
Introduction
The role of transposable elements (TEs) in tumorigenesis and tumor progression is, despite active research [1-4], still largely unknown. L1 elements (Large Interspersed Element-1, or LINE-1) are transposable elements that amplify in the genome by retrotransposition [2]. Retrotransposition takes place via an RNA intermediate, leaving the origin intact. L1 sequences are very common, comprising ~17% of human genome [5]. However, only about 100 L1 elements are full-length (~6 kb) [6, 7], containing a promoter and two open reading frames (ORF1, ORF2), and thus capable of retrotransposition. These open reading frames encode an endonuclease and a reverse transcriptase, which are necessary to copy and paste the L1 sequence elsewhere in the genome. In somatic cells, activity of L1 elements is repressed by hypermethylation and post-transcriptional mechanisms [8, 2]. In cancer cells, however, hypomethylation is a common early event that allows L1 retrotransposition activity via loss of promoter methylation [8-11].
L1 retrotranspositions have been reported in several tumor types including colorectal cancer [1]. Colorectal cancers (CRCs), like most other solid tumors, display a variety of chromosomal changes such as deletions, inversions, translocations, amplifications, and other genetic abnormalities in addition to point mutations [12, 13]. The majority of such changes are passengers and a result of stochastic events and genetic instability observed in tumors, but a number of changes also contribute tumorigenesis. L1 retrotransposition is a structural change that may modify expression of the targeted gene. Although insertions of L1 elements are often 5’ truncated, insertion of L1 sequence into an intron can for example truncate a transcript [14] or provide promoter for a novel transcript [15]. Furthermore, ORF2 endonuclease activity has been associated with aggressive prostate cancer phenotype [16] and excessive DNA double strand breaks [17]. In CRC, L1 retrotranspositions have been found to target genes including ODZ3, ROBO2, PTPRM, PCM1, and CDH11 that have a role in tumorigenesis [4].
Whole genome sequencing (WGS) is a state-of-the-art method for analyzing structural variations in the genome [12] and is greatly contributing to our understanding of cancer genomes. In this work, we focused on a specific locus - TTC28 intron 1 - that was recently reported to be the site for the most frequent structural change in CRC (~22% of cases; [18]). The change was interpreted in the study to be an inactivating translocation, thus depicting TTC28 as a prime candidate for a new key CRC gene. We here challenge this interpretation based on investigation of our WGS data obtained from 92 paired CRC and normal tissue samples. We characterize a strikingly frequent somatic L1 retrotransposition originating from the first intron of TTC28. We find one of such retrotransposition to be a common polymorphism in the Finnish population.
Results
Aberrant WGS signal in the first intron of TTC28 stems from an L1 retrotransposition
We initially identified an aberrant signal in 19 out of 92 (21%) WGS in the first intron of TTC28 (chr 22: 29,065,455-29,066,124, GRCh37; Figure 1), where paired-end mates were mapped discordantly but consistently to a chromosome different from chr 22. This signal matched the location and frequency of the signal reported in [18], where it was interpreted as a translocation. On a closer visual inspection of mapped sequence data, however, we interpreted the signal to stem from a retrotransposition of an L1 element (L1Base id 129, [19]; dbRIP id 2000144, [20]) in the first intron of TTC28, instead of a translocation. The visual inspection also yielded additional cases with the retrotransposition signal that were not detected initially, described in detail below.
L1 retrotranspositions originating from TTC28 are frequent in CRC
A total of 83 somatic L1 insertions originating from a specific L1 element residing in the first intron of TTC28 were observed in WGS data of 52 out of 92 (57%) CRC cases. Deep sequencing coverage (>40x) facilitated identification of multiple retrotransposition events in some of the cases. A total of 17 cases with two separate retrotransposition events were identified. In addition, we observed four cases that harbored three events, and two cases with four events. Insertion sites are illustrated in Figure 2 and reported in Supplementary Table 1. In 51 out of 83 retrotranspositions (61%), the insertion target was within an intron of a gene, implying significant intron preference (χ2=54.2, df=1, p=1.81e-13), compatible with previous literature on retrotranspositions [5]. All insertions which occurred within a gene hit a unique target except for two insertions targeting neuro-oncological ventral antigen 1 (NOVA1) (p=0.025).
Sanger sequencing of somatic retrotransposition events.
