
Introduction
Breast cancer is the most common cancer worldwide in women and contributed more than 25% of the total number of new cancer cases diagnosed in 2012 [1]. The mutation frequency of the tumor suppressor p53 is relatively low in breast cancer compared to other solid tumors [2]; however, this mutation is the second most frequent genetic alteration observed in 30-35% of breast cancer cases [3, 4]. A mutation in p53 is also associated with aggressive subtypes, i.e., 12% in luminal A, 32% in luminal B, 75% in HER2, and 84% in triple-negative tumors [3]. Previous studies have shown that p53 mutation is an independent marker of poor prognosis in breast cancers [5] and is also associated with the response to specific treatment regimens in breast cancer [6]. p53 functions as a transcription factor, and its target genes are involved in several pathways, including cell stemness, extracellular matrix maintenance, cell adhesion, and cell motility [7]. Thus, the dysregulation of p53 targets via p53 inactivation may be related to the poor prognosis of breast cancer patients. We have previously identified a number of p53 targets and elucidated the molecular mechanism by which p53 regulates apoptosis, the cell cycle, senescence, iron metabolism, and post-translational modifications [8-10]. However, the role of p53 in breast carcinogenesis has not been fully elucidated. Thus, the identification of p53 targets in breast tissues is important to understand the pathogenesis of breast cancer.
COL17A1 encodes Collagen XVII (COL17; formerly known as BP180 or BPAG2), a transmembrane protein. COL17 is an essential component of type I hemidesmosomes (HDs) and functions as a cell-matrix adhesion molecule [11]. COL17 is highly expressed in tissues with a prominent epithelial component, including the mammary gland [12]. Autoimmunity to COL17 and mutations in COL17A1 result in blistering skin diseases caused by a loss of attachment between the epidermis and the underlying basement membrane [13, 14]. Type I HDs were observed in normal epithelial cells but were lost in cancer cells, including invasive breast cancer [15], and pancreatic ductal epithelium [16]. COL17 has been previously reported as a down-regulated protein signature in stage II compared to premalignant cells and in premalignant cells compared to normal myoepithelial cells [17]. Although recent studies have suggested a role of COL17 in cell migration [18-20], its function in breast carcinogenesis has never been investigated. Here, we revealed COL17A1 as a novel downstream target gene of p53 that is suppressed in breast cancer tissues with a p53 mutation.
Results
Correlation of COL17A1 and p53 status
To identify p53 targets in breast tissues, we performed transcriptome analysis using non-tumorigenic breast epithelial cell lines with or without wild-type p53 (MCF10A p53+/+ and MCF10A p53−/−, respectively). MCF10A p53+/+ and MCF10A p53−/− cells were treated with 0.5 μg/ml of doxorubicin (trade name Adriamycin®, ADR). Total RNA was isolated at 12, 24, and 48 hours after ADR treatment and then subjected to cDNA microarray analysis. We identified 209 genes that were up-regulated more than 3-fold (P < 0.05) at 12, 24, or 48 hours after ADR treatment in MCF10A p53+/+ cells compared to MCF10A p53+/+ cells without ADR treatment or MCF10A p53−/− cells at any timepoint (Figure 1A). Second, we used the data obtained from a breast invasive carcinoma cohort (BRCA), TCGA. We selected 735 genes that were up-regulated more than two-fold in breast cancer tissues with wild-type p53 compared to those with mutant p53 (P < 0.05, Figure 1A). The 17 overlapping genes in the two analyses, including 7 reported p53 target genes, is shown in Table 1, and those results are displayed in Supplementary Figures 1 and 2. Among them, 16 genes except SYTL2 exhibited significant association even after Bonferroni correction (P < 0.05/209) using the number of genes those showed significant association in the first screening (MCF10A p53+/+ and MCF10A p53−/− cells), indicating low possibility of false positive associations. Therefore, this set of genes is likely to be regulated by p53 both in vitro and in vivo.
Table 1: List of 17 p53 target gene candidates from the screen (Figure 1A)
Gene |
Description |
MCF10A |
TCGA |
Accession No. |
Reference PMID* |
||
FCa |
P-valueb |
FCc |
P-valueb |
||||
ACER2* |
alkaline ceramidase 2 |
7 |
0.0320 |
2 |
1.83E-27 |
NM_001010887 |
26943039 |
BTG2* |
BTG anti-proliferation factor 2 |
17 |
0.0382 |
3 |
3.47E-44 |
NM_006763 |
11814693, 8944033 |
COL17A1 |
collagen type XVII alpha 1 chain |
7 |
0.0284 |
3 |
1.44E-04 |
NM_000494 |
|
CROT |
carnitine O-octanoyltransferase |
4 |
1.80E-05 |
2 |
5.56E-06 |
NM_001243745 |
|
EDA2R* |
ectodysplasin A2 receptor |
5 |
2.21E-05 |
2 |
1.51E-46 |
NM_001242310 |
19543321 |
FAM198B |
family with sequence similarity 198 member B |
78 |
0.0154 |
2 |
2.36E-14 |
NM_016613 |
|
GDF15* |
growth differentiation factor 15 |
52 |
0.0036 |
3 |
1.35E-12 |
NM_004864 |
17276395 |
GLS2* |
glutaminase 2 |
51 |
0.0438 |
3 |
3.41E-21 |
NM_013267 |
20351271 |
GREB1 |
growth regulation by estrogen in breast cancer 1 |
13 |
0.0260 |
8 |
8.25E-23 |
NM_014668 |
|
LAMA3 |
laminin subunit alpha 3 |
4 |
0.0405 |
3 |
4.88E-08 |
NM_198129 |
|
MICALCL |
MICAL C-terminal like |
4 |
0.0457 |
2 |
3.88E-10 |
NM_032867 |
|
NPY1R |
neuropeptide Y receptor Y1 |
4 |
0.0346 |
5 |
2.34E-13 |
NM_000909 |
|
SLC27A2 |
solute carrier family 27 member 2 |
4 |
0.0342 |
3 |
3.55E-09 |
NM_003645 |
|
SPATA18* |
spermatogenesis associated 18 |
45 |
0.0437 |
3 |
1.52E-37 |
NM_145263 |
21300779 |
SYTL2 |
synaptotagmin like 2 |
5 |
0.0337 |
3 |
0.0042 |
NM_032943 |
|
TNFRSF10C* |
TNF receptor superfamily member 10c (TRID, TRAIL-R3) |
162 |
0.0080 |
2 |
6.71E-23 |
NM_003841 |
10435597 |
TSPAN1 |
tetraspanin 1 |
8 |
0.0203 |
4 |
5.57E-09 |
NM_005727 |
|
aFold-change, the up-regulated fold expression after ADR in p53 wild-type cells (see calculation in Material and methods).
bTwo-tailed Student’s t-test P-value.
cFold-change, the increased fold expression in wild-type compared to mutant p53 tumors.
* Reported p53 downstream target gene and the reported reference study.
