
INTRODUCTION
During metastasis, cancerous cells have to migrate through the extracellular matrix (ECM) for intravasation into blood and lymphatic vessels. The early steps of metastasis involve the coordination of intracellular signals and the microenvironment to allow detachment of the cell from the primary site, and this is first demonstrated by a change in the cells shape. The cells become densely packed and have a reduced amount of protrusions. For stability, cells are linked to the ECM through cell adhesion molecules. In cancer, many cell adhesion molecules participate in this cell-matrix linkage [1].
Focal adhesions (FAs) are large protein complexes that connect the cell cytoskeleton to the ECM through integrins. Integrins are cell adhesion proteins with α and β transmembrane heterodimers that play a major role in converging signals from the cell membrane to the inside of the cell [2]. The cytosolic portions of both transmembrane heterodimers interact with a number of cytoskeletal proteins and signaling molecules. These heterodimers act as receptors for ECM proteins including Collagen, Laminin, Thrombospondins, Vitronectin and Fibronectin [3, 4]. The two major roles of integrins are to convey outside-in and inside-out signaling. For inside-out signaling, proteins containing FERM domains, such as Talins and Kindlins, bind to Integrin cytoplasmic tails to trigger conformational changes [5]. Many FA proteins contain these FERM domains which are discussed later on in this review. The conformational changes triggered by these FERM domains lead to integrin activation. In mature FAs, Integrins are active, erect, and bound to their ligand [6].
For outside-in signaling, the binding of the active Integrin to their respective immobilized ligand results in the recruitment of more adaptor proteins which results in a strengthened cytoskeleton at the FA [7]. Whilst bound to the ligands, Integrins act as force sensors and force transmitters [8, 9]. The ECM can exert forces on Integrins that transfer to the Integrin-associated FA proteins and expose new binding sites for further intramolecular interactions [9]. Furthermore, cells also respond to this altered tension by inducing changes in gene transcription [10]. Oppositely, the cytoskeleton of the FA generates forces that allow the Integrins to pull ECM proteins [11].The ability of Integrins to connect to the cytoskeleton and transmit forces speaks to its critical role in FA turnover. Because of this, tumor cell migration and invasion rely on Integrin activation/deactivation, as well as, their crosstalk with oncogenes. Integrin signaling is a well orchestrated but complicated phenomenon. Understanding the protein networks that regulate Integrin activation will give us a better understanding of how cells regulate their adhesion.Cell-matrix adhesion sites were first observed more than 40 years ago in chick heart fibroblasts by electron microscopy and interference reflexion microscopy [12–14]. These contacts were first identified to be 2-10 μm long, and 0.25-0.5 μm wide [14]. Since then, studies have found that FA size ranges from 0.25 to 10 μm, depending on cell mobility [15]. For example, rapidly moving cells such as Dictyostelium discoidem cells display small FAs [16], while cells with prominent FAs move slower [17, 18].
Intracellularly, FA proteins participate in a number of cell signaling pathways. Tyrosine phosphorylation is a key signaling event that leads to the recruitment of multiple proteins to FAs [19]. Although the kinases ILK, PAK, Abl, Csk, PYK2 and PKC are present in FAs [12], Focal Adhesion Kinase (FAK) and Src are the most dominant kinases in FA signaling [20]. Tyrosine phosphorylation by these kinases leads to a recruitment of cytoskeletal proteins, such as Paxillin [21], small GTPases, tyrosine phosphatases and other enzymes.
Cancer cells participate in different types of directed cell migration including: chemotaxis (chemoattractant gradient), haptotaxis (environmental gradient), electrotaxis (electrical attractant), and durotaxis (rigidity attractant) [22]. Environmental sensing by the cancer cells allows them to interpret various signals that are mediated by FAs. ECM generated stiffness induces and cooperates with cell-matrix forces applied by cells [23]. Cells are capable of sensing many characteristics of the ECM such as rigidity and anisotropy [7]. In fact, ECM composition is a key contributor of patient survival [24]. It has been shown that Collagen fibers straighten as the breast tumor progresses [25]. Detection of ECM rigidity by Integrins through Myosin II leads to downstream signaling cascades through FAs [26]. For example, the FAK/Paxillin/Vinculin signaling pathway transmits force information to allow the cell to migrate in different ECM rigidities [27]. In fact, a study showed that Null-FAK cells are unable to detect changes in ECM rigidity [28].
Most FA proteins contain multiple binding sites for other proteins; therefore supramolecular structures tend to form within these sites. The regulation of these protein-protein interactions plays a key role in the development and turnover of adhesion sites. Since it has been shown that the size and composition of the FA contributes to cellular functions such as migration and mechanosensing [29], it is important to study the different components of FA complexes. This review will describe some of the key proteins involved in FA signaling.
INITIATION OF FA ASSEMBLY
Mature FAs are dependent on Actin filament assembly at the leading edge, also referred to as Actin flow [30]. In order for this to happen, a number of proteins have to be recruited to the site. Tyrosine phosphorylation is a key signaling event that leads to the recruitment of multiple proteins to FAs [19]. Although not as common, serine phosphorylation also plays an important role in adhesion formation. Focal Adhesion Kinase (FAK) and Src are the most dominant kinases in FA signaling [20]. Other kinases have been shown to participate in this labyrinthine signaling pathway, but inactivation of FAK and Src averts FA assembly. Therefore, it is important to understand the importance of FAK in FA assembly.
FAK and Src
FAK, a non-receptor tyrosine kinase, is the central regulator of Integrin-mediated FA assembly. FAK was first found to be activated by the ECM or growth factors and its tyrosine phosphorylation is associated with the formation and turnover of FAs [31]. In fact, Null-FAK fibroblasts exhibit an abundance of FAs [32]. Immunohistochemical analysis revealed increased FAK expression in highly malignant human breast and colon cancers [33]. Since FAK is both a signaling kinase and an adaptor/scaffold protein, it is involved in various signaling pathways including those that contribute to cell migration and angiogenesis [34, 35].
In the past ten years, the molecular mechanisms of FAK regulation has been uncovered in detail. The N-terminus of FAK contains a FERM (Four-point-one, Ezrin, Radixin, Moesin) domain that is important for protein:phosphoinositide and protein:protein interactions [36, 37]. The C-terminus contains a four-helix bundle FAT (Focal Adhesion Targeting) domain that is critical for FAKs targeting to FAs through the protein Paxillin [38]. These FERM and FAT domains are the dominant protein:protein binding domains of FAK [39]. For example, Met interacts with the FERM domain to promote hepatocyte growth factor-mediated cell invasion [40]. The FERM domain also interacts with the FAK kinase domain to block the active site from accessing ATP and other substrate binding sites for activation [38, 41]. Mutation of the FERM domain at K38 interrupts the FERM:kinase domain interaction and leads to FAK activation [42]. In summary, the self-inactivation of FAK consists of the FERM domain directly interacting with the kinase domain.Posttranslational modifications are the major regulatory activators for FAK, mainly phosphorylation. The major autophosphorylation site during FA signaling is Tyr397 [34, 43]. Phosphorylation at this site establishes a motif that is recognized by proteins containing SH2 domains, including Src kinase [44]. Many proteins have been identified that contribute to FAK phosphorylation. Multiple factors, including increased p130RhoGEF expression, contribute to the localization of FAK to the elicited adhesion site [45]. In fact, a mutation in the pleckstrin homology (PH) domain of p130RhoGEF inhibits FA assembly [45]. As stated earlier, FAK may be activated by growth factors. Of those growth factors, platelet-derived growth factor (PDGF) treatment of cells increases autophosphorylation of FAK at Tyr397 [46]. Also, PDGF stimulation of the cells induced Akt-mediated phosphorylation of FAK at Ser695 and Thr700 to enhance Integrin signaling [46]. In the early stages of adhesion development, FAK responds to increased Integrin clustering due to ECM rigidity by autophosphoylation at tyrosine 397 and subsequent activation [47–50]. This autophosphorylation site creates a motif that allows recognition of proteins with SH2 domains, such as the p85 subunit of PI3K, Phospholipase Cγ (PLCγ) and the SRC-Family Kinases (SFKs) [34]. Even though this autophosphorylation is an indispensable event for FA signaling, FAK is phosphorylated at other sites as well.
The oncogene Migration and invasion enhancer 1 (MIEN1) regulates cell migration by inducing phosphorylation of FAK at Tyr-925 [51]. MIEN1 is located in the chromosomal region 17q12-21 and is typically amplified along with its neighboring gene ERBB2 in breast cancer [52]. Its many functions include regulating Actin cytoskeletal dynamics and influencing expression of ECM-degrading proteases and angiogenic factors [51, 53]. Down regulation of MIEN1 leads to a reduction in FAK phosphorylation at Tyr-925, as well as reduced phosphorylation of Akt, Erk1/2 and NF-κB [51]. Analysis of FAK activation is of paramount importance to any FA signaling study. Many FAK-activating proteins have been characterized, including p130RhoGEF [45], PDGF [46] and MIEN1 [51], however, the complexity of this signaling pathway suggests that multiple other factors are waiting to be discovered.
FAK autophosphorylation also leads to the activation of the tyrosine kinase Src [54]. As discussed earlier, PDGF is a growth factor that activates FAK. Epidermal growth factor (EGF) is another growth factor that activates FAK, as well as Src [55]. Src tyrosine kinase works alongside with active FAK to facilitate the phosphorylation of many FAK-associated proteins. Src-family kinases are essential for the formation and maturation of FAs and are highly activated in many human cancers [56, 57]. Genomic amplifications and activating mutations of Src are rare in human cancers so this hyperactivation is often induced by upstream phosphatases or kinases [57]. Src has several functional domains, including, a 14-carbon myristic acid moiety attached to a SH4 domain, a SH3 domain followed by an SH2 domain, an SH2 linker, kinase domain and C-terminal regulatory domain. Src is capable of interacting with the plasma membrane through its 14-carbon N-terminal fatty acid moiety [57, 58]. After activation, its SH3 domain interacts with Actin filaments to transmit signals from adhesion receptors to cytoskeletal proteins [59].
