
Introduction
Estrogens regulate growth, differentiation and development in many tissues including the female reproductive tract, bone, central nervous system, immune and cardiovascular systems [1-3]. The actions of estrogens are mediated by a group of estrogen receptors (ER). Aromatase, a member of the cytochrome P450 superfamily, catalyzes the final step in the biosynthesis of estrogen from androgen [4]. In women with breast cancer, the expression of aromatase is the highest in or near tumor sites [5, 6]. Anastrozole (Arimidex) is a potent nonsteroidal aromatase inhibitor, producing approximately 97% inhibition of estrogen biosynthesis [7].
Several studies have evaluated combination approaches using hormone therapy with other targeted agents in patients with breast and gynecologic cancers. The Bolero-2 trial (trial registration ID: NCT00863655), a large randomized Phase III study, compared the aromatase inhibitor exemestane alone to a combination of exemestane with everolimus, an mTOR inhibitor [8]. The combination improved progression-free survival (PFS) in patients with ER and/or progesterone receptor (PR)-positive advanced and metastatic breast cancer. Based on data from the Bolero-2 trial, in July 2012 the US Food and Drug Administration (FDA) approved everolimus in combination with exemestane for use in postmenopausal women with hormone receptor-positive, HER2/neu-negative, advanced breast cancer who have progressed on anastrozole or letrozole. Other studies have evaluated the aromatase inhibitor letrozole in combination with everolimus and have demonstrated responses in patients with metastatic endometrial carcinoma (trial registration ID: NCT01068249) [9] and breast cancer (trial registration ID: NCT00107016) [10].
Research into the mechanism of endocrine responsiveness and resistance in breast and gynecologic cancers has revealed that the PI3K/AKT/mTOR pathway becomes activated and utilized by cancer cells to bypass the effects of hormone therapy [11-13]. We therefore investigated the use of the aromatase inhibitor anastrozole in combination with everolimus in patients with ER and/or PR-positive breast and gynecologic tumors including ovarian and endometrial cancer (trial registration ID: NCT01197170) and including an analysis of molecular data.
Results
Patients
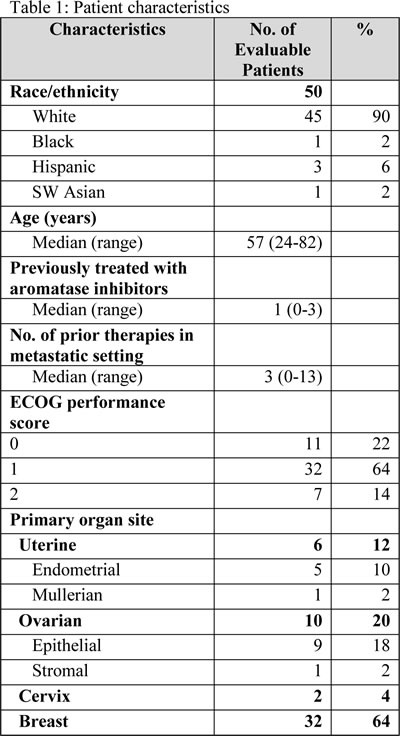
Fifty-five women with advanced or metastatic breast, ovarian, endometrial, and cervical malignancies were enrolled. All patients received at least one dose of treatment. All patients were considered eligible for toxicity evaluation. Fifty patients were considered eligible for response evaluation. Five patients were ineligible for response evaluation because they had not reached restaging at the time of analysis. Demographic and clinical characteristics of evaluable patients are summarized in Table 1. The median age of patients was 57 years (range: 24-82 years). The median number of prior therapies in the advanced or metastatic setting was 3 (range: 0-13). Twenty-three of the evaluable patients had received prior aromatase inhibitors. Fifteen deaths occurred, all attributed to disease progression. Expansion cohorts were opened for breast, endometrial, ovarian and cervical tumor types and for patients with PTEN loss and PIK3CA mutations.
Overall Survival and Time to Treatment Failure
The median survival has not been reached after a median follow up of 6.1 months. At the time of analysis, 37 of 55 (67%) were off study. The overall median TTF was 3.1 months (95% CI 2.1-4.1).
Dose Escalation, DLT and Tolerance
Seven patients were enrolled at dose level 1 and 48 at dose level 2. Two of 55 patients (4%) experienced a DLT. The two DLTs both occurred in expansion cohorts of dose level 2 and were grade 3 mucositis. The full federal drug administration (FDA) dose for each drug evaluated in dose level 2 (anastrozole 1 mg PO daily and everolimus 10 mg PO daily) was found to be safe and well tolerated.