The overlapping genes from the two analyses were further investigated for enriched biological process using Gene Ontology (GO) enrichment analysis (Figure 1B). Epidermis development and ectoderm development were the most and second most significant GO terms, respectively. COL17A1, LAMA3, and EDA2R were included in both terms. EDA2R (ectodysplasin A2 receptor, XEDAR) is a known p53 target in several cell types including cells in the breast [21, 22]. LAMA3 encodes a subunit of laminin-5 (Laminin-332) whose expression is altered by mutant p53 in a breast cell line [23]. A positive correlation between p53 level and the transcription level of COL17A1 has been suggested by high-throughput analyses [24, 25], although direct regulation of COL17A1 by p53 was not investigated previously.
Our screening indicated a strong correlation between COL17A1 and p53 (Figure 1C, Supplementary Figures 1 and 2). Next, we verified the induction of mRNA and protein expression of COL17A1 in MCF10A p53+/+ cells at several points of time after ADR treatment (Figure 1D and 1E). The p53 levels were gradually induced in accordance with time after ADR treatment (Figure 1E). Similarly, the COL17A1 levels increased with time after ADR treatment in a p53-dependent manner. The qPCR and western blot analyses supported the screening results that COL17A1 expression was regulated by p53.
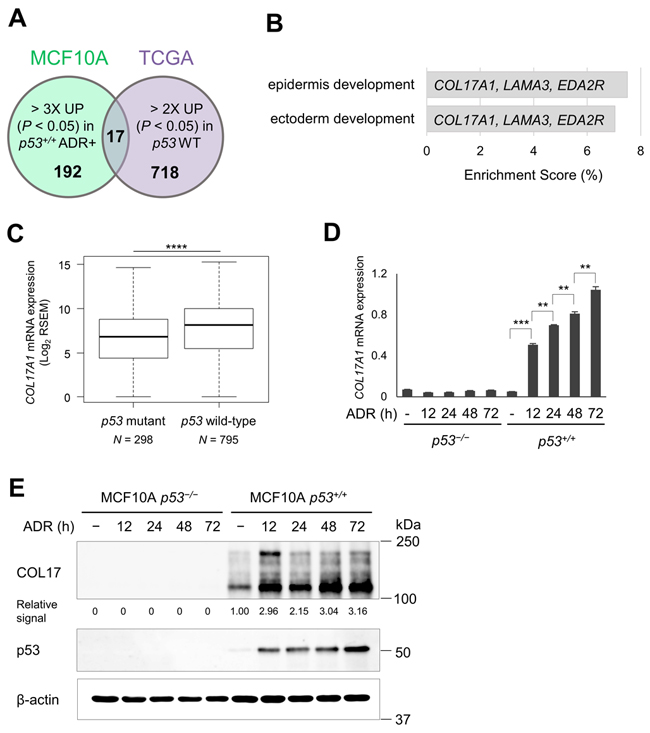
Figure 1: Correlation of COL17A1 and p53 status. A. The p53 targets were screened using MCF10A microarray analysis and invasive breast carcinoma (TCGA). A Venn diagram shows the number of genes that satisfied the inclusion criteria from each analysis and the 17 overlapping genes. Inclusion criteria: MCF10A, > 3-fold up-regulated with P < 0.05 by doxorubicin (ADR+) in p53 wild-type cells (p53+/+); TCGA, > 2-fold up-regulated with P < 0.05 in p53 wild-type (WT) tumors (see Material and methods for details). B. GO biological processes of the 17 overlapping gene set with P < 0.05 and enrichment score > 5% are shown in the bar chart with indicated participating genes. C. Differential mRNA expression of COL17A1 in p53 wild-type and mutant tumors. N, number of tissues. D. The MCF10A cells were treated with 0.5 μg/ml of ADR for 2 hours. Total RNA was isolated at 12, 24, 48 and 72 hours after ADR treatment. The qPCR result shows the relative mRNA expression of COL17A1 normalized by B2M in MCF10A cells bearing p53 wild-type (p53+/+) or knockout (p53−/−), with or without ADR; times (hours) indicate period after ADR treatment. E. Western blot analyses of COL17 and p53 in MCF10A cells treated as described in Figure 1D. β-actin is shown as a loading control. The relative signal of western blot represents protein level of combined 180-kDa and 120-kDa COL17 quantified by ImageJ and normalized by β-actin. Two-tailed Student’s t-test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.00024 (0.05/209).
The p53-dependent induction of COL17A1 in human cells and mouse tissues
Next, we further investigated the relationship between COL17A1 and p53 using human breast cancer cell lines and mouse breast tissues. ADR treatment in HBC4 human breast cancer cells induced an accumulation of p53 protein (Figure 2A). Although p53 protein was not increased in HBL-100 cells (Figure 2A), most likely due to a high basal expression of p53 as previously described [26], phosphorylation of p53 at Ser15, a marker of p53 activation [27], was clearly induced by ADR (Figure 2A). Next, we treated these cells with siRNA against p53 (sip53). Consistent with p53 expression, COL17A1 mRNA and protein expression levels were significantly induced by ADR, and this induction was diminished by sip53 in both cell types (Figure 2A and 2B). We detected both the 180-kDa full-length form of COL17 and its 120-kDa extracellular C-terminal domain (ectodomain) in the cell lysates, whereas only the 120-kDa form was detected in the media (Figure 2A). The ectodomain was generated by constitutive shedding of the full-length form and was released from the cell surface into media [28]. The notably decreased 180-kDa form, with a concomitant increased 120-kD ectodomain, was observed in HBC4 cells treated with ADR and siEGFP (Figure 2A), as observed in keratinocytes [29]. This increased ectodomain shedding can be activated by proinflammatory cytokines [30] which have been reported to be non-targeted induced in some siRNA-treated mammalian systems [31].
In the mouse experiment, 10-week-old p53+/+ and p53−/− mice were exposed to 10 Gy X-ray irradiation. Twenty-four hours after irradiation, mammary tissues were collected for qPCR analysis and immunohistochemistry. The COL17A1 mRNA and protein levels were significantly induced by X-ray in p53+/+ but not p53−/− mice (Figure 2C and 2D). In the mouse mammary gland, elevated COL17 protein levels were detected in myoepithelial cells surrounding secretory luminal cells (Figure 2E) regarding its role as a cell-matrix adhesion molecule. These data demonstrated that COL17A1 expression depends on p53 and is induced by ADR in human breast cell lines and by X-ray in mouse mammary tissues.