Though FAK autophosphorylation engenders Src activation, concurrent signaling events may occur. Although the precise order in which each activating event occurs is unknown, FAK and Src do work in concert to activate other proteins. For example, in response to extracellular Fibronectin, mechanical stretching of the substrate domain of Cas exposes Src-specific phosphorylation sites [60], and in order for this Src-mediated phosphorylation to occur, Cas must be bound to FAK [61]. Src activates various downstream signaling pathways through its interactions with FAK, epidermal growth factor receptors (EGFRs), hepatocyte growth factor receptor (C-Met or HGFR), Integrin cell adhesion receptors, G protein-coupled receptors, and other cytoskeleton components [62, 63].
Many protein:protein complexes are found at FA sites. It has been shown that recruitment of FA proteins such as Vinculin and Paxillin precedes tyrosine phosphorylation [19]. This justifies the importance of the localization of the tyrosine kinases FAK and Src to these nascent adhesion sites. These initial phosphorylation events lead to the activation of other kinases and phosphatases while creating a substrate for SH2-containing proteins. Although the kinases ILK, PAK, Abl, Csk, PYK2 and PKC are present [12], Focal Adhesion Kinase (FAK) and Src are the most dominant kinases in FA signaling [20]. The FAK-Src symbiotic relationship leads to the subsequent recruitment of many proteins to FA sites. FAK and Src bind different partners to uniquely regulate FA dynamics. Some studies only choose to assess FAK activation as a determinant of FA function. Other kinases such as ILK, PAK, Abl, Csk, PYK2 and PKC may be excluded in adhesion studies but it is imperative that Src kinase function be gauged, along with FAK.
PROTEIN RECRUITMENT
Immature FAs are detected under the microscope as “Actin dots” that are rich in Integrins, Paxillin and Vinculin [64]. Recruitment and concentration of Paxillin and Vinculin to Integrin clutches is necessary for FA development. Though both proteins have unique roles in adhesion signaling, they play a fundamental role in recruiting key proteins to the adhesion interactome. Their ultimate goals are to transmit signals back and forth from the ECM to Actin and its binding proteins. Formation of Actin filament stress fibers precedes FA formation [65].
Paxillin
Activation of the FA scaffold protein, Paxillin, occurs subsequently after kinase recruitment [66]. The active FAK-Src proteins phosphorylate Paxillin, and various other adhesion molecules [67]. Localization of active Src and FAK to FAs results in the phosphorylation of Paxillin at tyrosines 31 and 118 as shown in Figure 1 [68]. Paxillin is an adaptor/scaffolding protein that participates in a number of cell signaling pathways. The C-terminus contains two LIM (Lin11, Isl-1, Mec-3) domains that are necessary for targeting to FAs [69, 70]. These LIM domains are also important for binding to FA regulatory and structural proteins such as Tubulin and Protein Tyrosine Phosphatase Non-Receptor Type 12 (PTPN12) [71, 72].
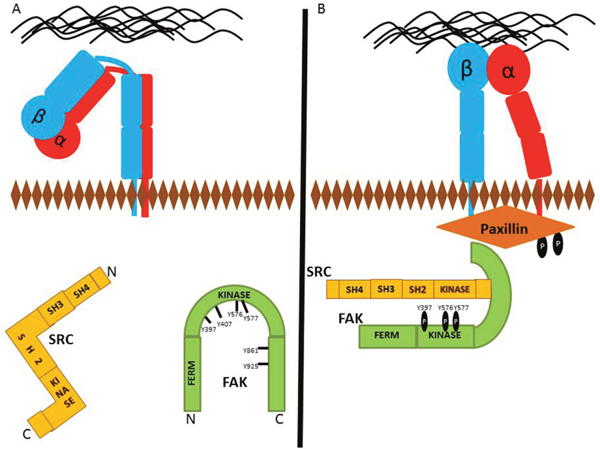
Figure 1: Protein recruitment to future FA site. (A) Detached FAs typically have inactivated Integrins, as well as inactive FAK and Src kinases. (B) FAK phosphorylation at Y397 leads to the activation of Src. In the early events of FA formation, localization of Src and FAK to FA sites results in Paxillin phosphorylation at tyrosines 118 and 31.
The N-terminus of Paxillin contains leucine and aspartate rich motifs that are critical for its interaction with FAK, Vinculin, PYK2 and a number of other FA proteins (Figure 2) [73]. The proline-rich region of Paxillin also binds to Src [74]. Furthermore, Paxillin contains many serine and threonine phosphorylation sites that are phosphorylated by a number of stimulants, including Receptor for Activated C Kinase 1 (RACK1), a Src kinase inhibitor, and the serine/threonine-protein kinase, p21-activated Kinase (PAK) [74]. In the early stages of FA formation, Paxillin experiences phosphorylation of tyrosine 118, and serines 85, 188 and 190 [75, 76]. Paxillin is one of the first proteins present in nascent adhesions and recruits many structural and regulatory proteins to the cell adhesion site [74, 77]. Since Paxillin has many direct binding partners, understanding the orchestration of signaling events will help to fathom the specificity Paxillin holds for each dynamic process. This makes Paxillin an attractive target for anti-metastatic drugs since its inhibition prevents the formation of FAs.
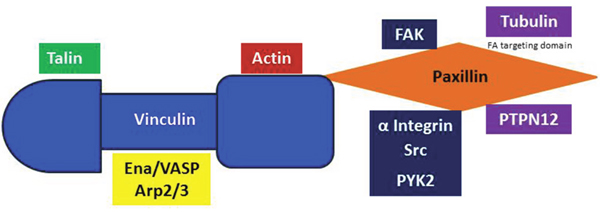
Figure 2: Vinculin and Paxillin binding. Vinculin is a direct binding partner of Paxillin, Talin, Arp2/3, Actin and the Ena/VASP proteins. Paxillin binds to PYK2, Integrin, Src, Tubulin and PTPN12. The LIM2 and LIM3 domains of Paxillin are necessary for FA targeting.
Vinculin
Vinculin is an F-Actin-binding protein that also appears in the early stages of adhesion formation [78]. Paxillin, F-actin and Talin are necessary for Vinculin recruitment to FA sites [78–82]. Vinculin is a direct binding partner of Paxillin and its interaction is important for coordinating force-induced signals (Figure 2) [27, 83]. Vinculin deficient cells are less stiff than normal cells and have lower traction forces due to a reduction in Vinculin’s tension sensing capabilities [84, 85]. Vinculin is a tension sensor that is recruited to the FA site in response to mechanical forces by conveying signals from the ECM to Actin filaments [86].
In addition to its role in mechanosensing, Vinculin directly interacts with a number of proteins that are important for Actin cytoskeletal dynamics. The α-helical Vinculin tail directly binds to Actin (Figure 2) [81, 87]. Immediately after binding, Vinculin dimerizes and begins to assemble bundles of Actin filaments [87, 88]. The polymerization of these filaments occurs by Vinculin-mediated recruitment of G-Actin to form a nucleus for polymerization [81]. Vinculin then recruits and directly binds to the Arp2/3 Actin polymerization complex [89]. Also, Vinculin directly interacts with molecules such as the membrane phospholipid, PIP2, and the cytoskeletal adaptor protein, Vinexin to increase FA size during the elongation phase of FA assembly [90, 91]. These interactions are necessary for Actin polymerization to occur.
Vinculin and Talin interactions are necessary for strong Integrin-cytoskeleton bonds. Vinculin knockdown in cells increases cell migration due to increased Paxillin and FAK phosphorylation, as well as, an increase in FA turnover [92, 93]. Vinculin has recently been discovered in prostate cancer exosomes [94]. Though there is a gap in knowledge about why Vinculin is present in exosomes, these secreted micro vesicles may be isolated from the serum of patients and identification of Vinculin could serve as another prognostic marker.
Vinculin is an indirect target of the common arthritis drug, Celecoxib. Celecoxib is a nonsteroidal anti-inflammatory drug that inhibits cyclooxygenase-2 (COX-2) [95]. This inhibitor was first published as a chemopreventative agent in colon carcinogenesis in 1998, just shortly after its synthesis [96]. Since then, the tumor preventative effects of Celecoxib have been seen in many other cancer models, including urinary bladder cancer [97], prostate cancers [98, 99], and breast cancer [100]. Recently, Celecoxib has been shown to exert anti-gastric cancer effects by down regulating the expression of 8 genes, including Vinculin [101]. The other 7 genes, Cysteine and glycine-rich protein 1 (CRP1) [102], Thrombospondin 1 (THBS1) [103], Myosin light chain 9 (MYL9) [104], Filamin A (FLNA) [105], Actinin alpha 1 (ACTN1) [106], Laminin subunit gamma 2 (LAMC2) [107] and Claudin 1 (CLDN1) [108] are also linked to cell adhesion. These research findings suggest that Celecoxib exerts its anti-cancer functions by down regulating the expression of cell adhesion molecules. This further stresses the importance of studying FAs and the therapeutic potential that could be obtained from those studies.