Twenty-five of 55 patients (45%) experienced at least one drug-related toxicity. Of the 36 reported drug-related toxicities, 25 (69%) were grade 1 or 2. The most common grade 1 and 2 drug-related toxicities included mucositis (6 patients), fatigue (4 patients), nausea/vomiting/anorexia, elevated cholesterol, pneumonitis, elevated triglycerides and elevated ALT (2 patients each). There were 11 grade 3 toxicities at least possibly related to treatment including mucositis (2 patients), pneumonitis, hypertension, hyperglycemia, hemoptysis, weakness, rash, low platelets, elevated AST and decreased ANC (1 patient each). A dose modification was required in four incidents for mucositis (2 patients), nausea (1 patient), and pneumonitis (1 patient). Two patients with pneumonitis (including one with grade 2 and one with grade 3 toxicity) were taken off study with resolution of toxicity.
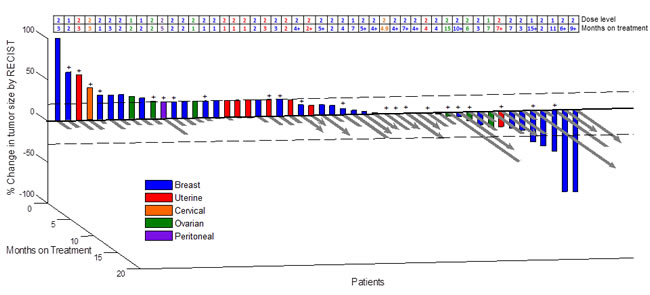
Fig 1: 3D Waterfall. Best radiologic response by RECIST and months on treatment (N = 50 patients). (+) Patients positive for alterations in PI3K/AKT1/mTOR pathway.
Response Data
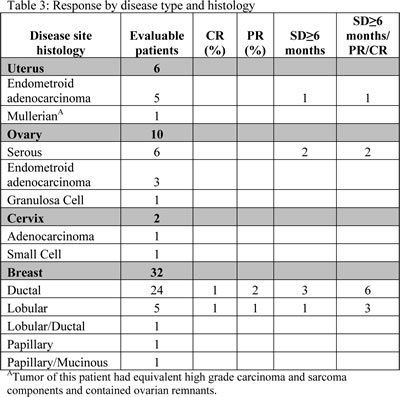
Twelve of 50 evaluable patients (24%) achieved SD ≥ 6 months/PR/CR, including 5 patients (10%) with PR/CR: 9 of 32 patients (28%) with breast cancer (cases 1, 2, 3, 4, 5, 6, 9, 10 and 12, Table 2); 2 of 10 patients (22%) with ovarian cancer (cases 7 and 11, Table 2); and 1 of 6 patients (17%) with endometrial cancer (case 8, Table 2). Neither of the 2 patients with cervical cancer achieved SD≥6 months/PR/CR. Five patients with breast cancer achieved a PR (cases 3, 4 and 5, Table 2) or CR (cases 1 and 2, Table 2). Three patients with PR included one patient with a 50% decrease in disease for 11 months (case 3, Table 2), one patient with a 44% decrease in disease for 2 months (case 4, Table 2) and, one with a 38% decrease in disease for 17+ months (case 5, Table 2). The two patients (4%) with CRs have ongoing responses at 9+ and 6+ months (cases 1 and 2, Table 2).

Prior Treatment with Aromatase Inhibitors and Response
Twenty-three of 50 evaluable patients (46%) had received at least one prior aromatase inhibitor in the advanced or metastatic setting. Five of the 23 patients (22%) who had been previously treated in the metastatic setting with an aromatase inhibitor achieved SD ≥ 6 months/PR/CR with the combination of anastrozole and everolimus, including 3 patients (13%) with PR/CR. Twenty of 32 patients (63%) with breast cancer had received prior aromatase inhibitors in the advanced or metastatic setting. Five of the 20 patients (25%) with breast cancer and prior aromatase inhibitor exposure achieved SD ≥ 6 months/PR/CR (3 patients with PR/CR).
