-induced tumors in mice of the strain of origin. Cancer Res. 1953; 13: 835-7.
63. Klein G, Sjogren HO, Klein E, Hellstrom KE. Demonstration of resistance against methylcholanthrene-induced sarcomas in the primary autochthonous host. Cancer Res. 1960; 20: 1561-72.
64. Gershon RK, Carter RL, Kondo K. Immunologic defenses against metastases: impairment by excision of an allotransplanted lymphoma. Science. 1968; 159: 646-8.
65. Deckers PJ, Edgerton BW, Thomas BS, Pilch YH. The adoptive transfer of concomitant immunity to murine tumor isografts with spleen cells from tumor-bearing animals. Cancer Res. 1971; 31: 734-42.
66. Vaage J. Concomitant immunity and specific depression of immunity by residual or reinjected syngeneic tumor tissue. Cancer Res. 1971; 31: 1655-62.
67. Bard DS, Pilch YH. The role of the spleen in the immunity to a chemically induced sarcoma in C3H mice. Cancer Res. 1969; 29: 1125-31.
68. North RJ, Bursuker I. Generation and decay of the immune response to a progressive fibrosarcoma. I. Ly-1+2- suppressor T cells down-regulate the generation of Ly-1-2+ effector T cells. J Exp Med. 1984; 159: 1295-311.
69. Salvadori S, Martinelli G, Zier K. Resection of solid tumors reverses T cell defects and restores protective immunity. J Immunol. 2000; 164: 2214-20.
70. Mullen CA, Rowley DA, Schreiber H. Highly immunogenic regressor tumor cells can prevent development of postsurgical tumor immunity. Cell Immunol. 1989; 119: 101-13.
71. Bursuker I, North RJ. Immunological consequences of tumor excision: from active immunity to immunological memory. Int J Cancer. 1986; 37: 275-81.
72. Berendt MJ, North RJ, Kirstein DP. The immunological basis of endotoxin-induced tumor regression. Requirement for T-cell-mediated immunity. J Exp Med. 1978; 148: 1550-9.
73. North RJ. The therapeutic significance of concomitant antitumor immunity. II. Passive transfer of concomitant immunity with Ly-1+2- T cells primes established tumors in T cell-deficient recipients for endotoxin-induced regression. Cancer Immunol Immunother. 1984; 18: 75-9.
74. Havell EA, Fiers W, North RJ. The antitumor function of tumor necrosis factor (TNF), I. Therapeutic action of TNF against an established murine sarcoma is indirect, immunologically dependent, and limited by severe toxicity. J Exp Med. 1988; 167: 1067-85.
75. Dye ES, North RJ. Macrophage accumulation in murine ascites tumors. I. Cytoxan-induced dominance of macrophages over tumor cells and the anti-tumor effect of endotoxin. J Immunol. 1980; 125: 1650-7.
76. Prehn RT, Main JM. Immunity to methylcholanthrene-induced sarcomas. J Natl Cancer Inst. 1957; 18: 769-78.
77. Prehn RT. Tumor immunogenicity: how far can it be pushed? Proc Natl Acad Sci U S A. 1993; 90: 4332-3.
78. Ostrand-Rosenberg S, Grusby MJ, Clements VK. Cutting edge: STAT6-deficient mice have enhanced tumor immunity to primary and metastatic mammary carcinoma. J Immunol. 2000; 165: 6015-9.
79. Barth RJ Jr, Mule JJ, Spiess PJ, Rosenberg SA. Interferon gamma and tumor necrosis factor have a role in tumor regressions mediated by murine CD8+ tumor-infiltrating lymphocytes. J Exp Med. 1991; 173: 647-58.
80. North RJ. The therapeutic significance of concomitant antitumor immunity. I. LY-1-2+ T cells from mice with a progressive tumor can cause regression of an established tumor in gamma-irradiated recipients. Cancer Immunol Immunother. 1984; 18: 69-74.
