
INTRODUCTION
Malignant adrenal tumours represent a severe medical condition. Among them, adrenocortical carcinoma (ACC) is a rare neoplasm with a poor prognosis since it is often diagnosed already at the metastatic stage. Decisive advances have been recently made in the identification of the genetic determinants of ACC pathogenesis [1–3] but the mechanisms driving its spread and metastasis remain largely unknown. Metastatic dissemination is a complex, dynamic and inefficient process involving multiple steps, which is responsible for more than 90% of fatal events in cancer patients [4]. Liver and lungs are the most common sites of ACC metastases. A cornerstone of the therapy for metastatic ACC is the use of mitotane, a derivative of the insecticide DDT [5], which can be associated to cytotoxic chemotherapy. However, current therapies for ACC are unable to significantly increase overall survival of patients [6]. A consensus exists that new, more active pharmacological tools are urgently needed for the treatment of metastatic ACC. Furthermore, modern therapies for those tumours should be tailored to the specific biological features of each cancer, as it is currently a standard for other types of cancer with the goal of a personalized therapy.
One major problem hindering preclinical experimentation of new therapies for ACC is represented by the lack of suitable animal models for metastatic disease. Current protocols envisage the use of human cell lines or patient-derived tumour fragments as xenografts implanted in immunodeficient mice [7, 8]. However, those xenografts have no metastatic potential and tests of new pharmacological therapies are restricted to targeting of the primary tumour [9–19]. Genetic models of ACC also exist but they are limited by their poor tendency to diffuse beyond the loco-regional stage [20, 21].
To overcome these limitations, in this study we tested several grafting procedures in order to establish a model of metastatic ACC.
RESULTS
Generation of the H295R/TR SF-1 GFP-luc cell line
Human ACC H295R/TR SF-1 cells [22] were transduced with a lentiviral vector and selected by cell sorting to generate a cell line expressing both firefly luciferase and GFP for bioluminescence and fluorescence detection. Luciferase activity from this cell line (H295R/TR SF-1 GFP-luc) is easily detectable in vitro from a low number of cells (Figure 1).
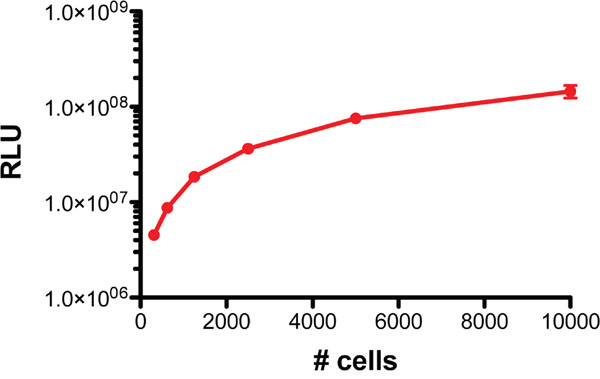
Figure 1: Luminescence of H295R/TR SF-1 GFP-luc cells. Various numbers of cells (1:2 dilutions starting from 104 to 312/well) were seeded in triplicate in a 96-well plate and luminescence measured by an in vitro luciferase assay.
Caudal vein injection
In the attempt to develop a metastatic mouse model of ACC, we first explored the potential of intravenous injection of cancer cells into the caudal vein [23]. However, out of ten mice receiving H295R/TR SF-1 GFP-luc cells using this procedure, only one displayed small foci detectable by bioluminescence at the level of kidneys and lung 43 days after the injection (Supplementary Figure 1).
Renal subcapsular grafts
Thirteen mice were grafted under the renal capsule using 2.7x106 H295R/TR SF-1 GFP-luc cells per animal. Bioluminescence imaging to detect primary tumour and potential metastases was performed 43 days after surgery just before the sacrifice of the animals. All mice showed a strong bioluminescent signal near the left kidney, that was comparable in intensity whether the mice were exposed on their ventral or dorsal face (Figure 2A, 2B). The intensity of the bioluminescent signal did not correlate with the weight of the tumor mass (r2 = 0.15), probably due to the high heterogeneity of the tumours revealed by histological analysis (Figure 2C). Imaging analysis did not reveal any potential metastatic site and no macroscopic metastasis could be observed at necropsy.