Molecular Analysis and Association with Response
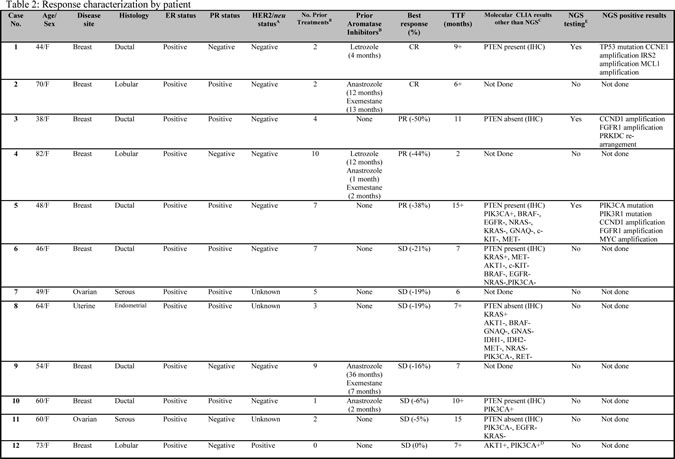
When archival cell blocks for patients were available, CLIA-certified molecular testing was performed for PI3K/AKT/mTOR pathway alterations. For the purposes of this study, we defined a PI3K/AKT/mTOR pathway alteration as one or more of the following: PIK3CA, AKT1, PTEN mutation, and/or PTEN loss (by IHC). Of the 12 patients who demonstrated SD ≥ 6 months/PR/CR, 8 had molecular testing (3 of 5 with PR/CR). Six of the 8 patients (75%) had at least one alteration in the PI3K/AKT/mTOR pathway including PIK3CA mutations (3 patients, one of whom also had an AKT1 mutation) and PTEN loss (IHC) (3 patients). The remaining two patients (25%) with molecular testing did not have a direct alteration in this pathway. Three patients who achieved PR/CR who also had molecular testing with NGS demonstrated additional alterations: amplifications in FGFR1 (encodes for fibroblast growth factor receptor 1, 2 patients), CCND1 (encodes for cyclin D1, also known as BCL1, 2 patients), CCNE1 (encodes for cyclin E1, 1 patient), IRS2 (encodes for insulin receptor substrate 2, 1 patient), MCL1 (myeloid leukemia cell gene, 1 patient) and MYC (myelocytomatosis viral oncogene, 1 patient) and, a re-arrangement in PRKDC (protein kinase DNA activated catalytic polypeptide, 1 patient).
A total of 35 patients had molecular testing for at least one of the following: PIK3CA, PTEN, or AKT1 mutation; and/or PTEN loss. Of the 35 patients tested, 22 (63%) were positive for at least one alteration in the PI3K/AKT/mTOR pathway. Of 35 patients tested for an alteration in the PI3K/AKT/mTOR pathway, 13 were negative. One of 13 patients (8%) attained SD ≥ 6 months/PR/CR (this patient had a CR). By comparison, 22 patients tested had a PI3K pathway alteration; 6 of these 22 patients (27%) had SD≥6 months/PR/CR (2 patients with PR/CR) (p = 0.16).
Discussion
Hormonal therapy is a mainstay of treatment for breast cancer and is an area of active investigation in gynecologic tumors. Strategies to augment response and overcome resistance to aromatase inhibitors are urgently needed. PI3K/AKT/mTOR pathway alterations are common in breast and gynecologic cancers [20-22]. Preclinical studies have shown that suppression of PTEN function, or activated AKT1 expression, caused by activating mutations in PIK3CA or AKT1, confers resistance to traditional chemotherapeutic drugs as well as hormonal based drugs, but results in sensitivity to mTOR inhibitors [23, 24]. Recent studies of breast cancer patients treated with everolimus in combination with exemestane [8] and of endometrial and breast cancer patients treated with everolimus in combination with letrozole [9, 10] have shown efficacy. Based on the Phase III study results, everolimus is now FDA-approved in combination with the aromatase inhibitor exemestane in patients with advanced, hormone receptor-positive breast cancer refractory to anastrozole and/or letrozole [8].
Our study indicates that the combination of anastrozole and everolimus can be given at full approved doses. Tolerance was excellent with the main side effects being grade 1 and 2 mucositis, fatigue, nausea/vomiting/anorexia and, grade 3 mucositis. Two patients experienced DLTs, mucositis in both cases. A dose reduction resulted in better tolerance in one patient and the other patient was taken off study. These results parallel those for exemestane [8] and letrozole [9, 10]; these hormone antagonists can be given safely with full dose everolimus (10 mg PO daily).
