
INTRODUCTION
Epidermal growth factor receptor (EGFR) activating mutations (e.g., L858R and exon 19 deletion) account for 30–60% of non-small cell lung cancer (NSCLC) cases in Asia [1–3]. NSCLC patients with EGFR activating mutations respond to first- and second-generation EGFR tyrosine kinase inhibitors (TKIs) [4–13]. However, drug resistance inevitably develops, with ~60% of events attributing to a secondary EGFR T790M gatekeeper mutation [14–16]. Osimertinib is an oral, irreversible, mutant-selective third-generation EGFR-TKI developed against NSCLC bearing EGFR activating mutation and T790M [17–19]. In the AURA and AURA3 studies, osimertinib was highly active in lung cancer patients with T790M mutation who had progressed during prior therapy with EGFR-TKIs [20, 21]. Among T790M-positive NSCLC patients, the median progression-free survival (PFS) was significantly longer with osimertinib than with platinum therapy plus pemetrexed (10.1 vs. 4.4 months; hazard ratio, 0.30; P < 0.001), and the objective response rate (ORR) was significantly better with osimertinib than with platinum therapy plus pemetrexed (71% vs. 31%; odds ratio, 5.39; P < 0.001) [21].
So far, only a few studies have been performed to understand potential mechanisms of resistance to osimertinib. EGFR C797S mutation has been demonstrated as a principle mechanism of acquired resistance [22, 23], presumable through abolishing the covalent binding between osimertinib and C797 residue. In addition, there are a limited number of case reports detailing the identification of EGFR L718Q, BRAF V600E and PIK3CA E545K mutations, as well as ERBB2 and MET amplification [23–26]. Little is known about alternative resistance mechanisms, the prevalence of each type of mechanisms, and the ethnic differences in resistance profiles. In this study, we present a case report on the first Chinese osimertinib NSCLC patient. We closely monitored the disease course of the patient, prospectively collected pretreatment and post-disease progression plasma specimens, and identified a standalone EGFR G796D mutation as a new resistance mechanism to osimertinib.
RESULTS
Medical history of patient246
Patient246 was a 56 year-old female diagnosed as stage IV lung adenocarcinoma (T4N2M1b), with multiple distant metastasis lesions including brain and bone (Figure 1, Supplementary Figure 1). She had received multiple lines of chemotherapy including 4 cycles of gemcitabine/cisplatin and 2 cycles of pemetrexed/carboplatin for 2 years and 5 months in total. After disease progression, she switched to gefitinib and showed a partial response within 1 month of treatment. At 13-month post-gefitinib, she relapsed and EGFR T790M mutation was detected from biopsy of progressed primary lesion (Figure 1). Through special approval of “named patient use” due to unavailability of osimertinib in Chinese market at the time, she became the first patient to receive osimertinib in China. After 6 weeks of treatment, scans demonstrated a partial response. However, she developed systemic progressive disease (PD) at ~6.5-month post-osimertinib and switched to radiotherapy subsequently (Figure 1).
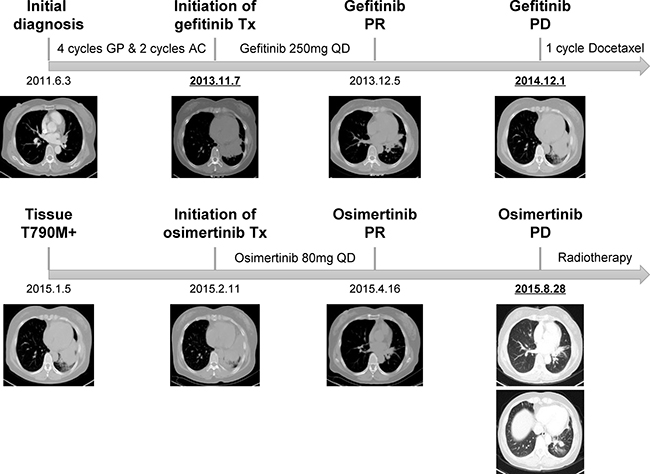
Figure 1: Medical history of patient246. The patient first underwent multiple lines of chemotherapy and gefitinib treatment. After the resistance biopsy was diagnosed as T790M-positive, she received osimertinib through “named patient use”. Abbreviations: GP, gemcitabine/cisplatin; AC, pemetrexed/carboplatin; Tx, treatment; PR, partial response; PD, progressive disease. The three time points of plasma sample collection are underlined and bolded.
