
INTRODUCTION
Anaplastic thyroid carcinoma (ATC) represents one of the most aggressive human malignancies [1]. Treatment of ATC is challenging, since it is an orphan disease and no established therapies offering a survival benefit are available. Thus prognosis remains very poor with a median survival of only 6 months [1].
Recently targeted therapies have been increasingly explored in ATC, based on the concept that molecular drivers of ATC may represent promising targets to be efficiently tackled. Genetic alterations in ATC enfold genes involved in the PI3K and MAPK/ERK pathway, receptor tyrosine kinases and tumour suppressor genes [2–5]. Activation of the PI3K cascade has been considered a hallmark for dedifferentiation of differentiated thyroid carcinoma (DTC) since activating mutations in this cascade are found with increasing prevalence in ATC compared to DTC [6], reaching 9–18% frequency for PI3KCA in ATC [5, 7]. Gene alterations in the MAPK/ERK pathway, e.g. BRAF mutations, have been identified in around 60% of papillary thyroid carcinoma (PTC) [8] and 15–45% of ATC [7, 9].
The most frequently mutated gene in ATC is p53 [7]. Mutations in p53 cause protein inactivation leading to inactivation of apoptosis and cell cycle progression [10]. Alterations of p53 are very rare in DTC, consistent with a late event in tumour dedifferentiation [11]. Knowledge of the genetic background of ATC has been broadened by the discovery of two human telomerase reverse transcriptase (TERT) mutations, the C228T and the C250T alterations, conferring increased telomerase activity [12]. These mutations have been suggested to further contribute to ATC aggressiveness [9]. Furthermore the group of Xing M et al. showed that coexisting BRAF V600E and TERT C228T alterations in papillary thyroid carcinoma (PTC) are associated with a significantly higher risk of recurrence and poorer outcome as well as older age and distant metastasis in ATC [9, 13].
Since the first identification of driver mutations in ATC, several case reports have shown response to targeted therapy in ATC patients. These included the application of inhibitors to BRAF [14–16], mTOR [17] and ALK [18]. In these patients the tumour mass reduction was related to the demonstration of the putative driver mutation followed by the consecutive targeted therapy. On the other hand, the multikinase-inhibitors sorafenib and pazobanib have failed to demonstrate survival benefit; however, it has to be emphasized that deductions from these studies are limited due to evaluation of only 20 and 15 ATC patients, respectively [19, 20]. In contrast axitinib, a VEGFR 1-3 inhibitor, was reported to confer disease stabilization over a period of 12 months in 2 patients [21]. In a phase II trial evaluating the EGFR inhibitor gefitinib disease stabilization was reported in 5 of 25 ATC patients [22].
Over the past 2 years, several groups have applied high throughput sequencing either by next generation sequencing (NGS) or whole genome sequencing to unravel the molecular signature of ATC. So far however, these studies were performed on ATC series comprising no more than 33 samples [7]. Hence, the prevalence of genetic alterations representing possible targets for ATC therapy that could be approached in clinical practice is still unknown. Furthermore, it remains particularly unclear whether screening for a set of mutations may help to define a subset of ATC patients, who could benefit from targeted therapy. These limitations in mind, we set out to comprehensively determine the prevalence of mutations in distinguished thyroid oncogenes and signalling pathways currently amendable to targeted drug therapy in a large cohort of ATC.
RESULTS
In this study the paraffin embedded tumour tissue of 118 ATC patients (57 males/61 females) with a median age at diagnosis of 65 years (ranging from 26 to 90 years) were analysed for mutations in hotspots of classical oncogenes. Patients characteristics are listed in Table 1.
Table 1: Patients characteristics
Number of tumours |
118 |
Female |
57 |
Male |
61 |
Derived from DTC* |
35 |
Derived from PDTC** |
4 |
de novo ATC*** |
64 |
Age at diagnosis [years] |
|
median |
65 |
range |
26–90 |
*DTC: Differentiated Thyroid Carcinoma. **PDTC: Poorly Differentiated Thyroid Carcinoma. ***ATC: Anaplastic Thyroid Carcinoma.
Mutation in the TERT promoter hotspots C250T and C228T were the most frequent alterations found in 86/118 (73%) tumour specimens, followed by p53 alterations in 65/118 (55%), RAS (HRAS, KRAS and NRAS) in 23/118 (19.5%), CDKN2A mutations in 20/118 (16.9%), PI3KCA in 14/118 (11.8%), BRAF in 13/118 (11%) and RET in 9/118 (7.6%) of ATC. EGFR, ERBB2, PDGFRA, RB1 and TSC2 were mutated in less than 2% of all ATC. In ALK, KIT and MET no mutations were found in the investigated exons. Identified mutations are listed in Supplementary Table 1.