Herein we report that 6 of 8 patients (75%) who experienced SD ≥ 6 months/PR/CR (3 patients with PR/CR) on this study and on whom molecular testing was performed, demonstrated at least one alteration in the PI3K/AKT/mTOR pathway. Six of 22 patients (27%) with molecular alterations in the PI3K/AKT/mTOR pathway achieved SD ≥ 6 months/PR/CR, including 2 patients (9%) with PR/CR. Our results support and expand on those previously reported [21, 25]. Indeed, 12 of 50 heavily pretreated patients (24%) with breast or gynecologic tumors achieved SD ≥ 6 months/PR/CR with anastrozole and everolimus, including 5 patients (10%) with PR/CR. These results are consistent with the SD ≥ 6 months/PR/CR rates of 20 to 30% previously reported by our group in patients treated with matched phase I therapy [21, 25]. Our response rates are slightly higher than those reported in Bolero-2 [8], but our patient numbers are also smaller. Further, some of our patients with breast cancer did not receive prior aromatase inhibitors. The number of patients with endometrial cancer are small, but the observed activity is lower than reported in the letrozole/everolimus combination in this tumor type [9]. Regarding correlation with molecular aberrations, we found that six of eight responders tested had pathway aberrations. These aberrations included abnormalities in PIK3CA, PTEN and AKT, consistent with the diversity of genes that can activate the PIK3CA/AKT/mTOR pathway. On the other hand, only 27% of patients with these pathway abnormalities responded. These data are consistent with data recently presented by Hortobagyi et al. [26], where patients with either no or only one molecular aberration in the PI3K/AKT/mTOR pathway had better outcomes with exemestane and everolimus than did patients with multiple aberrations. On the other hand, three of our patients with CR or PR showed multiple aberrations indicating that their presence does not rule out response.
A total of 3 patients who achieved PR/CR had NGS testing. These patients demonstrated numerous alterations including: CCNE1, CCND1, FGFR1, IRS2, MCL1, and MYC amplifications and a PRKDC re-arrangement, as well as TP53 mutations (cases 1, 3 and 5, Table 2). CCNE1 and CCND1 code for cyclin D1 and cyclin E1, respectively, which are proteins that help control the transition of cells from G1 to S phase during proliferation [27]. Both of these proteins are regulated by GSK-3β, which is directly phosphorylated by AKT [28]. MYC is a regulator gene that codes for a transcription factor involved in cell proliferation and is also directly regulated by GSK-3β [29]. Insulin receptor substrates (IRS1 and IRS2) are proteins that dock to IGFR receptors to recruit other factors such as the p85 regulatory subunit of PI3K, thereby leading to activation of the PI3K/AKT/mTOR pathway [30]. Fibroblast growth factor receptors (FGFR) bind to growth factors (FGF) involved in angiogenesis. Activation of FGFR induces PI3K and AKT activities through recruitment and tyrosine phosphorylation of the docking protein Gab1 that results in the activation of PI3K [31]. MCL1 codes for a protein that enhances cell survival by inhibiting apoptosis. Control of MCL1 stability by GSK-3β is an important mechanism for the regulation of apoptosis by AKT [32]. Tumor Protein 53 (TP53) codes for a tumor suppressor (p53) that regulates cell cycle. AKT influences the activity of p53 through phosphorylation of the p53-binding protein MDM2 [33]. Protein kinase, DNA-activated, catalytic polypeptide (PRKDC) codes for a catalytic subunit of the DNA-dependent protein kinase, a member of the PI3K family [34]. The presence of multiple gene aberrations may reflect increased genetic instability and poor patient prognosis [35]; however, the combination of anastrozole and everolimus was beneficial for each of these three patients. One of the patients with a durable CR (case 1, Table 2) had NGS testing and demonstrated a TP53 mutation as well as amplifications in CCNE1, IRS2, a protein important for insulin receptor signaling [36], and MCL1, a gene that plays a role in regulating cell-fate decisions [37]. The latter results suggest that CR can be achieved in the absence of a direct alteration in the PI3K/AKT/mTOR pathway, even when multiple molecular aberrations are present. However, some of the above genes may modulate the PIK3CA/AKT/mTOR axis perhaps explaining in part the response [31, 38-43]. Systems biology bioinformatics approaches will be needed to determine whether or not multiple aberrant signals in patients with metastatic tumors converge on pathways such as PI3K/AKT/mTOR pathway, and are hence actionable.
Our results suggest that patients can obtain salutary effects including CR (and PR) despite the presence of multiple alterations; however, should relapse occur in any of these patients, it might be explained by the additional alterations eventually leading to resistance. It is not surprising that patients with advanced disease have multiple molecular alterations, as this observation is consistent with previous literature reports of heterogeneity within and between tumors [44-46].