We successfully validated three somatic L1 insertions by Sanger sequencing. Insertion sites of the validated somatic events were in introns of SGIP1, NOVA1 and ARHGEF4. In each case, we identified two PCR products of varying length: one matching the expected wild type allele size and a larger one representing the allele containing also the L1 insertion. In two cases (SGIP1 and NOVA1), we were able to see sequence corresponding to the truncated 5’ end of the L1 element. In SGIP1 case, we observed 449 bp of L1 sequence originating from 5484 bp downstream from the start of ORF1. Similarly in NOVA1, 246 bp of inserted L1 sequence originating from 5472 bp downstream from ORF1 start was identified. For the ARHGEF4 case, we observed 235 bp of sequence from 6162 bp downstream from ORF1. Finally, in SGIP1 and ARHGEF4 cases, we also observed a poly(A) tail at the 3’ end.
L1 retrotransposition events involving TTC28 are also present in germline
In addition to the somatic retrotranspositions, two germline L1 retrotranspositions originating from TTC28 were observed in WGS data; at GABRA4 and rp11-136O12 (Figure 2). The retrotransposition from TTC28 to GABRA4 (Figure 1) was confirmed by Sanger sequencing and found to be present in 4/92 (4.3%) WGS sequences from CRC patients, in 3/90 (3.3%) additional CRC patients not used for WGS and in 9/90 (10%) anonymous Finnish blood donors, indicating that the aberration is a common polymorphism in Finns. An inversion of the 3’ end of the original L1 sequence was seen in the inserted site (Figure 1).
The four WGS cases sharing the germline retrotransposition at GABRA4 (chr 4) were found to share significantly longer haplotypes around the insertion locus than other cases, indicating a shared founder haplotype (t=2.6, p=0.046, 95% CI [74353,6150003]). As expected, evidence for shared haplotypes at TTC28 (chr 22) were not observed in cases sharing the TTC28-GABRA4-retrotransposition (t=-0.27, p=0.79). The 3/92 (3.3%) WGS cases with the germline L1 insertion at rp11-136O12 (chr 8) were found not to have a significantly longer haplotype (t=-0.27, p=0.81), suggesting more ancient or independent origins.
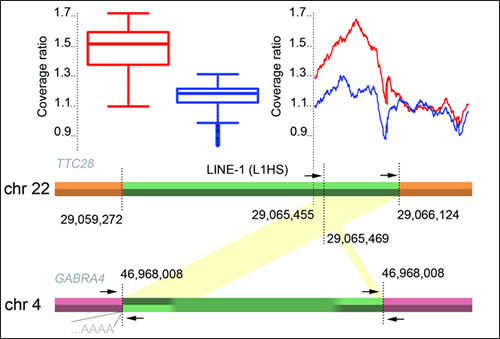
Figure 1: A schematic picture of the TTC28-GABRA4 retrotransposition. Primer locations (arrows), and relative mean coverage change in tumors are depicted: Mean coverages in cases with and without breakpoint signals were compared with the mean coverage of whole chromosome 22 across all tumors. Box plot illustrates the coverage ratios for the region 22:29,065,455-29,066,124 (GRCh37). Mean values for cases with a consistent discordant paired-end signal of at least three distally mapped mates are shown in red (n=54), the remaining cases in blue (n=38). Green region shows the originating L1 element in TTC28 and the inserted sequence in GABRA4. Mapping of discordant read-pairs are shown in yellow. Location of the L1 poly-A insertion is also shown.
Gain of L1 sequence from the TTC28 first intron in retrotransposition cases
Finally, we examined the copy number changes around the 3’ end of TTC28 L1 element (Figure 1), and found that these data too were compatible with L1 retrotransposition rather than translocation, in both germline and tumors displaying the aberrant WGS signal. Since the sequence in the 3’ end of the L1 element in TTC28 allowed unique mapping of paired-end reads, copy number analysis could be performed with WGS data. By comparing the tumor mean read coverage in the L1 3’ end region to the whole chromosome 22, we discovered that in tumors with TTC28 “breakpoints” the relative coverage was significantly increased compared to cases where no retrotransposition signal was observed (t=49, p<2.2x10-16, 95% CI [0.32, 0.34]; Figure 1). This result indicates gain of L1 sequence (Figure 1) in cases displaying the retrotransposition signal, suggesting that this sequence in chromosome 22 was present in more than two copies.