Identification of EGFR G796D mutation from plasma samples
To understand the mechanisms underlying resistance to osimertinib in patient246, we collected and profiled her plasma samples from three time points – pre-gefitinib, gefitinib PD and osimertinib PD (Figure 1). Using next-generation panel sequencing, we identified EGFR L858R mutation with mutant allele frequency (MAF) of 4.28% in the pre-gefitinib plasma sample (Figure 2). Interestingly, low MAF (0.61%) of EGFR G796D was also detected at this point. After gefitinib PD, T790M emerged to MAF of 1.85%, consistent with tissue testing results. Upon osimertinib PD, both L858R and T790M became undetectable, however, G796D increased to MAF of 1.91% (Figure 2). As control, none of the three types of mutations were detected in whole blood. These data suggested that G796D was a de novo mutation likely involved in resistance to osimertinib. The different dynamics of L858R, T790M and G796D during treatment journey also implicated that G796D might be a standalone mutation independent of L858R and T790M.
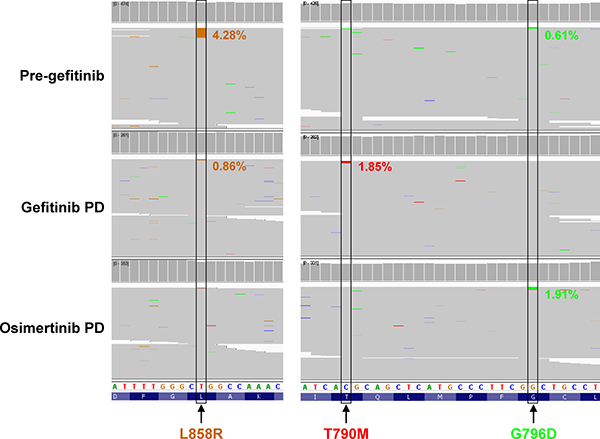
Figure 2: Dynamics of EGFR L858R, T790M and G796D mutations. IGV view of variant calls covering EGFR L858, T790 and G796 residues from panel sequencing of plasma samples at indicated time points. G796D mutation increased from 0.61% of reads at pre-gefitinib to 1.91% upon osimertinib PD, while T790M acquired at gefitinib resistance was cleared by osimertinib.
EGFR G796D mutation as the resistance mechanism to osimertinib
G796 is located adjacent to C797 residue, where osimertinib forms covalent bond with EGFR. Thus we hypothesized that G796D could drive resistance to osimertinib by interfering with osimertinib-EGFR interaction. Indeed, structural modeling of EGFR kinase domain in complex with osimertinib showed that the side chain of mutated D796 residue would clash into the molecular surface of osimertinib, lead to a steric and energetic repulsion, and result in the loss of binding affinity (Figure 3).
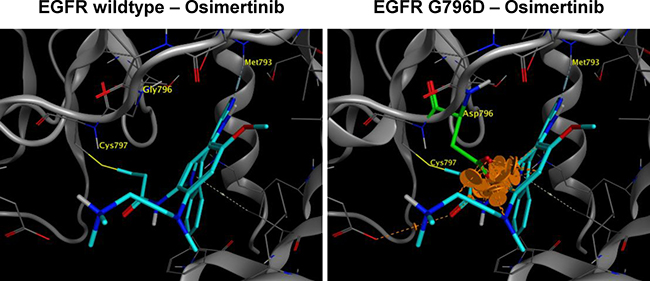
Figure 3: Structural modeling of EGFR G796D in complex with osimertinib. G796D mutation is expected to sterically interfere with binding of osimertinib. The hydrophilic side chain CH2COOH of mutated D796 residue will bump into the greasy core of osimertinib, which would either push the inhibitor out of the current binding mode, or distort the loop and affect the hinge binding. Either case could lower the binding affinity of osimertinib. The orange disks indicate the clashes between the side chain of the mutated reside and the core of osimertinib.