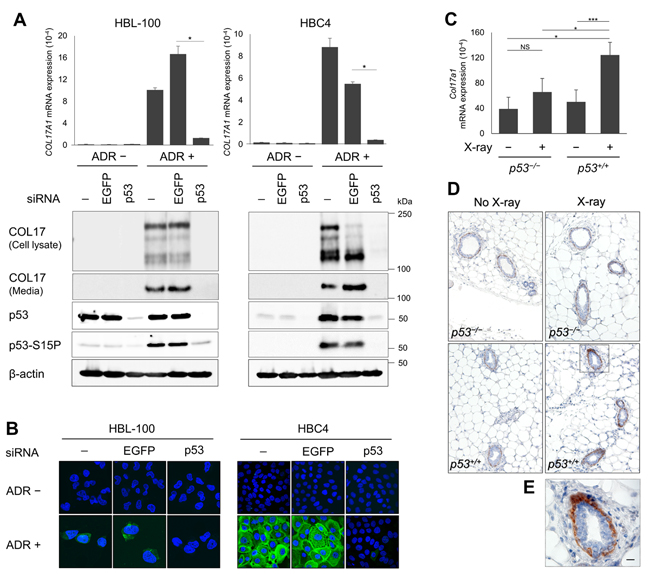
Figure 2: The p53-dependent induction of COL17A1 in human cells and mouse tissues. A. The qPCR and western blot results of COL17A1 in p53 wild-type breast cell lines, HBC4 and HBL-100, transfected with siRNA of either EGFP or p53, with or without doxorubicin (ADR) treatment. The cells were treated with ADR for 2 hours. ADR concentrations were 1 μg/mL for HBL-100 and 2 μg/mL for HBC4. Total RNA and protein were isolated at 48 hours after ADR treatment. siRNA of EGFP was used as a control. Relative mRNA expression of COL17A1 was normalized to ACTB; n = 3. Immunoblotting results of cell lysates with antibodies against COL17 (anti-Collagen XVII), p53, and p53-S15P (p53 phosphorylated at serine 15). Cell media were blotted with anti-Collagen XVII. β-actin is shown as a loading control. B. Immunocytochemistry of HBL-100 (40× magnification) and HBC4 (20× magnification) cells treated as described in Figure 2A. The cells were stained with DAPI (blue) and anti-Collagen XVII (green). C. Relative mRNA expression of Col17a1 normalized to Gapdh in mouse mammary tissues of p53 knockout (p53−/−) or p53 wild-type (p53+/+) mice; with or without X-ray irradiation; n = 3. The mice were sacrificed at 24 hours after X-ray. D. Immunohistochemistry results of mammary tissues staining with anti-Collagen XVII; n = 3 mice per group. Images were obtained at 10× magnification. E. The inset of Figure D, p53+/+ mice with X-ray at 40× magnification. Scale bar, 20 μm. Two-tailed Student’s t-test; *P < 0.05, **P < 0.01, ***P < 0.001; NS, P ≥ 0.05.
COL17A1 is a direct target of p53
To examine whether COL17A1 is a direct target of p53, we surveyed the genomic sequence of the COL17A1 gene and found two putative p53 binding sites, BS1 and BS2, within the first intron (Figure 3A). The DNA fragment was amplified using primers covering both BS1 and BS2 regions and then cloned into pGL4.24 vectors (BS) to evaluate the p53-dependent transcriptional activity using a reporter assay. The luciferase activity of the BS construct was markedly enhanced by co-transfection with wild-type p53 but not with mutant p53, an Arg-to-His substitution at p53 amino acid 175 (p53R175H) (Figure 3B). Next, we introduced mutations at each binding site, resulting in mutations of BS1 (MT1) and BS2 (MT2) (Figure 3A). MT1 and MT2 significantly reduced the luciferase activity compared to that of BS, although a high signal was still detected due to the existence of another non-mutated BS. The luciferase activity of MT2 when co-transfected with p53 was superior to that of MT1 (Figure 3B), suggesting that p53 binds strongly to BS1, most likely because BS1 contains long binding sequences (30 bases) and matched highly to the consensus p53 binding sequence (Figure 1A). Moreover, after two binding sites had been mutated (MT1+2), the luciferase activity was entirely drop to the control level (Figure 3B), suggesting the complete deletion of p53 binding. To further confirm whether p53 can bind to this DNA segment, we performed a chromatin immunoprecipitation (ChIP) assay using HBC4 cells with or without ADR treatment. After precipitation with an anti-p53 antibody, a DNA fragment containing p53 binding sites was quantified by qPCR. The p53 binding site in WAF1, a p53 target gene, was examined as a positive control region (Supplementary Figure 3). The qPCR result using COL17A1 BS primers indicated that the endogenous wild-type p53 significantly binds stronger to COL17A1 in ADR-treated cells compared to non-treated cells (Figure 3C). Taken together, our results suggest that p53 directly regulates COL17A1 through p53-binding sites located in the first intron.
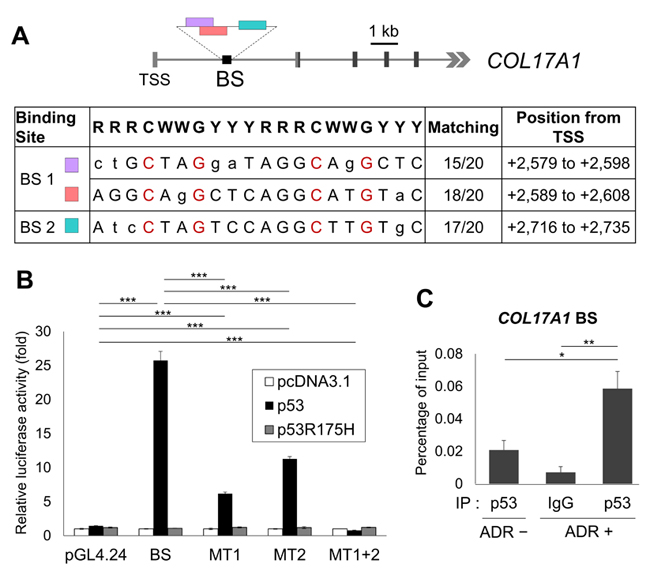
Figure 3: Identification of the p53 binding site. A. Genomic structure of the human COL17A1 gene indicates potential p53 binding sites (color boxes) in the BS construct (black box). Light gray box, untranslated region; dark gray box, translated region; TSS, transcription start site. The table shows the BS sequences aligned with the consensus p53 binding sequence (20 bases): R, purine; W, A or T; Y, pyrimidine. Identical nucleotides to the consensus are written in capital letters with the number of matching (n/20). The red-labeled nucleotides were mutated to thymine to examine the specificity of BS after mutagenesis. BS1 was mutated to MT1; BS2 was mutated to MT2; MT1+2 is double mutation of BS1 and BS2. B. Luciferase assay of the BS construct, the mutated BS construct (MT1, MT2, MT1+2), and the empty vector (pGL4.24) using H1299 cells co-transfected with control vector (pcDNA3.1), wild-type p53 (p53), or mutant p53 (p53R175H). Luciferase activity is indicated relative to the activity of the control vector (pcDNA3.1); n = 3. C. ChIP assay of non-treated or ADR-treated HBC4 cells of which DNA-protein complexes were then immunoprecipitated with the indicated antibodies followed by qPCR. Anti-mouse IgG was used as a negative control. The graphs show qPCR results indicating the amount of genomic fragments containing p53-binding sequences in COL17A1 using BS primers. Two-tailed Student’s t-test; *P < 0.05, **P < 0.01, ***P < 0.001.