81. Hakansson L, Adell G, Boeryd B, Sjogren F, Sjodahl R. Infiltration of mononuclear inflammatory cells into primary colorectal carcinomas: an immunohistological analysis. Br J Cancer. 1997; 75: 374-80.
82. Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, Meatchi T, Bruneval P, Cugnenc PH, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005; 353: 2654-66.
83. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, Rubin SC, Coukos G. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003; 348: 203-13.
84. Tsiatas ML, Gyftaki R, Liacos C, Politi E, Rodolakis A, Dimopoulos MA, Bamias A. Study of T lymphocytes infiltrating peritoneal metastases in advanced ovarian cancer: associations with vascular endothelial growth factor levels and prognosis in patients receiving platinum-based chemotherapy. Int J Gynecol Cancer. 2009; 19: 1329-34.
85. Kondo T, Nakazawa H, Ito F, Hashimoto Y, Osaka Y, Futatsuyama K, Toma H, Tanabe K. Favorable prognosis of renal cell carcinoma with increased expression of chemokines associated with a Th1-type immune response. Cancer Sci. 2006; 97: 780-6.
86. Khan H, Pillarisetty VG, Katz SC. The prognostic value of liver tumor T cell infiltrates. J Surg Res. 2014; 191: 189-95.
87. Ibrahim EM, Al-Foheidi ME, Al-Mansour MM, Kazkaz GA. The prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancer: a meta-analysis. Breast Cancer Res Treat. 2014; 148: 467-76.
88. Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN, Martino S, Wang M, Jones VE, Saphner TJ, Wolff AC, Wood WC, Davidson NE, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol. 2014; 32: 2959-66.
89. Hotta K, Sho M, Fujimoto K, Shimada K, Yamato I, Anai S, Konishi N, Hirao Y, Nonomura K, Nakajima Y. Prognostic significance of CD45RO+ memory T cells in renal cell carcinoma. Br J Cancer. 2011; 105: 1191-6.
90. Lee HE, Chae SW, Lee YJ, Kim MA, Lee HS, Lee BL, Kim WH. Prognostic implications of type and density of tumour-infiltrating lymphocytes in gastric cancer. Br J Cancer. 2008; 99: 1704-11.
91. Maluf PJ, Michelin MA, Etchebehere RM, Adad SJ, Murta EF. T lymphocytes (CD3) may participate in the recurrence of cervical intraepithelial neoplasia grade III. Arch Gynecol Obstet. 2008; 278: 525-30.
92. Carvalho MI, Pires I, Prada J, Queiroga FL. A role for T-lymphocytes in human breast cancer and in canine mammary tumors. Biomed Res Int. 2014; 2014: 130894.
93. Liu S, Lachapelle J, Leung S, Gao D, Foulkes WD, Nielsen TO. CD8+ lymphocyte infiltration is an independent favorable prognostic indicator in basal-like breast cancer. Breast Cancer Res. 2012; 14: R48.
94. Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, Platell C, Iacopetta B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol. 2009; 27: 186-92.
95. Shen Z, Zhou S, Wang Y, Li RL, Zhong C, Liang C, Sun Y. Higher intratumoral infiltrated Foxp3+ Treg numbers and Foxp3+/CD8+ ratio are associated with adverse prognosis in resectable gastric cancer. J Cancer Res Clin Oncol. 2010; 136: 1585-95.
96. Liu S, Foulkes WD, Leung S, Gao D, Lau S, Kos Z, Nielsen TO. Prognostic significance of FOXP3+ tumor-infiltrating lymphocytes in breast cancer depends on estrogen receptor and human epidermal growth factor receptor-2 expression status and concurrent cytotoxic T-cell infiltration. Breast Cancer Res. 2014; 16: 432.
97. Gooden MJ, de Bock GH, Leffers N, Daemen T, Nijman HW. The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br J Cancer. 2011; 105: 93-103.
98. Garnelo M, Tan A, Her Z, Yeong J, Lim CJ, Chen J, Lim KH, Weber A, Chow P, Chung A, Ooi LL, Toh HC, Heikenwalder M, et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut. 2017; 66:342-351.