To confirm that G796D could induce resistance to osimertinib, we generated stable lines of Ba/F3 cells carrying various EGFR mutants (Supplementary Figure 2). IL3-independent growth assay suggested that G796D was likely a mild oncogenic driver comparing to L858R and L858R/T790M mutations (Supplementary Figure 3). Osimertinib inhibited the growth of L858R or L858R/T790M lines with GI50 of 30–40 nM, but GI50 increased to 1.5–2 μM in G796D mutant line, with about 50-fold shift (Figure 4A). Moreover, p-EGFR level of G796D mutant line, and its downstream p-AKT and p-ERK1/2 levels were not modulated by osimertinib up to 1 μM concentration (Figure 4B). These cellular functional results suggested that G796D mutation rendered the resistance to osimertinib.
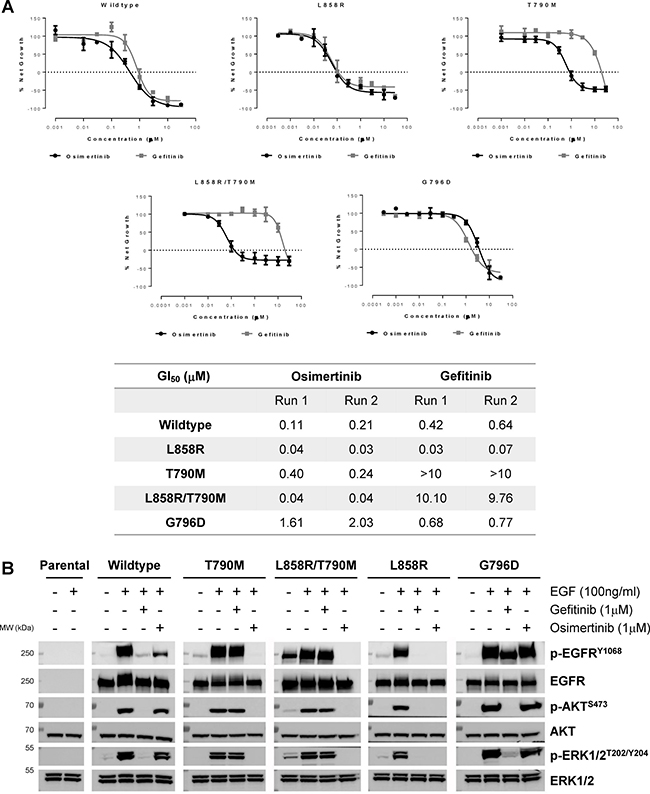
Figure 4: EGFR G796D confers resistance to osimertinib. (A) Upper: Ba/F3 cells harboring various EGFR mutants were treated with osimertinib or gefitinib at indicated concentrations. Viable cells were measured after 72 h of treatment and cell proliferation was calculated as % Net Growth = (Gday3, inh - G0)/(Gday3, ctl - G0) × 100%. Experiments were repeated twice, with mean ± S.D. plotted at each concentration. The curves were fitted using a nonlinear regression model with a sigmoidal dose response. Lower: GI50 (μM) of the two independent runs. (B) Ba/F3 cells expressing various EGFR mutants were pre-incubated with 1 μM gefitinib or osimertinib for 2 h followed by stimulation of EGF for 10 min in serum-free medium. Cell extracts were immunoblotted to detect phosphorylated or total EGFR, AKT and ERK1/2 levels.
DISCUSSION
Plasma cell-free DNA (cfDNA) samples have been shown to contain tumor-specific genomic alterations and utilized to dynamically monitor tumor response and relapse [27, 28]. Comparing with tissue testing, plasma testing is often associated with lower sensitivity but on the other hand, can circumvent the challenges in obtaining re-biopsy specimens and “false-negatives” resulted from tumor heterogeneity. The abundances of mutations in cfDNA are correlated with tumor burden, stage and metastasis status [29, 30]. And several studies have demonstrated that quantitative measurement of mutation abundances in cfDNA could be used to accurately reflect clinical response and emerge of resistance [31–33]. In this study, we used cfDNA to understand the resistance mechanisms of osimertinib.
Our dynamic genomic analysis of serial cfDNA samples from patient246 at time points of pre-gefitinib, gefitinib PD and osimertinib PD discovered EGFR G796D mutation as a new resistance mechanism to osimertinib. The MAF of G796D in cfDNA samples increased from 0.61% at pre-gefitinib to 1.91% upon osimeritinib PD. Meanwhile, the acquired T790M mutation following gefitinib PD was cleared to the undetectable level by osimeritinib, which is consistent with the reported data in some patients receiving second-line treatment of osimertinib or rociletinib, another third-generation EGFR-TKI [22, 23, 26, 34, 35]. This dynamic switch between G796D and T790M mutations suggested a clonal evolution event which featured diminishment of T790M-positive clones and outgrowth of pre-existing G796D-positive clones under selection pressure of osimertinib (Figure 5).