ACTIN ASSEMBLY
Actin is one of the most abundant proteins in eukaryotic cells. Polymerization of Actin into microfilaments is required for a sturdy cell-matrix adhesion point. This assembly requires three phases: a nucleation, elongation and steady state phase. During nucleation, Actin monomers are converted to stable multimers. These multimers are then rapidly elongated by adding monomers to both ends. Finally, the steady state phase is achieved once equilibrium is reached between the polymerization at one end and the disassembly at the opposite as shown in Figure 3. This three-step process requires many proteins but we will focus on the three most studied assembly pathways that include the Arp2/3 complex, the Ena/VASP family, Formins and Talin.
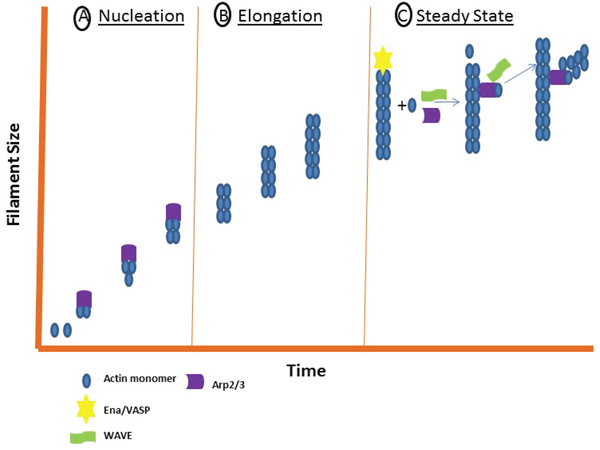
Figure 3: The three stages of filament assembly. Actin filament assembly requires three stages: a nucleation, elongation and steady state phase. (A) During nucleation, the Actin monomers are converted to stable multimers through nucleating proteins such as Arp2/3. (B) During elongation, monomers are rapidly added to each end. (C) During the steady state phase, WAVE and Ena/VASP interact to enhance Arp2/3 filament assembly.
Arp2/3 complex
Actin nucleation is the mechanism by which Actin monomers form into a stable multimer. Actin Related Protein 2 (Arp2) and Actin Related Protein 3 (Arp3) were the first Actin nucleators to be discovered [109]. These two proteins form a complex known as the Arp2/3 complex. For activation, this complex relies on nucleation promoting factors such as Wiskott-Aldrich Syndrome Protein (WASP) and WASP-family verprolin-homologous protein (WAVE) [110, 111]. Cortactin has also been shown to cooperate with N-WASP to activate the Arp2/3 complex [112, 113]. The precise mechanism that the Arp2/3 complex uses for Actin nucleation is heavily debated [109–111]. For example, one study showed that the Actin binding protein Caldesmon increases Arp2/3-mediated branching activity by promoting the binding of newly polymerized Actin to the Arp2/3 complex [114]. Another study showed that accelerated release of WASP proteins from the Actin-Arp2/3 complex accelerated branch forming [115], suggesting that WASP dissociates from the Actin-Arp2/3 complex prior to Actin filament growth.
Cells with HER2 gene amplification have increased co-expression of both WAVE2 and Arp2/3 [116]. This is of great significance because HER2 gene amplification is a poor prognostic indicator in early breast cancer. Furthermore, HER2 status plays a role in selecting the most effective treatment option for both early and advanced breast cancer. The reason why there is co-amplification of these genes is unclear, especially since the genes are all located on different chromosomes. Arp2/3 mRNA and protein expression are often upregulated in some other invasive cancer cells as well, including colorectal and lung adenocarcinoma’s [117–119]. Though these other invasive cancers are not associated with frequent HER2 amplification, its co-amplification patterns in breast cancer need to be further explored.
Two highly effective small molecule inhibitors, CK-666 and CK-869 bind the Arp 2/3 complex and reversibly inhibit Actin nucleation [120]. Both molecules have distinct binding sites with CK-666 blocking the movement of the complex proteins to their activated conformation [121]. CK-666 treatment of the human ovarian carcinoma cell line, A2780, resulted in a collapse of lamellipodia [122], while treatment of fibroblasts with CK-869 caused blebbing [123]. CK-636 is a similar inhibitor that has a thiopene rather than the fluorobenzene ring of CK-666 but has a much lower IC50 [120]. CK-869 alternatively binds to an off-hand cavity on Arp 3 that destabilizes its interaction with Arp 2 [121]. Its analog, CK-548, has ortho-hydroxy and meta-chlorine substituent’s where CK-869 has methoxy groups in the ortho and para positions [120]. CK-869 is acknowledged as the more effective inhibitor because a study showed that CK-548 does not effectively inhibit comet tail formation by Listeria [120]. CK-869 inhibition in the MCF10A mammary gland cell line disrupted lamellipodia formation and cell spreading [124].
The ability of Arp complex inhibitors to prevent cell adhesion speaks to the importance of adhesion assembly. Although the disruption of lamellipodia upon treatment with Actin nucleation inhibitors is universal, several studies have found no disruption of monolayer integrity or cytotoxicity of normal epithelial cells [124, 125]. Given that treatment of cells with these compounds prevents Actin branched networks, these inhibitors could serve as effective cancer therapeutics. Recruitment of key FA proteins such as FAK and Paxillin to FA sites is important but if assembly of the Actin filament is perturbed, FAs will not form.
Ena/VASP protein family
The nucleation-promoting factor and Arp2/3 activator, WAVE, directly binds to Ena/VASP to enhance Arp2/3 filament assembly (Figure 3) [126]. The Ena/VASP protein family regulates Actin assembly and has been implicated in fibroblast migration, T cell polarization and axon guidance [127]. These proteins are targeted to areas of Actin remodeling, such as the leading edges of invadopodia, lamellipodia, filopodia and Actin-dependent cell-cell contacts [127–129]. Actin proteins are non-symmetrical structures with two ends, each referred to as a “barbed end” and “pointed end” [130]. Polarized growth of the filament is regulated by proteins that cap their ends to block the assembly of polarized filaments [130]. Disassembly of the filaments occurs at the pointed end, while elongation occurs at the barbed end [130]. Ena/VASP regulatory molecules protect Actin filaments from capping proteins at their barbed ends [131, 132]. Also, Ena/VASP proteins increase the density of Arp2/3-dependent Actin filament branches during Actin polymerization [127].
Proteins from this family regulate many cell motility pathways and are associated with poor clinical outcome in breast cancer patients [133, 134]. Ena/VASP proteins are uniquely linked to Profilin-1 (Pfn1), a G-Actin binding protein that is down regulated in breast, hepatic and pancreatic adenocarcinomas [135–137]. In breast cancer, down regulation of Pfn1 in MDA-MB-231 cells leads to a highly motile phenotype that was orchestrated by Ena/VASP proteins [138]. In fact, silencing of Pfn1 in these cells enhanced the localization of VASP to their leading edge to enhance cell motility [138]. The targeting of Ena/VASP proteins to the leading edge and subsequent enhanced motility hints to its importance for adhesion dynamics in cancer cells.
Formins
Like Ena/VASP proteins, the Formin protein family is responsible for the formation of unbranched Actin filaments. The Formin family consists of fifteen genes that play a role in many cellular processes, including cell adhesion, migration, stress fiber formation, and development [139]. Formins are defined by their highly conserved C-terminal Formin homology 2 (FH2) domain, which nucleates Actin monomers [140]. The N-terminus contains a FH1 domain that recruits Profilin-Actin monomers to grow the filament [141].
FHOD1 is a Formin that is necessary for cell spreading, adhesion maturation, and the transduction of adhesive force [142]. It is the major Formin transcriptionally upregulated during the epithelial to mesenchymal transition (EMT) of oral squamous carcinoma cells (OSCC) [143]. EMT is a process by which non-motile epithelial cells gain an invasive, mesenchymal phenotype [144]. Knockdown of FHOD1 results in the repression of elongation, migration and invasion of SCC-43B OSCCs [143]. As a cell undergoes the EMT, it experiences an increase in Actin organization, motility and invasiveness mainly due to an increase in the expression of mesenchymal markers. The up-regulation of FHOD1 during this process suggests that it is a mesenchymal marker. This new fibroblast-like morphology has to have dynamic Actin assembly/disassembly capabilities. Arp2/3, Ena/VASP and Formins are crucial for Actin nucleation but require proteins like Talin to enhance the length of the FA.
Talin
Talin is a critical component of cell contacts and the primary determinant of FA length [145]. Talin binds to many adhesion proteins, including Actin, FAK, Vinculin, and Integrins to enhance the strength of FAs [146–149]. Talin acts as a linker between Actin and Integrins to allow the transmission of biochemical signals and force across the cell membrane [150, 151]. Specifically, the interaction between Talin and β-Integrin triggers a conformational change in the Integrin extracellular domain that promotes a stronger affinity to the ECM [152]. Talin binding to β Integrins through its FERM domain is what induces this conformational change in Integrins that promotes its affinity for the ECM [153, 154].
Not only is Talin’s role as a linker important for strong adhesions, but its role as an Integrin activator is significant as well [155]. In fact, downregulation of Talin leads to aberrant folding and processing of Integrins [156]. Talin overexpression is generally correlated with poor prognosis and this overexpression has been noted in triple negative breast cancers, nasopharyngeal carcinoma, and human hepatocellular carcinomas [157–159]. Interestingly, Talin 1 has been shown to have an opposite effect in prostate cancer because it impairs cell adhesion and motility [160]. Actin assembly is important for anchoring the cell, but the turnover of the FAs is essential to cell migration and invasion.
Actin filament disassembly is required for cell spreading and migration of cells. A key indicator of FA turnover is proteolysis of Integrins, FAK and Talin [161–163]. RhoGTPases promote FA turnover through FAK-mediated suppression [164]. Although much is known about Actin assembly in cancer, the opposite is evident for Actin disassembly. However, it is known that filaments grow until they are capped by proteins, such as those from the Ena/VASP family [165]. Extracellular cues, including substrate tension information instructs Actin assembly machinery on how long the particular FA should be. In the absence of such external signals, it is unknown which intrinsic signals regulate Actin assembly.