Mutations in classical thyroid oncogenes e.g. BRAF, PI3KCA and RAS were detected in 40/118 (33%) ATCs. Genetic variations in the BRAF gene were detected in 13 of 118 (11%) ATCs. In 9 ATCs the classical V600E mutation was present, while the G469A variation was detected in 2 tumours, and the G469V and the D594A mutation were found in 1 ATC each.
Genetic alterations in PI3KCA were detected in 14/118 (11.8%) ATCs and comprised the E545K variation in 10 tumours, the H1047R variation in 3 tumours and the N1044L variation in 1 tumour.
Genetic alterations in the HRAS, KRAS and NRAS genes were found in 23/118 (19.5%) of all ATCs. In HRAS the Q61R mutation was detected in 4/118 (3.4%) and the G60S mutation in 1/118 (0.9%) tumours. In KRAS 3 distinct mutations were identified: G12R in 7 (5.9%), H27L and G12V each in 1 (0.9%) ATC sample. In NRAS, the variation Q61R was found in 7 and the G61K mutation was detected in 2 ATCs.
Only 8 of 40 ATCs harboured single mutational events in the MAPK/ERK- and PI3K-pathways: 4 BRAF (two V600E, G469V and G469A), 3 PI3KCA (two E545K and H1047R) and 1 NRAS (Q61K) variation. In 11 ATCs concurrent mutations in BRAF, PI3KCA and RAS were found: 7 ATCs showed PI3KCA and RAS mutations, 2 BRAF and PI3KCA variations, 1 BRAF and RAS and 1 ATC had mutation in BRAF, PI3KCA and RAS. In the other 21 ATCs concurrent p53 and/or CDKN2A mutations or RET or ERBB2 mutations were detected (Figure 1).
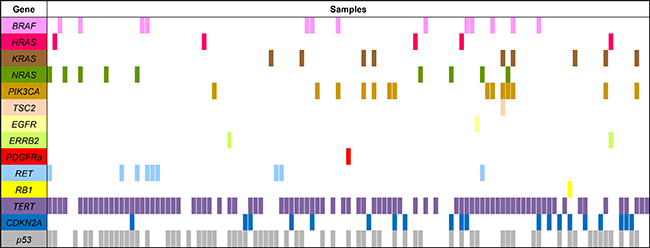
Figure 1: Mutations in genes that can be targeted by molecular drug therapy in 118 ATC primary tumour samples. P53 and TERT mutations were analyzed as a hallmark of ATC.
Mutations in the receptor tyrosine kinases EGFR, PDGFRA, ERBB2 and RET were identified in 13 ATC and involved EGFR (T790M, N = 1), PDGFRA (S667P, N = 1) and ERBB2 (L787P and G746S, N = 2). Variations in RET were detected in 9/118 (7.6%) ATC: 4 ATC with Y791F, 2 ATC with P596L and 1 ATC each with E818K or E595K.
Mutations in the TERT promotor (C250T and C228T) were detected in 73% of ATC. The C228T mutation was far more frequent and occurred in 82/118 compared to the C250T mutation in only 3/118 tumours. In 1 ATC sample both mutations were found by Sanger sequencing (Supplementary Table 1).
The tumour suppressor gene p53 was mutated in 65/118 (55%) tumour samples and in total 77 mutations occurred: R248Q in 7 ATCs; T125A, R209fs, P278S and R280T in 5 ATCs; S183* and R273C in 3 ATCs each; and C135F, Y220C, G266E, V272L, R282W and E285K in 2 ATCs each (Supplementary Table 1).
For the cell cycle regulating gene CDKN2A the most frequent mutation was the E88G missense mutation, detected in 8/118 (7%) of ATC samples. Further CDKN2A mutations were L65P, D84G and R98Q (in 2 ATCs each); and G83R, P72L and H66R in 1 ATC 1 each (Supplementary Table 1).
In 10/118 (8.5%) ATC no mutations were found in the analyzed genes. In 44/118 (37.3%) only one gene was altered: BRAF (V600E, G469V and G469A) in four ATC, PI3KCA and RET in three ATC each, NRAS in one ATC and p53 in 27 ATC.
Furthermore no ALK, KIT, MET or mTOR mutations were detected in the sequenced exons of the ATC series.