There are several limitations to our study. Whether the responses observed are due simply to the inhibition of the PI3K/AKT/mTOR pathway by everolimus or due to hormone modulation by aromatase inhibition alone (especially in those patients not previously treated with anastrozole) or contribution from both is unknown, as this was not a randomized study. However, it should be noted that the rate of SD ≥ 6 months/PR/CR was similar in patients who had failed prior aromatase inhibition therapy (5 of 23 patients, 22%) versus those who had not had aromatase inhibition (7 of 27 patients, 26%), which suggests that everolimus contributed to those responses. Another potential limitation of this analysis is that a majority of our patients had breast cancer, and that the combination of an aromatase inhibitor and an mTOR inhibitor has recently been demonstrated to be effective. However, we provide correlative molecular data and show responses in endometrial and ovarian cancers, as well as breast cancer. Further, we show that the activity in breast cancer is seen with anastrozole and therefore is not limited to exemestane (as used in previous studies) [8]. Additional limitations of our study stem from the fact that only a subset of patients had next generation sequencing. Furthermore, comparison of MD Anderson CLIA “hotspot” testing versus next generation sequencing was also performed in only limited number of patients. It is possible that other mutations exist in some of the patients tested by “hotspot” analysis, and were not discerned by this type of testing. As an example, a recent study in breast cancers has identified additional driver mutations in AKT1 that are not located in hotspot regions frequently tested [47]. We also had molecular data from only one time point; pre- and post-tissue analyses would be more informative to elucidate resistance patterns. Future investigations should emphasize molecular analysis so that biological correlates of clinical response can be identified. Finally, our patient numbers were relatively small. Therefore, these results must be interpreted in this context and considered hypothesis generating.
In conclusion, our study demonstrates that full doses of the aromatase inhibitor anastrozole and the mTOR inhibitor everolimus can be given for prolonged periods of time (longest so far = 17 months) with excellent tolerance. A subset of heavily pretreated patients with breast, ovarian and endometrial cancer achieved SD ≥ 6 months/PR/CR. SD ≥ 6 months/PR/CR was seen in 27% of patients with molecular alterations in the PI3K/AKT/mTOR pathway. Patients with multiple molecular alterations still benefited from therapy. Further exploration of this combination in larger cohorts of patients with breast and gynecologic tumors and in-depth analysis of molecular correlates is warranted.
Patients and Methods
Patients
The study enrolled adult patients with pathologically-confirmed advanced or metastatic cancer. Patients were required to have tumors that demonstrated ER and/or PR-positive disease (positivity by immunohistochemistry staining ≥ 1% based on reported guidelines for breast cancer) [14]. For women, only those who were postmenopausal or premenopausal and receiving a gonadotropin-releasing hormone agonist were eligible. All patients had an Eastern Cooperative Oncology Group (ECOG) performance status score of 0 to 2. Other criteria included adequate neutrophil counts (≥ 1,000/mL), platelets (≥ 50,000/mL), creatinine (≤ 2 × upper limit of normal), bilirubin (≤ 2.0), alanine aminotransferase (ALT) (≤ 3 × upper limit of normal with the exception of patients with liver metastasis in whom ALT ≤ 5 × upper limit of normal and bilirubin ≤ 3 × upper limit of normal). Patients provided written informed consent according to institutional guidelines. The study was approved by the Institutional Review Board (IRB).
Study Design and Treatment
This was a single institution, open-label, dose-escalation study, with a standard 3+3 design. The study allowed an additional (optional) three patients at any dose level in order to provide flexibility for enrollment and additional safety or correlative data. Endpoints were to establish safety and preliminarily assess antitumor efficacy and molecular correlates.
Each treatment cycle was 28-days with both drugs given daily. Cycles were consecutive with no treatment breaks. Patients were initially treated at dose level 1 (anastrozole 1 mg PO daily and everolimus 5 mg PO daily) and if no serious toxicities were observed the dose was escalated to dose level 2 (anastrozole 1 mg PO daily and everolimus 10 mg PO daily). Dose level 2 represents the full approved dose of each drug. Once the safety of dose level 2 was established, additional patients were enrolled at that level.
If a response was observed in a particular tumor subtype, study cohorts were expanded to include up to an additional 14 participants with that specific tumor type. For the purpose of adding up to 14 additional participants, a tumor (including, but not limited to tumor type, histologic subtype or genomic subtype) response was defined as one of the following: (1) stable disease (SD) ≥ 6 months; or (2) decrease in measurable tumors ≥ 20% by RECIST criteria.
Assessment of Safety and Efficacy
Treatment continued until unacceptable toxicity or disease progression occurred. Dose delays and reductions were at the discretion of the treating physician. Toxicities were graded based on the Common Terminology Criteria for Adverse Events (CTCAE v4.0). Dose limiting toxicities (DLTs) were defined as adverse events (AEs) related to study agents that occurred during the first cycle of treatment with an attribution of possible, probable, or definitely related to therapy and fulfilling one of the following requirements: grade 4 neutropenia lasting > 7 days, febrile neutropenia, platelet count <25,000/mm lasting >7 days, any grade 4 or toxicity, any grade 3 toxicity (excluding nausea, vomiting and diarrhea unless it persisted despite optimal treatment).