The role of COL17 in tumor metastasis suppression
Regarding the function of COL17 as a cell-matrix adhesion molecule [11], we examined its role in tumor metastasis using a highly invasive MDA-MB-231 cell line expressing mutant p53. We generated MDA-MB-231 cell stably expressing COL17 in the presence of doxycycline (Dox) and control cells using the tetracycline-regulated lentiviral expression system. In the absence of Dox, the Tet repressor suppresses COL17 expression (Figure 4A and 4B, Dox−). By contrast, after the addition of Dox into cell culture media for 48 hours, the 180-kDa form of COL17 is expressed, with the shed 120-kDa ectodomain form that is predominantly detected in the culture media (Figure 4A and 4B). These stable cell lines were used for subsequent experiments to investigate cell migration and invasion in vitro. The cells were plated onto a COL1-coated container using a lower percentage of serum to minimize cell proliferation, as previously described [32]. Twenty-four hours after the scratch, the COL17-expressing cells exhibited significantly decreased migration compared to the non-producing cells (Figure 4C). To strengthen this result, we performed an invasion assay using basement membrane matrix Matrigel-coated chambers to mimic the in vivo extracellular environment [33]. Cells were allowed to invade depending on their invasive potential through membrane pores to the lower chamber for 24 hours. The invading cells were fixed, stained, and quantified. The COL17-expressing cells exhibited nearly 50% repressed invasive ability compared to mock cells (Figure 4D), suggesting a role of COL17 in suppressing cell migration and invasion.
Next, we examined the effect of the secreted form of COL17 on cells imitating the natural cell-matrix environment surrounding the mammary glands. Parental MDA-MB-231 cells were plated using conditioned media harvested from the stable cell culture dishes. The cells cultured in the COL17-enriched media demonstrated a significantly lower percentage of invasion compared to the cells cultured in mock media (Figure 4E), indicating that the COL17 ectodomain regulated cancer cell invasion. Our results established a role of COL17 in suppressing breast tumor migration and invasion in vitro.
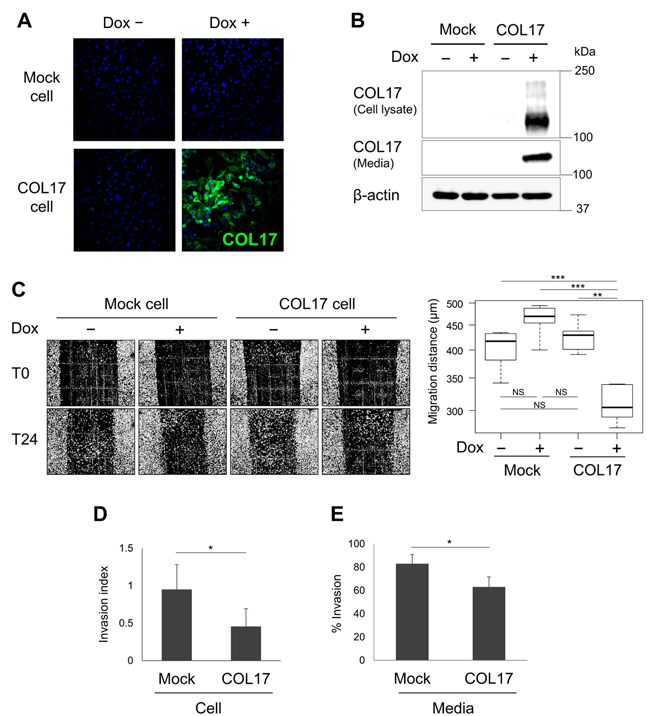
Figure 4: The role of COL17 in tumor metastasis suppression. MDA-MB-231 cell line stably overexpressing COL17 and mock cells were used for all experiments. Cells were not treated (Dox−) or treated with 1 μg/mL of doxycycline (Dox+) for 48 hours before analysis. A. Immunocytochemistry of the stable cells stained with anti-Collagen XVII. B. Western blot of the stable cell lysates and media extracts blotting with anti-Collagen XVII. C. Images of the scratch region taken immediately after the scratch (T0) and 24 hours later (T24) using a phase-contrast microscope with 10× magnification; n = 6 per group. Migration distances from T0 to T24 were measured at the reference points (n = 3) using ImageJ, and the average was calculated and used for the box plot analysis. D. Invasion assay of stable cells analyzed at 24 hours after plating. The number of invading cells was quantified and calculated as the % invasion through the Matrigel over control chambers. The invasion index displays a proportion of the % invasion for the Dox+/Dox− condition. E. Media assay using parental MDA-MB-231 cells plated with conditioned media harvested from the stable COL17 or mock cell culture dishes. The parental cells invasive ability was displayed as the % invasion through the Matrigel over the control. Two-tailed Student’s t-test; *P < 0.05, **P < 0.01, ***P < 0.001; NS, P ≥ 0.05.
COL17A1 depletion is associated with tumor progression and poor prognosis
We further analyzed the relationship between COL17A1 expression and tumor progression in breast cancer patients using TCGA data. The COL17A1 mRNA expression level was significantly decreased in metastatic tissues compared to primary tumors (Figure 5A) and even from the same patients (Figure 5B). Moreover, low COL17A1 expression was significantly associated with a shorter median survival time among patients with breast invasive carcinoma (Figure 5C). In summary, we elucidated the role of COL17A1 as a breast tumor metastasis suppressor by inhibition of cancer cell migration and invasion, resulting in a better prognosis of patients.
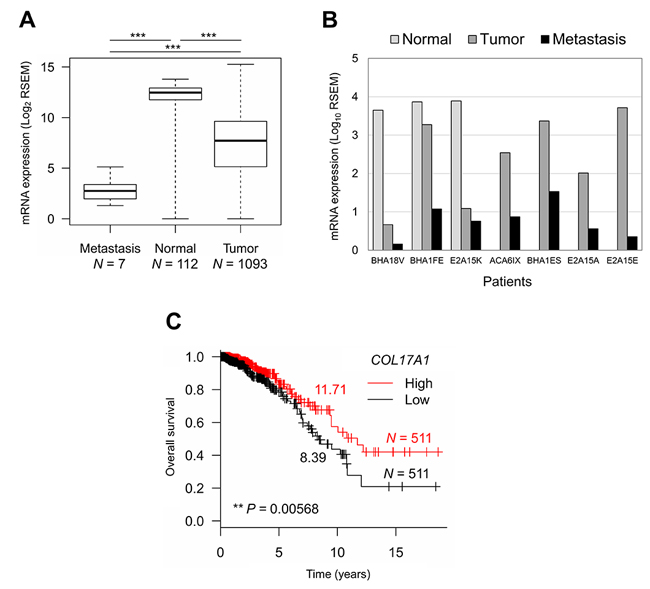
Figure 5: COL17A1 depletion associates with tumor progression and poor prognosis. A. and B. Differential mRNA expression of COL17A1 in metastatic, normal, and tumor tissues from all patients A. and from the seven patients with metastasis B. N, number of tissues. Mann-Whitney U-test; *P < 0.05, **P < 0.01, ***P < 0.001. C. Kaplan-Meier survival curve of patients with high (red) and low (black) expression of COL17A1 compared to median expression. Number, median survival time (years). N, number of patients. P-value was assessed by log-rank test.