THE CANCER CELLULAR ADHESOME
In recent years, novel proteins have been linked to FA regulation in cancer cells. Using proteomic analysis, a 2,413-protein Integrin adhesion complex has been identified [166]. These proteins are grouped into four axes of core adhesion machinery comprising the FAK-Paxillin, Talin-Vinculin, α-Actinin-Zyxin-VASP, and ILK-PINCH-Kindlin pathways [166]. The FAK-Paxillin, Talin-Vinculin, and α-Actinin-Zyxin-VASP have been well studied in cancer cells but not all constituents in the pathways have been discovered. Alterations in gene expression of these FA molecules can either lead to a change in the size of each cell-matrix contact, or cell death.
Targeting of FA proteins has been shown to sensitize cancer cells to treatment [167]. In MCF7 breast cancer cells, knockdown of FAK, Integrin linked kinase (ILK), Talin, and Zyxin leads to enlarged FAs [168]. Furthermore, Protein Phosphatase 1 Regulatory Subunit 12B (PPP1R12B), Homeodomain Interacting Protein Kinase 3 (HIPK3), Rac2, and Tropomyosin 1 (TPM1) inhibit Insulin-like Growth Factor 1 Receptor (IGF1R) driven migration of MCF7 cells [168]. These proteins enlarge FAs to anchor the cell to the matrix and reduce the rate of disassembly.
Inhibition of the serine/threonine kinase, NEK3 also leads to enlarged FAs in MCF7 breast cancer cells. The NimA related kinase (NEK) family is known for controlling cell cycle progression in response to genotoxic stress. NEK3, in particular, regulates Prolactin-mediated cytoskeletal reorganization in breast cancer cells [169]. Prolactin signaling also activates NEK3 at threonine 165 [170]. Inhibition of the NEK3 Thr-165 phosphorylation results in actin cytoskeleton reorganization and increased FA size [170]. Overexpression of NEK3-T165V in cancer cells led to a decrease in migration [170]. The coordination of adhesion assembly and disassembly is delicately orchestrated. The inhibition of MCF7 cell migration seems to be the consequence of enlarged FAs. This hints to an increase in the rate of Actin assembly and a decrease in the rate of Actin disassembly. Enlarged FAs are not always a consequence of the inhibition of cancer cell migration; some proteins can inhibit cell migration without enlarging the FAs.
Transformation-related protein 63 (p63) is a member of the p63/p53/p73 family of transcription factors that also induces cell cycle arrest and apoptosis [171]. This family of transcription factors contains three highly conserved domains: a transactivating domain, a DNA binding domain, and an oligomerization domain [171]. Alternative splicing of p63 brings forth two different isoforms: those with or without the full length transactivating domain [172]. Expression of the truncated isoform (ΔNp63) in basal-A triple-negative breast cancer (TNBC) cell lines resulted in a reduction of cell adhesion, which led to cell death [173]. Opposite effects have been demonstrated in squamous cell carcinomas of the esophagus (ESCC). Another group suggested that the amplification of TP63 that leads to the hyper-expression of ΔNp63 seen in ESCC is what promotes cell adhesion [174]. In fact, inhibition of p63 in the TE13 squamous esophageal carcinoma cell line altered expression of cell adhesion proteins [174]. P63 has both oncogenic and suppressive capabilities that all eventually lead to a regulation of cell adhesion dynamics. The ΔNp63 isoform in particular seems to control these effects since the full-length protein has not been shown to affect cell adhesion. Though ΔNp63 alters the expression of some of the key FA proteins, the exact mechanism by which it exerts its functions is unknown.
Approximately 15-20% of newly described breast cancer cases are triple-negative breast cancers (TNBCs). This subtype is highly invasive and has the greater probability to metastasize. A few pharmacological inhibitors have been developed for reducing TNBC cell adhesion. AMPI-109 selectively targets TNBC cells by activating pro-apoptotic caspases-3 and 7, DNA fragmentation, and PARP cleavage [175]. Studies performed in BT-20 TNBC cell lines showed that AMPI-109 treatment impairs cell migration and invasion by reducing levels of the oncogenic tyrosine phosphatase, PRL-3 [175]. Further investigations of this showed that knockdown of PRL-3 or AMPI-109 treatment deactivate the Src and ERK signaling pathways, which reduces RhoA and Rac1/2/3 GTPase protein expression [176]. On the contrary, PRL-3 over expression increased adherence to and degradation of TNBC cells to the ECM component Laminin [176]. In vivo animal models further demonstrate that PRL-3 inhibition reduces growth of TNBC tumors [177]. These studies were the first to link PRL-3 to the FA pathway in TNBC cells. The fact that the effectiveness of the AMPI-109 is in its ability to disrupt cell adhesion dynamics speaks to the importance of fully understanding all of the components of the FA-signaling pathway.
CLOSING REMARKS
Adhesion of a cell to the ECM regulates cancer cell morphology, migration, survival, and proliferation. The proteins involved in FA assembly serve as markers for standard Western Blot or Immunofluorescence techniques using lysed or fixed cells (Figure 4). However, live cell imaging allows us to study FA strength, turnover, and polymerization in real-time. The dynamics of the cytoskeleton near the edge of the cell can be visualized by using total internal reflection fluorescence (TIRF) microscopy. To use this, FA proteins are genetically tagged to fluorescent proteins then introduced into cells. Once the imaging has started, only the fluorophores within 100 nm from the bottom of the cell are excited [178]. This process allows one to visualize the attachment, assembly, and turnover of the FA.
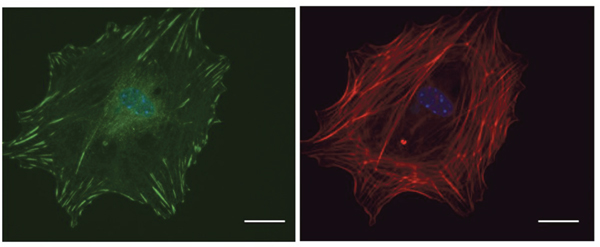
Figure 4: Microscopic visualization of FAs. Primary tumor cells isolated from MMTV-PyMT tumors were plated on a Fibronectin-coated cover slip and labeled with Vinculin (green) to visualize FAs and DAPI (blue) to visualize the nucleus. Phalloidin (red) staining is shown in the right panel. Scale bar: 10μm.
A migrating cell often experiences rapid re-organization of its FA sites. Understanding the spatial regulation of each site gives a more detailed picture of cell migration. Major advances to the field of optical microscopy now allow the precise visualization of a single fluorescent molecule [179–181]. To supplement these discoveries, efficient algorithms have been developed for single-particle tracking [182]. Super-resolution microscopy that is based on single molecule localizations includes photoactivation localization microscopy (PALM), stochastic optical reconstruction microscopy (STORM), ground state depletion microscopy followed by individual molecular return (GSDIM) and universal point accumulation imaging in the nanoscale topography (uPAINT) [183, 184]. iPALM is a unique form of super-resolution microscopy that combines PALM with simultaneous multi-phase interferometry of photons from individual fluorescent molecules [185]. This technology allows one to image fluorescence-tagged FA proteins with three-dimensional nanoscale resolution [186]. This imaging was used by Kanchanawong, et al to determine the three-dimensional localization of many FA proteins including FAK, Paxillin, Talin, Vinculin, and Actin [186]. Super resolution techniques such as iPALM continue to be useful for defining the molecular architecture of FAs.
Some other novel imaging techniques include measuring nanomechanics through atomic force microscopy (AFM) [187]. Time-lapse AFM of live cells permits the observation of the dynamic rearrangements that occur at the cell membrane and the underlying cytoskeleton. AFM allows for the measurement of Young’s (E) and shear (G) modulus [188]. Since mechanical tension sensed by Integrins directs the formation of FAs, AFM provides an excellent opportunity to measure cell traction force. The development of more novel imaging techniques would help our understanding of FA dynamics.
A cells interaction with its ECM regulates a number of cancer cell processes, including proliferation, migration, survival, and morphology. Specifically, Integrin-mediated adhesion regulates cell signaling, migration and cell contractile force. Alterations in the adhesion mechanics of cells may play a role in the development and progression of cancer. A diverse array of proteins participate in FA signaling, including tyrosine kinases, phosphatases, G-Actin binding proteins, and GTPases as shown in Figure 5. Significant progress has been made in identifying key proteins involved in adhesion signaling, as well as linking these incidents with oncogenic events. Although many proteins in the FA-signaling pathway have been discovered, many more undiscovered proteins may contribute to this phenomena. Fully understanding the unique mechanisms of Integrin-mediated cell adhesions in different cancer types could provide new diagnostic or prognostic markers. To facilitate this, development of novel high-resolution microscopy/spectroscopy methods could aid the field in understanding the complex molecular interactions that occur at specific FA points.
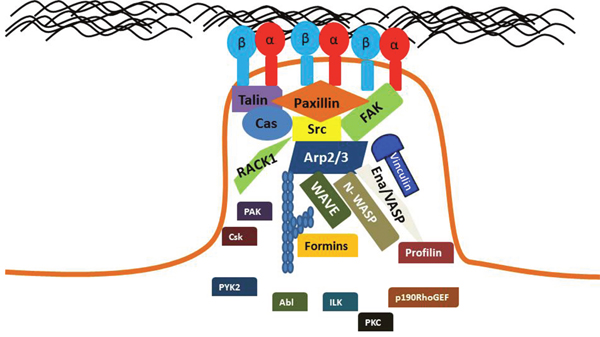
Figure 5: Key proteins are localized to mature FAs. Diverse proteins participate in FA signaling, including tyrosine kinases, phosphatases, G-Actin binding proteins, and GTPases. A mature FA will contain activate Integrins that are bound to their ECM ligand. FA signaling pathways regulate the activation of these Integrins.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Okegawa T, Pong RC, Li Y, Hsieh JT. The role of cell adhesion molecule in cancer progression and its application in cancer therapy. Acta biochimica Polonica. 2004; 51:445-457.
