
INTRODUCTION
Chronic kidney disease (CKD) is a severe complication of hypertension [1–3]. Glomerulosclerosis, renal tubule interstitial fibrosis and proteinuria are the main pathological characteristics of the hypertension-induced kidney injury [4–6]. Podocytes, as the terminally differentiated cells of glomerular filtration barrier, play a key role in maintenance of the glomerular filtration barrier so as to prevent proteinuria in CKD [7, 8]. In addition, podocytes damage or loss can result in renal fibrosis and CKD due to the accumulation of fibrosis factors [9]. Reducing fibrosis factors in kidneys, such as fibronectin and collagen type IV, could alleviate and even reverse renal fibrosis in hypertensive nephropathy.
SIRT3, a member of the evolutionary conserved family of NAD+-dependent deacetylases, is a key regulator of various processes and localized to the mitochondria matrix [10–12]. SIRT3 can regulate various processes through deacetylating its substrates at posttranslational modification [13–16]. Recently, some studies reported that SIRT3 blocks TGFβ1 signaling and aging-associated tissue fibrosis by deacetylating GSK3β [17]. In kidney, SIRT3 has been reported to protect cisplatin-induced acute kidney injury (AKI) [18, 19]. Moreover, AngII could downregulate SIRT3 mRNA expression in cultured tubular epithelial cells, and this effect might be prevented by an AT1 antagonist [20]. In addition, SIRT3 is upregulated by nicorandil and reduced by glibenclamide in dose-dependent manner in podocytes [15]. However, the function and possible molecular mechanism of SIRT3 in hypertensive nephropathy remains unclear.
Kruppel-like factors (KLFs) family includes 17 members and mediates cell growth and differentiation, metabolism and inflammation [21, 22]. KLF15 as a transcription factor, is widely expressed and enriched in kidney and liver [23, 24]. It has been reported that KLF15 regulates cardiac hypertrophy, adipogenesis, renal cells proliferation and differentiation [25, 26]. The previous report showed that KLF15 is required for restoring podocyte differentiation markers in mice and human podocytes under cell stress [27]. In addition, under high glucose, KLF15 could suppress mesangial cell proliferation and extracellular matrix (ECM) protein fibronectin expression via ERK1/2 MAPK signaling [28]. Moreover, previous evidence indicated that KLF15 is an essential negative regulatory factor for cardiac remodeling and renal interstitial fibrosis [22, 24, 29–31]. In cardiac fibroblasts, Transforming Growth Factor-β1 (TGFβ1) strongly reduces KLF15 expression, resulting in increased connective tissue growth factor (CTGF) [31]. Although KLF15 as a negative regulator of fibrosis contributes to the pathogenesis of various diseases, the molecule mechanism has not been fully understood. Whether SIRT3 could directly regulate and deacetylate KLF15 in hypertensive nephropathy requires further studies.
Honokiol (HKL), a low-molecular polyphenolic compound isolated from the bark of Magnolia officinalis (Houpo) [32–34], has been reported to activate SIRT3 and block cardiac hypertrophy [35]. What’s more, HKL exhibits a variety of pharmacological activities, such as anti-inflammatory, antioxidant, antitumor and anti-thrombosis properties [36–40]. A previous study revealed that HKL inhibits tubulointerstitial fibrosis in rat UUO model by decreasing pro-inflammatory factors and pro-fibrotic factors [41]. In our study, we would explore whether HKL alleviate renal damage via elevating the expression of SIRT3 and activating its targets in hypertensive nephropathy.
Therefore, we were aimed to explore the role and possible molecular mechanism of SIRT3 in hypertensive nephropathy. Here, data from our study provided strong evidence that SIRT3 improved renal function and fibrosis by deacetylating KLF15. And for the first time, we identified that SIRT3-KLF15 signaling was involved in the development and progression of hypertensive nephropathy.
RESULTS
SIRT3 improved renal function in AngII-induced hypertensive nephropathy
To investigate the role of SIRT3 in hypertension-mediated kidney injury, WT mice, SIRT3-KO mice and SIRT3-LV mice were all subjected to AngII infusion for 42 days. Firstly, we confirmed SIRT3 expression in mouse kidneys and found that SIRT3 decreased in mice subjected to chronic AngII infusion compared with the controls infused with saline (Figure 1A and 1B). Secondly, blood pressure markedly increased in those subjected to AngII, but the changes affected by SIRT3 were of no statistical significance (Figure 1C). Thirdly, the ratio of urine creatinine to albumin, blood urea nitrogen (BUN) and serum creatinine (Scr) were all elevated in mice after AngII infusion, and especially in the SIRT3-KO group (Figure 1D-1F). Besides, the tendency of glomerular filtration rate (GFR) was opposite to those of urine creatinine to albumin, BUN and Scr (Figure 1G). Taken together, our findings showed that SIRT3-KO mice developed aggravated hypertensive nephropathy and deteriorated renal function, indicating that SIRT3 may be involved in the signaling pathways mediating hypertensive kidney injury.