In ATC harbouring residues of DTC, BRAF and PI3KCA mutations were more prevalent than in de novo ATC samples (22.9% versus 6.3% and 22.9% versus 6.3%). As for TERT and p53 variations described as hallmarks in thyroid cancer dedifferentiation, only p53 mutation prevalence was higher in de novo ATC in comparison to ATC derived DTC (64.1% versus 34.3%).
DISCUSSION
Conventional radiochemotherapy in ATC does mostly not prolong survival, while single case reports have shown impressive tumour shrinkage after targeted drugs inhibiting single mutations in ATC [14, 17, 18]. Aiming to develop novel treatment strategies for ATC and to improve outcome, we have performed next generation sequencing of a large ATC series to clarify which mutational hotspots and/or signalling pathways are affected and may thus be considered for targeted drug therapy.
We found genetic alterations in the classical oncogenes (BRAF, RAS and PIK3CA) involved in thyroid tumourigenesis in 33% (40/118) of all ATC. Moreover, in 11% (13/118) of all ATC, mutations in targetable receptor tyrosine kinases (e.g. RET) were detected. In 40% (47/118) of all ATC mutations involved in the MAPK/ERK or PI3K pathways or receptor tyrosine kinase encoding genes were altered.
BRAF V600E is probably the most studied mutation in PTC and ATC. In our cohort a lower prevalence of BRAF V600E was detected than reported in the literature. Kunstman et al. [5] found a prevalence of 27% BRAF V600E mutations in their series and Landa et al. [7] detected mutations even in 45% of the investigated ATC samples. Several reasons may be considered: First the analysed cohorts by Kunstman and Landa only included 21 [5] and 33 ATC samples [7] and therefore the prevalence of BRAF mutation in these cohorts might be overrated. Secondly, ATC in our series may have developed de novo rather than evolved by dedifferentiation from DTC or PDTC. Hence we retrieved data from ATC primary report and found that only 29.6% of our samples had indication of concurrent or preceding DTC. In previous studies between 40–45% tumour specimens had concurrent DTC [5, 7].
Interestingly, besides BRAF V600E, 3 other BRAF variants were detected in our ATC series: G469V, G469A and D594A. These alterations are known oncogenes in malignant melanoma [23, 24] and non-small-cell lung cancer (NSCLC) [25, 26] but have so far not been reported in thyroid carcinoma. Since they were found in other human malignancies, we consider these alterations as likely oncogene also in the context of ATC.
In single case reports, ATC patients with BRAF V600E mutated tumours were treated with a BRAF-inhibitor [14–16]. According to our analysis, targeted BRAF treatment would only be an option in few of our ATC patients.
PIK3CA alterations have been reported in 4.5% [5] and 18% [7] of ATC using NGS. We found PI3KCA mutations in 11.8 % of our ATC samples, in particular the E545K PI3KCA alteration in the helical domain and the H1047R variation located in the kinase domain. Earlier reports identified the same mutations [5, 7] in ATC samples. Small molecule PI3K-inhibitors are approved for the treatment of lymphomas and chronic lymphatic leukaemia and are currently evaluated in solid tumours e.g. breast cancer [27] and NSCLC [28]. In these malignancies, activation of the PI3K-pathway (PI3KCA mutations or PTEN loss) was consistent with good response to PI3K-inhibitors [28, 29]. However, in a phase II study including glioblastoma patients no association between pathway activation in tumour tissues and efficacy of PI3K-inhibitor treatment was found [28]. The efficacy of PI3K-inhibitors in ATC has not been investigated in the clinical setting so far. However, since almost 12% of our ATC samples harboured activating PI3K cascade mutation in agreement with previous studies by Landa et al. [7] and Xing et al. [30] showing that activation of PI3K signalling is highly prevalent in ATC, we suggest that PI3K inhibitors could be worthwhile exploring in clinical studies.
RAS mutations were detected in 23 ATCs: Among them are mutations that have frequently been found in thyroid tumours [31]. The HRAS G60S and KRAS H27L found in 1 ATC each, have so far not been linked to thyroid cancer. RAS mutations are known events in benign and malignant thyroid tumours [32, 33]. In PTC they were shown to be mutually exclusive with BRAF mutations and are consistent with follicular variant PTC [11], but in ATC RAS mutations may occur concurrently with other mutations including BRAF V600E [7]. In RAS-mutated PTC the RAF-MEK-ERK cascade has been shown to confer cell proliferation, however with smaller transcriptional output than BRAF mutations [11]. This is explained by an ERK-induced negative feedback that disrupts RAF dimerization [34]. In RAS mutated ATC this feedback is mostly not observed [7] which could potentially render ATC with activating RAS mutations susceptible to downstream MEK-Inhibitors.