Discussion
This study presents a p53-target screening based on human breast cell lines and breast cancer tissues. Ten genes including COL17A1, CROT, FAM198B, GREB1, LAMA3, MICALCL, NPY1R, SLC27A2, SYTL2, and TSPAN1 are identified as p53 target gene candidates from this screening. GO analysis identified two enriched biological processes as significantly up-regulated by wild-type p53, epidermis development and ectoderm development, using a similar set of genes, i.e., COL17A1, LAMA3, and EDA2R. LAMA3 encodes Laminin-5, an extracellular matrix protein, which binds to COL17 and functions in cell-matrix adhesion [34]. An alteration in the expression patterns of cell adhesion molecules, including Laminin-5, by knockdown and mutations of p53 in MCF10A cells has been previously described, but COL17 has not been examined [23]. Here, we provide evidence that COL17 expression levels are elevated after the induction of endogenous wild-type p53 following ADR-treatment in human breast cells or X-ray in mouse mammary glands. The results from a reporter assay and a ChIP assay indicate binding sites of p53 in intron 1, indicating COL17A1 as a direct p53 target. In response to X-ray exposure, COL17 protein is expressed in the myoepithelial cells (basal cell) of the mammary gland, indicating its function in facilitating cell adhesion to the underlying basement membrane [15, 35]. Consistent with our result, gene expression profiling demonstrated that COL17A1 expression is high in human breast myoepithelial cells but low in luminal epithelial cells [36].
The loss of function and mutation of p53 not only prevent breast cancer cells from undergoing oncogene-induced senescence and apoptosis but also result in the disruption of metastasis-involved molecules [7, 37, 38]. To metastasize, cells must invade through the basement membrane, enter the vasculature (intravasate), survive in the absence of adhesion, exit the vasculature (extravasate) and establish a new tumor in a foreign microenvironment [39]. The alteration and loss of cell adhesion structures are involved in an early step in epithelial cancer progression [40]. COL17A1 expression is particularly low in metastatic breast cancer cells [17]. Conversely, MDA-MB-231 cells that genetically over-express COL17, exhibited significantly decreased invasive properties. The released COL17 ectodomain, also known as LAD-1, was detected in cell culture media, human skin extracts, and serum obtained from patients with an autoimmune blistering disorder [41]. The skin blistering of this disease is caused by autoantibodies against the COL17 ectodomain, which introduces its role in maintaining dermal-epidermal cohesion [42]. Recent studies have clarified its function in decreased keratinocyte migration by dampening mTOR signaling [18]. This study revealed the role of the 120-kDa COL17 ectodomain in the suppression of breast cancer cell invasion. Indeed, myoepithelial cells act as natural tumor suppressors by secreting various molecules that have inhibitory effects on tumor cell growth, invasion and angiogenesis [43]. Our results propose a new mechanism supporting myoepithelial cell function as a physical barrier to prevent the invasion of tumor cells from the duct to the stroma. An analysis of metastasis breast invasive carcinoma revealed that each patient demonstrates different expression levels of COL17A1 in different tissue types depending on the tumor progression. Moreover, patients bearing high COL17A1-expressing tumors exhibit a longer survival time compared with those bearing low COL17A1-expressing tumors. Taken together, our results implicate a role of COL17 in suppressing breast cancer cell migration and invasion, whereby a high level of COL17A1 expression leads to a better prognosis of patients with breast invasive carcinoma.
In this study, COL17A1 was identified as a p53 transcriptional target gene. Wild-type p53 suppresses breast cancer invasion in pre-metastatic steps by up-regulating the expression of COL17, resulting in the promotion of the adhesion between the basement membranes and myoepithelial cells. The COL17-mediated adhesion prevents the primary tumor cells from escaping into the surrounding breast tissues. By contrast, p53 loss or mutation diminished COL17 function, resulting in less adhesive basement membranes, which cause it to eventually break down and allow tumor cells to escape into the bloodstream to form metastasis in the secondary organ, thereby contributing to a worse prognosis of patients. Although the molecules involved in the p53-COL17 signaling pathway are essential to be elucidated, our findings indicated the regulation of breast cancer metastasis by the p53-COL17 pathway and potential roles of COL17 as a prognostic biomarker.
Materials and methods
Cell lines and treatments
The non-tumorigenic breast epithelial cell line MCF10A p53+/+ and its isogenic p53 knockout cell line (MCF10A p53−/−) were purchased from Sigma Aldrich (St. Louis, MO, USA). HBC4 cells were a gift from Dr. Takao Yamori (Japanese Foundation for Cancer Research, Tokyo, Japan). Human embryonic kidney cells transformed with the SV40 large T antigen (293FT) were provided in the ViraPower™ Lentiviral Expression System (Thermo Fisher Scientific). The other cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cell cultures were maintained under their depositors’ recommendations. Cells were transfected with plasmids using FuGENE6 (Promega, Madison, WI, USA). siRNA oligonucleotides (see sequences in Supplementary Table 1), commercially synthesized by Sigma Genosis (St Louis, MO, USA), were reverse transfected with Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s protocol. To induce p53, cells were continuously incubated with ADR for 2 hours on the day following cell plating. Based on the cytotoxic effects of the drug, MCF10A, HBL-100, and HBC4 cells were treated with 0.5, 1, and 2 μg/mL of ADR, respectively.
Microarray
Total RNA was isolated from MCF10A p53+/+ or MCF10A p53−/− cells at 12, 24, and 48 hours after ADR treatment, as well as from non-treated cells as described [44]. The integrity and purity of the RNA templates were determined using a 2100 Bioanalyzer (Agilent Technologies). Agilent’s One-Color Quick Amp labeling kit (Agilent Technologies) was used to generate fluorescent cRNA using cyanine 3-labeled targets according to the manufacturer’s instructions. Gene expression analysis was performed using SurePrint G3 8x60K microarray (Agilent Technologies). The fold-change after ADR in the MCF10A p53+/+ cells was calculated using the following equation:
![]()
An F-test and two-tailed Student’s t-test were performed to calculate the P-value. Genes demonstrating at least one probe satisfied the screening criteria, i.e., above 3-fold and P < 0.05, were included. The MCF10A microarray data is available from the NCBI GEO database (GSE98727).