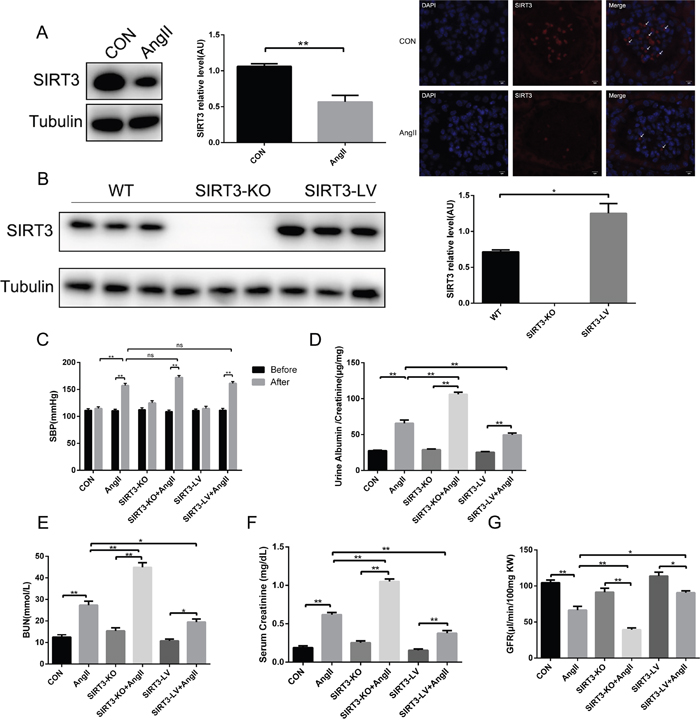
Figure 1: SIRT3 regulates renal function in hypertensive nephropathy. (A) Representative Western blot analysis and quantification of SIRT3 with saline or AngII treatment in wild type mice. Representative immunofluorescence of SIRT3 (red) with saline or AngII treatment in murine kidney. DAPI stained nucleus in blue. Bars=5μm. (n=6) (B) Representative Western blot analysis and quantification of SIRT3 in WT, SIRT3-KO and SIRT3-LV mice (n=6). (C) Systolic blood pressure of six groups (n=6). (D-G) Ratio of urine creatinine to albumin, blood urea nitrogen, serum creatinine and glomerular filtration rate in WT, SIRT3-KO and SIRT3-LV mice infused with either saline or AngII for 42 days (n=6). The data are presented as the means ± SEM of three independent experiments. *P<0.05, **P<0.01.
SIRT3 suppressed renal fibrosis in hypertensive nephropathy mice
Renal fibrosis is the key process of chronic kidney injury caused by hypertension. In order to preliminarily assess the kidney fibrosis, we weighed the kidney and body of each mouse in all groups at the end of the experiment and found that the ratio of kidney weight to body weight (KW/BW) decreased in AngII-infused mice, especially in the SIRT3-KO group (Figure 2A). The body weight and kidney weight were shown separately in Table 1. In addition, PAS and MASSON staining results showed that the areas of mesangial matrix and interstitial fibrosis increased in AngII-infused mice, of which the extent in SIRT3-KO mice was the greatest (Figure 2B and 2C). Furthermore, Western blot and immunohistochemistry confirmed that the expressions of fibronectin and collagen type IV increased in mice with chronic AngII treatment. And notably, SIRT3-LV mice exhibited lower level of fibrosis markers than WT and SIRT3-KO mice. (Figure 2D-2G). Altogether, SIRT3 may protect against renal fibrosis in hypertensive nephropathy.
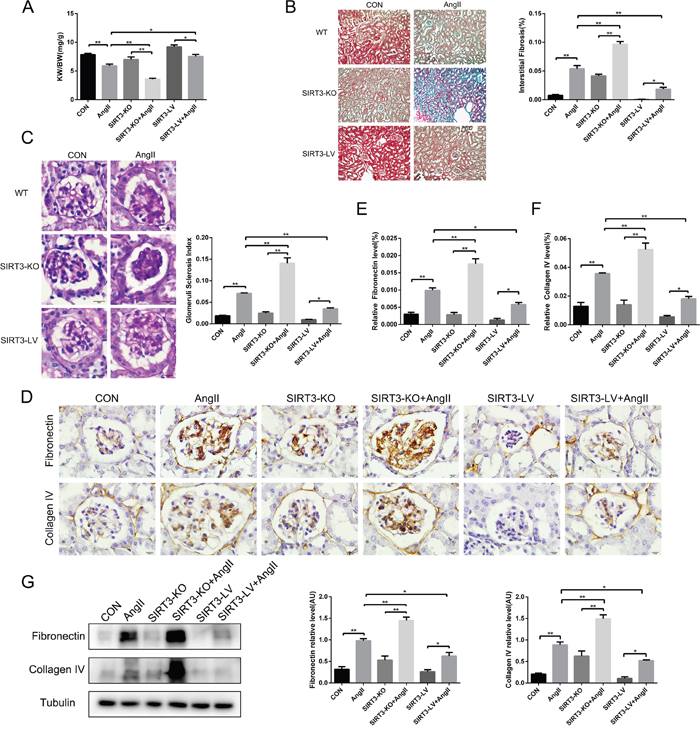
Figure 2: SIRT3 suppresses renal fibrosis in vivo. (A) Ratio of the kidney weight to body weight in WT, SIRT3-KO and SIRT3-LV mice infused with either saline or AngII for 42 days (n=6). (B) Masson’s trichrome staining showed the level of kidney fibrosis (green). Bars=50μm. The bar graph showing the quantification of kidney fibrosis (n=6). (C) Photomicrographs showing typical glomerular structure and quantification of glomerular sclerosis index in six different groups. Bars=10μm (n=6). (D-F) Immunohistochemistry analysis and quantification of fibronectin and collagen type IV in murine kidney. Bars=10μm (n=6). (G) Representative Western blot analysis and quantification of fibronectin and collagen type IV in murine kidney (n=6). The data are presented as the means ± SEM of three independent experiments. *P<0.05, **P<0.01.
Table 1: Body weight and kidney weight
Control |
AngII |
SIRT3-KO |
SIRT3-KO+AngII |
SIRT3-LV |
SIRT3-LV+AngII |
|
|---|---|---|---|---|---|---|
Body weight(g) |
24.92±0.37 |
25.24±0.52 |
25.20±0.40 |
25.09±0.46 |
24.34±0.33 |
24.70±0.33 |
Kidney weight(mg) |
195.86±3.69 |
149.10±10.21* |
176.58±8.91 |
89.45±3.88*# |
223.67±6.55 |
186.59±10.37*# |
Data are means± SEM from three independent experiments; n=6 mice/group.
*P<0.05, vs. the corresponding controls; #P<0.05, vs. the AngII group.