2. Takada Y, Ye X, Simon S. The integrins. Genome Biol. 2007; 8:215.
3. Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J Cell Sci. 2006; 119:3901-3903.
4. Plow EF, Haas TA, Zhang L, Loftus J, Smith JW. Ligand binding to integrins. J Biol Chem. 2000; 275:21785-21788.
5. Moser M, Legate KR, Zent R, Fassler R. The tail of integrins, talin, and kindlins. Science. 2009; 324:895-899.
6. Ivaska J. Unanchoring integrins in focal adhesions. Nat Cell Biol. 2012; 14:981-983.
7. Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009; 10:21-33.
8. Friedland JC, Lee MH, Boettiger D. Mechanically activated integrin switch controls alpha5beta1 function. Science. 2009; 323:642-644.
9. Moore SW, Roca-Cusachs P, Sheetz MP. Stretchy proteins on stretchy substrates: the important elements of integrin-mediated rigidity sensing. Dev Cell. 2010; 19:194-206.
10. Ingber DE. Tensegrity: the architectural basis of cellular mechanotransduction. Annu Rev Physiol. 1997; 59:575-599.
11. Klotzsch E, Smith ML, Kubow KE, Muntwyler S, Little WC, Beyeler F, Gourdon D, Nelson BJ, Vogel V. Fibronectin forms the most extensible biological fibers displaying switchable force-exposed cryptic binding sites. Proc Natl Acad Sci U S A. 2009; 106:18267-18272.
12. Zamir E, Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. J Cell Sci. 2001; 114:3583-3590.
13. Abercrombie M, Heaysman JE, Pegrum SM. The locomotion of fibroblasts in culture. IV. Electron microscopy of the leading lamella. Exp Cell Res. 1971; 67:359-367.
14. Izzard CS, Lochner LR. Cell-to-substrate contacts in living fibroblasts: an interference reflexion study with an evaluation of the technique. J Cell Sci. 1976; 21:129-159.
15. Horzum U, Ozdil B, Pesen-Okvur D. Step-by-step quantitative analysis of focal adhesions. MethodsX. 1:56-59.
16. Nagasaki A, Kanada M, Uyeda TQ. Cell adhesion molecules regulate contractile ring-independent cytokinesis in Dictyostelium discoideum. Cell Res. 2009; 19:236-246.
17. Huttenlocher A, Horwitz AR. Integrins in cell migration. Cold Spring Harb Perspect Biol. 3:a005074.
18. Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996; 84:359-369.
19. Kirchner J, Kam Z, Tzur G, Bershadsky AD, Geiger B. Live-cell monitoring of tyrosine phosphorylation in focal adhesions following microtubule disruption. J Cell Sci. 2003; 116:975-986.
20. Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004; 1692:103-119.
21. Burridge K, Turner CE, Romer LH. Tyrosine phosphorylation of paxillin and pp125FAK accompanies cell adhesion to extracellular matrix: a role in cytoskeletal assembly. J Cell Biol. 1992; 119:893-903.
22. Roussos ET, Condeelis JS, Patsialou A. Chemotaxis in cancer. Nat Rev Cancer. 2011; 11:573-587.
23. Krieg M, Arboleda-Estudillo Y, Puech PH, Kafer J, Graner F, Muller DJ, Heisenberg CP. Tensile forces govern germ-layer organization in zebrafish. Nat Cell Biol. 2008; 10:429-436.
24. Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 178:1221-1232.
25. Brabrand A, Kariuki, II, Engstrom MJ, Haugen OA, Dyrnes LA, Asvold BO, Lilledahl MB, Bofin AM. Alterations in collagen fibre patterns in breast cancer. A premise for tumour invasiveness? APMIS. 123:1-8.
26. Schiller HB, Hermann MR, Polleux J, Vignaud T, Zanivan S, Friedel CC, Sun Z, Raducanu A, Gottschalk KE, Thery M, Mann M, Fassler R. beta1- and alphav-class integrins cooperate to regulate myosin II during rigidity sensing of fibronectin-based microenvironments. Nat Cell Biol. 15:625-636.
27. Plotnikov SV, Pasapera AM, Sabass B, Waterman CM. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell. 151:1513-1527.
28. Wang HB, Dembo M, Hanks SK, Wang Y. Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc Natl Acad Sci U S A. 2001; 98:11295-11300.
29. Shemesh T, Geiger B, Bershadsky AD, Kozlov MM. Focal adhesions as mechanosensors: a physical mechanism. Proc Natl Acad Sci U S A. 2005; 102:12383-12388.
30. Alexandrova AY, Arnold K, Schaub S, Vasiliev JM, Meister JJ, Bershadsky AD, Verkhovsky AB. Comparative dynamics of retrograde actin flow and focal adhesions: formation of nascent adhesions triggers transition from fast to slow flow. PLoS One. 2008; 3:e3234.
31. Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003; 116:1409-1416.
32. Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004; 6:154-161.
33. Cance WG, Harris JE, Iacocca MV, Roche E, Yang X, Chang J, Simkins S, Xu L. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin Cancer Res. 2000; 6:2417-2423.
34. Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005; 6:56-68.
35. Zhao X, Guan JL. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2011; 63:610-615.
36. Barret C, Roy C, Montcourrier P, Mangeat P, Niggli V. Mutagenesis of the phosphatidylinositol 4,5-bisphosphate (PIP(2)) binding site in the NH(2)-terminal domain of ezrin correlates with its altered cellular distribution. J Cell Biol. 2000; 151:1067-1080.
37. Mangeat P, Roy C, Martin M. ERM proteins in cell adhesion and membrane dynamics: Authors' correction. Trends Cell Biol. 1999; 9:289.
38. Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007; 129:1177-1187.
39. Schlaepfer DD, Mitra SK. Multiple connections link FAK to cell motility and invasion. Curr Opin Genet Dev. 2004; 14:92-101.
40. Chen SY, Chen HC. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol Cell Biol. 2006; 26:5155-5167.
41. Cooper LA, Shen TL, Guan JL. Regulation of focal adhesion kinase by its amino-terminal domain through an autoinhibitory interaction. Mol Cell Biol. 2003; 23:8030-8041.
42. Cohen LA, Guan JL. Residues within the first subdomain of the FERM-like domain in focal adhesion kinase are important in its regulation. J Biol Chem. 2005; 280:8197-8207.
43. Cai X, Lietha D, Ceccarelli DF, Karginov AV, Rajfur Z, Jacobson K, Hahn KM, Eck MJ, Schaller MD. Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol Cell Biol. 2008; 28:201-214.
44. Schaller MD. Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim Biophys Acta. 2001; 1540:1-21.
45. Miller NL, Lawson C, Kleinschmidt EG, Tancioni I, Uryu S, Schlaepfer DD. A non-canonical role for Rgnef in promoting integrin-stimulated focal adhesion kinase activation. J Cell Sci. 2013; 126:5074-5085.
46. Higuchi M, Kihara R, Okazaki T, Aoki I, Suetsugu S, Gotoh Y. Akt1 promotes focal adhesion disassembly and cell motility through phosphorylation of FAK in growth factor-stimulated cells. J Cell Sci. 2013; 126:745-755.
47. Katsumi A, Orr AW, Tzima E, Schwartz MA. Integrins in mechanotransduction. J Biol Chem. 2004; 279:12001-12004.
48. Lo CM, Wang HB, Dembo M, Wang YL. Cell movement is guided by the rigidity of the substrate. Biophys J. 2000; 79:144-152.
49. Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999; 71:435-478.
50. Shi Q, Boettiger D. A novel mode for integrin-mediated signaling: tethering is required for phosphorylation of FAK Y397. Mol Biol Cell. 2003; 14:4306-4315.
51. Kpetemey M, Chaudhary P, Van Treuren T, Vishwanatha JK. MIEN1 drives breast tumor cell migration by regulating cytoskeletal-focal adhesion dynamics. Oncotarget. 2016; 7:54913-54924. doi: 10.18632/oncotarget.10798.
52. Staaf J, Jonsson G, Ringner M, Vallon-Christersson J, Grabau D, Arason A, Gunnarsson H, Agnarsson BA, Malmstrom PO, Johannsson OT, Loman N, Barkardottir RB, Borg A. High-resolution genomic and expression analyses of copy number alterations in HER2-amplified breast cancer. Breast Cancer Res. 2010; 12:R25.
53. Dasgupta S, Cushman I, Kpetemey M, Casey PJ, Vishwanatha JK. Prenylated c17orf37 induces filopodia formation to promote cell migration and metastasis. J Biol Chem. 2011; 286:25935-25946.
54. Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol. 2010; 11:633-643.
55. Long W, Yi P, Amazit L, LaMarca HL, Ashcroft F, Kumar R, Mancini MA, Tsai SY, Tsai MJ, O'Malley BW. SRC-3Delta4 mediates the interaction of EGFR with FAK to promote cell migration. Mol Cell. 2010; 37:321-332.
56. Li L, Okura M, Imamoto A. Focal adhesions require catalytic activity of Src family kinases to mediate integrin-matrix adhesion. Mol Cell Biol. 2002; 22:1203-1217.