Responses were assessed after three cycles (about 12 weeks) or earlier at the discretion of the treating physician. All radiological tests were assessed by an MD Anderson radiologist. In addition, results were reviewed in a departmental tumor measurement clinic and by the attending physician. RECIST criteria were used for progressive disease (PD), stable disease (SD), partial and complete responses (PR and CR).
Molecular Assays for Biological Markers: PIK3CA, AKT1, PTEN mutations, and PTEN loss
Clinical Laboratory Improvement Amendment (CLIA) certified mutational and/or immunohistochemistry assays were performed, when tissue was available, for PIK3CA, AKT1, and PTEN mutations, and PTEN expression. The tests were done within the Division of Pathology and Laboratory Medicine at MD Anderson. Archival formalin-fixed, paraffin-embedded tissue blocks or tissue from fine-needle aspiration or surgical biopsies were used for mutational analysis. DNA was extracted and analyzed using a polymerase chain reaction (PCR)-based DNA sequencing method for PIK3CA mutations in codons [c] 532-554 of exon 9 (helical domain) and c1011-1062 of exon 20 (kinase domain) [15], which included the mutation hot spot region of the PIK3CA proto-oncogene by Sanger sequencing following amplification of 276 bp and 198 bp amplicons, respectively, utilizing primers designed at MD Anderson. For AKT1 and PTEN mutations, similar methods were used. Codons 17, 1173, and 179 were examined for AKT1 mutations, and for PTEN, the entire coding sequence of exons 1 through 9 were examined as previously described [16]. PTEN loss was assessed using a Dako antibody (Carpinteria, CA, USA) as previously published [16, 17].
Evaluation of HER2/neu amplification, estrogen and progesterone receptor status
Under CLIA conditions, immunohistochemistry was used to measure of HER2/neu, estrogen and progesterone receptors. Estrogen and progesterone receptors were assessed using antibody 6F11 (Novocastra Laboratories, Ltd., Newcastle Upon Tyne, UK). Alternatively, fluorescence in situ hybridization (FISH) was used to measure the copy number of HER2/neu.
Next-Generation Sequencing
Genomic libraries were captured for 3230 exons in 182 cancer-related genes plus 37 introns from 14 genes often rearranged in cancer and sequenced to average median depth of 734× with 99% of bases covered >100× (Foundation Medicine, Cambridge, MA, USA).
Statistical Analysis
Descriptive statistics are provided for all endpoints using SPSS v.19 (Chicago, IL, USA). Continuous measurements were summarized using mean, standard deviation, median, range, number of patients, and percentages. Time to treatment failure (TTF) and overall survival were calculated using the method of Kaplan and Meier [18, 19] in months, from date of first treatment dose to the date of last treatment dose or death from any cause, whichever came first. Patients still on study at the time of data analysis were censored at the time of last assessment for TTF. For survival, patients still alive at the time of analysis were censored at that time point. A three-dimensional waterfall plot depicting best RECIST responses by percent and time on therapy is presented in Figure 1.
Acknowledgements:
The authors would like to acknowledge Mercy David and Uyen Vu for their data and patient care during this study.
Grant Support:
This study was not financial supported by any grants.
Conflicts of Interest:
The authors have no conflicts of interests to declare.
References
1. Harris HA. Estrogen receptor-beta: recent lessons from in vivo studies. Mol Endocrinol. 2007; 21(1):1-13.
2. Welboren WJ, Sweep FC, Span PN and Stunnenberg HG. Genomic actions of estrogen receptor alpha: what are the targets and how are they regulated? Endocr Relat Cancer. 2009; 16(4):1073-1089.
3. Zhao C, Dahlman-Wright K and Gustafsson JA. Estrogen receptor beta: an overview and update. Nucl Recept Signal. 2008; 6:e003.
4. Simpson ER, Mahendroo MS, Means GD, Kilgore MW, Hinshelwood MM, Graham-Lorence S, Amarneh B, Ito Y, Fisher CR, Michael MD and et al. Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endocr Rev. 1994; 15(3):342-355.
5. Bulun SE, Price TM, Aitken J, Mahendroo MS and Simpson ER. A link between breast cancer and local estrogen biosynthesis suggested by quantification of breast adipose tissue aromatase cytochrome P450 transcripts using competitive polymerase chain reaction after reverse transcription. J Clin Endocrinol Metab. 1993; 77(6):1622-1628.