SIRT3 prevented podocyte injury and decreased the expression of fibrosis factors in podocytes stimulated by AngII
Podocytes are the important cells in glomerulus, which produce a number of fibrosis factors under damage. Firstly, we observed foot processes were fused or effaced in AngII-infused mice and SIRT3 ablation aggravated podocyte injury (Figure 3A). Secondly, in order to further investigate the effect of SIRT3 in cultured MPC-5 podocytes, we have verified the podocytes by the immunofluorescence of WT-1 and synaptopodin, which are the markers of podocytes (Figure 3B). And next, we knocked down SIRT3 through siRNA transfection in cultured MPC-5 podocytes and they were stimulated by AngII (10-6 mol/L, 48h). The results showed that the expressions of fibronectin and collagen type IV were elevated obviously with siRNA transfection and increased more following AngII stimulation (Figure 3C-3F). In sum, the above results revealed that SIRT3 could protect podocytes from damage and ameliorate the fibrosis phenotype of MPC-5 podocytes.
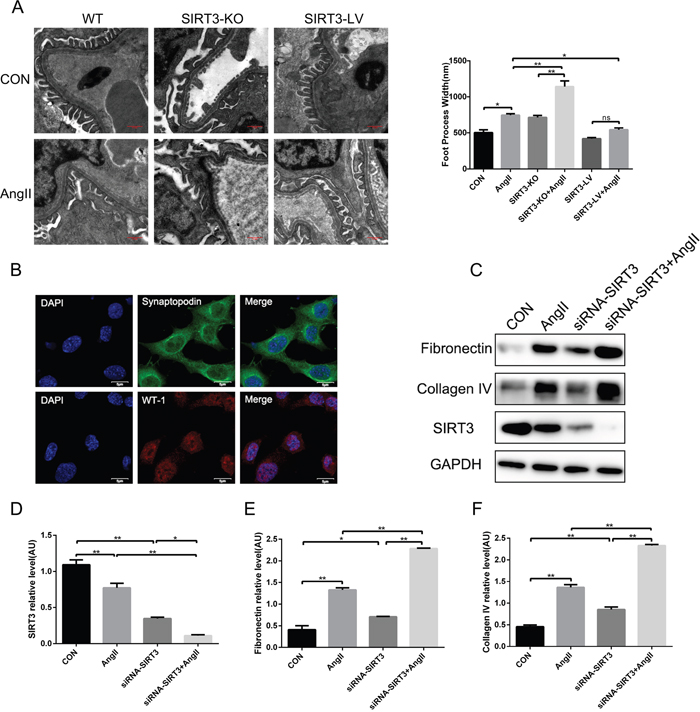
Figure 3: SIRT3 decreases the expression of fibrosis factors in podocytes. (A) Morphological changes in the podocyte foot process by electron microscopy and quantification of foot process width. Bars=0.5μm (n=6). (B) Immunofluorescence of synaptopodin (green) and WT-1(red). DAPI stained nucleus in blue. Bars=5μm (C-F) Representative Western blot analysis and quantification of SIRT3, fibronectin and collagen type IV in MPC-5 podocytes. The data are presented as the means ± SEM of three independent experiments. *P<0.05, **P<0.01.
SIRT3 interacted with KLF15 and deacetylated KLF15 in vitro
Previous evidence showed that KLF15 is an anti-fibrosis transcription factor. In this study, immunofluorescence showed a co-localization of SIRT3 and KLF15 in MPC-5 cells and they could interact directly with each other (Figure 4A and 4B). However, it was worth noting that the interaction between SIRT3 and KLF15 weakened after AngII infusion (Figure 4B). In addition, to make sure which are the upstream molecules of SIRT3 and KLF15, we measured the expression of them after siRNA transfection respectively in cultured MPC-5 cells by Western blot. The results showed that there was no change about the protein level of SIRT3 after KLF15 knockdown, whereas KLF15 expression was reduced following SIRT3 knockdown (Figure 4C). Therefore, we conjectured that KLF15 might be the target of SIRT3. Moreover, immunoprecipitation confirmed SIRT3 could deacetylate KLF15 at posttranslational modification in MPC-5 cells and the level of KLF15 acetylation was increased by AngII stimulation (Figure 4D). Furthermore, we knocked down KLF15 in MPC-5 cells, and found that the expressions of both fibronectin and collagen type IV were increased compared with the control cells (Figure 4E).
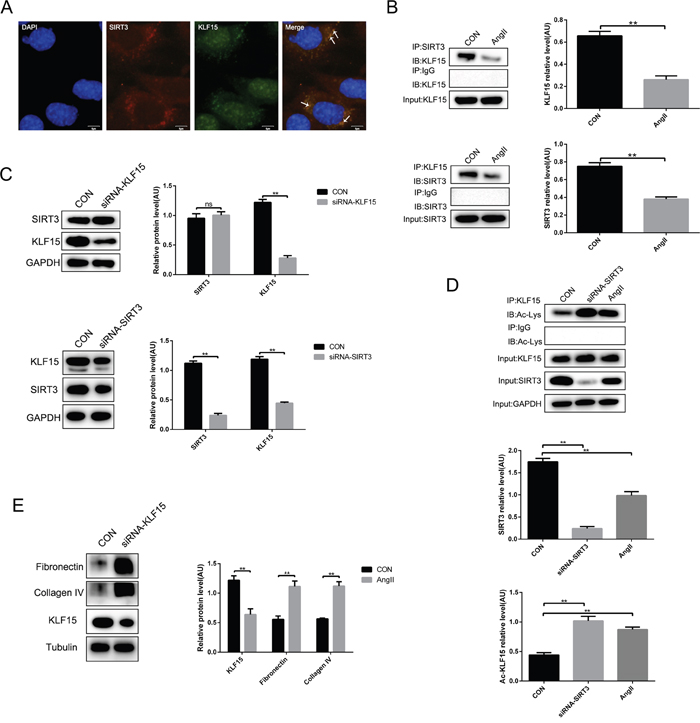
Figure 4: SIRT3 deacetylates KLF15 in podocytes. (A) Immunofluorescence of SIRT3 (red) and KLF15 (green). DAPI stained nucleus in blue. Bars=5μm. (B) Co-immunoprecipitation analysis and quantification of the interaction between SIRT3 and KLF15 in MPC-5 cells. (C) Western blot analysis and quantification of SIRT3 and KLF15 after siRNA-KLF15 or siRNA-SIRT3 transfection in vitro. (D) Immunoprecipitation analysis and quantification of the KLF15 acetylation with SIRT3 knockdown or AngII stimulation in podocytes. (E) Western blot analysis and quantification of fibronectin and collagen type IV after KLF15 knockdown in MPC-5 cells. The data are presented as the means ± SEM of three independent experiments.*P<0.05, **P<0.01.