57. Sen B, Johnson FM. Regulation of SRC family kinases in human cancers. J Signal Transduct. 2011; 2011:865819.
58. Brown MT, Cooper JA. Regulation, substrates and functions of src. Biochim Biophys Acta. 1996; 1287:121-149.
59. Bjorge JD, Jakymiw A, Fujita DJ. Selected glimpses into the activation and function of Src kinase. Oncogene. 2000; 19:5620-5635.
60. Sawada Y, Tamada M, Dubin-Thaler BJ, Cherniavskaya O, Sakai R, Tanaka S, Sheetz MP. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell. 2006; 127:1015-1026.
61. Zhang X, Moore SW, Iskratsch T, Sheetz MP. N-WASP-directed actin polymerization activates Cas phosphorylation and lamellipodium spreading. J Cell Sci. 2014; 127:1394-1405.
62. Bromann PA, Korkaya H, Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004; 23:7957-7968.
63. Ishizawar R, Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell. 2004; 6:209-214.
64. Zimerman B, Volberg T, Geiger B. Early molecular events in the assembly of the focal adhesion-stress fiber complex during fibroblast spreading. Cell Motil Cytoskeleton. 2004; 58:143-159.
65. Izzard CS. A precursor of the focal contact in cultured fibroblasts. Cell Motil Cytoskeleton. 1988; 10:137-142.
66. Hu YL, Lu S, Szeto KW, Sun J, Wang Y, Lasheras JC, Chien S. FAK and paxillin dynamics at focal adhesions in the protrusions of migrating cells. Sci Rep. 2014; 4:6024.
67. Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004; 23:7906-7909.
68. Wu JC, Chen YC, Kuo CT, Wenshin Yu H, Chen YQ, Chiou A, Kuo JC. Focal adhesion kinase-dependent focal adhesion recruitment of SH2 domains directs SRC into focal adhesions to regulate cell adhesion and migration. Sci Rep. 2015; 5:18476.
69. Brown MC, Perrotta JA, Turner CE. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J Cell Biol. 1996; 135:1109-1123.
70. Turner CE. Paxillin interactions. J Cell Sci. 2000; 113:4139-4140.
71. Cote JF, Turner CE, Tremblay ML. Intact LIM 3 and LIM 4 domains of paxillin are required for the association to a novel polyproline region (Pro 2) of protein-tyrosine phosphatase-PEST. J Biol Chem. 1999; 274:20550-20560.
72. Herreros L, Rodriguez-Fernandez JL, Brown MC, Alonso-Lebrero JL, Cabanas C, Sanchez-Madrid F, Longo N, Turner CE, Sanchez-Mateos P. Paxillin localizes to the lymphocyte microtubule organizing center and associates with the microtubule cytoskeleton. J Biol Chem. 2000; 275:26436-26440.
73. Brown MC, Curtis MS, Turner CE. Paxillin LD motifs may define a new family of protein recognition domains. Nat Struct Biol. 1998; 5:677-678.
74. Deakin NO, Turner CE. Paxillin comes of age. J Cell Sci. 2008; 121:2435-2444.
75. Bellis SL, Perrotta JA, Curtis MS, Turner CE. Adhesion of fibroblasts to fibronectin stimulates both serine and tyrosine phosphorylation of paxillin. Biochem J. 1997; 325:375-381.
76. Kwak TK, Lee MS, Ryu J, Choi YJ, Kang M, Jeong D, Lee JW. Cell adhesion-dependent serine 85 phosphorylation of paxillin modulates focal adhesion formation and haptotactic migration via association with the C-terminal tail domain of talin. J Biol Chem. 2012; 287:27499-27509.
77. Digman MA, Brown CM, Horwitz AR, Mantulin WW, Gratton E. Paxillin dynamics measured during adhesion assembly and disassembly by correlation spectroscopy. Biophys J. 2008; 94:2819-2831.
78. Carisey A, Tsang R, Greiner AM, Nijenhuis N, Heath N, Nazgiewicz A, Kemkemer R, Derby B, Spatz J, Ballestrem C. Vinculin regulates the recruitment and release of core focal adhesion proteins in a force-dependent manner. Curr Biol. 2013; 23:271-281.
79. Wang P, Ballestrem C, Streuli CH. The C terminus of talin links integrins to cell cycle progression. J Cell Biol. 2011; 195:499-513.
80. Zhang X, Jiang G, Cai Y, Monkley SJ, Critchley DR, Sheetz MP. Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat Cell Biol. 2008; 10:1062-1068.
81. Peng X, Nelson ES, Maiers JL, DeMali KA. New insights into vinculin function and regulation. Int Rev Cell Mol Biol. 2011; 287:191-231.
82. Burridge K, Mangeat P. An interaction between vinculin and talin. Nature. 1984; 308:744-746.
83. Turner CE, Glenney JR Jr, Burridge K. Paxillin: a new vinculin-binding protein present in focal adhesions. J Cell Biol. 1990; 111:1059-1068.
84. Xu W, Baribault H, Adamson ED. Vinculin knockout results in heart and brain defects during embryonic development. Development. 1998; 125:327-337.
85. Mierke CT, Kollmannsberger P, Zitterbart DP, Smith J, Fabry B, Goldmann WH. Mechano-coupling and regulation of contractility by the vinculin tail domain. Biophys J. 2008; 94:661-670.
86. Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, Yang MT, McLean MA, Sligar SG, Chen CS, Ha T, Schwartz MA. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 2010; 466:263-266.
87. Jockusch BM, Isenberg G. Interaction of alpha-actinin and vinculin with actin: opposite effects on filament network formation. Proc Natl Acad Sci U S A. 1981; 78:3005-3009.
88. Janssen ME, Kim E, Liu H, Fujimoto LM, Bobkov A, Volkmann N, Hanein D. Three-dimensional structure of vinculin bound to actin filaments. Mol Cell. 2006; 21:271-281.
89. DeMali KA, Barlow CA, Burridge K. Recruitment of the Arp2/3 complex to vinculin: coupling membrane protrusion to matrix adhesion. J Cell Biol. 2002; 159:881-891.
90. Kioka N, Sakata S, Kawauchi T, Amachi T, Akiyama SK, Okazaki K, Yaen C, Yamada KM, Aota S. Vinexin: a novel vinculin-binding protein with multiple SH3 domains enhances actin cytoskeletal organization. J Cell Biol. 1999; 144:59-69.
91. Gilmore AP, Burridge K. Regulation of vinculin binding to talin and actin by phosphatidyl-inositol-4-5-bisphosphate. Nature. 1996; 381:531-535.
92. Mierke CT, Kollmannsberger P, Zitterbart DP, Diez G, Koch TM, Marg S, Ziegler WH, Goldmann WH, Fabry B. Vinculin facilitates cell invasion into three-dimensional collagen matrices. J Biol Chem. 2010; 285:13121-13130.
93. Goldmann WH, Schindl M, Cardozo TJ, Ezzell RM. Motility of vinculin-deficient F9 embryonic carcinoma cells analyzed by video, laser confocal, and reflection interference contrast microscopy. Exp Cell Res. 1995; 221:311-319.
94. Kawakami K, Fujita Y, Kato T, Mizutani K, Kameyama K, Tsumoto H, Miura Y, Deguchi T, Ito M. Integrin beta4 and vinculin contained in exosomes are potential markers for progression of prostate cancer associated with taxane-resistance. Int J Oncol. 2015; 47:384-390.
95. Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, Graneto MJ, Lee LF, Malecha JW, Miyashiro JM, Rogers RS, Rogier DJ, Yu SS, et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl] benze nesulfonamide (SC-58635, celecoxib). J Med Chem. 1997; 40:1347-1365.
96. Kawamori T, Rao CV, Seibert K, Reddy BS. Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, against colon carcinogenesis. Cancer Res. 1998; 58:409-412.
97. Grubbs CJ, Lubet RA, Koki AT, Leahy KM, Masferrer JL, Steele VE, Kelloff GJ, Hill DL, Seibert K. Celecoxib inhibits N-butyl-N-(4-hydroxybutyl)-nitrosamine-induced urinary bladder cancers in male B6D2F1 mice and female Fischer-344 rats. Cancer Res. 2000; 60:5599-5602.
98. Johnson AJ, Song X, Hsu A, Chen C. Apoptosis signaling pathways mediated by cyclooxygenase-2 inhibitors in prostate cancer cells. Adv Enzyme Regul. 2001; 41:221-235.
99. Hsu AL, Ching TT, Wang DS, Song X, Rangnekar VM, Chen CS. The cyclooxygenase-2 inhibitor celecoxib induces apoptosis by blocking Akt activation in human prostate cancer cells independently of Bcl-2. J Biol Chem. 2000; 275:11397-11403.
100. Howe LR, Subbaramaiah K, Brown AM, Dannenberg AJ. Cyclooxygenase-2: a target for the prevention and treatment of breast cancer. Endocr Relat Cancer. 2001; 8:97-114.
101. Jin GH, Xu W, Shi Y, Wang LB. Celecoxib exhibits an anti-gastric cancer effect by targeting focal adhesion and leukocyte transendothelial migration-associated genes. Oncol Lett. 2016; 12:2345-2350.
102. Schmeichel KL, Beckerle MC. LIM domains of cysteine-rich protein 1 (CRP1) are essential for its zyxin-binding function. Biochem J. 1998; 331:885-892.
103. Sumagin R, Parkos CA. Epithelial adhesion molecules and the regulation of intestinal homeostasis during neutrophil transepithelial migration. Tissue Barriers. 2015; 3:e969100.
104. Park I, Han C, Jin S, Lee B, Choi H, Kwon JT, Kim D, Kim J, Lifirsu E, Park WJ, Park ZY, Kim DH, Cho C. Myosin regulatory light chains are required to maintain the stability of myosin II and cellular integrity. Biochem J. 2011; 434:171-180.