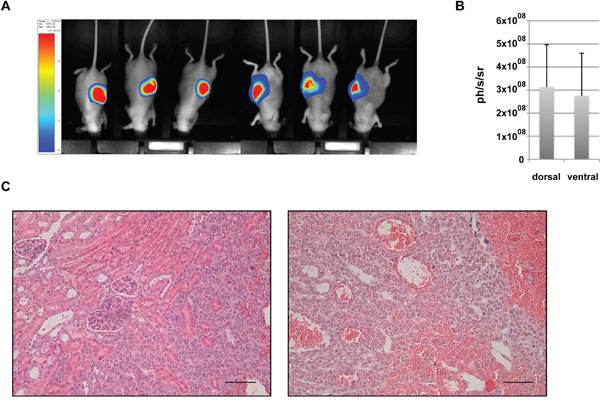
Figure 2: Analysis of renal subcapsular xenografts. (A) Representative bioluminescence images and (B) quantitative analysis of photon counts derived from renal subcapsular xenografts of H295R/TR SF-1 GFP-luc cells. (C) Representative images of HES-stained sections of renal subcapsular tumours at 52 days after cell graft. Note the large vascular spaces inside the xenograft tissue. Scale bars, 50 μm.
Intrasplenic grafts
Because of the lack of success of the previous protocols, we chose to inject H295R/TR SF-1 GFP-luc cells intrasplenically, to elicit formation of hepatic metastases. We injected 5x106 H295R cells into the spleen of 17 mice and monitored tumour burden by bioluminescence imaging. 47 days after the graft, 9 mice presented a bioluminescence signal indicative of a splenic tumour. Among those mice, 3 also had a detectable signal in the upper part of the abdomen, suggestive of a hepatic tumour, and one had a signal in the lower part of the abdomen (Figure 3A, 3B). To allow metastasis progression before sacrifice, mice underwent radical splenectomy 54 days after the graft. Only 6 mice (out of the 9 mice having bioluminescent signal) had a macroscopically evident splenic tumour, highlighting the sensitivity of bioluminescence imaging over macroscopic observation. Histological analysis of the splenic tumours showed nearly complete replacement of lymphoid tissue by xenograft (Figure 3C). Tumour burden was monitored by bioluminescence imaging and mice were sacrificed either when the identified hepatic tumour reached the luminescence of 6x108 ph/s/sr or 109 days after the graft. At the end of the experiment, 6 mice out of 17 had liver metastasis detected by imaging and confirmed by histology (Figure 3D), one had a pancreatic tumour and a tumour linked to the peritoneal muscle (both tumours very close to the injection site). Another mouse also presented a peritoneal tumour next to the injection site. Both primary tumours and liver metastases expressed the adrenocortical marker SF-1 and displayed a high Ki67 labelling index (data not shown).
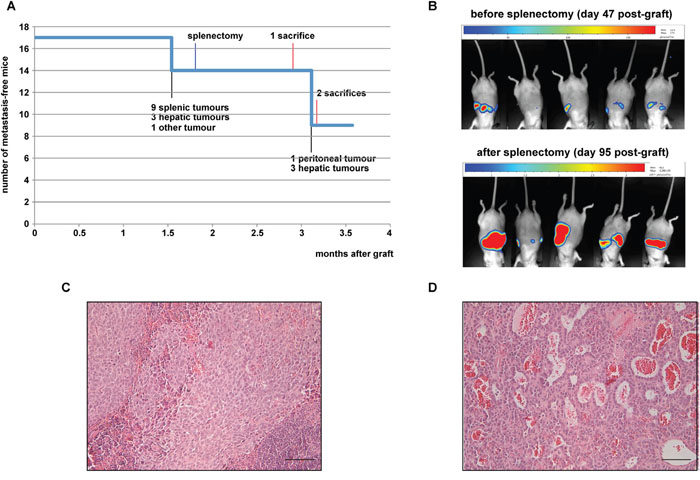
Figure 3: Analysis of intrasplenic xenografts. (A) Number of mice without metastases detectable by bioluminescence imaging in function of time after graft. Milestones of the experiment are indicated. (B) Representative bioluminescence images of mice 47 and 94 days after intrasplenic xenograft of H295R/TR SF-1 GFP-luc cells (before and after splenectomy, respectively). (C) A representative HES-stained section of a splenic tumour at 54 days after cell graft. (D) A representative HES-stained section of a hepatic metastasis 109 days after cell graft. Note the large vascular spaces inside the metastasis. Scale bars, 50 μm.