Murugan and Xing et al. [35] were the first to describe mutations in ALK exons 20 and 23 in 18 ATC. In our series no ALK mutations were detected in exons 20–25. In a recent report of our group [36], ALK mutations were exclusively detected in exons 1, 3, 16, 28 and 29, which were not addressed in the present NGS approach, suggesting that probably the entire ALK gene has to be sequenced to assess for genetic alterations in ATC.
We also analysed genetic variations in receptor tyrosine kinases EGFR, RET, PDGFRA and ERBB2 since targeted therapies for these tyrosine kinases are available for human malignancies. The EGFR T790M variation is well studied in lung cancer and is known to confer EGFR-inhibitor resistance [37]. In our ATC series EGFR mutations were shown to be rare events and in clinical practice mutational screening would be helpful only when treatment with an EGFR-inhibitor is planned.
Hereditary medullary thyroid carcinoma (MTC) is characterized by germline RET mutations [38] and in 40–60% of sporadic MTC somatic RET mutations are present [39, 40], which cause constitutive activation of the RET tyrosinkinase and in consequence MAPK/ERK and PI3K pathway activation. In follicular-cell derived thyroid carcinoma RET mutations are generally rare events [41]. In a previous report RET gene variations were found in 1/22 (4.5%) ATC investigated [5]. In our study we found RET mutations in 9/118 (7.6%) ATC: in 4 ATC the Y791F variation was detected, which is described in Multiple endocrine neoplasia type 2A (MEN2A) cases, but is interestingly not associated with MTC development [42]. The P596L, E818K (in 2 ATCs each) and the E595K RET variation (1 ATC) have so far not been described in association with thyroid cancer or other human malignancies and their biological relevance is not known.
Genetic variations in ERBB2 and PDGFRA were rare events (2/118 and 1/118). This is in accordance with previous reports [5, 7] and hence screening does not seem to be warranted in ATC.
Genetic variations in tumour suppressor genes are frequent events in cancer. Inactivating p53 mutations are a molecular hallmark of ATC. In agreement within this and recent large-scale analysis of ATC (5, 7) p53 was frequenty mutated in our cohort with genetic variations in 65 of 118 tumours. Somatic p53 mutations are very rare in DTC and it has been shown in mouse models that p53 inactivation contributes to the progression from PTC to ATC [43]. Less well studied are other genetic variations involved in cell cycle regulation like mutations in the cyclin-dependent kinases (CDK). Kunstman et al. reported single mutations in the CDKN1B and CDKN2C encoding genes [5]. In our study we found CDKN2A mutations in 23 ATC (19.5%). Thereby, the most frequent mutation was the E88G variation, detected in 14 ATC, which has not previously been described in ATC. Reports have shown that co-targeting CDK 4/6 and BRAF is efficient in BRAF mutated tumours to overcome BRAF-inhibitor resistance [44].
Weaknesses of our study are the lack of clinical data, rendering correlation of the genetic findings with disease course impossible. Furthermore, only hotspot regions of genes were sequenced, hence we cannot exclude that mutational events were present in other exons. The strength of our study is the application of a NGS which is already routinely applied in patient care at the Essen University Hospital’s Comprehensive Cancer Centre underlining its applicability in clinical practice. Moreover, our series presents the largest ATC cohort comprehensively analysed for mutations in targetable oncogenes so far and all tissues were classified as ATC by board certified pathologists of the German Thyroid Pathology Reference Centre at the University Hospital of Essen.
In summary we show, that mutations in hotspots of targetable oncogenes are present in approximately 33% of ATC. We therefore propose a panel of oncogenes (BRAF, PI3KCA and RAS) that should be screened in clinical practice in patients with newly discovered ATC in order to offer patients targeted therapy treatment as part of a highly individualized treatment approach where feasible and if desired by the patient. The clinical care of ATC patients urgently needs to be improved including central pathology review, national and international networks and accessibility to basket studies testing novel targeted and other therapies.
MATERIALS AND METHODS
We retrospectively analyzed a study cohort of 118 patients with ATC. FFPE tissue from primary tumour was used to perform NGS. All samples were reviewed by two board-certified pathologists and diagnosis was made according to the WHO classification [45]. When DTC co-occurred in tumour specimen, the respective ATC case was regarded to be derived from DTC.
After deparaffinization of FFPE tissue with deparaffinization solution (Qiagen, USA) and incubation with 20 μl proteinase K solution (20 mg/ml) over night at 56°C, DNA isolation from the primary tumour tissue was performed using QIAamp DNA FFPE tissue kit (Qiagen, USA) according to the manufacturer’s instructions. DNA concentrations were determined by Qubit® 2.0 Fluorometer dsDNA HS assay kit (LifeTechnologies, USA). All samples showed a concentration between 1–100 ng/μl.