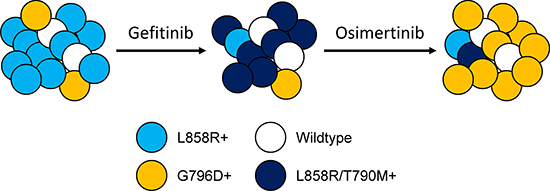
Figure 5: Tumor heterogeneity and clonal evolution. A conceptual model showing that the tumor was genomically heterogeneous, comprising of both L858R-positive and G796D-positive populations, with the former as the major clone. Upon gefitinib resistance, L858R-positive cells acquired the secondary T790M mutation and became the dominant driver of tumor growth. These L858R/T790M-positive tumor cells were then inhibited by osimertinib, whereas pre-existing G796D-positive clones, which are resistant to osimertinib, gradually made up the majority of the population.
Previously, G796C/D/R were demonstrated to render resistance to erlotinib and CI-1033 in Ba/F3 cell system through a random mutagenesis screening approach [36]. The impact was more dramatic on CI-1033, a pan-ErbB inhibitor that forms a covalent bond with EGFR C797 residue [37]. The GI50 increased 10- and 100-fold for erlotinib and CI-1033, respectively, in G796R mutant line compared with Ba/F3 cells expressing wildtype EGFR [36]. G796D could induce resistance to osimertinib by interfering with osimertinib-EGFR interaction due to steric hindrance around C797 residue, the covalent binding site for the inhibitor. We noted that G796D is resistant to gefitinib as well as erlotinib (Supplementary Figure 4), albeit to a less extent. However, G796D-positive cells remained as minority of population upon gefitinib PD. We reasoned that it was probably because G796D is a milder mutation comparing to L858R/T790M. Therefore, L858R/T790M-positive population presented as the dominant force of tumor growth at gefitinib resistance, and G796D-positve cells only emerged as majority after L858R/T790M-positive clones were diminished by osimertinib (Figure 5).
To our knowledge, all the EGFR mutations previously reported to drive resistance to osimertinib or rociletinib, such as L718Q, C797S and L798I, were identified with T790M concurrently [22–24, 34], while G796D is found in the absence of T790M context. Hence this is the sole case by far of EGFR mutation-dependent mechanism causing intrinsic resistance to osimertinib. G796A/D/S mutations were reported in TKI-naive lung adenocarcinoma patients, either alone or in combination with exon 19 deletion [38–40], supporting that it can exist as a de novo oncogenic mutation. Consistently, in our case, G796D was identified together with L858R at pre-gefitinib, as a minor, independent clone. It remains to be seen whether G796D can also present as an acquired resistance mutation.
In conclusion, we have identified a case of Chinese NSCLC patient with intrinsic EGFR G796D mutation that led to resistance to osimertinib. The loss of T790M-positive clones and selection of pre-existing G796D-positive clones suggest that genomic heterogeneity contributes to clonal evolution which ultimately results in the emergence of drug resistance during treatment journey. Our findings highlight the needs for development of new targeted therapies and combination strategies dynamically adjusted based on real-time monitored genomic analysis throughout the entirety of disease course.
MATERIALS AND METHODS
Patient information
Patient246 was diagnosed with advanced NSCLC at Shanghai Pulmonary Hospital in June 2011. The study was approved by the hospital research ethics committee and the patient had signed the informed consent form. Serial plasma samples were collected at time points of pre-gefitinib, gefitinib PD and osimertinib PD. Whole blood were obtained at osimertinib PD. Tumor responses were assessed according to Response Evaluation Criteria In Solid Tumors Version 1.1 (RECIST 1.1).
Extraction of genomic DNA and plasma cell-free DNA (cfDNA)
Genomic DNA from whole blood was extracted using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Plasma samples were prepared and stored as described previously [33]. cfDNA was extracted with QIAamp Circulating Nucleic Acid Kit (Qiagen) according to the manufacturer’s protocol.