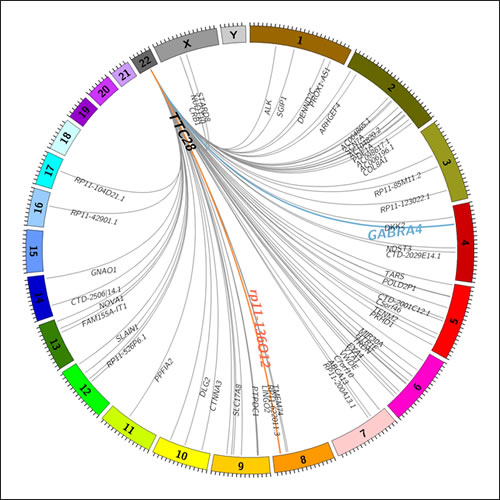
Figure 2: A Circos plot showing identified transposition events originating from TTC28 locus in 92 CRC cases. Germline transpositions to GABRA4 and rp11-136O12 loci are shown in blue and red, respectively. Somatic events are shown in grey. Insertion targets within genes are denoted by gene name.
Discussion
Transposable elements, such as L1, and their role in cancer are under active study [1, 2, 3]. We observed strikingly frequent L1 retrotranspositions in 57% of colorectal cancers originating from an L1 element in TTC28. In total, 83 somatic and 7 germline retrotranspositions that had originated from the element were observed. This remarkably active retrotransposition was the most frequent structural change detected in our WGS data consisting of 92 tumor-normal pairs. The retrotransposed sequence, belonging to the L1 subfamily TA-1nd, has earlier been found to be one of the most active L1 elements in a cultured cell retrotransposition assay [6].
Identification of retrotranspositions by WGS is often difficult due to the large number of almost identical copies of retroelements [1]. Detection of the particular retrotranspositions studied here and their origin in TTC28 was possible due to the distinct 3’ UTR of the element, which allowed identification of the source locus by short-read mapping. In many cases, however, accurate identification of retrotransposition source locus is not feasible using only short-read data. Besides shedding light on the activity of this particular transposable element in cancer, our result highlights the importance of careful analysis of WGS data and robust validation of candidate aberrations. Reliable detection of structural variants, such as events involving transposable element insertions, requires sufficient sequencing coverage. Here we were able to observe multiple retrotransposition targets per sample stemming from the same TTC28 locus due to sufficiently deep coverage.
The frequent breakpoint signals in TTC28 (22:29,065,671-29,066,377) observed by TCGA [18] and those in our study reside in the same narrow region and are very likely to reflect the same phenomenon. Our interpretation contradicts that presented in the TCGA study where TTC28 was depicted as a frequent target for inactivating translocations [18]. The interpretation by TCGA was based on WGS calls, as well as validation of some of the breakpoints by PCR and Sanger sequencing. In our study, we managed to validate both junction breakpoints related to the retrotransposition. Such validation is required to identify this particular structural change as a retrotransposition instead of a translocation involving only one breakpoint at the originating locus. Overall, we strongly suggest that the changes interpreted by the TCGA as inactivating translocations involving TTC28 are in fact L1 retrotranspositions, based on the evidence presented here.
Retrotranspositions may have oncogenic effects [21], or they can be neutral depending on their insertion sites. In our data, all insertions hit separate targets, except for two insertions targeting NOVA1. Given the large number of potential insertion sites, even two hits to a single gene was somewhat unexpected (p=0.025) and might reflect selective value. NOVA1 has been identified as a splicing factor playing a role in neuronal splicing program, but it is also expressed in fibroblasts [22]. Other splicing factors such as SRSF6 have been associated with colorectal cancer [23]. The possible role of NOVA1 in CRC remains to be studied. We also found that TTC28-GABRA4 retrotransposition is a common polymorphism in the Finnish population. Thus this particular event is not likely to be oncogenic.
To summarize, our study sheds light on the nature of TTC28 aberrations in CRC, as well as provides a valuable lesson in interpretation of WGS data. The TTC28 events that we observed are frequent, and some may be involved in tumorigenesis while others are likely to be neutral. Much work remains to be done to unravel the biological consequences of retrotranspositions in cancer. Accurate identification of these events is a prerequisite for success.
