
INTRODUCTION
Breast cancer affects women life drastically and nearly 1.7 million new cases worldwide are being identified every year since 2012 and it contributes more than 25% of the all kinds of newly identified cancer cases (http://www.cancer.org/) [1, 2]. Apart from a series of extrinsic factors promoting the occurrence, many genetic settings (intrinsic factors) drive breast cancer initiation and progression significantly. The activation of oncogenes and deactivation of tumor suppressor genes (TSGs) largely affect the maintenance and integrity of cells leading to tumorigenesis [3]. Although not all TSGs are vulnerable to mutations yet other genetic mechanisms indirectly interrupt their expressions and functions resulting in tumorigenesis [4]. In humans, several genes such as TP53, BRCA1, BRCA2, PTEN, ATM, p27, Skp2, RAD51, etc. are well known TSGs, which are involved in DNA repair and cellular mechanisms [5, 6]. TSGs are further classified into gatekeepers or caretakers based on their functions. Apart from tumor suppressors, a group of genes like PUM1, B2M, ACTB, RPL13A, LDHA, NONO, etc. are reported as housekeeping genes playing basic cellular functions (governing or preventing cell growth) and mutations in these genes promote cell proliferation [7]. In contrast, caretaker genes are mainly involved in the healthy maintenance of cells by encoding products, which stabilize the entire genome and protect genes from mutational events. Investigation of biological pathways affected by mutations of these genes will help us to understand the determinants of cancer initiation, progression, and other biological functions [8–10]. The advancement in the next generation sequencing and their allied computational techniques have paved way to identify large numbers of breast cancer gene mutations and their impacts [11]. In every cancer type, a set of significant gene mutations will strongly associate with tumorigenesis by being growth advantageous for the carcinogenic cells and those genes are known as driver genes [12]. In many breast cancer cases a significant numbers of somatic mutations as well as considerable number of germline mutations are found which are tumor enhancers and impose the risk of breast cancer tumorigenesis. Most of the driver mutations occur at somatic level, while a small number of mutations are passed on to lineages, which cause for 5 to 10% of all familial breast cancers types [13]. The most recurrently mutated published driver genes are AKT1, GATA3, PIK3CA, MAP3K1 and TP53 [14–17]. Apart from these genes, many other genes such as CBFB, RUNX1 are involved in somatic mutations in breast cancer. Deletion or translocation events in tumor suppressor genes such as AKT3 & MAGI3 genes lead to functional abnormalities and initiates breast tumorigenesis. Recent studies on breast cancer driver genes uncovered a list of genes such as CCND1, ERBB2, FGFR1, MYC, PIK3CA, PTEN, GATA3, MAP3K1, and RB1 etc., which are responsible for breast cancer [18, 19].
SIGNIFICANT DRIVER GENES ARE REAL MARKERS OF BREAST CANCER
Genetic mutations are rare and occur due to truncation, frame shift, insertions and deletions (indels), amplification and splicing abnormalities, etc. leading to loss or gain of functions. In breast cancer, over 30626 significant mutations are reported and many of them affect function of single gene or group of genes which leads to cancer progression. Its worthwhile to note that a specific genetic change cause for adverse effects i.e. neoplastic transformation. BRCA1 and BRCA2 genomic insertions, deletions or single nucleotide polymorphisms are also major founder mutational events and show high-risk in many breast cancer cases [20, 21]. While, a few gene mutations such as breast cancer gene 1 and 2 (BRCA1/2) instigate up to 25% breast cancer and also responsible for the highest number of mortalities [22, 23]. In addition to germline mutations, BRCA1/2, PTEN, CDH1, and STK11 gene mutations are associated with specific disorders such as Cowden syndrome, hereditary diffuse gastric cancer syndrome and Peutz-Jeghers syndrome (https://seer.cancer.gov/archive/csr/1975_2012/) [24–26]. The mutated tumor suppressor gene, PALB2 largely affects BRCA2, which increase the risk of BRCA1/2 based breast cancer [27–30]. Apart from aforementioned genes CDH1, STK11, PALB2, CHEK2, BRIP1, CDKN2A, CTNNB1, MLH1, MSH2, MSH6, NBN, RAD50, RAD51, TP53, etc. are having strong association with breast cancer. Frequent gene mutations resulting in variations in single nucleotide polymorphism (SNP), copy number variations, etc. exhibit significant impacts on tumor development, these kind of genes are called driver mutation genes (http://www.cancer.org/) [8, 31]. Among the known breast cancer genes, ATM gene abnormality causes the development of breast cancer RAD51C, and TP53 also play a strong role in the initiation and progression of breast cancer [32, 33]. The BRIP1 gene mutations lead to high risk of both breast and ovarian cancer, whereas MRE11A gene abnormality is linked to ataxia-telangiectasia along with cancer [34]. Mre11, Rad50, and Nbs1 form MRN complex, which facilitate DNA repair and also reported that NBN gene encoding Nbs1, has the strong association with breast cancer [35]. Somatic mutations and their role in breast cancer disposition have been revealed in earlier breast cancer related studies and it is also found that genes like ATM, PTEN, etc. play major role in several germline point mutations [36–38]. Along with point mutations, insertions, and deletions, a significant number of missense mutations occur in various genes, which raise the breast cancer susceptibility [39–41]. Among the aforementioned known breast cancer driver genes, a tumor suppressor (TSG), TP53, is the top-mutated gene, with nearly 100% risk of breast cancer [42, 43]. A germline mutation of TP53 also causes Li-Fraumili and Li Fraumeni-like syndromes that claim more than 40% of familial cancer [44, 45]. In addition, it also causes autosomal dominant disorders characterized by predisposition of several early inceptions of cancers, many of which are conveyed with homozygous mutant genotype with cancer relapse, and high probability of progressive and secondary cancer [46–48].
POSSIBLE DRIVER GENES MUTATIONS IN BREAST CANCER
Identification of cancer drivers is indeed the most challenging task in cancer research and many cancer drivers are predicted using several computational and statistical methods and validated with true expression levels in cancer [49]. The genetics home reference (https://ghr.nlm.nih.gov) published list of genes such as BARD1, BRCA1, BRCA2, CASP8, CHEK2, CTLA4, CYP19A1, FGFR2, H19, LSP1, MAP3K1, MRE11A, RAD51C, STK11, TERT, TOX3, XRCC2, GATA3, PIK3CA, AKT1, CDH1, RB1, TP53, PTEN and XRCC3, and suggested that these are the most susceptible genes involved in driver mutation and having a strong association with breast tumorigenesis [11]. The copy number variations (CNVs) and single nucleotide variations (SNVs) are major root of driver mutations in breast cancer [50]. Stephens et al. (2012) reported that the numbers of mutations in protein-coding genes are remarkably unique between individuals along with list of driver mutations responsible for tumorigenesis. Most frequently mutated genes in breast cancer are TP53, ERBB2, GATA3, FGFR1, CCND1 and PIK3CA [51]. A number of genes which are involved in breast tumorigenesis and act as potential drivers are ARID5B, CDH1, CTCF, HDAC9, KDM5B, NCOR2, SETD1A, SXL2, etc. Some of these genes encode proteins that control chromatin structure whereas other driver genes, such as ATR, and FANCA are mainly involved in DNA repair pathway. The recent whole genome sequencing (WGS) of 560 breast cancers samples identified 89 genes and 2433 breast cancer sequencing projects identified 40 breast cancer drivers genes from ER positive and ER negative breast cancer subtypes and many of these genes such as ARID1A, CTNND1, NUP107, CHD8, FANCI, CHD9, CTCF, KEAP1, PCDH18, LAMA2, HDAC9, ARFGEF1, MLLT4, FOXO3, CDKN2A, MAP3K1, GPS2, CTCF, CDH1, GATA3, AKT1, etc. have diversified functional change mutations [17, 52]. However, each published data on cancer drivers reported a distinct set of driver genes with few overlaps and so far, no standard approach is developed to identify and validate breast cancer driver genes [53].
CURRENT TRENDS IN SCREENING OF GENES INVOLVED IN BREAST CANCER DRIVER MUTATIONS
Driver gene mutations are necessary tool for the characterization of cancer phenotype, since they mainly affect gene expression followed by miscoding of amino acids, which provoke functional changes at protein level. However, passenger genes replicate many folds during DNA replication events without any extricating functional impacts [54, 55]. Voluminous methodologies have been employed to predict and identify breast cancer driver mutation genes, including computational identification, statistical testing, and, so on. Genetic mutational screening is one of the most widely used methods for the identification of mutations in germ cells based on looking at the family history of breast cancer [56, 57]. Driver mutation frequency is largely interrelated with breast cancer subtypes for example, TP53 mutation frequency is many folds higher in basal-like than other breast cancer subtypes [11]. Statistical analysis yield better results in driver genes identification, it also predicts high-frequency cancer driver genes using oncogenic tree model construction [58]. Thus, breast cancer driver prediction methodologies depend on key factors such as, the number of samples used for analysis, mutational patterns, frequency and function modifying mutations, etc. Several efficient tools exist to predict the mutation drivers, though each tool works using its own hypothesis/algorithms with diverse limitations. Accordingly, each driver mutation recognition protocol delivers distinctive results from one another. In this paper, we used several intensive computational driver gene identification approaches, tools, resources, etc. for the identification of most impressive driver mutation genes and their role in breast tumorigenesis.
COMPUTATIONAL APPROACHES FOR DISTINGUISHING BREAST CANCER DRIVER GENES MUTATION
Predicting breast cancer driver gene is a cumbersome task, as it generates a lot of false positive data and corroborating those results are most challenging. In this study, we used a dozen of computational driver gene identification approaches including online resources, offline and online tools to explore most potential breast cancer driver genes to avoid limitations of each approach. These include, the cBioportal (www:cbioportal.org/), The Cancer Genome Atlas (TCGA), International Cancer Genome Consortium (ICGC), 1000 Genomes, Catalogue of Somatic Mutations in Cancer (COSMIC), Human Cancer Database (http://db.cngb.org/cancer/), National Cancer Database (https://www.facs.org/quality-programs/cancer/ncdb), OASIS (http://oasis-genomics.org/) and many other useful cancer resources (Table 1). Apart from above-mentioned resources, Pan-Cancer (https://www.synapse.org/) developed by TCGA database is very efficient resource which provides analyzed cancer data, including mutation profiles, copy number variations, gene expression information, microRNA, etc. [59–62]. Included in Table 1 also several other methods that have been recently designed to find potential breast cancer driver genes by computational and statistical techniques, such as IntOGen, Driver DBV2, MutSigCV, etc.