Altogether, it is suggested that the molecular mechanism of SIRT3 improving hypertensive renal fibrosis might be regulating the acetylation status of KLF15 in MPC-5 cells. Moreover, the decreased interaction of SIRT3 and KLF15 resulted in the reducing of KLF15 deacetylation by AngII stimulation.
Honokiol alleviated hypertension-induced renal fibrosis by elevating the expression of SIRT3
To investigate the effect of honokiol in hypertension-mediated kidney injury, both AngII-infused WT mice and their controls were subjected to honokiol for 42 days by intraperitoneal injection. Firstly, renal function was improved by HKL treatment (Figure 5A). Secondly, PAS stain and MASSON stain showed that HKL decreased the extent of glomerulosclerosis and kidney fibrosis (Figure 5B and 5C). Thirdly, we found elevated SIRT3 expression and the decreased fibronectin and collagen type IV after HKL treatment. (Figure 5D and 5E). These findings revealed that HKL might alleviate hypertension-induced renal fibrosis through elevating SIRT3 expression.
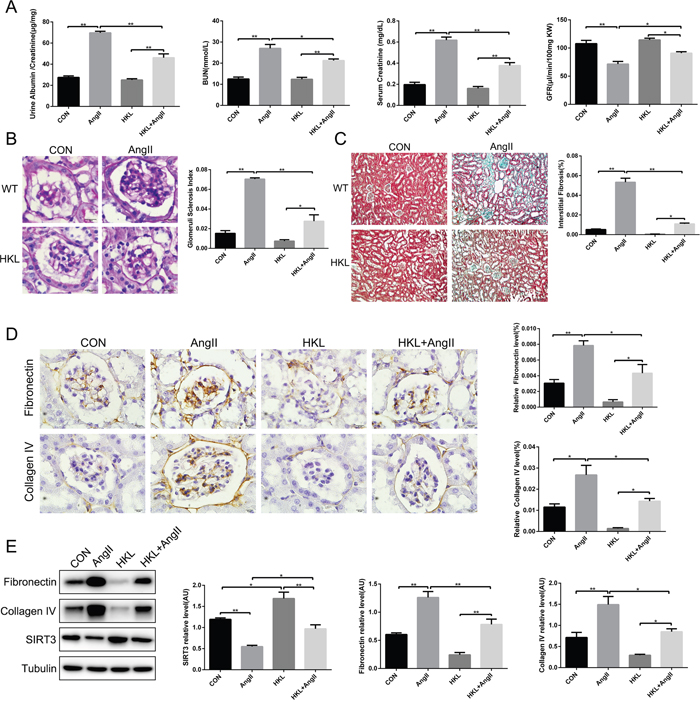
Figure 5: Honokiol alleviates hypertension-mediated kidney fibrosis by elevating SIRT3. (A) Ratio of urine creatinine to albumin, blood urea nitrogen, serum creatinine and glomerular filtration rate in control and AngII-infused mice with HKL treatment (n=6). (B) Photomicrographs showing typical glomerular structure and quantification of glomerular sclerosis index in four different groups. Bars=10μm (n=6). (C) Masson’s trichrome stain and quantification of kidney fibrosis (green). Bars=50μm (n=6). (D) Immunohistochemistry analysis and quantification of fibronectin and collagen type IV in murine kidney with HKL treatment and AngII infusion. Bars=10μm (n=6). (E) Representative Western blot analysis and quantification of SIRT3, fibronectin and collagen type IV in kidney with HKL treatment and AngII infusion (n=6). The data are presented as the means ± SEM of three independent experiments.*P<0.05, **P<0.01.
Honokiol activated SIRT3-KLF15 signaling and protected podocytes against fibrosis by AngII infusion
In vitro, MPC-5 cells were incubated with AngII (10-6 mol/L, 48h) following HKL stimulation (10μM, 1h). The results showed HKL treatment decreased fibrosis factors but increased SIRT3 expression (Figure 6A). What’s more, to further validate the activated status of SIRT3-KLF15 signaling affected by HKL, immunoprecipitation and Western blot confirmed, that the expression and the deacetylation status of KLF15 were elevated by HKL treatment (Figure 6B and 6C). These results suggested that HKL can alleviate fibrosis via activating the SIRT3-KLF15 signaling in podocytes.
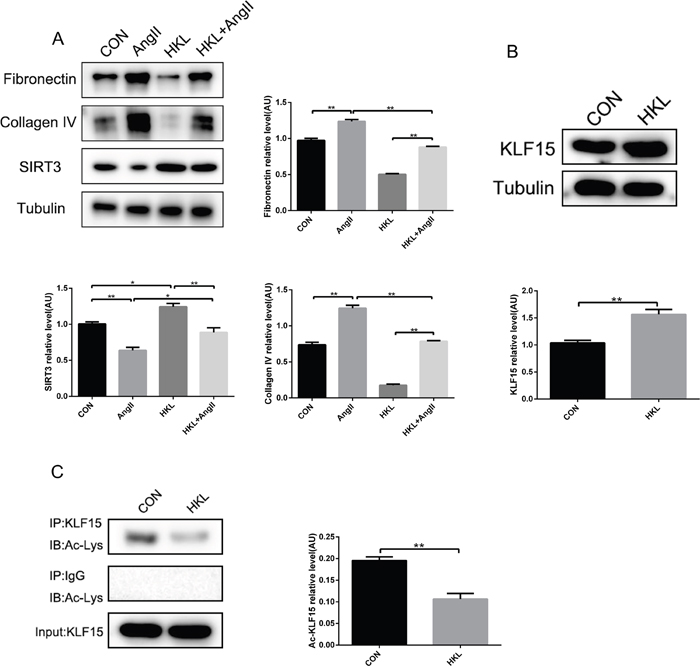
Figure 6: Honokiol activates SIRT3-KLF15 signaling. (A) Representative Western blot analysis and quantification of SIRT3, fibronectin and collagen type IV in MPC-5 cells with HKL treatment and AngII infusion. (B) Representative Western blot analysis and quantification of KLF15 with HKL treatment in MPC-5 cells. (C) Immunoprecipitation analysis and quantification of the KLF15 acetylation with HKL treatment in podocytes. The data are presented as the means ± SEM of three independent experiments.*P<0.05, **P<0.01.