105. Sato T, Ishii J, Ota Y, Sasaki E, Shibagaki Y, Hattori S. Mammalian target of rapamycin (mTOR) complex 2 regulates filamin A-dependent focal adhesion dynamics and cell migration. Genes Cells. 2016; 21:579-593.
106. Hamill KJ, Hiroyasu S, Colburn ZT, Ventrella RV, Hopkinson SB, Skalli O, Jones JC. Alpha actinin-1 regulates cell-matrix adhesion organization in keratinocytes: consequences for skin cell motility. J Invest Dermatol. 2015; 135:1043-1052.
107. Navdaev A, Heitmann V, Desantana Evangelista K, Morgelin M, Wegener J, Eble JA. The C-terminus of the gamma 2 chain but not of the beta 3 chain of laminin-332 is indirectly but indispensably necessary for integrin-mediated cell reactions. Exp Cell Res. 2008; 314:489-497.
108. Tsukita S, Furuse M. The structure and function of claudins, cell adhesion molecules at tight junctions. Ann N Y Acad Sci. 2000; 915:129-135.
109. Goley ED, Welch MD. The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol. 2006; 7:713-726.
110. Padrick SB, Doolittle LK, Brautigam CA, King DS, Rosen MK. Arp2/3 complex is bound and activated by two WASP proteins. Proc Natl Acad Sci U S A. 2011; 108:E472-479.
111. Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. 2007; 8:37-48.
112. Helgeson LA, Nolen BJ. Mechanism of synergistic activation of Arp2/3 complex by cortactin and N-WASP. Elife. 2013; 2:e00884.
113. Uruno T, Liu J, Zhang P, Fan Y, Egile C, Li R, Mueller SC, Zhan X. Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat Cell Biol. 2001; 3:259-266.
114. Jensen MH, Morris EJ, Huang R, Rebowski G, Dominguez R, Weitz DA, Moore JR, Wang CL. The conformational state of actin filaments regulates branching by actin-related protein 2/3 (Arp2/3) complex. J Biol Chem. 2012; 287:31447-31453.
115. Smith BA, Padrick SB, Doolittle LK, Daugherty-Clarke K, Correa IR Jr, Xu MQ, Goode BL, Rosen MK, Gelles J. Three-color single molecule imaging shows WASP detachment from Arp2/3 complex triggers actin filament branch formation. Elife. 2013; 2:e01008.
116. Yokotsuka M, Iwaya K, Saito T, Pandiella A, Tsuboi R, Kohno N, Matsubara O, Mukai K. Overexpression of HER2 signaling to WAVE2-Arp2/3 complex activates MMP-independent migration in breast cancer. Breast Cancer Res Treat. 2011; 126:311-318.
117. Otsubo T, Iwaya K, Mukai Y, Mizokami Y, Serizawa H, Matsuoka T, Mukai K. Involvement of Arp2/3 complex in the process of colorectal carcinogenesis. Mod Pathol. 2004; 17:461-467.
118. Semba S, Iwaya K, Matsubayashi J, Serizawa H, Kataba H, Hirano T, Kato H, Matsuoka T, Mukai K. Coexpression of actin-related protein 2 and Wiskott-Aldrich syndrome family verproline-homologous protein 2 in adenocarcinoma of the lung. Clin Cancer Res. 2006; 12:2449-2454.
119. Wang W, Wyckoff JB, Goswami S, Wang Y, Sidani M, Segall JE, Condeelis JS. Coordinated regulation of pathways for enhanced cell motility and chemotaxis is conserved in rat and mouse mammary tumors. Cancer Res. 2007; 67:3505-3511.
120. Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD, Hartman J, Sakowicz R, Pollard TD. Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature. 2009; 460:1031-1034.
121. Hetrick B, Han MS, Helgeson LA, Nolen BJ. Small molecules CK-666 and CK-869 inhibit actin-related protein 2/3 complex by blocking an activating conformational change. Chem Biol. 2013; 20:701-712.
122. Paul NR, Allen JL, Chapman A, Morlan-Mairal M, Zindy E, Jacquemet G, Fernandez del Ama L, Ferizovic N, Green DM, Howe JD, Ehler E, Hurlstone A, Caswell PT. alpha5beta1 integrin recycling promotes Arp2/3-independent cancer cell invasion via the formin FHOD3. J Cell Biol. 2015; 210:1013-1031.
123. Rotty JD, Wu C, Bear JE. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat Rev Mol Cell Biol. 2013; 14:7-12.
124. Beckham Y, Vasquez RJ, Stricker J, Sayegh K, Campillo C, Gardel ML. Arp2/3 inhibition induces amoeboid-like protrusions in MCF10A epithelial cells by reduced cytoskeletal-membrane coupling and focal adhesion assembly. PLoS One. 2014; 9:e100943.
125. Ilatovskaya DV, Chubinskiy-Nadezhdin V, Pavlov TS, Shuyskiy LS, Tomilin V, Palygin O, Staruschenko A, Negulyaev YA. Arp2/3 complex inhibitors adversely affect actin cytoskeleton remodeling in the cultured murine kidney collecting duct M-1 cells. Cell Tissue Res. 2013; 354:783-792.
126. Havrylenko S, Noguera P, Abou-Ghali M, Manzi J, Faqir F, Lamora A, Guerin C, Blanchoin L, Plastino J. WAVE binds Ena/VASP for enhanced Arp2/3 complex-based actin assembly. Mol Biol Cell. 2015; 26:55-65.
127. Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003; 19:541-564.
128. Reinhard M, Halbrugge M, Scheer U, Wiegand C, Jockusch BM, Walter U. The 46/50 kDa phosphoprotein VASP purified from human platelets is a novel protein associated with actin filaments and focal contacts. EMBO J. 1992; 11:2063-2070.
129. Rottner K, Behrendt B, Small JV, Wehland J. VASP dynamics during lamellipodia protrusion. Nat Cell Biol. 1999; 1:321-322.
130. Shekhar S, Pernier J, Carlier MF. Regulators of actin filament barbed ends at a glance. J Cell Sci. 2016; 129:1085-1091.
131. Barzik M, Kotova TI, Higgs HN, Hazelwood L, Hanein D, Gertler FB, Schafer DA. Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J Biol Chem. 2005; 280:28653-28662.
132. Di Modugno F, DeMonte L, Balsamo M, Bronzi G, Nicotra MR, Alessio M, Jager E, Condeelis JS, Santoni A, Natali PG, Nistico P. Molecular cloning of hMena (ENAH) and its splice variant hMena+11a: epidermal growth factor increases their expression and stimulates hMena+11a phosphorylation in breast cancer cell lines. Cancer Res. 2007; 67:2657-2665.
133. Di Modugno F, Bronzi G, Scanlan MJ, Del Bello D, Cascioli S, Venturo I, Botti C, Nicotra MR, Mottolese M, Natali PG, Santoni A, Jager E, Nistico P. Human Mena protein, a serex-defined antigen overexpressed in breast cancer eliciting both humoral and CD8+ T-cell immune response. Int J Cancer. 2004; 109:909-918.
134. Di Modugno F, Mottolese M, Di Benedetto A, Conidi A, Novelli F, Perracchio L, Venturo I, Botti C, Jager E, Santoni A, Natali PG, Nistico P. The cytoskeleton regulatory protein hMena (ENAH) is overexpressed in human benign breast lesions with high risk of transformation and human epidermal growth factor receptor-2-positive/hormonal receptor-negative tumors. Clin Cancer Res. 2006; 12:1470-1478.
135. Janke J, Schluter K, Jandrig B, Theile M, Kolble K, Arnold W, Grinstein E, Schwartz A, Estevez-Schwarz L, Schlag PM, Jockusch BM, Scherneck S. Suppression of tumorigenicity in breast cancer cells by the microfilament protein profilin 1. J Exp Med. 2000; 191:1675-1686.
136. Wu N, Zhang W, Yang Y, Liang YL, Wang LY, Jin JW, Cai XM, Zha XL. Profilin 1 obtained by proteomic analysis in all-trans retinoic acid-treated hepatocarcinoma cell lines is involved in inhibition of cell proliferation and migration. Proteomics. 2006; 6:6095-6106.
137. Gronborg M, Kristiansen TZ, Iwahori A, Chang R, Reddy R, Sato N, Molina H, Jensen ON, Hruban RH, Goggins MG, Maitra A, Pandey A. Biomarker discovery from pancreatic cancer secretome using a differential proteomic approach. Mol Cell Proteomics. 2006; 5:157-171.
138. Bae YH, Ding Z, Zou L, Wells A, Gertler F, Roy P. Loss of profilin-1 expression enhances breast cancer cell motility by Ena/VASP proteins. J Cell Physiol. 2009; 219:354-364.
139. Faix J, Grosse R. Staying in shape with formins. Dev Cell. 2006; 10:693-706.
140. Breitsprecher D, Goode BL. Formins at a glance. J Cell Sci. 2013; 126:1-7.
141. Higgs HN. Formin proteins: a domain-based approach. Trends Biochem Sci. 2005; 30:342-353.
142. Iskratsch T, Yu CH, Mathur A, Liu S, Stevenin V, Dwyer J, Hone J, Ehler E, Sheetz M. FHOD1 is needed for directed forces and adhesion maturation during cell spreading and migration. Dev Cell. 2013; 27:545-559.
143. Gardberg M, Kaipio K, Lehtinen L, Mikkonen P, Heuser VD, Talvinen K, Iljin K, Kampf C, Uhlen M, Grenman R, Koivisto M, Carpen O. FHOD1, a formin upregulated in epithelial-mesenchymal transition, participates in cancer cell migration and invasion. PLoS One. 2013; 8:e74923.