Intrasplenic grafts followed by splenectomy
To increase tumour burden we then decided to increase the number of grafted cells. However, to avoid the development of tumours around the injection site (pancreas or peritoneum), we decided to splenectomize all mice during the initial surgery. We grafted 22 mice with 107 H295R/TR SF-1 GFP-luc cells through the intrasplenic route and subsequently we ligatured and resected the spleen 10 minutes after injection. At 1 month after cell graft, 20 mice (91%) presented tumours in the upper part of the abdomen detectable by bioluminescence imaging. At the end of the experiment 2 months after graft, all mice had bioluminescence detectable and macroscopically visible hepatic tumours (Figure 4A-4C). A significant linear correlation existed between liver weight at necropsy and the intensity of bioluminescent signals (Figure 4D). No other kind of tumour was observed at necropsy. Histological analysis of these tumours revealed strong metastatic invasion of hepatic tissue (Figure 4E). Metastatic nodules retained their adrenocortical differentiation, as shown by SF-1 staining, and displayed a high Ki67 labelling index, in contrast to the surrounding liver tissue (Supplementary Figure 2).
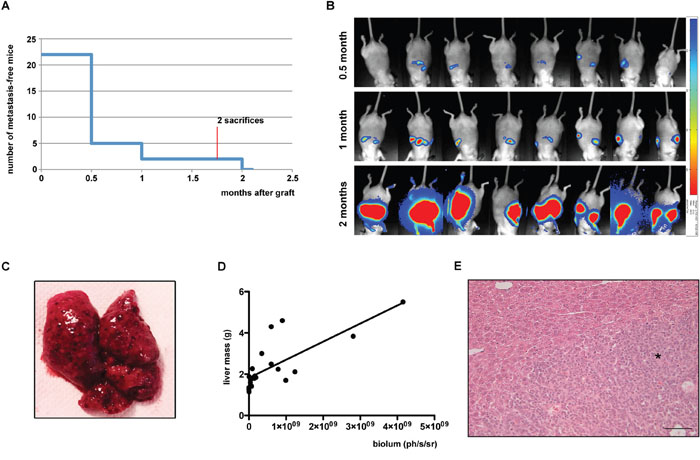
Figure 4: Analysis of intrasplenic xenografts complemented by splenectomy. (A) Number of mice without metastases detectable by bioluminescence imaging in function of time after graft. (B) Representative in vivo images from bioluminescence imaging of mice 0.5, 1 or 2 months after intrasplenic xenograft of H295R/TR SF-1 GFP-luc cells followed by splenectomy. (C) Macroscopic image of a liver from a mouse injected with H295R/TR SF-1 GFP-luc cells in the spleen, showing multiple metastatic nodules. (D) Correlation between liver mass at necropsy and intensity of bioluminescence signals measured 2 months after graft. r2= 0.5793, p<0.0001. (E) HES-stained section of a hepatic metastasis 2 months after cell graft. Metastatic tissue is indicated with an asterisk. Scale bar, 50 μm.
DISCUSSION
Until very recently, H295R cells and its derivatives were the only differentiated human ACC cell line available for in vitro and xenograft studies. New perspectives to better appreciate ACC heterogeneity may now be offered by the recently described SJ-ACC3 xenografts [24] and MUC-1 xenografts and cell line [19]. Nevertheless, H295R cells remain the workhorse among human ACC models. While retaining steroidogenic capacities, these cells have multiple features of a highly malignant phenotype, including among others a very complex karyotype (our unpublished observations), TP53 [22] and CTNNB1 [25] mutations. However, these characteristics do not translate into the potential of this cell line to spontaneously metastasize when grown as xenografts in the highly vascularized environment under the renal capsule. The metastatic process requires multiple steps, from the capacity of the tumour cells to reach the blood and lymphatic circulation to the ability to extravasate and colonize peripheral organs [26]. H295R cells are clearly defective in one or more of these essential steps for tumour spread and metastasis, which will be worthwhile to precisely identify in future studies. We acknowledge that intrasplenic graft is an experimental model of metastasis and not a spontaneous one. As such, it eludes the stages of local invasion and intravasation. However, intrasplenic graft is a validated experimental model of liver metastasis, representative of the later stages of the malignant process: circulation to a distant organ, arrest and extravasation, followed by establishment of micrometastasis and metastatic colonization. Since the two last steps are often considered as rate-limiting in the malignant process [27, 28], the establishment of a model recapitulating those steps appears valuable for the understanding of human disease.