Taken together, the findings revealed that SIRT3 ameliorated renal function and protected against renal fibrosis in AngII-induced hypertensive nephropathy. What’s important is that SIRT3 might deacetylate KLF15 and activate its anti-fibrosis function in podocytes. In addition, HKL treatment alleviated kidney injury via activating the SIRT3-KLF15 signaling (Figure 7).
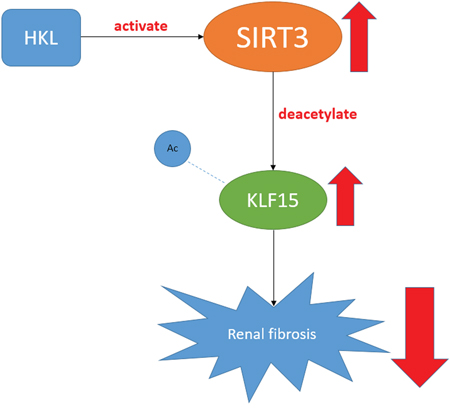
Figure 7: A figure shows the SIRT3-KLF15 signaling pathway.
DISCUSSION
Renal fibrosis involves in the process of hypertension. The recent studies mainly focused on the effect of SIRT3 in acute kidney injury and diabetic nephropathy. In this study, we studied the function and molecular mechanism of SIRT3 in hypertensive nephropathy. First of all, on account of the apoptosis of kidney inherent cells, glomerulus and kidney tubules atrophy in the progression of renal fibrosis, the kidney weight of SIRT3-KO hypertensive mice reduced at a largest extent as shown in Table 1. It is suggested that SIRT3 may play a protective role in hypertensive kidney injury. Moreover, SIRT3 ablation aggravated renal function impairment and renal fibrosis in hypertensive nephropathy. However, SBP was not affected by SIRT3. It reveals that SIRT3 may directly regulate its targets rather than changing the blood pressure to prevent kidney injury. Therefore, we further investigated the targets and molecular mechanism in vitro.
Podocytes are an important part of the glomerular filtration membrane in kidney and its foot processes interlinked by slit diaphragms serve to prevent the filtration of protein from the blood [42–45]. Podocyte injury is associated with proteinuria and consequent kidney fibrosis that progresses to chronic kidney disease [46–48]. Therefore, we made a preliminary observation of the morphological changes in podocyte foot process in murine kidney. We found that foot processes were fused or effaced in AngII-infused mice and the changes were even worse in SIRT3-KO group. It is suggested that SIRT3 may prevent podocyte damage in hypertensive nephropathy. Next, we further investigated the target and molecular mechanism of SIRT3 involved in renoprotective effect in podocytes. For one thing, we found that SIRT3 could bound to KLF15 in cultured MPC-5 cells. Moreover, SIRT3, as a NAD+-dependent deacetylase, also regulated the acetylation status of KLF15 at posttranslational modification. In addition, we observed a co-localization of SIRT3 and KLF15 in the cytoplasm. Taken together, we speculate that SIRT3 may deacetylate KLF15 in the cytoplasm and the deacetylated KLF15 is translocated to nuclear to activate its anti-fibrosis function. For another, the results showed that the interaction between SIRT3 and KLF15 reduced and the acetylated KLF15 increased by AngII stimulation. However, we didn’t investigate whether the co-localization of SIRT3 and KLF15 decreased with AngII stimulation. In a word, we concluded that the SIRT3-KLF15 signaling activation suppressed renal fibrosis and ameliorated renal function in hypertensive nephropathy. To our knowledge, this study is the first to describe a role of SIRT3 in hypertensive nephropathy via deacetylating KLF15.
We also investigated HKL effect in AngII-induced kidney injury. The results showed that HKL increased SIRT3 level and improved AngII-induced renal damage in vivo and in vitro. In addition, we further observed that HKL not only upregulated the expression of KLF15 but also increased the KLF15 deacetylation. It indicated that HKL might serve as a SIRT3-KLF15 signaling agonist to prevent hypertensive nephropathy.
In conclusion, for the first time, our findings demonstrate that SIRT3 plays a renoprotective role by deacetylating KLF15 and the SIRT3-KLF15 signaling may be a novel pathway to contribute to prevent hypertensive nephropathy. What’s more, it is revealed that honokiol alleviates hypertension-induced renal damage as a SIRT3-KLF15 signaling agonist. HKL might provide a new approach to combating renal damage and fibrosis in hypertensive nephropathy.
MATERIALS AND METHODS
Ethics statement
The animal experimental protocol and animal care procedures complied with the Animal Management Rules of the Ministry of Public Health, People’s Republic of China (documentation No 55, 2001) and were approved by the Animal Care Committee of Shandong University.
Materials and reagents
Mini-osmotic pumps were purchased from DURECT Corporation (Model 2006, Cupertino, CA). Angiotensin II (AngII), Honokiol (HKL), FITC-inulin, Bovine Serum Albumin (BSA) were purchased from Sigma Aldrich (Sigma Aldrich, USA). Mouse urine creatinine assay kit was purchased from R&D Systems (R&D, USA). Mouse urine albumin, BUN and Scr assay kit were purchased from AssayPro Corporation (AssayPro, USA). Antibodies against SIRT3 (28kDa), acetylated lysine, Tubulin and GAPDH were purchased from Cell Signaling Technology (CST, USA). Antibodies against KLF15, fibronectin, collagen type IV, synaptopodin, WT-1 were purchased from abcam (Abcam, USA).