TCGA analysis
The mRNA expression of p53 target gene candidates, the p53 mutation status, and clinical data were obtained from TCGA project by cBioPortal [45, 46]. For the differential expression analysis, 1093 tumor tissues were categorized according to p53 mutation status and were subjected to a box plot analysis. The fold-change in wild-type p53 tumors was calculated using the following equation:

An F-test and two-tailed Student’s t-test were performed to calculate the P-value as described [44]. Genes that satisfied the screening criteria, i.e., above 2-fold and P < 0.05, were included. A survival analysis was performed using the log-rank test stratified by the expression level of COL17A1 in tumors (above or below the median expression level). Box plot and survival analyses were performed using the EZR software program [47].
Gene Ontology (GO) enrichment analysis
The resultant 17 gene set was submitted for Gene Ontology analyses using the DAVID Functional Annotation Tool [https://david.ncifcrf.gov/home.jsp]. The enrichment score for biological process was calculated using the chi-squared test.
RNA extraction and quantitative PCR (qPCR)
Total RNA was isolated from cells and tissues using RNeasy Plus Mini Kits (Qiagen) according to the manufacturer’s instructions. Complementary DNA molecules were synthesized using the SuperScript III reverse transcriptase (Invitrogen). qPCR was performed using SYBR Green Master Mix and a Light Cycler 480 (Roche). The primer sequences are shown in Supplementary Table 1.
Western blot analysis
Cells were harvested by scraping and were lysed using chilled RIPA buffer (Thermo Fisher Scientific) containing 1 mM PMSF, 0.1 mM DTT, and 0.1% Protease Inhibitor Cocktail Set III (Calbiochem). The cell lysates were sonicated using a Bioruptor UCD-200 (Cosmobio, Tokyo, Japan) and centrifuged at 15,000 × g for 15 minutes at 4°C. Protein concentrations were measured using a BCA™ protein assay (Thermo Fisher Scientific). For media extract, cell culture media were changed to antibiotic-free media at 12 hours before harvest. Proteins were precipitated in chilled acetone, incubated for 1 hour at −80°C, and centrifuged at 15,000 × g for 15 minutes at 4°C. The cell lysates and precipitated protein from the media were added to SDS sample buffer (Bio-Rad), boiled for 5 minutes, and separated using SDS-PAGE. The proteins were transferred onto nitrocellulose membranes, which were subsequently blocked in 5% milk. The membranes were incubated with primary antibodies according to the manufacturers’ protocols. The membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 hour at room temperature. The immunoblots were developed using Amersham™ ECL (GE Healthcare), and images were acquired using the luminescent image analyzer LAS-4000 mini (Fujifilm, GE Healthcare). The western blot signal representing protein level was obtained by image quantification using ImageJ software.
Immunohistochemistry and immuno-cytochemistry
For immunocytochemistry, cells were plated onto glass coverslips, fixed with 4% paraformaldehyde, and permeabilized in 0.2% Triton-X in PBS. The samples were blocked with 3% BSA, stained with anti-Collagen XVII, incubated with HRP-conjugated secondary antibodies, and counterstained with DAPI. The mounted coverslips were visualized using a confocal microscope (Olympus FluoView FV1000). For immunohistochemistry, paraffin sections of mouse mammary tissues were stained using anti-Collagen XVII according to the manufacturer’s protocol. For visualization, the sections were incubated with HRP-labeled polymer anti-rabbit (Dako) and DAB (Dako) was used as a chromogen. Then, the samples were counterstained with hematoxylin.
Antibodies
Anti-Collagen XVII (ab184996, Abcam) was used for all human and mouse experiments. Anti-actin (AC15, Sigma-Aldrich), anti-p53 (OP43, Merck Millipore), and anti-p53-S15P (9284, Cell Signaling) were used in the western blot analyses.
Animal experiments
The p53 knockout C57BL/6J mice were provided by RIKEN BioResource Center (Ibaraki, Japan) [48]. The mice were maintained under specific pathogen-free conditions and were handled according to the Guidelines for Animal Experiments of the University of Tokyo. The mouse genotypes were confirmed by PCR analysis. Primer sequences are shown in Supplementary Table 1. The p53 wild-type and knockout female mice at 10 weeks of age were exposed to 10 Gy of X-rays using an X-ray irradiation system (MBR-1520R-3, Hitachi). At 24 hours after irradiation, the mice were sacrificed for mammary tissue extraction.
Gene reporter assay
A DNA fragment that includes the potential p53 binding sites of COL17A1 was amplified and subcloned into the pGL4.24 [luc2P/minP] vector (Promega). To create a mutant vector, point mutations were introduced at the 4th and 14th nucleotides (C to T mutations) and the 7th and 17th nucleotides (G to T mutations) within the consensus p53 binding site using site-directed mutagenesis. Reporter assays were performed using the Dual-Glo Luciferase Assay System (Promega) according to the manufacturer’s protocol. The H1299 cells were co-transfected with the analyzed constructs and the control vector (pcDNA3.1), wild-type p53 (p53), or mutant p53 (p53R175H). The primer sequences for amplification and site-directed mutagenesis are shown in Supplementary Table 1.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using EZ-Magna ChIP G Chromatin Immunoprecipitation Kits (Merck Millipore, Darmstadt, Germany) according to the manufacturer’s protocol. Briefly, HBC4 cells with or without ADR treatment were cross-linked with 1% formaldehyde for 10 minutes, washed with PBS, and lysed in nuclear lysis buffer. The lysate was then sonicated using Bioruptor UCD-200 (Cosmo Bio, Tokyo, Japan) to shear the DNA into fragments of approximately 200-1000 bp. The supernatant from 1 × 106 cells was used for each immunoprecipitation with anti-p53 antibody (OP140, Merck Millipore) or mouse IgG (SC-2025, Santa Cruz). Before immunoprecipitation, 1% of the supernatant was removed as “input”. Column-purified DNA was quantified by qPCR using primers for p53 binding site in WAF1 and COL17A1 BS primers (Supplementary Table 1).
Stable cell line generation
The full-length COL17A1 genomic DNA fragment containing Kozak sequence was cloned into the entry vector (pENTR™3C). The primer sequences are shown in Supplementary Table 1. The COL17A1 fragment was integrated into the destination vector (pLenti6.3/TO/V5-DEST) using Gateway LR Clonase II enzyme mix and then transformed into Stbl3 cells to generate the expression clone. Lentiviruses containing COL17A1 expression construct, empty vector, or TetR vector (pLenti3.3/TR) were produced in 293FT cells using ViraPower™ Packaging Mix according to the manufacturer’s protocol. The virus-containing media were harvested and used to determine the viral titer by qPCR. The TetR construct was co-transduced at 10 MOI into MDA-MB-231 cells with either the COL17A1 construct for COL17A1-expressing cells or empty vector for mock cells. The transduced cells were selected and continuously cultured in media containing Blasticidin and Geneticin® for 3 weeks. Stable cells were maintained using Leibovitz’s L-15 medium in a humidified 37°C incubator without CO2.