A total amount of 45 ng DNA was used to perform multiplex-pcr (four primer pools with 10 ng/primer pool + 10% excess). Multiplex PCR and purification were performed with the GeneRead DNAseq Custom Panel V2, GeneRead DNAseq Panel PCR Kit V2 (Qiagen, USA) and Agencourt® AMPure® XP Beads (Beckmann, USA), followed by measurement of total DNA amount by Qubit® 2.0 Fluorometer dsDNA HS assay kit. The library preparation was performed with NEBNext Ultra DNA Library Prep Set for Illumina (New England Biolabs, USA), according to the manufacturer’s recommendations by using 24 different indices per run. The pooled library was sequenced on MiSeq (Illumina; 2 × 150 bases paired-end run) and analyzed by Cancer Research Workbench (CLC Bio, Qiagen, USA).
For targeted sequencing a customized ATC-panel was designed containing regions of interest. The ATC-panel contains hot-spot regions out of 17 genes. The analyzed exons are listed in Supplementary Table 2. The regions were covered by a total of 136 amplicons. In all runs an average coverage of approximately 8000 × was obtained.
Within CLC Cancer Research Workbench demultiplexed paired-end sequencing data was mapped to human genome (version hg19). A local realignment was performed to reach better quality (especially for regions with small insertions or deletions). All reads, which were mapped outside of targeted-regions, got deleted after mapping process. Within target regions information from different databases (Cosmic, Clinvar, dbSNP, 1000 genome project, HapMap) was annotated.
Performing a second filtering-step all reference-variants and variants found in dbSNP common, 1000 genome project and HapMap were deleted. All variants found in Cosmic, Clinvar or those which weren’t annotated in any database were listed. Therefore, an allele-frequency of minimum 5% and coverage of at least 100 mapped-reads were the selection-parameters.
For detection of the two known TERT promotor mutations on chromosome 5 position g.1295250C > T and g.1295228C > T (reference genome hg19) Sanger sequencing was performed. The promoter region of TERT was amplified by PCR, followed by direct sequencing. PCR reaction was carried out with oligonucleotides TERT fwd (CCTGCCCCTTCACCTTCCAG) and TERT rev (AGGACGCAGCGCTGCCTGAA) in a total volume of 50 μl with HotStar Taq Mastermix (Qiagen) according to the recommendations of the supplier. Amplification was performed in a Biometra T3000 Thermocycler (Biometra, Göttingen) with 10 min of initial enzyme activation at 95°C followed by 45 cycles of denaturation at 94°C for 30 sec, oligonucleotide annealing at 62°C for 60 sec and extension at 72°C for 30 sec. Sequences of both, forward and reverse strand were analyzed on an ABI 3500 Genetic Analyser (LifeTechnologies) using BigDye Terminator chemistry v3.1 (LifeTechnologies).
Abbreviations
ATC: Anaplastic Thyroid Carcinoma; DTC: Differentiated Thyroid Carcinoma; MTC: Medullary Thyroid Carcinoma; NGS: Next Generation Sequencing; NSCLC: Non-small Cell Lung Cancer; PTC: Papillary Thyroid Carcinoma; TERT: Telomerase Reverse Transcriptase.
Authors’ contributions
Vera Tiedje, Saskia Ting, Kurt Werner Schmid and Dagmar Fuehrer made substantial contributions to conception, design and interpretation of the data and writing of the manuscript.
Vera Tiedje, Saskia Ting, Thomas Herold were responsible for the acquisition and analysis of the data.
Sarah Synoracki, Soeren Latteyer, Lars C Moeller, Denise Zwanziger and Martin Stuschke participated in the analysis and interpretation of the data.
ACKNOWLEDGMENTS
We would like to thank Thomas Marx and Elisabeth Jeske for their substantial contribution to this study.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
No funding to declare.
REFERENCES
1. Smallridge RC, Ain KB, Asa SL, Bible KC, Brierley JD, Burman KD, Kebebew E, Lee NY, Nikiforov YE, Rosenthal MS, Shah MH, Shaha AR, Tuttle RM, et al. American Thyroid Association guidelines for management of patients with anaplastic thyroid cancer. Thyroid. 2012; 22:1104–39. doi: 10.1089/thy.2012.0302.
2. Liu Z, Hou P, Ji M, Guan H, Studeman K, Jensen K, Vasko V, El-Naggar AK, Xing M. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J Clin Endocrinol Metab. 2008; 93:3106–16. doi: 10.1210/jc.2008–0273.