Table 1: List of driver identification methods used to incorporates the prediction of breast cancer driver genes, their working principle and supporting references
Driver Identification Method |
Driver Gene Identification Principle |
Citations |
|---|---|---|
IntOGen |
Identifies alterations at transcriptomics level, CN gain and losses in tumor sample. It also integrates OncodriveFM for the identification of accumulation mutations, background mutation rate and OncodriveCLUST for mutation cluster identifications. Further, SIFT, Polyphen and Mutation Assessor are used to predict the impact of mutations. |
|
SIFT |
Amino acids substitutions and their deleterious impacts prediction. It find the homologous sequences using PSI-BLAST followed by picking sequences with specific diversity and calculating the SIFT scores. |
[66] |
PolyPhen-2 |
Analyzes non-synonymous SNP using multiple sequence alignment and structure information followed by predicting the probabilistic damaging variants with confidence prediction and at last interpret the results with mutational impact. |
[67] |
Mutation Assessor |
Predicts mutational impact by calculating functional impact score derived from addition of conservation score and specificity score. |
[68] |
Driver DBv2 |
Uses large exome and RNAseq datasets to predict the driver genes using several incorporated tools. |
[69] |
Active Driver |
It identifies significant mutations of cancer genes in active sites of proteins such as mutations in signaling proteins or domains or regulatory elements. It uses gene-centric logistic regression model including multiple factors to estimate mutation significance. |
[70] |
Dendrix |
This algorithm discovers driver genes with high coverage and high specificity using mutation data. |
|
MDPFinder |
It combines mutation and expression data to validate the driver genes and their mutated pathways. |
[73] |
Simon |
It identifies functional mutation impact on proteins, variations in background mutation frequency and genetic code redundancy among tumors. |
[74] |
NetBox |
It identifies the driver genes by comparing genes and performing network analysis on human interaction Network (HIN) data. |
[75] |
MutSigCV |
It uses overall mutation rates and distribution patterns and analyzes background mutation rates with patient specific as well as gene specific mutation rates. Finally it includes expression levels and replication periods. |
[76] |
MEMo |
It identifies the driver genes based on recurrently mutated genes among tumor data with consistent mutational specificity. |
[77] |
e-Driver |
It manipulates internal distribution of somatic functional missense mutations amongst functional domains by relating mutation rates with other regions of same protein. |
[78] |
DawnRank |
Uses gene expression data to construct gene network and rank them based on impact and it analyzes somatic alteration data to identify personalized driver alterations. |
[79] |
DriverNet |
Driver genes are identified based on genomic aberration states of various patients, genes, gene expression data and it further takes biological pathway data into account and builds the network driver genes. |
[80] |
MSEA |
It predicts cancer driver genes based on patterns of mutation hotspot. |
[81] |
iPAC |
Identifies non-random somatic mutations in protein using tertiary protein structure information. |
[82] |
CoMDP |
It uses mutation data to identify driver genes and their pathways. It also predicts genes with other multiple co-occurring biologically significant pathways. |
[83] |
Mutation modeling and evaluating number of deleterious mutations in breast cancer are also employed to predict potential driver genes and massive statistical testing is carried out to predict the prompt driver genes and their functional domains [84]. Computational modeling, gene pathway and network analysis are other feasible techniques proposed to identify most probable driver genes [49]. DrGaP is a tool that predicts driver genes and their signaling pathway using statistical analysis [76]. Apart from aforesaid tools, many viable techniques, tools, and databases provide significant driver gene mutations, and mutational significance of genes involved in single/multiple cellular pathways, etc. The OASIS web portal is also one of comprehensive resource providing tons of information about somatic mutation, gene expression, copy number alteration, etc. from normal, tumor cases, and cell lines (http://oasis-genomics.org/) [77]. This web portal, fetch primary genetic and metabolic pathway analyses data from Pan-Cancer project, COSMIC, BIOCARTA (http://www.biocarta.com), KEGG (http://www.genome.jp/kegg/pathway.html), etc. [78].
Apart from abovementioned resources for driver gene identification, we have also validated the genetic interactions through various approaches. To test the capability of identifying the driver genes in genetic interaction level, we constructed FunCoup (functional Coupling) database to explore the functional relationship between genes and their functions [79]. Genetic network is most significant method to derive genetic as well as functionally associated genes using Genemania web server and it predicts gene functions by integrating several functionally associated networks [81]. The consequent level of network analysis is performed using MUFFINN (MUtations For Functional Impact on Network Neighbors). MUFFINN is one of the efficient programs for identifying most common driver genes by mutation frequency and most linked pathway neighbors [82, 83]. MUFFINN uses a pathway-centric approach and it also identifies the top 1000, 500, 100 interacting gene clusters along with network constructed using HumanNet and String (Search Tool for the Retrieval of Interacting Genes/Proteins) Database [85, 86]. To further validate the identified driver genes and their genetic interaction network construction we used FunRich (Functional Enrichment) program. FunRich analyzes genes and their interacting partners based on comprehensive information obtained from various renowned databases with strong annotations [87]. SIFT algorithm is used for functional impact and validation of identified driver genes. SIFT is one of the most powerful algorithms used to identify and evaluate detrimental effects of genetic variations in driver genes and their impacts at protein level. PolyPhen2 is another potential tool, which predicts the probabilities of amino acid substitutions and its collective impacts on structural and functional tendency [88, 89]. Thus, every driver gene prediction approach has some representative strength to identify the real cancer driver genes and this ends with the major concerns. For example, a frequency-based approach always needs large number of samples to possibly identify the rarely mutated cancer drivers [90].
Nevertheless, through this study, we established that driver gene identification is purely based on mutations in key genes, which are really driven, by functional mutations. Every Driver DB associated algorithms are working in a distinctive way and it yields various outputs. For example, algorithms such as Active Driver focus on phosphorylation and kinase domain site. Similarly Dendrix, MDP Finder, Oncodrive-FM and MutSigCV predict based on mutational specificity, high impact mutational accumulation and patient-specific mutations respectively. Hence, Driver DB associated tools provide comprehensive ways of predicting drivers based on several criteria such as recurring mutations, accumulation of mutation with high functional impact, mutual exclusivity and the spectrum of mutation, gene expression data, background mutation rate, etc. resulting in 956 breast cancer drivers identified from various breast cancer subtypes (detailed list of driver genes is given in Supplementary Table 1). The driver genes are filtered out and further shortlisted based on more than one Driver DB associated tools, which report the genes with hotspots of mutation, missense mutation, etc. From the initial filtering 452 genes were obtained and further redundant genes were removed leaving 195 driver genes, which are chosen for further analysis (Figure 1). The ICGC database (https://icgc.org/icgc) is used to fetch the detailed mutations data including chromosomal location, type of mutation, codon alterations, and amino acid variations and cancer subtypes of identified driver genes are retrieved, analyzed and tabulated in Supplementary Table 2. Similarly, IntOGen integrate results with various mutation-calling protocol such as OncodriveFM & OncodriveCLUST, and it identifies genes responsible for functional mutations and mutational impacts at protein level.
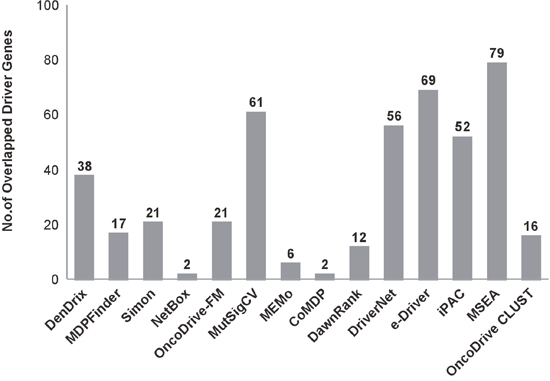
Figure 1: Total number of breast cancer driver genes identified using various computational methods.
In this study, a list of top candidate genes were identified through our approaches, by incorporating a selective list of efficient driver gene prediction tools and resources which were proved earlier with other type of cancer gene prediction. We used the TCGA and ICGC breast cancer data to identify frequency and type of mutations, and we found a number of new genes such as FLG, DNAH14, NBPF12, RYR2, ARHGAP35, OBSCN, CLTC, etc. are highly mutated in breast cancer, along with some well-known driver genes, like TP53, PIK3CA, MLL3, PTEN, GATA3, ARID1A. We further categorized the identified breast cancer driver genes into four major types based on the mutation percentage of each driver used for this study (Figure 2A). The analysis further extended to find the mutations frequency of breast cancer genes among the nine BRCA projects available in cBioPortal (www.cbioportal.org). We identified driver genes (TP53 gene 36.11% followed by PIK3CA 27.78%, MLL3 15.78%, TTN, FLG, DNAH14, GATA3, ERBB2, RYR2, HRNR, NBPF12, RUNX1, NOTCH2, OBSCN) and overall average mutation rate is calculated and given in Figure 2B. Investigation is further continued to validate candidate driver genes and their mutation profiles in breast invasive carcinoma samples obtained from TCGA-Pan-Cancer data resource (https://www.synapse.org). Genes were chosen for further analysis based on the most frequent functional mutations such as splice site mutations, missense mutations, frame shift insertion and deletions mutations, In-frame insertion and deletions, etc. Result of this analysis identified 61,466 functionally significant gene mutations and all genes are further screened for most potential driver genes discovery. Moreover, the analysis extended to confirm the identified drivers genes and their mutational impact at functional level by using PolyPhen-2 programs for predicting the high-impact deleterious mutations along with high IntOGen variant impact score. To validate the most probable breast cancer driver genes, the results of IntOGen prediction, COSMIC, CBioPortal breast cancer data, OASIS data portal were used. As a result of intensive filtering and analysis, 63 driver genes were short-listed. The TCGA breast cancer projects were used to calculate the average mutations of individual high-confidence driver genes by chromosome-wise (Figure 3) and individual project wise (Supplementary Figure 1). Nevertheless, a significant percentage and type of mutations diversity (Missense, truncation, amplification, etc.) are found among the breast cancer projects, due to heterogeneity, individual gene mutations, and patient specific clinical factors, etc.