Next-generation panel sequencing
Indexed Illumina NGS libraries were prepared from plasma cfDNA and fragmented genomic DNA using KAPA Hyper Prep Kit (Kapa Biosystems, Woburn, MA, USA) according to the manufacturer’s protocol. Ligation was performed at 4°C overnight using adapters from SeqCap Adapter Kit (Roche NimbleGen, Madison, WI, USA). The indexed libraries were captured using a customized SeqCap EZ Choice Library (Roche NimbleGen) covering 403 genes (Supplementary Table 1). Hybrid selection, washing, recovery, amplification and purification of captured multiplex DNA samples were performed using SeqCap EZ system (Roche NimbleGen). The amplified products were then purified with QIAquick PCR Purification Kit (Qiagen), and multiplexed libraries sequenced on the Illumina HiSeq X10 platform with 150-bp paired-end reads according to the manufacturer’s protocol. The raw average sequence depth was >7000× for plasma cfDNA samples and ~2800× for the genomic DNA from whole blood.
Bioinformatics data analysis
BWA [41] was employed for mapping the pair-end reads to human reference genome hg19, samblaster [42] for marking duplicate reads, and VarDict [43] for detection of single nucleotide variants (SNVs) and small insertions and deletions (indels). SNVs and small indels were called with the criteria of allele frequency ≥ 0.5% and at least two supporting reads after deduplication. Variant calls of interest were manually inspected in IGV [44, 45].
Structural modeling
G796 was mutated to aspartic acid based on the optimal trajectory for the side chain using the EGFR-osimertinib co-crystal structure (PDB code: 4ZAU) and “Mutate Residue” method in Maestro (Schrodinger LLC., New York, NY, USA).
Cell proliferation assay
Generation of Ba/F3 stable lines expressing various EGFR mutants was described in Supplementary Materials and Methods. For MTS assay, Ba/F3 cells were prepared in RPMI1640 with 10% FBS and 10 ng/ml EGF, seeded in 96-well plates at 30,000 cells per well, and incubated overnight. The next day, compounds at different concentrations were added to the assay plates, and cells were incubated for additional 72 hours. Following compound treatment, 20 μl/well of CellTiter 96® AQueous One Solution Reagent (Promega, Madison, WI, USA) was added, incubated at room temperature for 2 hours, then 25 μl/well of 10% SDS was added to stop the reaction. The absorbance was measured at 490 nm wavelength using 650 nm as reference on Tecan Spark 20M (Tecan Group Ltd., Zurich, Switzerland). The data was processed and plotted using GraphPad Prism 6 (GraphPad Software Inc., San Diego, CA, USA).
Modulation of signal pathways
Ba/F3 cells harboring various EGFR mutants were starved in RPMI1640 for 4 hours, pre-treated with 1 uM gefitinib or osimertinib for 2 hours, and then stimulated by 100 ng/ml EGF for 10 minutes in serum-free medium. Cells were collected, washed once in cold PBS and lysed in 2×SDS lysis buffer [100 mM Tris pH6.8, 4% SDS, 20% glycerol, and 1× protease and phosphatase inhibitors (Pierce | Thermo Fisher Scientific, Waltham, MA, USA)]. The lysates were boiled at 100°C for 10 minutes, and protein concentration was quantified by BCA Protein Assay Kit (Pierce). Equal amount of protein was loaded onto SDS-PAGE gel, transferred to nitrocellulose membrane using iBolt (Invitrogen, Carlsbad, CA, USA), and subjected to immunoblotting analysis with indicated antibodies according to manufacturers’ instructions.
ACKNOWLEDGMENTS
We are grateful to the patient and her family for providing the samples in this study. We thank Dr. Tony S. Mok (the Chinese University of Hong Kong and Prince of Wales Hospital) for the prescription of osimertinib to the patient through special approval of “named patient use”. We acknowledge Chunlei Huang, Jiangang Fu, Haiyan Xu, Jiangchun Pi, Xin Zhang and Zhijie Ding (AstraZeneca) for technical support, Changting Liu and Xinying Su (AstraZeneca) for sample management, and Zhengwei Dong (Shanghai Pulmonary Hospital) and Hulin Han (AstraZeneca) for pathology consultation.