In many types of epithelial cancer, cells transition to a mesenchymal-like phenotype (EMT) is of high relevance for invasion and metastasis [29]. It has been reported that loss of β-catenin induces reversal of EMT in H295R cells, as shown by downregulation of vimentin, N-cadherin and Slug upon CTNNB1 knockdown [30]. However, mesoderm-derived tissues such as the adrenal cortex normally express vimentin, while N-cadherin was reported to be downregulated in ACC compared to adrenocortical adenomas [31]. It thus remains uncertain whether a process related to EMT really exists and has relevance for metastatic dissemination in ACC.
In this study, by comparison of different procedures, we have developed a protocol to induce liver metastasis in immunodeficient mice grafted with the human H295R/TR SF-1 GFP-luc ACC cell line. This method, based upon intrasplenic injection followed by splenectomy, produced metastases in the liver with 100% efficiency, whose development could be followed over time by bioluminescence measurements. Early splenectomy reduces the manipulation of animals, eliminating the need for a second surgery, and avoids the development of primary splenic tumours, which allows time for liver metastases to develop. The number of engrafted cells may be an important factor for the success of intrasplenic injection compared to the other tested routes for tumour cells administration, eliminating the number of cells or injection volume constraints imposed by caudal vein injection and subcapsular renal graft, respectively On the other hand, one limitation of our model is that no metastases were observed in the other organs mainly targeted by ACC metastasis, i.e. lungs and bone. However, caudal vein injection of tumor cells was not efficient in producing lung metastasis and experimental metastases to the bone are usually obtained by intracardiac injection, a technically demanding and high-risk procedure. Conversely, the method described here can be easily performed with no loss of animals caused by complications of surgery. We expect that the availability of this mouse xenograft model of metastatic ACC will greatly improve the possibilities for preclinical testing of new treatments for advanced-stage disease.
MATERIALS AND METHODS
Cell culture
H295R/TR SF-1 cells, derived from the H295R cell line [32], were generated in our laboratory [22] and reauthenticated by STR profiling (Promega PowerPlex 21 PCR Kit) immediately prior to use for this work. Cell were cultured in a humidified atmosphere containing 5% CO2 at 37°C in DMEM/F12 complemented with 2% NuSerum (BD Biosciences), 1% ITS+ (BD Biosciences), blasticidin (5 μg/ml, Cayla InvivoGen) zeocin (100 μg/ml, Cayla InvivoGen), and penicillin/streptomycin (Invitrogen), as described [22].
Lentiviral infection and production of H295R/TR SF-1 GFP-luc cells
The pRRL-GFP/Luc2 vector [33] was packaged into lentiviral particles and used to infect H295R/TR SF-1 cells in the exponential phase of growth seeded in a 6-well plate at a density of 1x106 cells/well. The following day, culture medium was replaced by fresh medium containing polybrene (Sigma Aldrich #H9268) at a final concentration of 4 μg/ml. After 1 hour, cells were incubated for 24 hours with complete culture medium (1.5 ml/well) containing 5x106 virions and 4 μg/ml polybrene. The following day the medium was replaced and the efficiency of infection was determined by monitoring the expression level of GFP under an inverted fluorescent microscope. Cells were amplified by sequential passage and then purified by FACS. Before cell sorting, cells were filtered through a 70 μM strainer to eliminate all remaining clumps and resuspended at a concentration of 8x106/ml. About 24 millions cells were sorted using a FACSARIA III instrument (BD Biosciences). A highly pure sorting mode (4-way purity sorting) was chosen. Sorted cells expressing the highest levels of GFP fluorescence (around 4% of the initial population) were collected in polypropylene tubes containing FBS, plated in 25 cm2 cell culture flasks and amplified. GFP expression in the polyclonal population obtained was confirmed by fluorescence microscopy. To measure luciferase expression, various numbers of cells were seeded in the wells of white 96-well plates (Corning) in triplicate in 100 μl of culture medium per well. After cell lysis, luciferase activity was measured using the Luciferase Assay System (Promega #E1500) on a GloMax instrument (Promega).