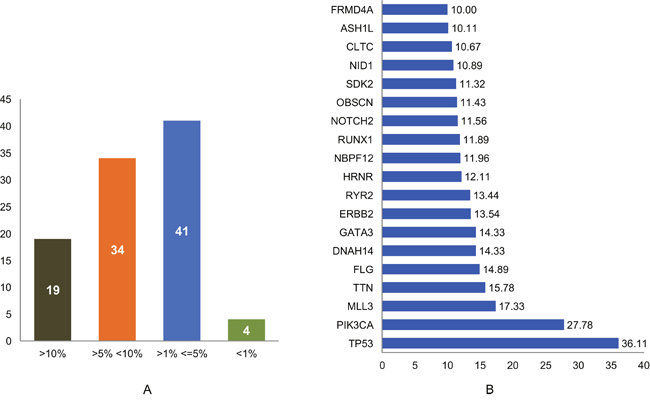
Figure 2: (A) Identified driver genes classified based on their Mutation percentage; (B) High percentage of mutations (>10%) are observed in the identified 63 breast cancer driver genes through the analysis of 9 breast cancer patients data analysis using cBioPortal.
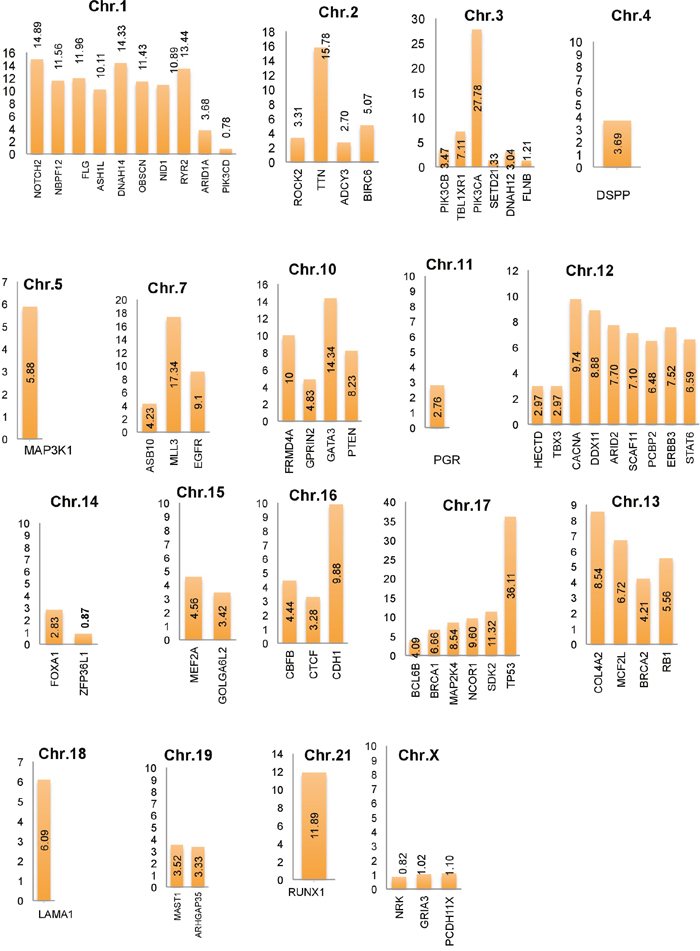
Figure 3: Average breast cancer gene mutations identified using cBioPortal projects (4162 breast cancer samples) along with identified top candidate driver genes and their respective chromosomes locations.
FUNCTIONAL EFFECTS OF BREAST CANCER DRIVERS IN TUMORIGENESIS
Several driver gene prediction tools exist to evaluate potential driver genes based on their functional mutations and impact and also use transcriptomics data to reveal potential driver genes at the protein level [91]. Although numerous computational techniques identify and classify the driver genes based on the mutations and functional impact, yet in vitro and in vivo assays are necessary for further validation. In breast cancer, many known genes are considered to be an effective driver genes including BRCA1/2, TP53, PIK3CA, GATA3, etc., which govern the most cancer pathways. Besides identified and known driver genes, many novel genetic elements are actively involved in breast cancer metabolic pathway. In this study, we have identified and propose numerous novel breast cancer driver genes, which are validated using various computational techniques. From the list of identified driver genes, titin (TTN) gene is one of the important genes with an average of 15.78% mutation rate in breast cancer while earlier studies also revealed that TTN is highly mutated in other cancers [92, 93]. Similarly, filaggrin (FLG) gene is a highly mutated driver gene, which had an average mutation of 14.89%. FLG gene mutations are found in several other cancer types such as non-melanoma skin cancer, head and neck cancer, lung cancer, colorectal cancer, uterine cancer, prostate cancer, etc. [94]. Hence, the FLG gene may also have a strong association with multiple carcinomas. The obscurin (OBSCN) gene is identified as one of most frequent driver genes in all our analysis, and an average mutation rate is 11.44%. OBSCN gene is vastly mutated in various cancer types and this gene mutation leads to giant obscurins protein loss followed by high susceptibility of breast epithelial cells to DNA damaging elements [92, 95]. Earlier studies revealed that OBSCN gene stimulated survival of breast epithelial cell and prevented cell apoptosis [96]. Hence, OBSCN gene is one of the potential breast cancer drivers and also has a strong association with other cancer types.
In addition to aforementioned genes, few driver genes may act as tumor suppressor, oncogenes, gatekeepers, and caretakers, etc. AT-Rich Interaction Domain 2 (ARID2) as a tumor suppressor gene is frequently mutated driver gene identified in all our analysis (Mutation average is 7.7%). ARID2, as a variant gene of SWI/SNF complex, mutation has strong associations with huge number of cancers especially in hepatocellular carcinoma, gastric cancers and breast cancer [97–99]. Rho-Associated Coiled-Coil Containing Protein Kinase 2 (ROCK2) is another important driver gene identified in this analysis, although its overall mutation frequencies are comparatively low (3.32%). Previous research on ROCK2 gene and its relevance to breast cancer are proven and a critical amino acid mutation (T431N) is identified as the high-risk factor in breast cancer metastasis. In addition to the above-mentioned functions of identified high-confidence driver genes (excluding published driver genes), we performed an intensive literature search to corroborate and strengthen our approach. The data including genes involved in various cancers, functions, and pathways along with supporting citations are tabulated (Table 2).
Table 2: Identified top candidate breast cancer driver genes (other than known driver genes) and their functional backgrounds
Identified Driver Genes |
Cancer Type |
Related pathway |
Known Functions |
References |
|---|---|---|---|---|
ADCY3 |
Gastric cancer |
cAMP/PKA/CREB pathway |
Increased cell migration, invasion, and proliferation, which are characteristic of cancer. |
|
ARHGAP35 |
Osteosarcoma, Breast cancer, |
Regulation of RhoA activity and focal adhesion and migration |
Human glucocorticoid receptor DNA binding factor |
|
ARID2 |
Hepatocellular carcinoma/melanoma |
Chromatin Remodeling |
Activating ligand dependent transcription by nuclear receptor |
|
ASB10 |
Glioblastoma multiform, Ovarian Cancer |
Cytokine signaling |
Ubiquitination and Ubiquitin protein ligase binding |
[106] |
ASH1L |
Liver cancer; |
Tight junction and lysine degradation |
Chromatin regulator; Site specific lysine methylation on histone and other proteins |
|
BCL6B |
Breast cancer; |
P53, MAPK and cancer related pathways |
Nucleic acid binding |
|
BIRC6 |
Breast cancer; |
Apoptosis and Autophagy |
miRNA dependent apoptosis induction |
[112] |
CACNA1C |
Breast cancer, Gastric, colorectal, pancreatic, leukemia, brain, skin, prostate cancer |
Circadian entrainment and NFAT and Cardiac Hypertrophy |
High alteration in Ca2+ ion it accelerates cell proliferation, migration and up-regulation in breast cancer |
|
COL4A2 |
Cardiovascular disease and intracerebral hemorrhage, glaucoma, etc. |
Interleukin-3, 5 and GM-CSF signaling and Pathways in cancer. |
Regulation of angiogenesis and tumor growth |
|
DDX11 |
Breast cancer, Fanconi Anemia |
Golgi and subsequent modification and unfolded protein response |
Genome stability |
[118] |
DNAH12 |
Prostate cancer |
Respiratory electron transport, ATP synthesis chemiosmotic coupling and uncoupling protein for heat production |
ATP binding and |
[119] |
DNAH14 |
Ovarian cancer |
Respiratory electron transport, ATP synthesis chemiosmotic coupling and uncoupling protein for heat production |
ATP binding and |
[120] |
DSPP |
Oral squamous cell carcinomas; |
ECM proteoglycan and degradation of the extracellular matrix organization |
Vital factor in dentinogenesis; |
|
FLG |
Nonmelanoma cancer, head and neck, colorectal, breast, ovarian, prostate cancer |
AhR pathways |
Calcium ion binding |
[94] |
FLNB |
Breast Cancer; Ovarian cancer; Colorectal cancer |
MMP-9 and ERK pathway |
RAS induced tumor growth |
|
FRMD4A |
Gastric cancer; |
- |
Protein Binding |
|
GOLGA6L2 |
Breast cancer; |
- |
Protein coding |
[127] |
GPRIN2 |
Rett Syndrome; |
- |
Neurite outgrowth |
|
GRIA3 |
Pancreatic Cancer; Breast cancer |
glutamate receptor signaling pathway |
excitatory synaptic transmission |
|
HECTD4 |
Esophageal, non-small-cell lung and head and neck cancer |
Protein modification and Ubiquitination |
Ubiquitin-protein transferase activity |
[131] |
LAMA1 |
Breast cancer |
Cancer and Integrin pathway |
Receptor binding |
|
MAST1 |
Breast Cancer |
- |
Ion/ATP/protein binding |
[133] |
MCF2L |
Breast cancer |
Rho/Rac signaling and p75 NTR-receptor-mediated signaling pathways |
Rho-guanyl-nucleotide exchange factor activity |
[134] |
MEF2A |
Breast cancer |