3. Liu D, Yang C, Bojdani E, Murugan AK, Xing M. Identification of RASAL1 as a major tumor suppressor gene in thyroid cancer. J Natl Cancer Inst. 2013; 105:1617–27. doi: 10.1093/jnci/djt249.
4. Smith N, Nucera C. Personalized therapy in patients with anaplastic thyroid cancer: targeting genetic and epigenetic alterations. J Clin Endocrinol Metab. 2015; 100:35–42. doi: 10.1210/jc.2014–2803.
5. Kunstman JW, Juhlin CC, Goh G, Brown TC, Stenman A, Healy JM, Rubinstein JC, Choi M, Kiss N, Nelson-Williams C, Mane S, Rimm DL, Prasad ML, et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum Mol Genet. 2015; 24:2318–29. doi: 10.1093/hmg/ddu749.
6. Garcia-Rostan G, Zhao H, Camp RL, Pollan M, Herrero A, Pardo J, Wu R, Carcangiu ML, Costa J, Tallini G. ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J Clin Oncol. 2003; 21:3226–35. doi: 10.1200/JCO.2003.10.130.
7. Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, Dogan S, Ricarte-Filho JC, Krishnamoorthy GP, Xu B, Schultz N, Berger MF, Sander C, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest. 2016; 126:1052–66. doi: 10.1172/JCI85271.
8. Jin L, Chen E, Dong S, Cai Y, Zhang X, Zhou Y, Zeng R, Yang F, Pan C, Liu Y, Wu W, Xing M, Zhang X, et al. BRAF and TERT promoter mutations in the aggressiveness of papillary thyroid carcinoma: a study of 653 patients. Oncotarget. 2016; 7:18346–55. doi: 10.18632/oncotarget.7811.
9. Shi X, Liu R, Qu S, Zhu G, Bishop J, Liu X, Sun H, Shan Z, Wang E, Luo Y, Yang X, Zhao J, Du J, et al. Association of TERT promoter mutation 1,295,228 C > T with BRAF V600E mutation, older patient age, and distant metastasis in anaplastic thyroid cancer. J Clin Endocrinol Metab. 2015; 100:E632–7. doi: 10.1210/jc.2014–3606.
10. Prives C, Lowe SW. Cancer: Mutant p53 and chromatin regulation. Nature. 2015; 525:199–200. doi: 10.1038/nature15212.
11. Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014; 159:676–90. doi: 10.1016/j.cell.2014.09.050.
12. Liu T, Wang N, Cao J, Sofiadis A, Dinets A, Zedenius J, Larsson C, Xu D. The age- and shorter telomere-dependent TERT promoter mutation in follicular thyroid cell-derived carcinomas. Oncogene. 2014; 33:4978–84. doi: 10.1038/onc.2013.446.
13. Xing M, Liu R, Liu X, Murugan AK, Zhu G, Zeiger MA, Pai S, Bishop J. BRAF V600E and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence. J Clin Oncol. 2014; 32:2718–26. doi: 10.1200/JCO.2014.55.5094.
14. Rosove MH, Peddi PF, Glaspy JA. BRAF V600E inhibition in anaplastic thyroid cancer. N Engl J Med. 2013; 368:684–5. doi: 10.1056/NEJMc1215697.
15. Lim AM, Taylor GR, Fellowes A, Cameron L, Lee B, Hicks RJ, McArthur GA, Angel C, Solomon B, Rischin D. BRAF Inhibition in BRAFV600E-Positive Anaplastic Thyroid Carcinoma. J Natl Compr Canc Netw. 2016; 14: 249–54.
16. Marten KA, Gudena VK. Use of vemurafenib in anaplastic thyroid carcinoma: a case report. Cancer Biol Ther. 2015; 16:1430–3. doi: 10.1080/15384047.2015.1071734.
17. Wagle N, Grabiner BC, Van Allen EM, Amin-Mansour A, Taylor-Weiner A, Rosenberg M, Gray N, Barletta JA, Guo Y, Swanson SJ, Ruan DT, Hanna GJ, Haddad RI, et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N Engl J Med. 2014; 371: 1426–33. doi: 10.1056/NEJMoa1403352.
18. Godbert Y, Henriques de Figueiredo B, Bonichon F, Chibon F, Hostein I, Perot G, Dupin C, Daubech A, Belleannee G, Gros A, Italiano A, Soubeyran I. Remarkable Response to Crizotinib in Woman With Anaplastic Lymphoma Kinase-Rearranged Anaplastic Thyroid Carcinoma. J Clin Oncol. 2015; 33:e84–7. doi: 10.1200/JCO.2013.49.6596.