6. Miller WR and O’Neill J. The importance of local synthesis of estrogen within the breast. Steroids. 1987; 50(4-6):537-548.
7. Geisler J, King N, Dowsett M, Ottestad L, Lundgren S, Walton P, Kormeset PO and Lonning PE. Influence of anastrozole (Arimidex), a selective, non-steroidal aromatase inhibitor, on in vivo aromatisation and plasma oestrogen levels in postmenopausal women with breast cancer. Br J Cancer. 1996; 74(8):1286-1291.
8. Baselga J, Campone M, Piccart M, Burris HA, 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, Deleu I, Perez A, Bachelot T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012; 366(6):520-529.
9. Slomovitz BM, Brown J, Johnston TA, Mura D, Levenback C, Wolf J, Adler KR, Lu H and Coleman RL. A phase II study of everolimus and letrozole in patients with recurrent endometrial carcinoma. J Clin Oncol. 2011; 29(suppl; abstr 5012).
10. Baselga J, Semiglazov V, van Dam P, Manikhas A, Bellet M, Mayordomo J, Campone M, Kubista E, Greil R, Bianchi G, Steinseifer J, Molloy B, Tokaji E, Gardner H, Phillips P, Stumm M, et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol. 2009; 27(16):2630-2637.
11. Nicholson RI, Hutcheson IR, Hiscox SE, Knowlden JM, Giles M, Barrow D and Gee JM. Growth factor signalling and resistance to selective oestrogen receptor modulators and pure anti-oestrogens: the use of anti-growth factor therapies to treat or delay endocrine resistance in breast cancer. Endocr Relat Cancer. 2005; 12 Suppl 1:S29-36.
12. Zhao S, Chen X, Lu X, Yu Y and Feng Y. Epidermal growth factor receptor signaling enhanced by long-term medroxyprogesterone acetate treatment in endometrial carcinoma. Gynecol Oncol. 2007; 105(1):45-54.
13. Jegg AM, Ward TM, Iorns E, Hoe N, Zhou J, Liu X, Singh S, Landgraf R and Pegram MD. PI3K independent activation of mTORC1 as a target in lapatinib-resistant ERBB2+ breast cancer cells. Breast Cancer Res Treat. 2012.
14. Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M, Hicks DG, Lester S, Love R, Mangu PB, McShane L, Miller K, et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010; 28(16):2784-2795.
15. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B and Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004; 304(5670):554.
16. Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, Horlings H, Berns K, Hung MC, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008; 68(15):6084-6091.
17. Janku F, Broaddus R, Bakker R, Hong D, Stepanek V, Naing A, Falchook G, Fu S, Wheler JJ, Piha-Paul SA, Moulder S, Luthra R, Tsimberidou A and Kurzrock R. PTEN assessment and PI3K/mTOR inhibitors: Importance of simultaneous assessment of MAPK pathway aberrations. J Clin Oncol. 2012; 30:suppl; abstr 10510.
18. Kaplan EL and Meier P. Nonparametric estimation from incomplete observations. J Amer Statist Assn. 1958; 53:457-481.
19. Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemotherapy Reports. 1966; 50(3):163-170.
20. Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, Hong DS, Naing A, Falchook GS, Moroney JW, Piha-Paul SA, Wheler JJ, Moulder SL, Fu S and Kurzrock R. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther. 2011; 10(3):558-565.
21. Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna I, Luthra R, Lee JJ, Lu KH and Kurzrock R. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012; 30(8):777-782.
22. Chandarlapaty S, Sakr RA, Giri D, Patil S, Heguy A, Morrow M, Modi S, Norton L, Rosen N, Hudis CA and King TA. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin Cancer Res. 2012.
23. Sokolosky ML, Stadelman KM, Chappell WH, Abrams SL, Martelli AM, Stivala F, Libra M, Nicoletti F, Drobot LB, Franklin RA, Steelman LS and McCubrey JA. Involvement of Akt-1 and mTOR in sensitivity of breast cancer to targeted therapy. Oncotarget. 2011; 2(7):538-550.
24. Steelman LS, Navolanic PM, Sokolosky ML, Taylor JR, Lehmann BD, Chappell WH, Abrams SL, Wong EW, Stadelman KM, Terrian DM, Leslie NR, Martelli AM, Stivala F, Libra M, Franklin RA and McCubrey JA. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity to mTOR inhibitors. Oncogene. 2008; 27(29):4086-4095.
25. Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Falchook GS, Fu S, Piha-Paul SA, Naing A, Janku F, Luthra R, Ye Y, Wen S, Berry DA and Kurzrock R. Personalized Medicine in a Phase I Clinical Trials Program:The MD Anderson Cancer Center Initiative. Clin Cancer Res. 2012.