99. Gunduz N, Fisher B, Saffer EA. Effect of surgical removal on the growth and kinetics of residual tumor. Cancer Res. 1979; 39: 3861-5.
100. Ben-Eliyahu S. The promotion of tumor metastasis by surgery and stress: immunological basis and implications for psychoneuroimmunology. Brain Behav Immun. 2003; 17: S27-36.
101. Zinkernagel RM. Localization dose and time of antigens determine immune reactivity. Semin Immunol. 2000; 12: 163-71; discussion 257-344.
102. Wherry EJ, Barber DL, Kaech SM, Blattman JN, Ahmed R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc Natl Acad Sci U S A. 2004; 101: 16004-9.
103. Raimondi G, Zanoni I, Citterio S, Ricciardi-Castagnoli P, Granucci F. Induction of peripheral T cell tolerance by antigen-presenting B cells. II. Chronic antigen presentation overrules antigen-presenting B cell activation. J Immunol. 2006; 176: 4021-8.
104. Wiesel M, Walton S, Richter K, Oxenius A. Virus-specific CD8 T cells: activation, differentiation and memory formation. APMIS. 2009; 117: 356-81.
105. Huang X, Yang Y. The fate of effector CD8 T cells in vivo is controlled by the duration of antigen stimulation. Immunology. 2006; 118: 361-71.
106. British Thoracic Society, Society of Cardiothoracic Surgeons of Great Britain and Ireland Working Party. BTS guidelines: guidelines on the selection of patients with lung cancer for surgery. Thorax. 2001; 56: 89-108.
107. Kim PS, Ahmed R. Features of responding T cells in cancer and chronic infection. Curr Opin Immunol. 2010; 22: 223-30.
108. Fisher B, Gebhardt M, Saffer E. Further observations on the inhibition of tumor growth by C. parvum with cyclophosphamide. VII. Effect of treatment prior to primary tumor removal on the growth of distant tumor. Cancer. 1979; 43: 451-8.
109. Fisher SA, Cleaver A, Lakhiani DD, Khong A, Connor T, Wylie B, Lesterhuis WJ, Robinson BW, Lake RA. Neoadjuvant anti-tumor vaccination prior to surgery enhances survival. J Transl Med. 2014; 12: 245.
110. Grinshtein N, Bridle B, Wan Y, Bramson JL. Neoadjuvant vaccination provides superior protection against tumor relapse following surgery compared with adjuvant vaccination. Cancer Res. 2009; 69: 3979-85.
111. Ferrer JF, Mihich E. Antitumor effects of kethoxal-bis(thiosemicarbazone) and 6-mercaptopurine in neonatally thymectomized mice. Proc Soc Exp Biol Med. 1967; 124: 939-44.
112. Mihich E. Combined effects of chemotherapy and immunity against leukemia L1210 in DBA-2 mice. Cancer Res. 1969; 29: 848-54.
113. Mihich E. Modification of tumor regression by immunologic means. Cancer Res. 1969; 29: 2345-50.
114. van der Most RG, Currie A, Robinson BW, Lake RA. Cranking the immunologic engine with chemotherapy: using context to drive tumor antigen cross-presentation towards useful antitumor immunity. Cancer Res. 2006; 66: 601-4.
115. Zhang L, Feng D, Yu LX, Tsung K, Norton JA. Preexisting antitumor immunity augments the antitumor effects of chemotherapy. Cancer Immunol Immunother. 2013; 62: 1061-71.
116. Tsung K, Norton JA. An immunological view of chemotherapy. Immunotherapy. 2015; 7: 941-3.
117. Zitvogel L, Apetoh L, Ghiringhelli F, Andre F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success? J Clin Invest. 2008; 118: 1991-2001.
118. Nowak AK, Lake RA, Marzo AL, Scott B, Heath WR, Collins EJ, Frelinger JA, Robinson BW. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J Immunol. 2003; 170: 4905-13.