Scratch assay
Stable cells were treated with 1 μg/mL doxycycline (Dox) for 48 hours before cell plating. The cells (1.2 × 105) were seeded onto the COL1-coated 24-well plates using 1% FBS-containing culture media with or without Dox. Twenty-four hours after plating, the confluent cells were scraped using a CELL Scratcher (Iwaki, Japan), washed and replaced with media. The culture plates were marked as reference points close to the scratch. Images of the scratched area at the reference points were recorded immediately after the scratch and 24 hours later using a phase-contrast microscope with 10× magnification. The distances of the scratched area were determined and measured using ImageJ software [49]. The average migration distance of each well was calculated and subjected to a box plot analysis using R programming.
Invasion assay
Stable cells were treated with 1 μg/mL Dox for 48 hours before cell plating. The cells (7.5 × 104) were seeded into the 24-well BioCoat Matrigel invasion chambers (Corning) or control inserts according to the manufacturer’s protocol. The upper chamber filled with serum-free DMEM was placed in a well containing 10% FBS DMEM with the same condition of Dox, i.e., presence or absence. For the cell assay, stable cell lines were used with fresh media in both the upper chamber and the lower well. For the media assay, parental MDA-MB-231 cells were seeded into the chamber using conditioned media harvested from the stable cell culture dishes; and the chambers were placed in fresh media. After 24 hours of incubation, the cells invading to the bottom surface of the chamber membrane were fixed with 4% paraformaldehyde for 30 minutes and stained with 0.1% crystal violet for 2 h. The invading cells were counted using CELLCOUNTER software [50] and calculated as the percent invasion through the Matrigel relative to the control. The invasion index is displayed as the ratio of % invasion of Dox+ over those of Dox− condition.
Abbreviations
ADR, Adriamycin® (doxorubicin); COL17, Collagen XVII; Dox, doxycycline; GO, Gene Ontology; HD, hemidesmosome; TCGA, The Cancer Genome Atlas; p53, tumor protein 53
Author contributions
Matsuda K. conceived the project; Yodsurang V., Tanikawa C., and Matsuda K. designed the experiments; Yodsurang V., Yi Lo PH., Miyamoto T., Makoto H., and Tanikawa C. conducted the experiments; Yodsurang V. and Matsuda K. wrote the manuscript; Miyamoto T. and Matsuda K. proofread the manuscript.
Acknowledgments
We are grateful for the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan for the academic scholarship to V. Yodsurang. We thank Dr. Jinichi Mori for his helpful advice, Liu Yuyu for the ImageJ programming, and Satomi Takahashi and Misato Oshima for the technical assistances. This work was supported in part by a grant from the Japan Society for the Promotion of Science and the Ministry of Education, Culture, Sports, Science and Technology of Japan to K. Matsuda and C. Tanikawa, a grant from the Japan Agency for Medical Research and Development to K. Matsuda and C. Tanikawa, a grant from the Ministry of Health, Labor and Welfare of Japan to K. Matsuda, and a grant from the Takeda Science Foundation to K. Matsuda and C. Tanikawa.
Conflicts of interest
The authors declare no conflicts of interest.
Funding
This work was supported in part by a grant from the Japan Society for the Promotion of Science and the Ministry of Education, Culture, Sports, Science and Technology of Japan to K. Matsuda and C. Tanikawa, a grant from the Japan Agency for Medical Research and Development to K. Matsuda and C. Tanikawa, a grant from the Ministry of Health, Labor and Welfare of Japan to K. Matsuda, and a grant from the Takeda Science Foundation to K. Matsuda and C. Tanikawa.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2013.
2. Gasco M, Shami S, Crook T. The p53 pathway in breast cancer. Breast Cancer Res. 2002; 4: 70-6.
3. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490: 61-70.
4. Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, Tsui DW, Liu B, Dawson SJ, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016; 7: 11479.
5. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010; 2: a001008.
6. Bertheau P, Lehmann-Che J, Varna M, Dumay A, Poirot B, Porcher R, Turpin E, Plassa LF, de Roquancourt A, Bourstyn E, de Cremoux P, Janin A, Giacchetti S, et al. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast. 2013 (Suppl 2); 22: S27-9.
7. Powell E, Piwnica-Worms D, Piwnica-Worms H. Contribution of p53 to metastasis. Cancer Discov. 2014; 4: 405-14.
8. Mori J, Tanikawa C, Funauchi Y, Lo PH, Nakamura Y, Matsuda K. Cystatin C as a p53-inducible apoptotic mediator that regulates cathepsin L activity. Cancer Sci. 2016; 107: 298-306.
9. Koguchi T, Tanikawa C, Mori J, Kojima Y, Matsuda K. Regulation of myo-inositol biosynthesis by p53-ISYNA1 pathway. Int J Oncol. 2016; 48: 2415-24.
10. Tanikawa C, Zhang YZ, Yamamoto R, Tsuda Y, Tanaka M, Funauchi Y, Mori J, Imoto S, Yamaguchi R, Nakamura Y, Miyano S, Nakagawa H, Matsuda K. The Transcriptional Landscape of p53 Signalling Pathway. EBioMedicine. 2017; 20:109-19.
11. Borradori L, Sonnenberg A. Structure and function of hemidesmosomes: more than simple adhesion complexes. J Invest Dermatol. 1999; 112: 411-8.
12. Aho S, Uitto J. 180-kD bullous pemphigoid antigen/type XVII collagen: tissue-specific expression and molecular interactions with keratin 18. J Cell Biochem. 1999; 72: 356-67.
13. McGrath JA, Gatalica B, Christiano AM, Li K, Owaribe K, McMillan JR, Eady RA, Uitto J. Mutations in the 180-kD bullous pemphigoid antigen (BPAG2), a hemidesmosomal transmembrane collagen (COL17A1), in generalized atrophic benign epidermolysis bullosa. Nat Genet. 1995; 11: 83-6.
14. Schumann H, Hammami-Hauasli N, Pulkkinen L, Mauviel A, Kuster W, Luthi U, Owaribe K, Uitto J, Bruckner-Tuderman L. Three novel homozygous point mutations and a new polymorphism in the COL17A1 gene: relation to biological and clinical phenotypes of junctional epidermolysis bullosa. Am J Hum Genet. 1997; 60: 1344-53.
15. Bergstraesser LM, Srinivasan G, Jones JC, Stahl S, Weitzman SA. Expression of hemidesmosomes and component proteins is lost by invasive breast cancer cells. Am J Pathol. 1995; 147: 1823-39.
16. Laval S, Laklai H, Fanjul M, Pucelle M, Laurell H, Billon-Gales A, Le Guellec S, Delisle MB, Sonnenberg A, Susini C, Pyronnet S, Bousquet C. Dual roles of hemidesmosomal proteins in the pancreatic epithelium: the phosphoinositide 3-kinase decides. Oncogene. 2014; 33: 1934-44.
17. Geiger T, Madden SF, Gallagher WM, Cox J, Mann M. Proteomic portrait of human breast cancer progression identifies novel prognostic markers. Cancer Res. 2012; 72: 2428-39.