Methods
Study samples
In total, 92 familial CRC cases from 89 families (tumor and normal DNA) fulfilling the following criteria were included in this study: (i) at least one CRC case in a first degree relative, (ii) negative for any known high penetrance CRC mutation, and (iii) availability of sufficient amount of DNA, and (iv) microsatellite stable tumors. Seventy-nine of these 92 cases, and the additional 90 cases, used in the validation are part of a previously described population-based collection of Finnish CRC cases [24, 25]. The rest of the CRC cases (n=13) are part of an unpublished sample series from two Finnish hospitals. Finnish blood donor DNA samples (n=90) obtained from the Finnish Red Cross Blood Transfusion Service were used as controls.
The study has been reviewed and approved by the Ethics Committee of the Hospital district of Helsinki and Uusimaa (HUS). Signed informed consent or authorization from the National Supervisory Authority for Welfare and Health has been obtained for all the study participants.
Whole-genome sequencing of 92 CRC tumor and normal DNA samples
Whole-genome sequencing was performed on the Illumina HiSeq 2000 platform with paired-end reads of length 100 bp. Each normal and tumor DNA sample was sequenced to at least 40x median coverage. Sequencing data quality was evaluated with FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc). Paired-end sequencing data was aligned using BWA 0.6.2 [26] against the 1000 Genomes Project Phase 2 reference assembly, which is derived from the GRCh37 reference [27]. Default BWA parameters were used, except for -n 0.06, -q 5 for bwa aln and -a 800 for bwa sampe. PCR duplicates were removed with samtools [28]. Local realignment was performed by GATK around known indel sites in 1000 Genomes and Mills gold standard sets, and 1000 Genomes Phase 1 indels, in addition to preliminary indel calls created using GATK UnifiedGenotyper [29]. Base quality scores were recalibrated with GATK.
Identification of somatic structural changes
Somatic structural aberrations were identified in WGS with DELLY [30] and custom scripts. DELLY is a computational method to detect deletions, tandem duplications, inversions and translocations in whole-genome paired-end sequencing data using paired-end and split-read signatures. Structural changes with respect to the reference genome were identified independently in tumor and normal samples. In tumors, a minimum mapping quality of 20 and at least five supporting reads were required to make a call. To make normal calls for subsequent somatic filtering, mapping quality threshold was not used and only two supporting reads were required, resulting in a highly sensitive call set. To identify somatic retrotransposition events, the following filtering approach was adopted. Breakpoints of translocation calls in each normal sample were first flanked by 500 bp in both directions. Any translocation called in a tumor sample where a breakpoint of the translocation was in the combined flanked regions of respective normal sample was removed. This process yielded a set of somatic translocation calls for each tumor-normal sample pair. Each translocation call was annotated with genes containing either of translocation breakpoints. A similar filtering approach to somatic translocation identification was followed in our previous whole-genome study [31].
Calling retrotransposition events at TTC28 in WGS data
The most frequently involved gene in DELLY translocation calls was TTC28 on chromosome 22. Breakpoints of translocation calls involving TTC28 were further investigated by manual inspection of aligned paired-end sequences using RikuRator genome analysis software (manuscript under preparation). Here it was observed that the presumed translocation calls corresponded to insertions of sequence originating from the TTC28 locus elsewhere in the genome, instead of a translocation. Whole-genome sequences of all tumor and normal samples at the breakpoint position were visually inspected in RikuRator. An L1 insertion to a specific locus was called when at least three paired-end reads supported the insertion. This inspection also revealed the germline insertions that were initially removed by the above somatic filtering.
Copy number analysis
Copy number changes around the 3’ end of the L1 element in TTC28 were examined by first calculating the sequencing coverage in tumors around the 3’ end of the L1 element (chromosome 22:29,065,455-29,066,124, GRCh37; Figure 1). This region of 669 bp was chosen based on the GABRA4 germline case, where it is copied in the retrotransposition (Figure 1). In particular, the region is unique to the reference genome (GRCh37), allowing discordant read and copy number analysis of the specific locus. For each tumor sample, the ratio of mean coverages in the region and chromosome 22 was computed. A t-test was employed to assess whether there is a significant difference in coverage ratios between cases with either a germline or somatic retrotransposition, and cases with no detected retrotransposition.