26. Hortobagyi GN, Piccart-Gebhart MJ, Rugo HS, Burris HA, Campone M, Noguchi S, Perez A, Deleu I, Shtivelband M, Provencher L, Masuda N, Dakhil SR, Anderson I, Chen D, Damasck A, Huang A, et al. Correlation of molecular alterations with efficacy of everolimus in hormone receptor-positive, HER2-negative advanced breast cancer: Results from BOLERO-2. J Clin Oncol. 2013; 31(18_suppl):LBA509.
27. Sauer K and Lehner CF. The role of cyclin E in the regulation of entry into S phase. Prog Cell Cycle Res. 1995; 1:125-139.
28. Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, Franklin RA and McCubrey JA. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003; 17(3):590-603.
29. Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR and Sears R. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004; 6(4):308-318.
30. LeRoith D and Roberts CT, Jr. The insulin-like growth factor system and cancer. Cancer Lett. 2003; 195(2):127-137.
31. Ong SH, Hadari YR, Gotoh N, Guy GR, Schlessinger J and Lax I. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc Natl Acad Sci U S A. 2001; 98(11):6074-6079.
32. Maurer U, Charvet C, Wagman AS, Dejardin E and Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006; 21(6):749-760.
33. Petrelli F and Barni S. A Pooled Analysis of 2618 Patients Treated with Trastuzumab Beyond Progression for Advanced Breast Cancer. Clin Breast Cancer. 2012.
34. Moroney JW, Schlumbrecht MP, Helgason T, Coleman RL, Moulder S, Naing A, Bodurka DC, Janku F, Hong DS and Kurzrock R. A phase I trial of liposomal doxorubicin, bevacizumab, and temsirolimus in patients with advanced gynecologic and breast malignancies. Clin Cancer Res. 2011; 17(21):6840-6846.
35. Cuny M, Kramar A, Courjal F, Johannsdottir V, Iacopetta B, Fontaine H, Grenier J, Culine S and Theillet C. Relating genotype and phenotype in breast cancer: an analysis of the prognostic significance of amplification at eight different genes or loci and of p53 mutations. Cancer Res. 2000; 60(4):1077-1083.
36. Lee YH and White MF. Insulin receptor substrate proteins and diabetes. Arch Pharm Res. 2004; 27(4):361-370.
37. Yang-Yen HF. Mcl-1: a highly regulated cell death and survival controller. J Biomed Sci. 2006; 13(2):201-204.
38. Aguirre D, Boya P, Bellet D, Faivre S, Troalen F, Benard J, Saulnier P, Hopkins-Donaldson S, Zangemeister-Wittke U, Kroemer G and Raymond E. Bcl-2 and CCND1/CDK4 expression levels predict the cellular effects of mTOR inhibitors in human ovarian carcinoma. Apoptosis. 2004; 9(6):797-805.
39. Heinonen H, Nieminen A, Saarela M, Kallioniemi A, Klefstrom J, Hautaniemi S and Monni O. Deciphering downstream gene targets of PI3K/mTOR/p70S6K pathway in breast cancer. BMC Genomics. 2008; 9:348.
40. Bouchard C, Marquardt J, Bras A, Medema RH and Eilers M. Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J. 2004; 23(14):2830-2840.
41. Welcker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, Clurman BE and Roberts JM. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol Cell. 2003; 12(2):381-392.
42. Metz HE and Houghton AM. Insulin receptor substrate regulation of phosphoinositide 3-kinase. Clin Cancer Res. 2011; 17(2):206-211.
43. Kuo ML, Chuang SE, Lin MT and Yang SY. The involvement of PI 3-K/Akt-dependent up-regulation of Mcl-1 in the prevention of apoptosis of Hep3B cells by interleukin-6. Oncogene. 2001; 20(6):677-685.
44. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366(10):883-892.
45. Li J, Wang K, Jensen TD, Li S, Bolund L and Wiuf C. Tumor heterogeneity in neoplasms of breast, colon, and skin. BMC Res Notes. 2010; 3:321.
46. Russnes HG, Navin N, Hicks J and Borresen-Dale AL. Insight into the heterogeneity of breast cancer through next-generation sequencing. J Clin Invest. 2011; 121(10):3810-3818.
47. Yi KH, Axtmayer J, Gustin JP, Rajpurohit A and Lauring J. Functional analysis of non-hotspot AKT1 mutants found in human breast cancers identifies novel driver mutations: implications for personalized medicine. Oncotarget. 2013; 4(1):29-34.