P38 MAPK signaling |
Neuronal differentiation and survival |
|
NBPF12 |
Neuroblastoma; small cell lung cancer neurogenetic diseases |
- |
CHEA Transcription factor binding site |
|
NID1 |
Gastrointestinal cancer |
Non-integrin membrane-ECM interactions and Degradation of the extracellular matrix |
Act as cross-linker with other extracellular matrix |
|
NRK |
Breast cancer |
TNF-alpha-induced signaling pathway |
Receptor signaling protein serine/threonine kinase activity and ATP binding |
[142] |
OBSCN |
Highly mutated in various cancers including breast cancer |
RhoA signaling |
Structural and regulatory functions |
|
PCBP2 |
Hepatocellular cancer; Familial breast cancer; lymphocytic leukemia, colorectal cancer |
RIG-I/MDA5 mediated induction of IFN-alpha/beta pathway and mRNA splicing pathways |
Transcriptional role |
[144] |
PCDH11X |
Esophageal carcinoma, breast cancer, Prostate cancer |
- |
Cell adhesion |
|
PGR |
Breast and Ovarian cancer |
oestrogen-mediated pathways |
Tumor repressing mechanism |
|
PIK3CB |
Oral-squamous cell carcinoma, breast cancer and other wide range of cancer |
Involved in AKT, PTEN and PIK3CA pathways |
Cell cycle growth regulation |
[149] |
PIK3CD |
Breast, Ovarian and colon cancers |
PIK signaling |
Transcription binding factor |
|
ROCK2 |
Breast, lung, ovarian, intestinal cancer |
RhoA signaling |
Actin cytoskeleton organization, Adhesion, migration, Proliferation and apoptosis. |
|
RYR2 |
Breast Cancer, Lung Cancer, Bladder cancer |
cAMP-dependent PKA activation |
Calcium ion binding, Calcium/calmodulin binding |
|
SCAF11 |
Lung adenocarcinoma, various cancers |
Apoptosis |
Protein/zinc ion/poly(A)RNA binding |
[158] |
SDK2 |
Non-small cell lung cancer |
- |
Adhesion, Promotes synaptic connectivity |
|
STAT6 |
Breast cancer, Lung cancer |
Integrin, Interleukin-3,5 and GM-CSF signaling pathway |
IL-4 mediate cell growth regulator, inhibit |
[125, 182, 183] |
TTN |
Colorectal, testis, gastric, breast, ovarian, renal cancers |
Platelet activation, Signaling and aggregation pathway |
Chromosome condensation and segregation |
Through the OASIS web portal, METABRIC, and BRCA-TCGA data were used for the identification and analysis of mutation profiles of 63 top driver genes. The details of mutational profiles with gene classifications of 63 top candidate breast cancer genes are given in Table 3. Among these drivers, we found 13 tumor suppressor genes (TSGs), ten oncogenes (OG), six gatekeepers and one gene had both OG and TSG features. More copy number loss mutations are observed in (>7%) genes such as CDH1, CBFB, CTCF, BCL6B, MAP2K4, TP53, NCOR1, PGR, RB1, and BRCA2 genes. The large proportion of driver mutations occurred in protein coding exonic as well as in intergenic regions and these regions are considered as most significant genetic fragments and the actual insights of those regions are functionally significant. Subsequently most of the intergenic nucleotide bases are the regulator of adjacent genes and still many intergenic regions and functional roles remain uncertain. Thus, intergenic regions might be responsible for genetic variations that cause tumorigenesis and further insight on those intergenic regions of sequences may enlighten driver genes transforming mutations with a good understanding of tumorigenesis process [162]. In breast cancer, driver mutations also emphasize the functional impact at the protein level. Many somatic driver mutations observed in breast cancers are tumor dependent and may vary from tumor to tumor. In order to increase the reliability of prediction, six different approaches such as Cancer Genome Consortium prediction, MuSiC, OncodriveFM, OncodriveCLUST, Active Driver, MutSig were used to ensure the confidence level of potential breast cancer drivers.
Table 3: Mutation profiles of identified top candidate BRCA driver genes
BRCA Drivers |
Substitution % |
InDel % |
Amplification % |
Copy Gain % |
Copy Loss % |
Deletion (%) |
Expression Outliers High % |
|---|---|---|---|---|---|---|---|
CDH1* |
3.2 |
0.1 |
- |
1.4 |
16.5 |
2.9 |
- |
CBFB^ |
1.7 |
- |
- |
1 |
16.4 |
1.3 |
- |
CTCF |
1.9 |
- |
0.1 |
1.1 |
16 |
2.9 |
|
MAP2K4* |
2.4 |
0.4 |
0.2 |
0.4 |
12.2 |
2.5 |
- |
BCL6B |
0.1 |
- |
0.1 |
0.4 |
12.2 |
1.4 |
- |
TP53* |
6.8 |
0.4 |
- |
0.6 |
12 |
1.5 |
- |
NCOR1+ |
3 |
0.3 |
0.2 |
0.8 |
11.3 |
1.5 |
- |
PGR |
0.5 |
- |
0.2 |
2 |
10.8 |
2.8 |
51.4 |
RB1* |
1.3 |
- |
- |
0.8 |
9.3 |
1.8 |
- |
BRCA2* |
1.3 |
- |
0.2 |
1.8 |
7.4 |
1.2 |
66.5 |
MCF2L |
0.5 |
- |
0.4 |
4 |
6.7 |
2.5 |
- |
COL4A2 |
0.5 |
- |
0.3 |
4.1 |
6.5 |
2 |
- |
GOLGA6L2 |
0.1 |
- |
0.3 |
2.2 |
6.2 |
1.7 |
- |
ARID1A* |
1.4 |
0.1 |
0.1 |
0.3 |
5.6 |
0.8 |
- |
SETD2* |
1 |
- |
- |
0.8 |
5.1 |
0.5 |
- |
PIK3CD |
0.4 |
- |
0.2 |
0.8 |
5.1 |
1.2 |
1.2 |
DNAH12 |
1 |
0.1 |
0.1 |
0.6 |
4.9 |
0.5 |
15.8 |
FLNB |
1.2 |
0.3 |
0.1 |
0.8 |
4.8 |
0.6 |
- |
ZFP36L1 |
0.5 |
0.1 |
0.1 |
1.4 |
4.7 |
1 |
- |
LAMA1 |
1 |
- |
0.1 |
2.7 |
4.3 |
1 |
- |
BRCA1* |
1 |
- |
0.2 |
4.7 |
4.2 |
0.1 |
52.2 |
MAP3K1 |
3.5 |
0.1 |
- |
2.2 |
4.1 |
0.8 |
- |
PTEN* |
1.7 |
0.4 |
- |
0.7 |
3.9 |
0.9 |
- |
ASB10 |
0.3 |
- |
0.2 |
3.9 |
2.7 |
0.8 |
- |
NRK+ |
0.9 |
- |
0.1 |
1 |
2.5 |
0.3 |
- |
DSPP |
1.2 |
0.3 |
0.2 |
1.1 |
2.3 |
0.4 |
- |
MEF2A |
0.1 |
- |
1.2 |
3.8 |
2.3 |
0.3 |
- |
PCDH11X |
0.8 |
- |
0.9 |
2.2 |
0.3 |
- |
|
CACNA1C |
1.3 |
- |
0.9 |
5.1 |
2.1 |
0.3 |
- |
GRIA3 |
1.2 |
0.1 |
0.1 |
1.1 |
2.1 |
0.2 |
- |
TTN |
13.7 |
- |
0.1 |
2 |
2 |
- |
- |
FOXA1 |
1.3 |
- |
0.8 |
4.7 |
2 |
0.4 |
84.2 |
TBX3 |
0.9 |
- |
- |
1.6 |
1.9 |
0.2 |
- |
NOTCH2^ |
0.8 |
- |
0.8 |
4 |
1.8 |
0.2 |
- |
ARHGAP35^ |
0.8 |
- |
0.1 |
1.9 |
1.8 |
0.1 |
- |
HECTD4 |
1.2 |
- |
0.1 |
1.6 |
1.5 |
0.2 |
- |
MAST1 |
1 |
- |
0.2 |
3.4 |
1.4 |
0.1 |
22 |
RUNX1^ |
1.2 |
0.1 |
0.2 |
3.5 |
1.4 |
0.1 |
- |
ADCY3+ |
0.3 |
- |
0.1 |
1.9 |
1.4 |
0.1 |
- |
ROCK2 |
0.5 |
- |
0.3 |
1.9 |
1.2 |
0.3 |
- |
PCBP2+ |
0.3 |
- |
0.1 |
2.2 |
1.1 |
0.1 |
- |
ARID2* |
0.5 |
- |
0.2 |
2.4 |
1 |
0.3 |
- |
SDK2 |
1 |
- |
1.4 |
11.6 |
1 |
0.4 |
- |
OBSCN* |
1.7 |
- |
1.4 |
32.9 |
1 |
0.1 |
- |
SCAF11 |
0.4 |
- |
0.2 |
2.4 |
1 |
0.2 |
- |
STAT6 |
0.1 |
- |
0.2 |
2.3 |
0.9 |
- |
- |
GATA3* |
0.9 |
2.4 |
0.9 |
7.3 |
0.8 |
0.2 |
81.1 |
NID1 |
1 |
0.1 |
1.6 |
33.3 |
0.8 |
0.2 |
- |
ERBB3^ |
1.3 |
- |
0.1 |
2.4 |
0.8 |
0.1 |
- |
DDX11+ |
0.4 |
- |
0.8 |
4.4 |
0.8 |
0.3 |
- |
FRMD4A* |
0.5 |
- |
0.8 |
6.5 |
0.8 |
0.1 |
- |
PIK3CB |
0.6 |
- |
0.3 |
4.8 |
0.8 |
- |
- |
BIRC6 |
1.3 |
- |
0.1 |
2.4 |
0.7 |
0.1 |
- |
EGFR^ |
0.6 |
- |
0.8 |
4.5 |
0.7 |
0.3 |
- |
RYR2 |
3.9 |
- |
3.5 |
33.1 |
0.6 |
0.3 |
5.9 |
DNAH14 |
0.5 |
0.1 |
1.3 |
33.7 |
0.4 |
0.1 |
79.8 |
TBL1XR1 |
0.5 |
- |
0.7 |
8.2 |
0.4 |
- |
- |
PIK3CA^ |
32.1 |
0.6 |
0.9 |
8 |
0.3 |
- |
0.1 |
GPRIN2 |
0.1 |
0.3 |
6.4 |
2.9 |
0.3 |
- |
- |
NBPF12 |
0.3 |
- |
4.5 |
45 |
0.2 |
0.1 |
- |
ASH1L |
1 |
0.1 |
1.4 |
31.2 |
0.1 |
- |
- |
MLL3* |
0.8 |
- |
0.8 |
4 |
1.8 |
0.2 |
- |
FLG |
4.4 |
- |
2 |
30.9 |
- |
0.1 |
- |
Identified driver genes are categorized with Tumor suppressor (* with bold caption); Oncogene (^ with bold caption); Gatekeeper (+ with bold caption).