119. Kepp O, Tesniere A, Zitvogel L, Kroemer G. The immunogenicity of tumor cell death. Curr Opin Oncol. 2009; 21: 71-6.
120. Linardou H, Dahabreh IJ, Bafaloukos D, Kosmidis P, Murray S. Somatic EGFR mutations and efficacy of tyrosine kinase inhibitors in NSCLC. Nat Rev Clin Oncol. 2009; 6: 352-66.
121. Zhang L, Feng D, Hu Y, Tsung K, Norton JA. IL-12 augments antitumor responses to cycled chemotherapy. J Immunother. 2015; 38: 137-44.
122. Tracy S, Mukohara T, Hansen M, Meyerson M, Johnson BE, Janne PA. Gefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res. 2004; 64: 7241-4.
123. Takeuchi K, Ito F. EGF receptor in relation to tumor development: molecular basis of responsiveness of cancer cells to EGFR-targeting tyrosine kinase inhibitors. FEBS J. 2010; 277: 316-26.
124. Kushwah R, Wu J, Oliver JR, Jiang G, Zhang J, Siminovitch KA, Hu J. Uptake of apoptotic DC converts immature DC into tolerogenic DC that induce differentiation of Foxp3+ Treg. Eur J Immunol. 2010; 40: 1022-35.
125. Nam SW, Han JY, Kim JI, Park SH, Cho SH, Han NI, Yang JM, Kim JK, Choi SW, Lee YS, Chung KW, Sun HS. Spontaneous regression of a large hepatocellular carcinoma with skull metastasis. J Gastroenterol Hepatol. 2005; 20: 488-92.
126. Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, Formenti SC. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004; 58: 862-70.
127. Golden EB, Chhabra A, Chachoua A, Adams S, Donach M, Fenton-Kerimian M, Friedman K, Ponzo F, Babb JS, Goldberg J, Demaria S, Formenti SC. Local radiotherapy and granulocyte-macrophage colonystimulating factor to generate abscopal responses in patients with metastatic solid tumours: a proof-of-principle trial. Lancet Oncol. 2015; 16: 975-803.
128. Wissniowski TT, Hansler J, Neureiter D, Frieser M, Schaber S, Esslinger B, Voll R, Strobel D, Hahn EG, Schuppan D. Activation of tumor-specific T lymphocytes by radio-frequency ablation of the VX2 hepatoma in rabbits. Cancer Res. 2003; 63: 6496-500.
129. den Brok MH, Sutmuller RP, van der Voort R, Bennink EJ, Figdor CG, Ruers TJ, Adema GJ. In situ tumor ablation creates an antigen source for the generation of antitumor immunity. Cancer Res. 2004; 64: 4024-9.
130. Zerbini A, Pilli M, Penna A, Pelosi G, Schianchi C, Molinari A, Schivazappa S, Zibera C, Fagnoni FF, Ferrari C, Missale G. Radiofrequency thermal ablation of hepatocellular carcinoma liver nodules can activate and enhance tumor-specific T-cell responses. Cancer Res. 2006; 66: 1139-46.
131. Nishikawa H, Kimura T, Kita R, Osaki Y. Radiofrequency ablation for hepatocellular carcinoma. Int J Hyperthermia. 2013; 29: 558-68.
132. Yao FY, Ferrell L, Bass NM, Watson JJ, Bacchetti P, Venook A, Ascher NL, Roberts JP. Liver transplantation for hepatocellular carcinoma: expansion of the tumor size limits does not adversely impact survival. Hepatology. 2001; 33: 1394-403.
133. Yao FY. Liver transplantation for hepatocellular carcinoma: beyond the Milan criteria. Am J Transplant. 2008; 8: 1982-9.
134. Ravaioli M, Grazi GL, Piscaglia F, Trevisani F, Cescon M, Ercolani G, Vivarelli M, Golfieri R, D’Errico Grigioni A, Panzini I, Morelli C, Bernardi M, Bolondi L, Pinna AD. Liver transplantation for hepatocellular carcinoma: results of down-staging in patients initially outside the Milan selection criteria. Am J Transplant. 2008; 8: 2547-57.