19. Savvides P, Nagaiah G, Lavertu P, Fu P, Wright JJ, Chapman R, Wasman J, Dowlati A, Remick SC. Phase II trial of sorafenib in patients with advanced anaplastic carcinoma of the thyroid. Thyroid. 2013; 23:600–4. doi: 10.1089/thy.2012.0103.
20. Bible KC, Suman VJ, Menefee ME, Smallridge RC, Molina JR, Maples WJ, Karlin NJ, Traynor AM, Kumar P, Goh BC, Lim WT, Bossou AR, Isham CR, et al. A multiinstitutional phase 2 trial of pazopanib monotherapy in advanced anaplastic thyroid cancer. J Clin Endocrinol Metab. 2012; 97:3179–84. doi: 10.1210/jc.2012-1520.
21. Cohen EE, Tortorici M, Kim S, Ingrosso A, Pithavala YK, Bycott P. A Phase II trial of axitinib in patients with various histologic subtypes of advanced thyroid cancer: long-term outcomes and pharmacokinetic/pharmacodynamic analyses. Cancer Chemother Pharmacol. 2014; 74:1261–70. doi: 10.1007/s00280-014-2604-8.
22. Pennell NA, Daniels GH, Haddad RI, Ross DS, Evans T, Wirth LJ, Fidias PH, Temel JS, Gurubhagavatula S, Heist RS, Clark JR, Lynch TJ. A phase II study of gefitinib in patients with advanced thyroid cancer. Thyroid. 2008; 18: 317–23. doi: 10.1089/thy.2007.0120.
23. Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol. 2005; 36:486–93. doi: 10.1016/j.humpath.2005.03.015.
24. Porcelli L, Guida G, Tommasi S, Guida M, Azzariti A. Metastatic melanoma cells with BRAF G469A mutation: nab-paclitaxel better than vemurafenib? Cancer Chemother Pharmacol. 2015; 76:433–8. doi: 10.1007/s00280-015-2796-6.
25. Luk PP, Yu B, Ng CC, Mercorella B, Selinger C, Lum T, Kao S, O’Toole SA, Cooper WA. BRAF mutations in non-small cell lung cancer. Transl Lung Cancer Res. 2015; 4: 142–8. doi: 10.3978/j.issn.2218-6751.2014.08.08.
26. Paik PK, Arcila ME, Fara M, Sima CS, Miller VA, Kris MG, Ladanyi M, Riely GJ. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. 2011; 29:2046-51. doi: 10.1200/JCO.2010.33.1280.
27. Mayer IA, Abramson V, Formisano L, Balko JM, Estrada MV, Sanders M, Juric D, Solit D, Berger MF, Won H, Li Y, Cantley LC, Winer EP, et al. A Phase Ib Study of Alpelisib (BYL719), a PI3Kalpha-specific Inhibitor, with Letrozole in ER+/HER2-Negative Metastatic Breast Cancer. Clin Cancer Res. 2016; 23:26–34. doi: 10.1158/1078-0432.CCR-16-0134.
28. Vansteenkiste JF, Canon JL, Braud FD, Grossi F, De Pas T, Gray JE, Su WC, Felip E, Yoshioka H, Gridelli C, Dy GK, Thongprasert S, Reck M, et al. Safety and Efficacy of Buparlisib (BKM120) in Patients with PI3K Pathway-Activated Non-Small Cell Lung Cancer: Results from the Phase II BASALT-1 Study. J Thorac Oncol. 2015; 10: 1319–27. doi: 10.1097/JTO.0000000000000607.
29. Schmid P, Pinder SE, Wheatley D, Macaskill J, Zammit C, Hu J, Price R, Bundred N, Hadad S, Shia A, Sarker SJ, Lim L, Gazinska P, et al. Phase II Randomized Preoperative Window-of-Opportunity Study of the PI3K Inhibitor Pictilisib Plus Anastrozole Compared With Anastrozole Alone in Patients With Estrogen Receptor-Positive Breast Cancer. J Clin Oncol. 2016; 34:1987–94. doi: 10.1200/JCO.2015.63.9179.
30. Liu R, Liu D, Trink E, Bojdani E, Ning G, Xing M. The Akt-specific inhibitor MK2206 selectively inhibits thyroid cancer cells harboring mutations that can activate the PI3K/Akt pathway. J Clin Endocrinol Metab. 2011; 96:E577–85. doi: 10.1210/jc.2010-2644.