GENE INTERACTION NETWORK ANALYSIS: IDENTIFYING DIRECT AND INDIRECT INTERACTING PARTNERS OF BREAST CANCER DRIVERS
The genetic network analysis is performed to explore more direct and indirect partners of breast cancer driver genes using FunCoup (Functional Coupling) package [85]. The FunCoup analysis is used to construct focused gene networks for driver genes and indirect genetic partners for further validation of hub (driver) genes. We constructed gene network using Genemania web server and found genetically as well as functionally associated genes among the identified driver genes to illustrate the close relationship among selected driver genes. Identified driver genes and their interaction network is constructed in Genemania server by combining several interaction network groups (n=572) obtained from various studies such as co-expression (180), co-localization (10), genetic (199), pathway related (43), physical (75), predicted interactions (9), and genes sharing protein domains (56), eventually these combined information provide more insight on molecular, functional and pathway level interaction among genes and it sorted all network groups based on Genemania score, false discovery rate (FDR) for further construction and validation of gene network (Figure 4) [85, 86]. The subsequent network analysis is further extended to cross-validate resulting driver genes using two more methods, MUFFINN and FUNRICH, which are commonly used methods to identify common driver genes by mutation frequency and most linked pathway neighbors in functional networks [66, 67]. We used top 100 neighbor genes from golden-standard databases used in MUFFINN and their mutation occurrences, and refine them for further network construction. The 63 driver genes are consistently identified through all approaches. Thus the data further confirm that these genes are most commonly mutated genes and their most damaging missense mutations flaunting highly deleterious functional impacts.
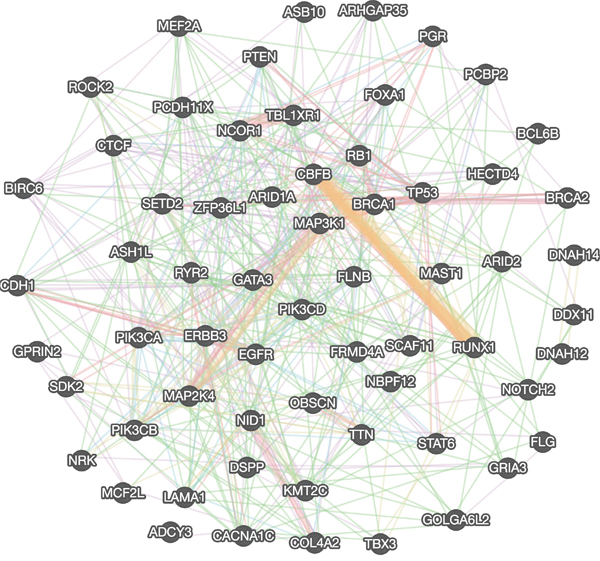
Figure 4: Genetic interaction network of identified top candidate breast cancer driver genes.
DISCUSSION AND FUTURE ASPECTS
In this study, using a combination of various methodologies, we have analyzed overall 41,948 significant mutations: including 26,448 missense mutations, 1,935 frameshift mutations (InDels), 832 in splice site mutations, as well as 115 and 563 in-frame insertion and in-frame deletion mutations, respectively. As a result of these analyses, we have top listed 63 driver genes, which have a strong correlation with breast cancer subtypes: Luminal A (28.06%), Luminal B (22.01%), basal (19.86%), Her2 (15.82%) and normal (14.23%) breast cancer types. Genes with functionally damaging mutations come after their worst impacts are taken as top candidate (63genes) driver genes. Our data indicate that 24 genes overlap with previously published well-known breast cancer driver genes, whereas the remaining 39 genes that are either not previously highlighted or reported as potential breast cancer drivers (Figure 5). Although recent studies on driver gene identification have developed a vast array of algorithms and resources, yet individual groups follow their own protocols with specific limitations.
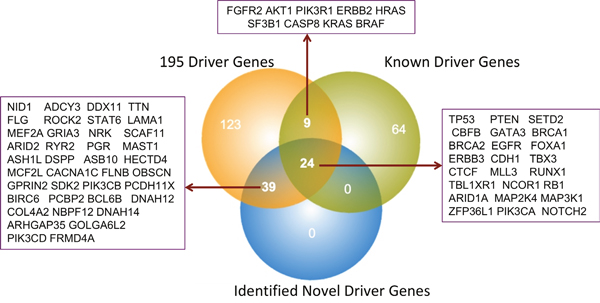
Figure 5: Overall comparisons between published and identified BRCA driver genes.
Hence, identifying the most potential driver genes are still challenging and also requires the integration of all the results from various tools for comprehensive evaluation. Most of the prior studies on driver gene identification mainly focused to find the driver gene by integrating several computational approaches for filtering out driver genes and their pathways related information relevant to breast cancer. Nevertheless, they failed to validate the identified driver genes with mutation analysis and their impact at transcriptome level. In addition, use of breast cancer patient mutation, mRNAseq expression, and methylation data for the further validation is also lacking in previous studies. In order to provide comprehensive information on breast cancer driver genes we used TCGA-Pan-Cancer breast cancer normal and patient clinical samples, COSMIC mutation data, and methylation as well mRNAseq expression data in the combination with other methodologies (Table 1). This comprehensive information helped us to avoid false positive genes come up during analysis.
For some well-known genes, many functional studies have been carried out. For example, others and we have extensively performed functional studies for p53 and BRCA1 [30, 163–166]. However the final evidence for the majority of other genes, especially for the 39 genes that have not been heighted before as cancer driver requires functional study at various levels, both in vitro and in vivo, such as gene knockout, knockin, gene overexpression, protein-protein interaction, protein modification, activation and inactivation, and etc. The validation of the mutations affecting regulatory network can be especially changeling. In this case, gene knockout or overexpression may be followed by RNA-sequencing, proteomics and/or epigenetic modifications to uncover alternations of downstream signaling pathways. These studies are vital to perceive the underlying mechanism related to functions of these genes and they will also allow researchers to better understand the tumor heterogeneity, cancer signaling pathway, genetic and epigenetic modifications.
In addition, all the data we have discussed are obtained from sequencing DNA isolated from bulk of each cancer. It is known that genetic instability within individual cancer could generate intratumoral heterogeneity, and that epigenetic modifications may further increase the heterogeneity. These events could significantly affect many aspects of tumorigenesis, including clonal expansion, metastasis, recurrence, drug resistance, and switch off cancer driver during the course of cancer progression. Thus, the use of bulk DNA for sequencing could certainly overshadow the intratumoral heterogeneity. This weakness can be overcome by sequencing DNA isolated from single cancer cells as illustrated by some recent studies [167–170]. Our future efforts will be delivered to analyze the data obtained from the sequencing of single cells, hence, facilitating the discovery of additional therapeutic druggable targets at single cell level for cancer therapies at a personalized fashion.
ACKNOWLEDGMENTS
We thank members of the Prof. Deng Research laboratory for critical discussions. This work is supported, in part by FDCT 094/2015/A3 for CXD, the Chair Professor Grant (CPG) and Startup Research Grant (SRG) granted to CXD by University of Macau, Macau SAR, China.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest.
REFERENCES
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136:E359–86.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7–30.
3. Xu X, Qiao W, Linke SP, Cao L, Li WM, Furth PA, Harris CC, Deng CX. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat Genet. 2001; 28:266–71.
4. Osborne C, Wilson P, Tripathy D. Oncogenes and tumor suppressor genes in breast cancer: potential diagnostic and therapeutic applications. Oncologist. 2004; 9:361–77.
5. Burke W, Petersen G, Lynch P, Botkin J, Daly M, Garber J, Kahn MJ, McTiernan A, Offit K, Thomson E, Varricchio C. Cancer Genetics Studies Consortium. Recommendations for follow-up care of individuals with an inherited predisposition to cancer. I. Hereditary nonpolyposis colon cancer. JAMA. 1997; 277:915–19.
6. Kerangueven F, Essioux L, Dib A, Noguchi T, Allione F, Geneix J, Longy M, Lidereau R, Eisinger F, Pébusque MJ, Jacquemeir J, Bonaiti-Pellie C, Sobol H, Birnbaum D. Loss of heterozygosity and linkage analysis in breast carcinoma: indication for a putative third susceptibility gene on the short arm of chromosome 8. Oncogene. 1995; 10:1023–26.
7. Kılıç Y, Çelebiler AC, Sakızlı M. Selecting housekeeping genes as references for the normalization of quantitative PCR data in breast cancer. Clin Transl Oncol. 2014; 16:184–90.
8. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013; 339:1546–58.
9. Kalari S, Pfeifer GP. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv Genet. 2010; 70:277–308.
10. Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C, Widaa S, Hinton J, Fahey C, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010; 463:893–98.
11. Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70.
12. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009; 458:719–24.
13. Turnbull C, Rahman N. Genetic predisposition to breast cancer: past, present, and future. Annu Rev Genomics Hum Genet. 2008; 9:321–45.
14. Takaku M, Grimm SA, Wade PA. Gata3 in breast cancer: tumor suppressor or oncogene? Gene Expr. 2015; 16:163–68.
15. Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007; 318:1108–13.
16. Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, Moorhead M, Chaudhuri S, Tomsho LP, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010; 466:869–73.
17. Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, Van Loo P, Ju YS, Smid M, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016; 534:47–54.
18. Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006; 314:268–74.
19. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012; 486:405–09.
20. Petrij-Bosch A, Peelen T, van Vliet M, van Eijk R, Olmer R, Drüsedau M, Hogervorst FB, Hageman S, Arts PJ, Ligtenberg MJ, Meijers-Heijboer H, Klijn JG, Vasen HF, et al. BRCA1 genomic deletions are major founder mutations in Dutch breast cancer patients. Nat Genet. 1997; 17:341–45.
21. Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res. 2006; 8:R38.
22. Gabai-Kapara E, Lahad A, Kaufman B, Friedman E, Segev S, Renbaum P, Beeri R, Gal M, Grinshpun-Cohen J, Djemal K, Mandell JB, Lee MK, Beller U, et al. Population-based screening for breast and ovarian cancer risk due to BRCA1 and BRCA2. Proc Natl Acad Sci USA. 2014; 111:14205–10.