CONFLICTS OF INTEREST
Hu M, Bai Y, Zhu XH, Lu XS, Wang Z and Yang ZF are employees of AstraZeneca. Yang ZF holds stocks in AstraZeneca. Zheng D reported lecture fees from AstraZeneca, Eli Lilly, Novartis and Pfizer. Ni J and Xu JF reported lecture fees from AstraZeneca, Eli Lilly, F. Hoffman-La Roche and Pfizer. No conflicts of interest were disclosed by the other authors.
GRANT SUPPORT
This work was supported with grant from Shanghai Municipal Science and Technology Commission (to Ni J; 134119a6500).
REFERENCES
1. An SJ, Chen ZH, Su J, Zhang XC, Zhong WZ, Yang JJ, Zhou Q, Yang XN, Huang L, Guan JL, Nie Q, Yan HH, Mok TS, et al. Identification of enriched driver gene alterations in subgroups of non-small cell lung cancer patients based on histology and smoking status. PLoS One. 2012; 7:e40109.
2. Lai Y, Zhang Z, Li J, Sun D, Zhou Y, Jiang T, Han Y, Huang L, Zhu Y, Li X, Yan X. EGFR mutations in surgically resected fresh specimens from 697 consecutive Chinese patients with non-small cell lung cancer and their relationships with clinical features. Int J Mol Sci. 2013; 14:24549–24559.
3. Shi Y, Li J, Zhang S, Wang M, Yang S, Li N, Wu G, Liu W, Liao G, Cai K, Chen L, Zheng M, Yu P, et al. Molecular Epidemiology of EGFR Mutations in Asian Patients with Advanced Non-Small-Cell Lung Cancer of Adenocarcinoma Histology - Mainland China Subset Analysis of the PIONEER study. PLoS One. 2015; 10:e0143515.
4. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010; 362:2380–2388.
5. Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, Asami K, Katakami N, Takada M, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010; 11:121–128.
6. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009; 361:947–957.
7. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, Porta R, Cobo M, Garrido P, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012; 13:239–246.
8. Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, Su WC, Bennouna J, Kato T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013; 31:3327–3334.
9. Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY, Xu CR, Massey D, Kim M, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014; 15:213–222.
10. Wu YL, Zhou C, Liam CK, Wu G, Liu X, Zhong Z, Lu S, Cheng Y, Han B, Chen L, Huang C, Qin S, Zhu Y, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol. 2015; 26:1883–1889.
11. Yang JC, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, Zhou C, Hu CP, O’Byrne K, Feng J, Lu S, Huang Y, Geater SL, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015; 16:141–151.
12. Shi Y, Wang L, Han B, Li W, Yu P, Liu Y, Ding C, Song X, Yong MZ, Ren X, Feng JF, Zhang H, Chen G, et al. First-line icotinib versus cisplatine/pemetrexed plus pemetrexed maintenance therapy in lung adenocarcinoma patients with sensitizing EGFR mutation (CONVINCE). American Society of Clinical Oncology Annual Meeting; June 3–7, 2016; Chicago, IL, USA [Abstract 9041]. 2016.
13. Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, Lu S, Zhang L, Hu C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011; 12:735–742.
14. Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014; 11:473–481.
15. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005; 352:786–792.
16. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, Riely GJ. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013; 19:2240–2247.
17. Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009; 462:1070–1074.
18. Ward RA, Anderton MJ, Ashton S, Bethel PA, Box M, Butterworth S, Colclough N, Chorley CG, Chuaqui C, Cross DA, Dakin LA, Debreczeni JE, Eberlein C, et al. Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J Med Chem. 2013; 56:7025–7048.
19. Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014; 4:1046–1061.
20. Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015; 372:1689–1699.
21. Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, Shepherd FA, He Y, Akamatsu H, Theelen WS, Lee CK, Sebastian M, Templeton A, et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N Engl J Med. 2017; 376:629–640.
22. Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015; 21:560–562.
23. Oxnard GR. Mechanisms of Acquired Resistance to AZD9291 in EGFR T790M Positive Lung Cancer. IASLC 16th World Conference on Lung Cancer; September 6–9, 2015; Denver, CO, USA [ID 1365]. 2015.
24. Bersanelli M, Minari R, Bordi P, Gnetti L, Bozzetti C, Squadrilli A, Lagrasta CA, Bottarelli L, Osipova G, Capelletto E, Mor M, Tiseo M. L718Q Mutation as New Mechanism of Acquired Resistance to AZD9291 in EGFR-Mutated NSCLC. J Thorac Oncol. 2016; 11:e121–123.