144. Serrano-Gomez SJ, Maziveyi M, Alahari SK. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer. 2016; 15:18.
145. Liu J, Wang Y, Goh WI, Goh H, Baird MA, Ruehland S, Teo S, Bate N, Critchley DR, Davidson MW, Kanchanawong P. Talin determines the nanoscale architecture of focal adhesions. Proc Natl Acad Sci U S A. 2015; 112:E4864-4873.
146. Hemmings L, Rees DJ, Ohanian V, Bolton SJ, Gilmore AP, Patel B, Priddle H, Trevithick JE, Hynes RO, Critchley DR. Talin contains three actin-binding sites each of which is adjacent to a vinculin-binding site. J Cell Sci. 1996; 109:2715-2726.
147. Chen HC, Appeddu PA, Parsons JT, Hildebrand JD, Schaller MD, Guan JL. Interaction of focal adhesion kinase with cytoskeletal protein talin. J Biol Chem. 1995; 270:16995-16999.
148. Bass MD, Patel B, Barsukov IG, Fillingham IJ, Mason R, Smith BJ, Bagshaw CR, Critchley DR. Further characterization of the interaction between the cytoskeletal proteins talin and vinculin. Biochem J. 2002; 362:761-768.
149. Horwitz A, Duggan K, Buck C, Beckerle MC, Burridge K. Interaction of plasma membrane fibronectin receptor with talin--a transmembrane linkage. Nature. 1986; 320:531-533.
150. Jiang G, Giannone G, Critchley DR, Fukumoto E, Sheetz MP. Two-piconewton slip bond between fibronectin and the cytoskeleton depends on talin. Nature. 2003; 424:334-337.
151. Calderwood DA, Ginsberg MH. Talin forges the links between integrins and actin. Nat Cell Biol. 2003; 5:694-697.
152. Calderwood DA. Integrin activation. J Cell Sci. 2004; 117:657-666.
153. Garcia-Alvarez B, de Pereda JM, Calderwood DA, Ulmer TS, Critchley D, Campbell ID, Ginsberg MH, Liddington RC. Structural determinants of integrin recognition by talin. Mol Cell. 2003; 11:49-58.
154. Calderwood DA, Zent R, Grant R, Rees DJ, Hynes RO, Ginsberg MH. The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J Biol Chem. 1999; 274:28071-28074.
155. Nayal A, Webb DJ, Horwitz AF. Talin: an emerging focal point of adhesion dynamics. Curr Opin Cell Biol. 2004; 16:94-98.
156. Albiges-Rizo C, Frachet P, Block MR. Down regulation of talin alters cell adhesion and the processing of the alpha 5 beta 1 integrin. J Cell Sci. 1995; 108:3317-3329.
157. Fang KP, Dai W, Ren YH, Xu YC, Zhang SM, Qian YB. Both Talin-1 and Talin-2 correlate with malignancy potential of the human hepatocellular carcinoma MHCC-97 L cell. BMC Cancer. 2016; 16:45.
158. Singel SM, Cornelius C, Batten K, Fasciani G, Wright WE, Lum L, Shay JW. A targeted RNAi screen of the breast cancer genome identifies KIF14 and TLN1 as genes that modulate docetaxel chemosensitivity in triple-negative breast cancer. Clin Cancer Res. 2013; 19:2061-2070.
159. Xu YF, Ren XY, Li YQ, He QM, Tang XR, Sun Y, Shao JY, Jia WH, Kang TB, Zeng MS, Liu N, Ma J. High expression of Talin-1 is associated with poor prognosis in patients with nasopharyngeal carcinoma. BMC Cancer. 2015; 15:332.
160. Zhang W, Mao YQ, Wang H, Yin WJ, Zhu SX, Wang WC. MiR-124 suppresses cell motility and adhesion by targeting talin 1 in prostate cancer cells. Cancer Cell Int. 2015; 15:49.
161. Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR, Huttenlocher A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat Cell Biol. 2004; 6:977-983.
162. Flevaris P, Stojanovic A, Gong H, Chishti A, Welch E, Du X. A molecular switch that controls cell spreading and retraction. J Cell Biol. 2007; 179:553-565.
163. Chan KT, Bennin DA, Huttenlocher A. Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK). J Biol Chem. 2010; 285:11418-11426.
164. Ren XD, Kiosses WB, Sieg DJ, Otey CA, Schlaepfer DD, Schwartz MA. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J Cell Sci. 2000; 113:3673-3678.
165. Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003; 112:453-465.
166. Horton ER, Byron A, Askari JA, Ng DH, Millon-Fremillon A, Robertson J, Koper EJ, Paul NR, Warwood S, Knight D, Humphries JD, Humphries MJ. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat Cell Biol. 2015; 17:1577-1587.
167. Eke I, Cordes N. Focal adhesion signaling and therapy resistance in cancer. Semin Cancer Biol. 2015; 31:65-75.
168. Fokkelman M, Balcioglu HE, Klip JE, Yan K, Verbeek FJ, Danen EH, van de Water B. Cellular adhesome screen identifies critical modulators of focal adhesion dynamics, cellular traction forces and cell migration behaviour. Sci Rep. 2016; 6:31707.
169. Miller SL, Antico G, Raghunath PN, Tomaszewski JE, Clevenger CV. Nek3 kinase regulates prolactin-mediated cytoskeletal reorganization and motility of breast cancer cells. Oncogene. 2007; 26:4668-4678.
170. Harrington KM, Clevenger CV. Identification of NEK3 Threonine 165 as a Novel Regulatory Phosphorylation Site that Modulates Focal Adhesion Remodeling Necessary for Breast Cancer Cell Migration. J Biol Chem. 2016; 291:21388-21406.
171. Graziano V, De Laurenzi V. Role of p63 in cancer development. Biochim Biophys Acta. 2011; 1816:57-66.
172. Chakrabarti R, Wei Y, Hwang J, Hang X, Andres Blanco M, Choudhury A, Tiede B, Romano RA, DeCoste C, Mercatali L, Ibrahim T, Amadori D, Kannan N, et al. DeltaNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat Cell Biol. 2014; 16:1004-1015, 1001-1013.
173. Nekulova M, Holcakova J, Gu X, Hrabal V, Galtsidis S, Orzol P, Liu Y, Logotheti S, Zoumpourlis V, Nylander K, Coates PJ, Vojtesek B. DeltaNp63alpha expression induces loss of cell adhesion in triple-negative breast cancer cells. BMC Cancer. 2016; 16:782.
174. Thepot A, Hautefeuille A, Cros MP, Abedi-Ardekani B, Petre A, Damour O, Krutovskikh V, Hainaut P. Intraepithelial p63-dependent expression of distinct components of cell adhesion complexes in normal esophageal mucosa and squamous cell carcinoma. Int J Cancer. 2010; 127:2051-2062.
175. Gari HH, Gearheart CM, Fosmire S, DeGala GD, Fan Z, Torkko KC, Edgerton SM, Lucia MS, Ray R, Thor AD, Porter CC, Lambert JR. Genome-wide functional genetic screen with the anticancer agent AMPI-109 identifies PRL-3 as an oncogenic driver in triple-negative breast cancers. Oncotarget. 2016; 7:15757-15771. doi: 10.18632/oncotarget.7462.
176. Gari HH, DeGala GD, Ray R, Lucia MS, Lambert JR. PRL-3 engages the focal adhesion pathway in triple-negative breast cancer cells to alter actin structure and substrate adhesion properties critical for cell migration and invasion. Cancer Lett. 2016; 380:505-512.
177. den Hollander P, Rawls K, Tsimelzon A, Shepherd J, Mazumdar A, Hill J, Fuqua SA, Chang JC, Osborne CK, Hilsenbeck SG, Mills GB, Brown PH. Phosphatase PTP4A3 Promotes Triple-Negative Breast Cancer Growth and Predicts Poor Patient Survival. Cancer Res. 2016; 76:1942-1953.
178. Mattheyses AL, Simon SM, Rappoport JZ. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J Cell Sci. 2010; 123:3621-3628.
179. Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods. 2006; 3:793-795.
180. Schutz GJ, Kada G, Pastushenko VP, Schindler H. Properties of lipid microdomains in a muscle cell membrane visualized by single molecule microscopy. EMBO J. 2000; 19:892-901.
181. Serge A. The Molecular Architecture of Cell Adhesion: Dynamic Remodeling Revealed by Videonanoscopy. Front Cell Dev Biol. 2016; 4:36.
182. Chenouard N, Smal I, de Chaumont F, Maska M, Sbalzarini IF, Gong Y, Cardinale J, Carthel C, Coraluppi S, Winter M, Cohen AR, Godinez WJ, Rohr K, et al. Objective comparison of particle tracking methods. Nat Methods. 2014; 11:281-289.
183. Godin AG, Lounis B, Cognet L. Super-resolution microscopy approaches for live cell imaging. Biophys J. 2014; 107:1777-1784.
184. Paszek MJ, DuFort CC, Rossier O, Bainer R, Mouw JK, Godula K, Hudak JE, Lakins JN, Wijekoon AC, Cassereau L, Rubashkin MG, Magbanua MJ, Thorn KS, et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature. 2014; 511:319-325.
185. Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006; 313:1642-1645.
186. Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, Waterman CM. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010; 468:580-584.
187. Klotzsch E, Stiegler J, Ben-Ishay E, Gaus K. Do mechanical forces contribute to nanoscale membrane organisation in T cells? Biochim Biophys Acta. 2015; 1853:822-829.
188. Haase K, Pelling AE. Investigating cell mechanics with atomic force microscopy. J R Soc Interface. 2015; 12:20140970.