23. Campeau PM, Foulkes WD, Tischkowitz MD. Hereditary breast cancer: new genetic developments, new therapeutic avenues. Hum Genet. 2008; 124:31–42.
24. Nelen MR, Padberg GW, Peeters EA, Lin AY, van den Helm B, Frants RR, Coulon V, Goldstein AM, van Reen MM, Easton DF, Eeles RA, Hodgsen S, Mulvihill JJ, et al. Localization of the gene for Cowden disease to chromosome 10q22-23. Nat Genet. 1996; 13:114–16.
25. Hemminki A, Tomlinson I, Markie D, Järvinen H, Sistonen P, Björkqvist AM, Knuutila S, Salovaara R, Bodmer W, Shibata D, de la Chapelle A, Aaltonen LA. Localization of a susceptibility locus for Peutz-Jeghers syndrome to 19p using comparative genomic hybridization and targeted linkage analysis. Nat Genet. 1997; 15:87–90.
26. Pharoah PD, Guilford P, Caldas C. International Gastric Cancer Linkage Consortium. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology. 2001; 121:1348–53.
27. Easton DF, Ford D, Bishop DT. Breast Cancer Linkage Consortium. Breast and ovarian cancer incidence in BRCA1-mutation carriers. Am J Hum Genet. 1995; 56:265–71.
28. Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, Weber B, Lenoir G, Chang-Claude J, Sobol H, Teare MD, Struewing J, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am J Hum Genet. 1998; 62:676–89.
29. Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, Manoukian S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003; 72:1117–30.
30. Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007; 25:1329–33.
31. Ashworth A, Lord CJ, Reis-Filho JS. Genetic interactions in cancer progression and treatment. Cell. 2011; 145:30–38.
32. Cremona CA, Behrens A. ATM signalling and cancer. Oncogene. 2014; 33:3351–60.
33. Masson AL, Talseth-Palmer BA, Evans TJ, Grice DM, Hannan GN, Scott RJ. Expanding the genetic basis of copy number variation in familial breast cancer. Hered Cancer Clin Pract. 2014; 12:15.
34. Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006; 38:1239–41.
35. Buslov KG, Iyevleva AG, Chekmariova EV, Suspitsin EN, Togo AV, Kuligina ES, Sokolenko AP, Matsko DE, Turkevich EA, Lazareva YR, Chagunava OL, Bit-Sava EM, Semiglazov VF, et al. NBS1 657del5 mutation may contribute only to a limited fraction of breast cancer cases in Russia. Int J Cancer. 2005; 114:585–89.
36. Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell. 2007; 11:103–05.
37. Nusbaum R, Vogel KJ, Ready K. Susceptibility to breast cancer: hereditary syndromes and low penetrance genes. Breast Dis. 2006-2007; 27:21–50.
38. Rouleau E, Jesson B, Briaux A, Nogues C, Chabaud V, Demange L, Sokolowska J, Coulet F, Barouk-Simonet E, Bignon YJ, Bonnet F, Bourdon V, Bronner M, et al. Rare germline large rearrangements in the BRCA1/2 genes and eight candidate genes in 472 patients with breast cancer predisposition. Breast Cancer Res Treat. 2012; 133:1179–90.
39. Sommer SS, Jiang Z, Feng J, Buzin CH, Zheng J, Longmate J, Jung M, Moulds J, Dritschilo A. ATM missense mutations are frequent in patients with breast cancer. Cancer Genet Cytogenet. 2003; 145:115–20.
40. Katagiri T, Kasumi F, Yoshimoto M, Nomizu T, Asaishi K, Abe R, Tsuchiya A, Sugano M, Takai S, Yoneda M, Fukutomi T, Nanba K, Makita M, et al. High proportion of missense mutations of the BRCA1 and BRCA2 genes in Japanese breast cancer families. J Hum Genet. 1998; 43:42–48.
41. Vallon-Christersson J, Cayanan C, Haraldsson K, Loman N, Bergthorsson JT, Brøndum-Nielsen K, Gerdes AM, Møller P, Kristoffersson U, Olsson H, Borg A, Monteiro AN. Functional analysis of BRCA1 C-terminal missense mutations identified in breast and ovarian cancer families. Hum Mol Genet. 2001; 10:353–60.
42. Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, Friend SH. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990; 250:1233–38.
43. Gasco M, Shami S, Crook T. The p53 pathway in breast cancer. Breast Cancer Res. 2002; 4:70–76.
44. Giacomazzi J, Selistre SG, Rossi C, Alemar B, Santos-Silva P, Pereira FS, Netto CB, Cossio SL, Roth DE, Brunetto AL, Zagonel-Oliveira M, Martel-Planche G, Goldim JR, et al. Li-Fraumeni and Li-Fraumeni-like syndrome among children diagnosed with pediatric cancer in Southern Brazil. Cancer. 2013; 119:4341–49.
45. Kouidou S, Malousi A, Maglaveras N. Li-Fraumeni and Li-Fraumeni-like syndrome mutations in p53 are associated with exonic methylation and splicing regulatory elements. Mol Carcinog. 2009; 48:895–902.
46. Borges LM, Ayres FM. R337H mutation of the TP53 gene as a clinical marker in cancer patients: a systematic review of literature. Genet Mol Res. 2015; 14:17034–43.
47. Yang X, Hu Z, Wu J, Liu G, Di G, Chen C, Hou Y, Huang X, Liu Z, Shen Z, Shao Z. [Germline mutations of TP53 gene among Chinese families with high risk for breast cancer] [Article in Chinese]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2015; 32:761–65.
48. Macedo GS, Araujo Vieira I, Brandalize AP, Giacomazzi J, Inez Palmero E, Volc S, Rodrigues Paixão-Côrtes V, Caleffi M, Silva Alves M, Achatz MI, Hainaut P, Ashton-Prolla P. Rare germline variant (rs78378222) in the TP53 3′ UTR: evidence for a new mechanism of cancer predisposition in Li-Fraumeni syndrome. Cancer Genet. 2016; 209:97–106.
49. Torkamani A, Schork NJ. Identification of rare cancer driver mutations by network reconstruction. Genome Res. 2009; 19:1570–78.
50. Shlien A, Malkin D. Copy number variations and cancer. Genome Med. 2009; 1:62.
51. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012; 486:400–04.
52. Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, Tsui DW, Liu B, Dawson SJ, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016; 7:11479.
53. Tokheim CJ, Papadopoulos N, Kinzler KW, Vogelstein B, Karchin R. Evaluating the evaluation of cancer driver genes. Proc Natl Acad Sci USA. 2016; 113:14330–35.
54. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, Vastrik I, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007; 446:153–58.
55. Haber DA, Settleman J. Cancer: drivers and passengers. Nature. 2007; 446:145–46.
56. De Carvalho DD, Sharma S, You JS, Su SF, Taberlay PC, Kelly TK, Yang X, Liang G, Jones PA. DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell. 2012; 21:655–67.
57. Liu Y, Hu Z. Identification of collaborative driver pathways in breast cancer. BMC Genomics. 2014; 15:605.
58. Szabo A, Boucher K. Estimating an oncogenetic tree when false negatives and positives are present. Math Biosci. 2002; 176:219–36.
59. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1.
60. Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, Kok CY, Jia M, Ewing R, Menzies A, Teague JW, Stratton MR, Futreal PA. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010; 38:D652–57.
61. Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA, and 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010; 467:1061–73.
62. Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Tamborero D, Schroeder MP, Jene-Sanz A, Santos A, Lopez-Bigas N. IntOGen-mutations identifies cancer drivers across tumor types. Nat Methods. 2013; 10:1081–82.
63. Cheng WC, Chung IF, Chen CY, Sun HJ, Fen JJ, Tang WC, Chang TY, Wong TT, Wang HW. DriverDB: an exome sequencing database for cancer driver gene identification. Nucleic Acids Res. 2014; 42:D1048–54.
64. Chen Y, Hao J, Jiang W, He T, Zhang X, Jiang T, Jiang R. Identifying potential cancer driver genes by genomic data integration. Sci Rep. 2013; 3:3538.
65. Tamborero D, Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Kandoth C, Reimand J, Lawrence MS, Getz G, Bader GD, Ding L, Lopez-Bigas N. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci Rep. 2013; 3:2650.
66. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012; 40:W452-7.
67. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010; 7:248–49.
68. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011; 39:e118.
69. Chung IF, Chen CY, Su SC, Li CY, Wu KJ, Wang HW, Cheng WC. DriverDBv2: a database for human cancer driver gene research. Nucleic Acids Res. 2016; 44:D975–79.
70. Reimand J, Bader GD. Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol Syst Biol. 2013; 9:637.
71. Vandin F, Upfal E, Raphael BJ. De novo discovery of mutated driver pathways in cancer. Genome Res. 2012; 22:375–85.
72. Leiserson MD, Blokh D, Sharan R, Raphael BJ. Simultaneous identification of multiple driver pathways in cancer. PLOS Comput Biol. 2013; 9:e1003054.
73. Zhao J, Zhang S, Wu LY, Zhang XS. Efficient methods for identifying mutated driver pathways in cancer. Bioinformatics. 2012; 28:2940–47.
74. Youn A, Simon R. Identifying cancer driver genes in tumor genome sequencing studies. Bioinformatics. 2011; 27:175–81.
75. Cerami E, Demir E, Schultz N, Taylor BS, Sander C. Automated network analysis identifies core pathways in glioblastoma. PLoS One. 2010; 5:e8918.
76. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499:214–18.
77. Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012; 22:398–406.
78. Porta-Pardo E, Godzik A. e-Driver: a novel method to identify protein regions driving cancer. Bioinformatics. 2014; 30:3109–14.
79. Hou JP, Ma J. DawnRank: discovering personalized driver genes in cancer. Genome Med. 2014; 6:56.
80. Bashashati A, Haffari G, Ding J, Ha G, Lui K, Rosner J, Huntsman DG, Caldas C, Aparicio SA, Shah SP. DriverNet: uncovering the impact of somatic driver mutations on transcriptional networks in cancer. Genome Biol. 2012; 13:R124.
81. Jia P, Wang Q, Chen Q, Hutchinson KE, Pao W, Zhao Z. MSEA: detection and quantification of mutation hotspots through mutation set enrichment analysis. Genome Biol. 2014; 15:489.