Genotyping and haplotype analysis
Each sample was genotyped on the Illumina HumanOmni2.5-8 BeadChip platform containing 2,379,855 markers. SNP calling was performed using Illumina GenomeStudio. Shared haplotypes around L1 breakpoints in germline cases were identified in genotyping data by extending a candidate haplotype from a given position to both 5’ and 3’ directions. Extension was terminated when a pair of opposing homozygotes was found such that the sites were separated by less than 2000 bp. Prior to analysis, the three first-degree relatives were removed, leaving 89 cases to be studied. A t-test was used to assess whether the haplotypes at GABRA4 and TTC28 loci found in this manner were significantly longer in cases sharing a GABRA4-targeting retrotransposition than in other cases. The other germline L1 insertion target, rp11-136O12, was tested identically to GABRA4.
Insertion site validation by Sanger sequencing
One germline L1 insertion site candidate (GABRA4) was validated by Sanger sequencing in four samples. Primer pairs comprising each insertion junction were designed using Primer3Plus ([32]; http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/); primer sequences and PCR conditions are available upon request. Fragments were amplified with AmpliTaqGoldVR (Applied Biosystems, Foster City, CA) and the PCR products were purified using the ExoSAP-IT PCR purification kit (USB, Cleveland, OH). Sequencing reactions were performed using the Big Dye Terminator v.3.1 kit (Applied Biosystems) and electrophoresis was run on 3730xl DNA Analyzer (Applied Biosystems) at FIMM Genome and Technology Centre, Finland. The sequence graphs were manually analyzed using FinchTV v.1.4 (Geospiza, Seattle, WA).
Additional three somatic L1 insertion candidates (SGIP1, NOVA1, and ARHGEF4) were successfully validated by Sanger sequencing. Primers flanking the insertion breakpoints were designed, as previously described. The PCR was performed using Expand Long Template PCR system (Roche, Basel, Switzerland). The PCR products were run in standard low-melting agarose gel. DNA band corresponding to the allele with the L1 insertion, was extracted from the agarose gel using QIAquick Gel extraction Kit (Qiagen, Hilden, Germany). The sequencing reaction and the analysis of the sequences were performed as previously described.
Permutation testing of NOVA1 significance
Significance of NOVA1 somatic hit recurrence was assessed with a permutation test. In each permutation, the target of each of 83 somatic events was randomly reassigned to either a gene (61% chance) or intergenic region (59%). In case of an event targeting a gene, the target gene was uniformly selected from 51573 genes, including protein-coding genes, short and long non-coding RNAs and pseudogenes (Ensembl 71.37). A total of 1000000 permutations were performed. An empirical p-value was derived as the fraction of permutations where any gene was hit two or more times.
Acknowledgements
We thank Sini Nieminen, Sirpa Soisalo, Inga-Lill Svedberg, and Iina Vuoristo for technical assistance. This work was supported by grants from the Academy of Finland (Finnish Centre of Excellence Program 2012-2017, and personal grant #137680 for OK), EU FP7 project SYSCOL, the Finnish Cancer Society, and the Sigrid Juselius Foundation. We acknowledge the computational resources provided by the ELIXIR node hosted at CSC - IT Center for Science for ICT (cloud) resources and Institute for Molecular Medicine Finland (FIMM).
References
1. Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette III LJ, Lohr JG, Harris CC, Ding L, Wilson RK, Wheeler DA, Gibbs RA, Kucherlapati R, et al. The Cancer Genome Atlas Research Network. Landscape of somatic retrotransposition in human cancers. Science. 2012; 337(6097):967-971.
2. Belancio VP, Roy-Engel AM, Deininger PL. All y’all need to know ‘bout retroelements in cancer. Seminars in cancer biology. 2010; 20(4):200-210.
3. Gualtieri A, Andreola F, Sciamanna I, Sinibaldi-Vallebona P, Serafino A, Spadafora C. Increased expression and copy number amplification of L1 and SINE B1 retrotransposable elements in murine mammary carcinoma progression. Oncotarget. 2013; 4(11):1882-1893.
4. Solyom S, Ewing AD, Rahrmann EP, Doucet T, Nelson HH, Burns MB, Harris RS, Sigmon DF, Casella A, Erlanger B, Wheelan S, Upton KR, Shukla R, et al. Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res. 2012; 22(12):2328-38.
5. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, et al. Initial sequencing and analysis of the human genome. Nature. 2001; 409(6822):860-921.
6. Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH Jr. Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci USA. 2003; 100(9):5280-5.
7. Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, Badge RM, Moran JV. LINE-1 Retrotransposition Activity in Human Genomes. Cell. 2010; 141(7):1159-1170.
8. Hata K, Sakaki Y. Identification of critical CpG sites for repression of L1 transcription by DNA methylation. Gene. 1997; 189(2):227-234.
9. Szpakowski S, Sun X, Lage JM, Dyer A, Rubinstein J, Kowalski D, Sasaki C, Costa J, Lizardi PM. Loss of epigenetic silencing in tumors preferentially affects primate-specific retroelements. Gene. 2009; 448(2):151-167.
10. Chalitchagorn K, Shuangshoti S, Hourpai N, Kongruttanachok N, Tangkijvanich P, Thong-ngam D, Voravud N, Sriuranpong V, Mutirangura A. Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis. Oncogene. 2004; 23(54):8841-8846.
11. Suter CM, Martin DI, Ward RL. Hypomethylation of L1 retrotransposons in colorectal cancer and adjacent normal tissue. Int J Colorectal Dis. 2004; 19(2):95-101.
12. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz Jr. LA, Kinzler KW. Cancer Genome Landscapes. Science. 2013; 339(6127):1546-1558.
13. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997; 386:623-627.
14. Han JS, Szak ST, Boeke JD. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature. 2004; 429(6989):268-274.
15. Wheelan SJ, Aizawa Y, Han JS, Boeke JD. Gene-breaking: A new paradigm for human retrotransposon-mediated gene evolution. Genome Res. 2005; 15:1073-1078.
16. Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, Zhang J, Rose DW, Fu XD, Glass CK, Rosenfeld MG. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009; 139(6):1069-83.
17. Gasior SL, Wakeman TP, Xu B, Deininger PL. The human LINE-1 retrotransposon creates DNA double-strand breaks. J Mol Biol. 2006; 357(5):1383-93.
18. The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487(7407):330-337.
19. Penzkofer T, Dandekar T, Zemojtel T. L1Base: from functional annotation to prediction of active L1 elements. Nucl. Acids Res. 2005; 33(suppl 1):D498-D500.
20. Wang J, Song L, Grover D, Azrak S, Batzer MA, Liang P. dbRIP: a highly integrated database of retrotransposon insertion polymorphisms in humans. Hum Mutat. 2006; 27(4):323-329.
21. Rodić N, Burns KH. Long interspersed element-1 (LINE-1): passenger or driver in human neoplasms? PLoS Genet. 2013; 9(3):e1003402.
22. Mallinjoud P, Villemin JP, Mortada H, Polay Espinoza M, Desmet FO, Samaan S, Chautard E, Tranchevent LC, Auboeuf D. Endothelial, epithelial, and fibroblast cells exhibit specific splicing programs independently of their tissue of origin. Genome Res. 2014. Published in Advance.
23. Cohen-Eliav M, Golan-Gerstl R, Siegfried Z, Andersen CL, Thorsen K, Ørntoft TF, Mu D, Karni R. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J Pathol. 2013; 229(4):630-9.
24. Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomäki P, Chadwick RB, Kääriäinen H, Eskelinen M, Järvinen H, Mecklin JP, de la Chapelle A. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med. 1998; 338(21):1481-7.
25. Salovaara R, Loukola A, Kristo P, Kääriäinen H, Ahtola H, Eskelinen M, Härkönen N, Julkunen R, Kangas E, Ojala S, Tulikoura J, Valkamo E, Järvinen H, Mecklin JP, Aaltonen LA, de la Chapelle A. Population-based molecular detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol. 2000; 18(11):2193-200.
26. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009; 25(14):1754-1760.
27. The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012; 491(7422):56-65.
28. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009; 25:2078-2079.
29. Van der Auwera GA, Carneiro M, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella K, Altshuler D, et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Current Protocols in Bioinformatics. 2013; 43:11.10.1-11.10.33.
30. Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012; 28:i333-i339.
31. Mehine M, Kaasinen E, Mäkinen N, Katainen R, Kämpjärvi K, Pitkänen E, Heinonen H.-R., Bützow R, Kilpivaara O, Kuosmanen A, Ristolainen, H, Gentile M, Sjöberg J, et al. New England Journal of Medicine. 2013; 369:42-53.
32. Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM: Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Research. 2007; 35:W71-W74.