Animal model
129 wild-type (WT) male mice of 8 weeks old were purchased from Department of Laboratory Animal Science of Peking University (Beijing, China) and SIRT3-knockout (SIRT3-KO) mice were purchased from the Jackson Laboratories (USA). SIRT3-overexpression (SIRT3-LV) mice were obtained by injection to the 129 WT mice SIRT3-overexpression-lentivirus (JIKAI GENE, Shanghai, China) via caudal vein. HKL were intraperitoneally injected in 129 WT mice for 42 days (25 mg/Kg, once a day) as the HKL treatment group. All experimental mice anesthetized with isoflurane (1%) were implanted subcutaneously corresponding osmotic pumps beforehand filled with AngII or saline for 42 days. The osmotic pumps infused with appropriate doses of AngII in sterile saline (2000 ng/kg per min) or saline according to the directions were placed in sterile 0.9% saline at 37°C for 60 hours to prime. Together, the experimental animals were randomly assigned to eight groups: WT+saline group, WT+AngII group, SIRT3-KO+saline group, SIRT3-KO+AngII group, SIRT3-LV+saline group, SIRT3-LV+AngII group, WT+HKL+saline group, WT+HKL+AngII group. Mice were sacrificed at the end of the experiment and the kidneys were used to assay for renal fibrosis. In addition, Blood pressure were measured by Tail-Cuff way (BP2010A, softron, Japan) before modeling and death.
Renal function
Urine albumin and creatinine, BUN, serum creatinine were detected by the ELISSA kit according to the direction of the kit and GFR was measured as previously described using fluorescein isothiocyanate label inulin (FITC-inulin, Sigma) [49].
Transmission electron microscopy
Mice were perfused with PBS and then immediately fixed in 2.5% glutaraldehyde for electron microscopy as previously described and the images were observed using a transmission electron microscope (TEM, H-7000FA, Hitachi, Tokyo, Japan) [27, 50]. Podocyte effacement was quantified as previously described [51].
Histology and immunohistochemistry
Periodic acid Schiff (PAS) staining and MASSON trichrome staining were performed to evaluate the level of glomerulosclerosis and renal fibrosis. The glomerulosclerotic index and collagen volume fraction was evaluated blindly by an automated image analysis system (Image-Pro Plus, Version 6.0, USA). The antibodies anti-fibronectin (1:200 dilution) and anti-collagen type IV (1:400 dilution) were used to immunohistochemical staining and photomicrographs were quantified also using Image-Pro Plus 6.0.
Cell culture
Conditionally immortalized mouse podocytes (MPC-5) were donated by Dr. Peter Mundel of Harvard Medical School (Boston, MA), and cultured with DMEM (Gibco) containing 10% fetal bovine serum (FBS, Gibco) at 33°C to propagate. To induce differentiation, podocytes were incubated at 37°C for 14 days and used for experiments in this study.
Immunofluorescence
To identify podocytes, immunofluorescence assay with synaptopodin or WT-1 acted as the marker protein of differentiation maturation podocytes was necessary. Cultured podocytes were fixed with 4% paraformaldehyde, treated with 0.5% Triton X–100, blocked with 1%BSA and then incubated with antibodies anti- synaptopodin or anti- WT-1. All imaging analyses were used in laser scanning confocal microscope after the incubating with the secondary antibodies.
Transfection and treatment
To knockdown SIRT3 or KLF15 in vitro, the podocytes were transfected with 50 nM of small interfering RNA (siRNA) with Lipofectamine 2000 (Invitrogen) for 48h. In addition, podocytes were stimulated by AngII (10-6M) for 48h following HKL (10μM) treatment 1h.
Western blot
Cultured cells or isolated kidney cortex were lysed by the addition of lysis buffer containing 50 mM Tris-buffer (pH 7.4), 150 mM NaCl, 0.5 mM EDTA, 0.5% NP40, and 1% Triton X-100, Phenylmethanesulfonyl fluoride. Protein samples were subjected to electrophorese on 10% SDS/PAGE gels before the disconnected proteins transferring to PVDF membrane in a designated time and then blocked with 5% skim milk in 1X TBST for 2h. The blocked membrane were incubated with corresponding antibodies at 4°C overnight. The membrane were washed with 1X TBST in the second day and then incubated with the secondary antibody in 1:5000 dilution. After washing with 1X TBST, the protein band were showed by enhanced chemiluminescence (Millipore) and an Image Quant LAS4000 chemiluminescence reader (GE, USA).
Immunoprecipitation
200μg proteins of cell lysates were used with Protein A+G Agarose and IgG to remove nonspecific binding according to manufacturer’s instructions. SIRT3 or KLF15 was immunoprecipitated with their corresponding antibodies and then the samples were performed immunoblotting according to the above.
Statistical analysis
Data were shown as mean ± SEM. Statistical analysis was performed with GraphPad Prism 6 (La Jolla, CA, USA) and using unpaired t test to analyze data between two groups. To assess differences among groups, ANOVA followed by Tukey post-hoc test were used. P<0.05 was considered significant.
ACKNOWLEDGMENTS
This work was supported by the National 973 Basic Research Program of China (No.2012CB722406) and the National Natural Science Foundation of China (No. 81070076, No.81170135).
CONFLICTS OF INTEREST
The authors declared no conflicts of interest.
REFERENCES
1. Xia Y, Entman ML, Wang Y. Critical role of CXCL16 in hypertensive kidney injury and fibrosis. Hypertension. 2013; 62:1129-37.
2. Nair AR, Ebenezer PJ, Saini Y, Francis J. Angiotensin II-induced hypertensive renal inflammation is mediated through HMGB1-TLR4 signaling in rat tubulo-epithelial cells. Exp Cell Res. 2015; 335:238-47.
3. Burlaka I, Nilsson LM, Scott L, Holtback U, Eklof AC, Fogo AB, Brismar H, Aperia A. Prevention of apoptosis averts glomerular tubular disconnection and podocyte loss in proteinuric kidney disease. Kidney Int. 2016; 90:135-48.
4. Yu SY, Qi R, Zhao H. Losartan reverses glomerular podocytes injury induced by AngII via stabilizing the expression of GLUT1. Mol Biol Rep. 2013; 40:6295-301.