82. Ryslik GA, Cheng Y, Cheung KH, Modis Y, Zhao H. Utilizing protein structure to identify non-random somatic mutations. BMC Bioinformatics. 2013; 14:190.
83. Zhang J, Wu LY, Zhang XS, Zhang S. Discovery of co-occurring driver pathways in cancer. BMC Bioinformatics. 2014; 15:271.
84. Greenman C, Wooster R, Futreal PA, Stratton MR, Easton DF. Statistical analysis of pathogenicity of somatic mutations in cancer. Genetics. 2006; 173:2187–98.
85. Schmitt T, Ogris C, Sonnhammer EL. FunCoup 3.0: database of genome-wide functional coupling networks. Nucleic Acids Res. 2014; 42:D380–88.
86. Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, Maitland A, Mostafavi S, Montojo J, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010; 38:W214-20.
87. Benito-Martin A, Peinado H. FunRich proteomics software analysis, let the fun begin! Proteomics. 2015; 15:2555–56.
88. Carter H, Chen S, Isik L, Tyekucheva S, Velculescu VE, Kinzler KW, Vogelstein B, Karchin R. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 2009; 69:6660–67.
89. Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016; 11:1–9.
90. Pon JR, Marra MA. Driver and passenger mutations in cancer. Annu Rev Pathol. 2015; 10:25–50.
91. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012; 486:395–99.
92. Tan H, Bao J, Zhou X. Genome-wide mutational spectra analysis reveals significant cancer-specific heterogeneity. Sci Rep. 2015; 5:12566.
93. Azim HA Jr, Nguyen B, Brohée S, Zoppoli G, Sotiriou C. Genomic aberrations in young and elderly breast cancer patients. BMC Med. 2015; 13:266.
94. Skaaby T, Husemoen LL, Thyssen JP, Meldgaard M, Thuesen BH, Pisinger C, Jørgensen T, Carlsen K, Johansen JD, Menné T, Szecsi PB, Stender S, Linneberg A. Filaggrin loss-of-function mutations and incident cancer: a population-based study. Br J Dermatol. 2014; 171:1407–14.
95. Shriver M, Stroka KM, Vitolo MI, Martin S, Huso DL, Konstantopoulos K, Kontrogianni-Konstantopoulos A. Loss of giant obscurins from breast epithelium promotes epithelial-to-mesenchymal transition, tumorigenicity and metastasis. Oncogene. 2015; 34:4248–59.
96. Perry NA, Shriver M, Mameza MG, Grabias B, Balzer E, Kontrogianni-Konstantopoulos A. Loss of giant obscurins promotes breast epithelial cell survival through apoptotic resistance. FASEB J. 2012; 26:2764–75.
97. Cornen S, Guille A, Adélaïde J, Addou-Klouche L, Finetti P, Saade MR, Manai M, Carbuccia N, Bekhouche I, Letessier A, Raynaud S, Charafe-Jauffret E, Jacquemier J, et al. Candidate luminal B breast cancer genes identified by genome, gene expression and DNA methylation profiling. PLoS One. 2014; 9:e81843.
98. Zhao H, Wang J, Han Y, Huang Z, Ying J, Bi X, Zhao J, Fang Y, Zhou H, Zhou J, Li Z, Zhang Y, Yang X, et al. ARID2: a new tumor suppressor gene in hepatocellular carcinoma. Oncotarget. 2011; 2:886–91. doi: 10.18632/oncotarget.355.
99. Aso T, Uozaki H, Morita S, Kumagai A, Watanabe M. Loss of arid1a, arid1b, and arid2 expression during progression of gastric cancer. Anticancer Res. 2015; 35:6819–27.
100. Hong SH, Goh SH, Lee SJ, Hwang JA, Lee J, Choi IJ, Seo H, Park JH, Suzuki H, Yamamoto E, Kim IH, Jeong JS, Ju MH, et al. Upregulation of adenylate cyclase 3 (ADCY3) increases the tumorigenic potential of cells by activating the CREB pathway. Oncotarget. 2013; 4:1791–803. doi: 10.18632/oncotarget.1324.
101. Son HS, Shin YM, Park KK, Seo KW, Yoon KY, Jang HK, Lee SH, Yang SI, Kim JH. Correlation between her2 overexpression and clinicopathological characteristics in gastric cancer patients who have undergone curative resection. J Gastric Cancer. 2014; 14:180–86.
102. Ouyang Q, Tian C, Gao J, Huang J, Fu H, He J, Yang J. HER2-positive double primary tumor of gastric and breast cancer occur synchronously in a patient: A case report. Mol Clin Oncol. 2016; 4:719–22.
103. Zhao, J, Xu H, He M, Wang Z, Wu Y. Rho gtpase-activating protein 35 rs1052667 polymorphism and osteosarcoma risk and prognosis. Biomed Res Int. 2014; 2014:396947.
104. Wolf RM, Draghi N, Liang X, Dai C, Uhrbom L, Eklöf C, Westermark B, Holland EC, Resh MD. p190RhoGAP can act to inhibit PDGF-induced gliomas in mice: a putative tumor suppressor encoded on human chromosome 19q13.3. Genes Dev. 2003; 17:476–87.
105. Manceau G, Letouzé E, Guichard C, Didelot A, Cazes A, Corté H, Fabre E, Pallier K, Imbeaud S, Le Pimpec-Barthes F, Zucman-Rossi J, Laurent-Puig P, Blons H. Recurrent inactivating mutations of ARID2 in non-small cell lung carcinoma. Int J Cancer. 2013; 132:2217–21.
106. Dunn GP, Cheung HW, Agarwalla PK, Thomas S, Zektser Y, Karst AM, Boehm JS, Weir BA, Berlin AM, Zou L, Getz G, Liu JF, Hirsch M, et al. In vivo multiplexed interrogation of amplified genes identifies GAB2 as an ovarian cancer oncogene. Proc Natl Acad Sci USA. 2014; 111:1102–07.
107. Fujimoto A, Furuta M, Totoki Y, Tsunoda T, Kato M, Shiraishi Y, Tanaka H, Taniguchi H, Kawakami Y, Ueno M, Gotoh K, Ariizumi S, Wardell CP, et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016; 48:500–09.
108. Zhu L, Li Q, Wong SH, Huang M, Klein BJ, Shen J, Ikenouye L, Onishi M, Schneidawind D, Buechele C, Hansen L, Duque-Afonso J, Zhu F, et al. Ash1l links histone h3 lysine 36 dimethylation to mll leukemia. Cancer Discov. 2016; 6:770–83.
109. Liu L, Kimball S, Liu H, Holowatyj A, Yang ZQ. Genetic alterations of histone lysine methyltransferases and their significance in breast cancer. Oncotarget. 2015; 6:2466–82. doi: 10.18632/oncotarget.2967.
110. Zhong BL, Bian LJ, Wang GM, Zhou YF, Chen YY, Peng F. Identification of key genes involved in HER2-positive breast cancer. Eur Rev Med Pharmacol Sci. 2016; 20:664–72.
111. Xu L, Li X, Chu ES, Zhao G, Go MY, Tao Q, Jin H, Zeng Z, Sung JJ, Yu J. Epigenetic inactivation of BCL6B, a novel functional tumour suppressor for gastric cancer, is associated with poor survival. Gut. 2012; 61:977–85.
112. Crippa E, Folini M, Pennati M, Zaffaroni N, Pierotti MA, Gariboldi M. miR-342 overexpression results in a synthetic lethal phenotype in BRCA1-mutant HCC1937 breast cancer cells. Oncotarget. 2016; 7:18594–604. doi: 10.18632/oncotarget.7617.
113. Wang CY, Lai MD, Phan NN, Sun Z, Lin YC. Meta-analysis of public microarray datasets reveals voltage-gated calcium gene signatures in clinical cancer patients. PLoS One. 2015; 10:e0125766.
114. Yu W, Wang P, Ma H, Zhang G, Yulin Z, Lu B, Wang H, Dong M. Suppression of T-type Ca2+ channels inhibited human laryngeal squamous cell carcinoma cell proliferation running title: roles of T-type Ca2+ channels in LSCC cell proliferation. Clin Lab. 2014; 60:621–28.
115. Buchanan PJ, McCloskey KD. CaV channels and cancer: canonical functions indicate benefits of repurposed drugs as cancer therapeutics. Eur Biophys J. 2016; 45:621–33.
116. Petitclerc E, Boutaud A, Prestayko A, Xu J, Sado Y, Ninomiya Y, Sarras MP Jr, Hudson BG, Brooks PC. New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J Biol Chem. 2000; 275:8051–61.
117. Kuo DS, Labelle-Dumais C, Gould DB. COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum Mol Genet. 2012; 21:R97–110.
118. Wu Y, Shin-ya K, Brosh RM Jr. FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol. 2008; 28:4116–28.
119. Tao S, Wang Z, Feng J, Hsu FC, Jin G, Kim ST, Zhang Z, Gronberg H, Zheng LS, Isaacs WB, Xu J, Sun J. A genome-wide search for loci interacting with known prostate cancer risk-associated genetic variants. Carcinogenesis. 2012; 33:598–603.
120. Braun R, Finney R, Yan C, Chen QR, Hu Y, Edmonson M, Meerzaman D, Buetow K. Discovery analysis of TCGA data reveals association between germline genotype and survival in ovarian cancer patients. PLoS One. 2013; 8:e55037.
121. Joshi R, Tawfik A, Edeh N, McCloud V, Looney S, Lewis J, Hsu S, Ogbureke KU. Dentin sialophosphoprotein (DSPP) gene-silencing inhibits key tumorigenic activities in human oral cancer cell line, OSC2. PLoS One. 2010; 5:e13974.
122. Jain A, McKnight DA, Fisher LW, Humphreys EB, Mangold LA, Partin AW, Fedarko NS. Small integrin-binding proteins as serum markers for prostate cancer detection. Clin Cancer Res. 2009; 15:5199–207.
123. Bandaru S, Zhou AX, Rouhi P, Zhang Y, Bergo MO, Cao Y, Akyürek LM. Targeting filamin B induces tumor growth and metastasis via enhanced activity of matrix metalloproteinase-9 and secretion of VEGF-A. Oncogenesis. 2014; 3:e119.
124. Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, Gertler FB. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011; 7:e1002218.
125. Fan Y, Li D, Qian J, Liu Y, Feng H, Li D. Increased expression of FERM domain-containing 4A protein is closely associated with the development of rectal cancer. Exp Ther Med. 2016; 11:421–26.