25. Ho CC, Liao WY, Lin CA, Shih JY, Yu CJ, Yang JC. Acquired BRAF V600E Mutation as Resistant Mechanism after Treatment with Osimertinib. J Thorac Oncol. 2017; 12:567–572.
26. Planchard D, Loriot Y, Andre F, Gobert A, Auger N, Lacroix L, Soria JC. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann Oncol. 2015; 26:2073–2078.
27. Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013; 10:472–484.
28. Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014; 32:579–586.
29. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014; 6: 224ra224.
30. Sacher AG, Paweletz C, Dahlberg SE, Alden RS, O’Connell A, Feeney N, Mach SL, Janne PA, Oxnard GR. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. 2016; 2:1014–1022.
31. Oxnard GR, Paweletz CP, Kuang Y, Mach SL, O’Connell A, Messineo MM, Luke JJ, Butaney M, Kirschmeier P, Jackman DM, Janne PA. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014; 20:1698–1705.
32. Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, Shrager JB, Loo BW Jr, Alizadeh AA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014; 20:548–554.
33. Zheng D, Ye X, Zhang MZ, Sun Y, Wang JY, Ni J, Zhang HP, Zhang L, Luo J, Zhang J, Tang L, Su B, Chen G, et al. Plasma EGFR T790M ctDNA status is associated with clinical outcome in advanced NSCLC patients with acquired EGFR-TKI resistance. Sci Rep. 2016; 6: 20913.
34. Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, Kurtz DM, Stehr H, Scherer F, Karlovich CA, Harding TC, Durkin KA, Otterson GA, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016; 7: 11815.
35. Piotrowska Z, Niederst MJ, Karlovich CA, Wakelee HA, Neal JW, Mino-Kenudson M, Fulton L, Hata AN, Lockerman EL, Kalsy A, Digumarthy S, Muzikansky A, Raponi M, et al. Heterogeneity Underlies the Emergence of EGFRT790 Wild-Type Clones Following Treatment of T790M-Positive Cancers with a Third-Generation EGFR Inhibitor. Cancer Discov. 2015; 5:713–722.
36. Avizienyte E, Ward RA, Garner AP. Comparison of the EGFR resistance mutation profiles generated by EGFR-targeted tyrosine kinase inhibitors and the impact of drug combinations. Biochem J. 2008; 415:197–206.
37. Fry DW, Bridges AJ, Denny WA, Doherty A, Greis KD, Hicks JL, Hook KE, Keller PR, Leopold WR, Loo JA, McNamara DJ, Nelson JM, Sherwood V, et al. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc Natl Acad Sci USA. 1998; 95:12022–12027.
38. Boldrini L, Ali G, Gisfredi S, Ursino S, Baldini E, Melfi F, Lucchi M, Comin CE, Maddau C, Tibaldi C, Camacci T, Servadio A, Mussi A, et al. Epidermal growth factor receptor and K-RAS mutations in 411 lung adenocarcinoma: a population-based prospective study. Oncol Rep. 2009; 22:683–691.
39. Sugio K, Uramoto H, Onitsuka T, Mizukami M, Ichiki Y, Sugaya M, Yasuda M, Takenoyama M, Oyama T, Hanagiri T, Yasumoto K. Prospective phase II study of gefitinib in non-small cell lung cancer with epidermal growth factor receptor gene mutations. Lung Cancer. 2009; 64:314–318.
40. Beau-Faller M, Prim N, Ruppert AM, Nanni-Metellus I, Lacave R, Lacroix L, Escande F, Lizard S, Pretet JL, Rouquette I, de Cremoux P, Solassol J, de Fraipont F, et al. Rare EGFR exon 18 and exon 20 mutations in non-small-cell lung cancer on 10 117 patients: a multicentre observational study by the French ERMETIC-IFCT network. Ann Oncol. 2014; 25:126–131.
41. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010; 26:589–595.
42. Faust GG, Hall IM. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics. 2014; 30:2503–2505.
43. Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, McEwen R, Johnson J, Dougherty B, Barrett JC, Dry JR. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016; 44:e108.
44. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013; 14:178–192.
45. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011; 29:24–26.