5. Schiessl IM, Hammer A, Kattler V, Gess B, Theilig F, Witzgall R, Castrop H. Intravital Imaging Reveals Angiotensin II-Induced Transcytosis of Albumin by Podocytes. J Am Soc Nephrol. 2016; 27:731-44.
6. Lv LL, Liu BC. Role of non-classical renin-angiotensin system axis in renal fibrosis. Front Physiol. 2015; 6:117.
7. Reiser J, Altintas MM. Podocytes. F1000Res. 2016; 5:F1000 Faculty Rev-114.
8. Bartlett CS, Jeansson M, Quaggin SE. Vascular Growth Factors and Glomerular Disease. Annu Rev Physiol. 2016; 78:437-61.
9. Wang XX, Edelstein MH, Gafter U, Qiu L, Luo Y, Dobrinskikh E, Lucia S, Adorini L, D'Agati VD, Levi J, Rosenberg A, Kopp JB, Gius DR, et al. G Protein-Coupled Bile Acid Receptor TGR5 Activation Inhibits Kidney Disease in Obesity and Diabetes. J Am Soc Nephrol. 2016; 27:1362-78.
10. Perico L, Morigi M, Benigni A. Mitochondrial Sirtuin 3 and Renal Diseases. Nephron. 2016; 134:14-9.
11. Kitada M, Kume S, Koya D. Role of sirtuins in kidney disease. Curr Opin Nephrol Hypertens. 2014; 23:75-9.
12. Kitada M, Kume S, Takeda-Watanabe A, Kanasaki K, Koya D. Sirtuins and renal diseases: relationship with aging and diabetic nephropathy. Clin Sci (Lond). 2013; 124:153-64.
13. Yang H, Zuo XZ, Tian C, He DL, Yi WJ, Chen Z, Zhang PW, Ding SB, Ying CJ. Green Tea Polyphenols Attenuate High-Fat Diet-Induced Renal Oxidative Stress through SIRT3-Dependent Deacetylation. Biomed Environ Sci. 2015; 28:455-9.
14. Zhuo L, Fu B, Bai X, Zhang B, Wu L, Cui J, Cui S, Wei R, Chen X, Cai G. NAD Blocks High Glucose Induced Mesangial Hypertrophy via Activation of the Sirtuins-AMPK- mTOR Pathway. Cell Physiol Biochem. 2011; 27:681-90
15. Shiraishi T, Tamura Y, Taniguchi K, Higaki M, Ueda S, Shima T, Nagura M, Nakagawa T, Johnson RJ, Uchida S. Combination of ACE inhibitor with nicorandil provides further protection in chronic kidney disease. Am J Physiol Renal Physiol. 2014; 307:F1313-22.
16. Koyama T, Kume S, Koya D, Araki S, Isshiki K, Chin-Kanasaki M, Sugimoto T, Haneda M, Sugaya T, Kashiwagi A, Maegawa H, Uzu T. SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic Biol Med. 2011; 51:1258-67.
17. Sundaresan NR, Bindu S, Pillai VB, Samant S, Pan Y, Huang JY, Gupta M, Nagalingam RS, Wolfgeher D, Verdin E, Gupta MP. SIRT3 blocks aging-associated tissue fibrosis in mice by deacetylating and activating GSK3β. Mol Cell Biol. 2015; 36:678-92.
18. Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D, Novelli R, Remuzzi G, Benigni A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest. 2015; 125:715-26.
19. Ugur S, Ulu R, Dogukan A, Gurel A, Yigit IP, Gozel N, Aygen B, Ilhan N. The renoprotective effect of curcumin in cisplatin-induced nephrotoxicity. Ren Fail. 2015; 37:332-6.
20. Benigni A, Corna D, Zoja C, Sonzogni A, Latini R, Salio M, Conti S, Rottoli D, Longaretti L, Cassis P, Morigi M, Coffman TM, Remuzzi G. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Invest. 2009; 119:524-30.
21. Gao X, Huang L, Grosjean F, Esposito V, Wu J, Fu L, Hu H, Tan J, He C, Gray S, Jain MK, Zheng F, Mei C. Low-protein diet supplemented with ketoacids reduces the severity of renal disease in 5/6 nephrectomized rats: a role for KLF15. Kidney Int. 2011; 79:987-96.
22. Gao X, Wu G, Gu X, Fu L, Mei C. Kruppel-like factor 15 modulates renal interstitial fibrosis by ERK/MAPK and JNK/MAPK pathways regulation. Kidney Blood Press Res. 2013; 37:631-40.
23. Mallipattu SK, Liu R, Zheng F, Narla G, Ma'ayan A, Dikman S, Jain MK, Saleem M, D'Agati V, Klotman P, Chuang PY, He JC. Kruppel-like factor 15 (KLF15) is a key regulator of podocyte differentiation. J Biol Chem. 2012; 287:19122-35.
24. Yu Y, Ma J, Xiao Y, Yang Q, Kang H, Zhen J, Yu L, Chen L. KLF15 is an essential negative regulatory factor for the cardiac remodeling response to pressure overload. Cardiology. 2015; 130:143-52.
25. Ivy JR, Drechsler M, Catterson JH, Bodmer R, Ocorr K, Paululat A, Hartley PS. Klf15 Is Critical for the Development and Differentiation of Drosophila Nephrocytes. PLoS One. 2015; 10:e0134620.
26. Hong Q, Li C, Xie Y, Lv Y, Liu X, Shi S, Ding R, Zhang X, Zhang L, Liu S, Chen X. Kruppel-like Factor-15 Inhibits the Proliferation of Mesangial Cells. Cell Physiol Biochem. 2012; 29:893-904.
27. Mallipattu SK, Guo Y, Revelo MP, Roa-Peña L, Miller T, Ling J, Shankland SJ, Bialkowska AB, Ly V, Estrada C, Jain MK, Lu Y, Ma'ayan A, et al. Krüppel-Like Factor 15 Mediates Glucocorticoid-Induced Restoration of Podocyte Differentiation Markers. J Am Soc Nephrol. 2017; 28:166-184.