126. Moleirinho S, Tilston-Lunel A, Angus L, Gunn-Moore F, Reynolds PA. The expanding family of FERM proteins. Biochem J. 2013; 452:183–93.
127. Darcy DG, Chiaroni-Clarke R, Murphy JM, Honeyman JN, Bhanot U, LaQuaglia MP, Simon SM. The genomic landscape of fibrolamellar hepatocellular carcinoma: whole genome sequencing of ten patients. Oncotarget. 2015; 6:755–70. doi: 10.18632/oncotarget.2712.
128. Artuso R, Papa FT, Grillo E, Mucciolo M, Yasui DH, Dunaway KW, Disciglio V, Mencarelli MA, Pollazzon M, Zappella M, Hayek G, Mari F, Renieri A, et al. Investigation of modifier genes within copy number variations in Rett syndrome. J Hum Genet. 2011; 56:508–15.
129. Ripka S, Riedel J, Neesse A, Griesmann H, Buchholz M, Ellenrieder V, Moeller F, Barth P, Gress TM, Michl P. Glutamate receptor GRIA3—target of CUX1 and mediator of tumor progression in pancreatic cancer. Neoplasia. 2010; 12:659–67.
130. Hamed M, Spaniol C, Zapp A, Helms V. Integrative network-based approach identifies key genetic elements in breast invasive carcinoma. BMC Genomics. 2015; 16:S2.
131. Cha S, Park AY, Kang C. A genome-wide association study uncovers a genetic locus associated with thoracic-to-hip ratio in koreans. PLoS One. 2015; 10:e0145220.
132. Piccinni SA, Bolcato-Bellemin AL, Klein A, Yang VW, Kedinger M, Simon-Assmann P, Lefebvre O. Kruppel-like factors regulate the Lama1 gene encoding the laminin alpha1 chain. J Biol Chem. 2004; 279:9103–14.
133. Robinson DR, Kalyana-Sundaram S, Wu YM, Shankar S, Cao X, Ateeq B, Asangani IA, Iyer M, Maher CA, Grasso CS, Lonigro RJ, Quist M, Siddiqui J, et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat Med. 2011; 17:1646–51.
134. Melchor L, Saucedo-Cuevas LP, Muñoz-Repeto I, Rodríguez-Pinilla SM, Honrado E, Campoverde A, Palacios J, Nathanson KL, García MJ, Benítez J. Comprehensive characterization of the DNA amplification at 13q34 in human breast cancer reveals TFDP1 and CUL4A as likely candidate target genes. Breast Cancer Res. 2009; 11:R86.
135. Clocchiatti A, Di Giorgio E, Ingrao S, Meyer-Almes FJ, Tripodo C, Brancolini C. Class IIa HDACs repressive activities on MEF2-depedent transcription are associated with poor prognosis of ER+ breast tumors. FASEB J. 2013; 27:942–54.
136. Zhao M, New L, Kravchenko VV, Kato Y, Gram H, di Padova F, Olson EN, Ulevitch RJ, Han J. Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol. 1999; 19:21–30.
137. Vandepoele K, Van Roy N, Staes K, Speleman F, van Roy F. A novel gene family NBPF: intricate structure generated by gene duplications during primate evolution. Mol Biol Evol. 2005; 22:2265–74.
138. Petroziello J, Yamane A, Westendorf L, Thompson M, McDonagh C, Cerveny C, Law CL, Wahl A, Carter P. Suppression subtractive hybridization and expression profiling identifies a unique set of genes overexpressed in non-small-cell lung cancer. Oncogene. 2004; 23:7734–45.
139. Ulazzi L, Sabbioni S, Miotto E, Veronese A, Angusti A, Gafà R, Manfredini S, Farinati F, Sasaki T, Lanza G, Negrini M. Nidogen 1 and 2 gene promoters are aberrantly methylated in human gastrointestinal cancer. Mol Cancer. 2007; 6:17.
140. Martino-Echarri E, Fernández-Rodríguez R, Rodríguez-Baena FJ, Barrientos-Durán A, Torres-Collado AX, Plaza-Calonge MC, Amador-Cubero S, Cortés J, Reynolds LE, Hodivala-Dilke KM, Rodríguez-Manzaneque JC. Contribution of ADAMTS1 as a tumor suppressor gene in human breast carcinoma. Linking its tumor inhibitory properties to its proteolytic activity on nidogen-1 and nidogen-2. Int J Cancer. 2013; 133:2315–24.
141. Pujuguet P, Simian M, Liaw J, Timpl R, Werb Z, Bissell MJ. Nidogen-1 regulates laminin-1-dependent mammary-specific gene expression. J Cell Sci. 2000; 113:849–58.
142. Yanagawa T, Denda K, Inatani T, Fukushima T, Tanaka T, Kumaki N, Inagaki Y, Komada M. Deficiency of x-linked protein kinase nrk during pregnancy triggers breast tumor in mice. Am J Pathol. 2016; 186:2751–60.
143. Ford-Speelman DL, Roche JA, Bowman AL, Bloch RJ. The rho-guanine nucleotide exchange factor domain of obscurin activates rhoA signaling in skeletal muscle. Mol Biol Cell. 2009; 20:3905–17.
144. Braconi C, Valeri N, Kogure T, Gasparini P, Huang N, Nuovo GJ, Terracciano L, Croce CM, Patel T. Expression and functional role of a transcribed noncoding RNA with an ultraconserved element in hepatocellular carcinoma. Proc Natl Acad Sci USA. 2011; 108:786–91.
145. Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol. 2009; 1:a003129.
146. Chen MW, Vacherot F, De La Taille A, Gil-Diez-De-Medina S, Shen R, Friedman RA, Burchardt M, Chopin DK, Buttyan R. The emergence of protocadherin-PC expression during the acquisition of apoptosis-resistance by prostate cancer cells. Oncogene. 2002; 21:7861–71.
147. Lancaster JM, Berchuck A, Carney ME, Wiseman R, Taylor JA. Progesterone receptor gene polymorphism and risk for breast and ovarian cancer. Br J Cancer. 1998; 78:277.
148. Smith RA, Lea RA, Curran JE, Weinstein SR, Griffiths LR. Expression of glucocorticoid and progesterone nuclear receptor genes in archival breast cancer tissue. Breast Cancer Res. 2003; 5:R9–12.
149. Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011; 13:224.
150. Kok K, Geering B, Vanhaesebroeck B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem Sci. 2009; 34:115–27.
151. Kok K, Nock GE, Verrall EA, Mitchell MP, Hommes DW, Peppelenbosch MP, Vanhaesebroeck B. Regulation of p110delta PI 3-kinase gene expression. PLoS One. 2009; 4:e5145.
152. Cizkova M, Vacher S, Meseure D, Trassard M, Susini A, Mlcuchova D, Callens C, Rouleau E, Spyratos F, Lidereau R, Bièche I. PIK3R1 underexpression is an independent prognostic marker in breast cancer. BMC Cancer. 2013; 13:545.
153. Morgan-Fisher M, Wewer UM, Yoneda A. Regulation of ROCK activity in cancer. J Histochem Cytochem. 2013; 61:185–98.
154. Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR. The catalogue of somatic mutations in cancer (cosmic). Curr Protoc Hum Genet. 2008; Chapter 10:Unit 10.11.
155. Lochhead PA, Wickman G, Mezna M, Olson MF. Activating ROCK1 somatic mutations in human cancer. Oncogene. 2010; 29:2591–98.
156. Van den Eynden J, Fierro AC, Verbeke LP, Marchal K. SomInaClust: detection of cancer genes based on somatic mutation patterns of inactivation and clustering. BMC Bioinformatics. 2015; 16:125.
157. Cazier JB, Rao SR, McLean CM, Walker AK, Wright BJ, Jaeger EE, Kartsonaki C, Marsden L, Yau C, Camps C, Kaisaki P, Taylor J, Catto JW, et al. Whole-genome sequencing of bladder cancers reveals somatic CDKN1A mutations and clinicopathological associations with mutation burden. Nat Commun. 2014; 5:3756.
158. Seo JS, Ju YS, Lee WC, Shin JY, Lee JK, Bleazard T, Lee J, Jung YJ, Kim JO, Shin JY, Yu SB, Kim J, Lee ER, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012; 22:2109–19.
159. Ehrich M, Field JK, Liloglou T, Xinarianos G, Oeth P, Nelson MR, Cantor CR, van den Boom D. Cytosine methylation profiles as a molecular marker in non-small cell lung cancer. Cancer Res. 2006; 66:10911–18.
160. Brynczka C, Labhart P, Merrick BA. NGF-mediated transcriptional targets of p53 in PC12 neuronal differentiation. BMC Genomics. 2007; 8:139.
161. Anglim PP, Alonzo TA, Laird-Offringa IA. DNA methylation-based biomarkers for early detection of non-small cell lung cancer: an update. Mol Cancer. 2008; 7:81.
162. Consortium EP. ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74.
163. Chang S, Wang RH, Akagi K, Kim KA, Martin BK, Cavallone L, Haines DC, Basik M, Mai P, Poggi E, Isaacs C, Looi LM, Mun KS, et al. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat Med. 2011; 17:1275–82.
164. Stracquadanio G, Wang X, Wallace MD, Grawenda AM, Zhang P, Hewitt J, Zeron-Medina J, Castro-Giner F, Tomlinson IP, Goding CR, Cygan KJ, Fairbrother WG, Thomas LF, et al. The importance of p53 pathway genetics in inherited and somatic cancer genomes. Nat Rev Cancer. 2016; 16:251–65.
165. Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007; 445:661–65.
166. Welcsh PL, King MC. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genet. 2001; 10:705–13.
167. Brouwer A, De Laere B, Peeters D, Peeters M, Salgado R, Dirix L, Van Laere S. Evaluation and consequences of heterogeneity in the circulating tumor cell compartment. Oncotarget. 2016; 7:48625–43. doi: 10.18632/oncotarget.8015.
168. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366:883–92.
169. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011; 472:90–94.
170. Kim KT, Lee HW, Lee HO, Song HJ, Jeong E, Shin S, Kim H, Shin Y, Nam DH, Jeong BC, Kirsch DG, Joo KM, Park WY. Application of single-cell RNA sequencing in optimizing a combinatorial therapeutic strategy in metastatic renal cell carcinoma. Genome Biol. 2016; 17:80.