18. Jackow J, Loffek S, Nystrom A, Bruckner-Tuderman L, Franzke CW. Collagen XVII Shedding Suppresses Re-Epithelialization by Directing Keratinocyte Migration and Dampening mTOR Signaling. J Invest Dermatol. 2016; 136: 1031-41.
19. Loffek S, Hurskainen T, Jackow J, Sigloch FC, Schilling O, Tasanen K, Bruckner-Tuderman L, Franzke CW. Transmembrane collagen XVII modulates integrin dependent keratinocyte migration via PI3K/Rac1 signaling. PLoS One. 2014; 9: e87263.
20. Qiao H, Shibaki A, Long HA, Wang G, Li Q, Nishie W, Abe R, Akiyama M, Shimizu H, McMillan JR. Collagen XVII participates in keratinocyte adhesion to collagen IV, and in p38MAPK-dependent migration and cell signaling. J Invest Dermatol. 2009; 129: 2288-95.
21. Brosh R, Sarig R, Natan EB, Molchadsky A, Madar S, Bornstein C, Buganim Y, Shapira T, Goldfinger N, Paus R, Rotter V. p53-dependent transcriptional regulation of EDA2R and its involvement in chemotherapy-induced hair loss. FEBS Lett. 2010; 584: 2473-7.
22. Tanikawa C, Furukawa Y, Yoshida N, Arakawa H, Nakamura Y, Matsuda K. XEDAR as a putative colorectal tumor suppressor that mediates p53-regulated anoikis pathway. Oncogene. 2009; 28: 3081-92.
23. Zhang Y, Yan W, Chen X. Mutant p53 disrupts MCF-10A cell polarity in three-dimensional culture via epithelial-to-mesenchymal transitions. J Biol Chem. 2011; 286: 16218-28.
24. Allen MA, Andrysik Z, Dengler VL, Mellert HS, Guarnieri A, Freeman JA, Sullivan KD, Galbraith MD, Luo X, Kraus WL, Dowell RD, Espinosa JM. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife. 2014; 3: e02200.
25. Janky R, Verfaillie A, Imrichova H, Van de Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K, Naval Sanchez M, Potier D, Svetlichnyy D, Kalender Atak Z, Fiers M, et al. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol. 2014; 10: e1003731.
26. Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E, Marks J, Sukumar S. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature. 2000; 405: 974-8.
27. Loughery J, Cox M, Smith LM, Meek DW. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res. 2014; 42: 7666-80.
28. Hirako Y, Usukura J, Uematsu J, Hashimoto T, Kitajima Y, Owaribe K. Cleavage of BP180, a 180-kDa bullous pemphigoid antigen, yields a 120-kDa collagenous extracellular polypeptide. J Biol Chem. 1998; 273: 9711-7.
29. Franzke CW, Tasanen K, Schacke H, Zhou Z, Tryggvason K, Mauch C, Zigrino P, Sunnarborg S, Lee DC, Fahrenholz F, Bruckner-Tuderman L. Transmembrane collagen XVII, an epithelial adhesion protein, is shed from the cell surface by ADAMs. EMBO J. 2002; 21: 5026-35.
30. Kariko K, Bhuyan P, Capodici J, Weissman D. Small interfering RNAs mediate sequence-independent gene suppression and induce immune activation by signaling through toll-like receptor 3. J Immunol. 2004; 172: 6545-9.
31. Franzke CW, Bruckner-Tuderman L, Blobel CP. Shedding of collagen XVII/BP180 in skin depends on both ADAM10 and ADAM9. J Biol Chem. 2009; 284: 23386-96.
32. Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007; 2: 329-33.
33. Hall DM, Brooks SA. In vitro invasion assay using matrigel: a reconstituted basement membrane preparation. Methods Mol Biol. 2014; 1070: 1-11.
34. Van den Bergh F, Eliason SL, Giudice GJ. Type XVII collagen (BP180) can function as a cell-matrix adhesion molecule via binding to laminin 332. Matrix Biol. 2011; 30: 100-8.
35. Lakhani SR, O’Hare MJ. The mammary myoepithelial cell—Cinderella or ugly sister? Breast Cancer Res. 2001; 3: 1-4.
36. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, et al. Molecular portraits of human breast tumours. Nature. 2000; 406: 747-52.
37. Arjonen A, Kaukonen R, Mattila E, Rouhi P, Hognas G, Sihto H, Miller BW, Morton JP, Bucher E, Taimen P, Virtakoivu R, Cao Y, Sansom OJ, et al. Mutant p53-associated myosin-X upregulation promotes breast cancer invasion and metastasis. J Clin Invest. 2014; 124: 1069-82.
38. Chander H, Brien CD, Truesdell P, Watt K, Meens J, Schick C, Germain D, Craig AW. Toca-1 is suppressed by p53 to limit breast cancer cell invasion and tumor metastasis. Breast Cancer Res. 2014; 16: 3413.
39. Vargo-Gogola T, Rosen JM. Modelling breast cancer: one size does not fit all. Nat Rev Cancer. 2007; 7: 659-72.
40. Davis TL, Goldman AJ, Cress AE. Suppression and alteration of adhesion structures in human epithelial cancer progression. In: Cress AE and Nagle RB, eds. Cell Adhesion and Cytoskeletal Molecules in Metastasis. Dordrecht: Springer Netherlands. 2006. pp. 19-46.
41. Marinkovich MP, Taylor TB, Keene DR, Burgeson RE, Zone JJ. LAD-1, the linear IgA bullous dermatosis autoantigen, is a novel 120-kDa anchoring filament protein synthesized by epidermal cells. J Invest Dermatol. 1996; 106: 734-8.
42. Nishie W, Lamer S, Schlosser A, Licarete E, Franzke CW, Hofmann SC, Jackow J, Sitaru C, Bruckner-Tuderman L. Ectodomain shedding generates Neoepitopes on collagen XVII, the major autoantigen for bullous pemphigoid. J Immunol. 2010; 185: 4938-47.
43. Pandey PR, Saidou J, Watabe K. Role of myoepithelial cells in breast tumor progression. Front Biosci (Landmark Ed). 2010; 15: 226-36.
44. Miyamoto T, Tanikawa C, Yodsurang V, Zhang YZ, Imoto S, Yamaguchi R, Miyano S, Nakagawa H, Matsuda K. Identification of a p53-repressed gene module in breast cancer cells. Oncotarget. 2017 Jul 26. https://doi.org/10.18632/oncotarget.19608. [Epub ahead of print].
45. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2: 401-4.
46. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6: pl1.
47. Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013; 48: 452-8.
48. Tsukada T, Tomooka Y, Takai S, Ueda Y, Nishikawa S, Yagi T, Tokunaga T, Takeda N, Suda Y, Abe S, et al. Enhanced proliferative potential in culture of cells from p53-deficient mice. Oncogene. 1993; 8: 3313-22.
49. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012; 9: 671-5.
50. Li X, Yang H, Huang H, Zhu T. CELLCOUNTER: novel open-source software for counting cell migration and invasion in vitro. Biomed Res Int. 2014; 2014: 863564.