28. Zhang J, Zhong HB, Lin Y, Yao W, Huang JY. KLF15 suppresses cell proliferation and extracellular matrix expression in mesangial cells under high glucose. Int J Clin Exp Med. 2015; 8:20330-6.
29. Wang Y, Mitch WE. Proteins and renal fibrosis: low-protein diets induce Kruppel-like factor-15, limiting renal fibrosis. Kidney Int. 2011; 79:933-4.
30. Chin MT. KLF15 and cardiac fibrosis: the heart thickens. J Mol Cell Cardiol. 2008; 45:165-7.
31. Wang B, Haldar SM, Lu Y, Ibrahim OA, Fisch S, Gray S, Leask A, Jain MK. The Kruppel-like factor KLF15 inhibits connective tissue growth factor (CTGF) expression in cardiac fibroblasts. J Mol Cell Cardiol. 2008; 45:193-7.
32. Li N, Xie H, Li L, Wang J, Fang M, Yang N, Lin H. Effects of honokiol on sepsis-induced acute kidney injury in an experimental model of sepsis in rats. Inflammation. 2014; 37:1191-9.
33. Fang CY, Chen SJ, Wu HN, Ping YH, Lin CY, Shiuan D, Chen CL, Lee YR, Huang KJ. Honokiol, a Lignan Biphenol Derived from the Magnolia Tree, Inhibits Dengue Virus Type 2 Infection. Viruses. 2015; 7:4894-910.
34. Chiang CK, Sheu ML, Hung KY, Wu KD, Liu SH. Honokiol, a small molecular weight natural product, alleviates experimental mesangial proliferative glomerulonephritis. Kidney Int. 2006; 70:682-9.
35. Pillai VB, Samant S, Sundaresan NR, Raghuraman H, Kim G, Bonner MY, Arbiser JL, Walker DI, Jones DP, Gius D, Gupta MP. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat Commun. 2015; 6:6656.
36. Kim KR, Park KK, Chun KS, Chung WY. Honokiol inhibits the progression of collagen-induced arthritis by reducing levels of pro-inflammatory cytokines and matrix metalloproteinases and blocking oxidative tissue damage. J Pharmacol Sci. 2010; 114:69–78.
37. Cheng S, Castillo V, Eliaz I, Sliva D. Honokiol suppresses metastasis of renal cell carcinoma by targeting KISS1/KISS1R signaling. Int J Oncol. 2015; 46:2293-8.
38. Li W, Wang Q, Su Q, Ma D, An C, Ma L, Liang H. Honokiol suppresses renal cancer cells' metastasis via dual-blocking epithelial-mesenchymal transition and cancer stem cell properties through modulating miR-141/ZEB2 signaling. Mol Cells. 2014; 37:383-8.
39. Prasad R, Kappes JC, Katiyar SK. Inhibition of NADPH oxidase 1 activity and blocking the binding. Oncotarget. 2016; 7:7899-912. doi: 10.18632/oncotarget.6860.
40. Zhang Q, Zhao W, Ye C, Zhuang J, Chang C, Li Y, Huang X, Shen L, Li Y, Cui Y, Song J, Shen B, Eliaz I, et al. Honokiol inhibits bladder tumor growth by suppressing EZH2miR-143 axis. Oncotarget. 2015; 6:37335-48. doi: 10.18632/oncotarget.6135.
41. Chiang CK, Sheu ML, Lin YW, Wu CT, Yang CC, Chen MW, Hung KY, Wu KD, Liu SH. Honokiol ameliorates renal fibrosis by inhibiting extracellular matrix and pro-inflammatory factors in vivo and in vitro. Br J Pharmacol. 2011; 163:586-97.
42. Kim SH, Kim HJ, Kim CW. GLCCI1 is a novel component associated with the PI3K signaling pathway in podocyte foot processes. Exp Mol Med. 2016; 48:e233.
43. He FF, Chen S, Su H, Meng XF, Zhang C. Actin-associated Proteins in the Pathogenesis of Podocyte Injury. Curr Genomics. 2013; 14:477-84.
44. Bi J, Pellenz CD, Krendel M. Visualization of cytoskeletal dynamics in podocytes using adenoviral vectors. Cytoskeleton (Hoboken). 2014; 71:145-56.
45. Andeen NK, Nguyen TQ, Steegh F, Hudkins KL, Najafian B, Alpers CE. The phenotypes of podocytes and parietal epithelial cells may overlap in diabetic nephropathy. Kidney Int. 2015; 88:1099-107.
46. Dong L, Pietsch S, Englert C. Towards an understanding of kidney diseases associated with WT1 mutations. Kidney Int. 2015; 88:684-90.
47. Fukusumi Y, Miyauchi N, Hashimoto T, Saito A, Kawachi H. Therapeutic target for nephrotic syndrome: Identification of novel slit diaphragm associated molecules. World J Nephrol. 2014; 3:77-84.
48. Liu Y, Liang W, Yang Y, Pan Y, Yang Q, Chen X, Singhal PC, Ding G. IQGAP1 regulates actin cytoskeleton organization in podocytes through interaction with nephrin. Cell Signal. 2015; 27:867-77.
49. Liao TD, Yang XP, Liu YH, Shesely EG, Cavasin MA, Kuziel WA, Pagano PJ, Carretero OA. Role of inflammation in the development of renal damage and dysfunction in angiotensin II-induced hypertension. Hypertension. 2008; 52:256-63.
50. Mallipattu SK, Liu R, Zhong Y, Chen EY, D'Agati V, Kaufman L, Ma'ayan A, Klotman PE, Chuang PY, He JC. Expression of HIV transgene aggravates kidney injury in diabetic mice. Kidney Int. 2013; 83:626-34.
51. Mallipattu SK, Gallagher EJ, LeRoith D, Liu R, Mehrotra A, Horne SJ, Chuang PY, Yang VW, He JC. Diabetic nephropathy in a nonobese mouse model of type 2 diabetes mellitus. Am J Physiol Renal Physiol. 2014; 306:F1008-17.