31. Jung CK, Little MP, Lubin JH, Brenner AV, Wells SA Jr, Sigurdson AJ, Nikiforov YE. The increase in thyroid cancer incidence during the last four decades is accompanied by a high frequency of BRAF mutations and a sharp increase in RAS mutations. J Clin Endocrinol Metab. 2014; 99: E276–85. doi: 10.1210/jc.2013-2503.
32. Rossi M, Buratto M, Tagliati F, Rossi R, Lupo S, Trasforini G, Lanza G, Franceschetti P, Bruni S, Degli Uberti E, Zatelli MC. Relevance of BRAF(V600E) mutation testing versus RAS point mutations and RET/PTC rearrangements evaluation in the diagnosis of thyroid cancer. Thyroid. 2015; 25:221–8. doi: 10.1089/thy.2014.0338.
33. Giordano TJ. Follicular cell thyroid neoplasia: insights from genomics and The Cancer Genome Atlas research network. Curr Opin Oncol. 2016; 28:1–4. doi: 10.1097/CCO.0000000000000248.
34. Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci USA. 2009; 106: 4519–24. doi: 10.1073/pnas.0900780106.
35. Murugan AK, Xing M. Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res. 2011; 71: 4403–11. doi: 10.1158/0008-5472.CAN-10-4041.
36. Latteyer S, Tiedje V, Konig K, Ting S, Heukamp LC, Meder L, Schmid KW, Fuhrer D, Moeller LC. Targeted next-generation sequencing for TP53, RAS, BRAF, ALK and NF1 mutations in anaplastic thyroid cancer. Endocrine. 2016; 54:733–741. doi: 10.1007/s12020-016-1080-9.
37. Tan CS, Cho BC, Soo RA. Next-generation epidermal growth factor receptor tyrosine kinase inhibitors in epidermal growth factor receptor -mutant non-small cell lung cancer. Lung Cancer. 2016; 93:59–68. doi: 10.1016/j.lungcan.2016.01.003.
38. Frank-Raue K, Raue F. Hereditary Medullary Thyroid Cancer Genotype-Phenotype Correlation. Recent Results Cancer Res. 2015; 204:139–56. doi: 10.1007/978-3-319-22542-5_6.
39. Elisei R, Cosci B, Romei C, Bottici V, Renzini G, Molinaro E, Agate L, Vivaldi A, Faviana P, Basolo F, Miccoli P, Berti P, Pacini F, et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab. 2008; 93: 682–7. doi: 10.1210/jc.2007-1714.
40. Tiedje V, Ting S, Walter RF, Herold T, Worm K, Badziong J, Zwanziger D, Schwid KW, Fuhrer D. Prognostic markers and response to vandetanib therapy in sporadic medullary thyroid cancer patients. Eur J Endocrinol. 2016; 175: 173–80. doi: 10.1530/EJE-16-0252.
41. Yip L, Nikiforova MN, Yoo JY, McCoy KL, Stang MT, Armstrong MJ, Nicholson KJ, Ohori NP, Coyne C, Hodak SP, Ferris RL, LeBeau SO, Nikiforov YE, et al. Tumor genotype determines phenotype and disease-related outcomes in thyroid cancer: a study of 1510 patients. Ann Surg. 2015; 262:519–25. doi: 10.1097/SLA.0000000000001420.
42. Toledo RA, Hatakana R, Lourenço DM Jr, Lindsey SC, Camacho CP, Almeida M, Lima JV Jr, Sekiya T, Garralda E, Naslavsky MS, Yamamoto GL, Lazar M, Meirelles O, et al. Comprehensive assessment of the disputed RET Y791F variant shows no association with medullary thyroid carcinoma susceptibility. Endocr Relat Cancer. 2015; 22:65–76. doi: 10.1530/ERC-14-0491.
43. McFadden DG, Vernon A, Santiago PM, Martinez-McFaline R, Bhutkar A, Crowley DM, McMahon M, Sadow PM, Jacks T. p53 constrains progression to anaplastic thyroid carcinoma in a Braf-mutant mouse model of papillary thyroid cancer. Proc Natl Acad Sci USA. 2014; 111: E1600–9. doi: 10.1073/pnas.1404357111.
44. Yadav V, Chen SH, Yue YG, Buchanan S, Beckmann RP, Peng SB. Co-targeting BRAF and cyclin dependent kinases 4/6 for BRAF mutant cancers. Pharmacol Ther. 2015; 149: 139–49. doi: 10.1016/j.pharmthera.2014.12.003.
45. DeLellis RA, Lloyd RV, Heitz PU, Eng C, editors. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Endocrine Organs. Lyon: IARC Press; 2004.