Animal experiments
All mouse husbandry and experimental procedures were performed in accordance with protocols 2–091009 and 14-041 approved by the local Committees for Animal Experimentation and followed the ARRIVE guidelines of the National Centre for the Replacement, Refinement, and Reduction of Animals in Research (London, UK). They were carried out using female MRI Nude mice (Janvier) aged 6 to 16 weeks. Animals had free access to water and standard laboratory chow, and were housed according to institutional rules with 12:12 h dark/light cycles. For procedures involving surgery, mice were anesthetized by inhalation of isoflurane and were given meloxicam (2 mg/kg s.c.) analgesia after the surgical intervention. For 1 week after surgery, animals were given a broad-spectrum antibiotic (enrofloxacin 100 μg/ml) in drinking water supplemented with sucrose.
Caudal vein injection
1x106 H295R/TR SF-1 GFP-luc cells resuspended in 100 μl of a 0.9% NaCl solution were injected into the mouse caudal vein using a 29G needle.
Renal subcapsular graft
H295R/TR SF-1 GFP-luc cells were trypsinized, centrifuged and gently resuspended in 30 μl ice-cold PBS per injection. An equal volume of ice-cold Matrigel (high concentration, catalog number 354248, BD Biosciences) was added. Mice were anesthetized and the left kidney was exposed by a retroperitoneal incision and gently lifted to allow the injection of the cells under the renal capsule. The kidney was kept humid during the procedure to avoid renal capsule rupture. 2.7x106 cells were injected using a 29G needle. The needle was slowly inserted under the renal capsule with the bevelled edge facing up. Air was first injected to separate the capsule from the kidney, and consequently 40 μl of cell suspension was injected. The needle was left immobile for 1 minute to allow Matrigel polymerization, then turned to face down the bevel, and carefully withdrawn from the kidney to prevent reflux of the injected cells.
Intra-splenic injection
5 -10x106 H295R cells were resuspended in 60 μL PBS and injected at the inferior pole of the spleen using a 29G needle. The bevel of the needle was observed through the splenic capsule to avoid injecting the cells under the spleen. Whitening of the spleen and blood vessels was observed upon injection. When stated, mice underwent splenectomy through ligature of the pancreas and splenic vessels and complete resection of the spleen.
Bioluminescence imaging
At various times after the beginning of the different protocols, after isoflurane anaesthesia mice were injected intraperitoneally with 150 mg/kg body weight of endotoxin-free beetle luciferin, potassium salt (Promega) solution in PBS 8 minutes prior to imaging. Bioluminescence measurements were performed using a PhotonIMAGER instrument (Biospace Lab). Data were acquired for 11 minutes and analyzed using the M3 vision software (Biospace Lab).
Immunohistochemistry
Immunohistochemistry was performed as described [34], using the anti SF-1 (clone N1665, Perseus Proteomics) and anti Ki67 (clone SP6, Thermo Fisher) antibodies.
Statistical analysis
Linear regression was calculated using the GraphPad Prism software v. 5.0d.
Author contributions
Aurélie Morin, Judith Favier and Enzo Lalli designed the study. Aurélie Morin, Estelle Robidel, Rémy Castellano and Emmanuelle Josselin performed mouse experiments. Carmen Ruggiero, Mabrouka Doghman-Bouguerra and Atze T. Das performed cell culture experiments. Judith Favier and Enzo Lalli obtained funding. Aurélie Morin and Enzo Lalli wrote the manuscript and prepared figures, receiving inputs from all other authors. All authors had full access to all the data and had final responsibility for the decision to submit for publication.
ACKNOWLEDGMENTS
We thank Constanze Hantel for advice on mouse xenograft protocols, Julie Cazareth for cell sorting and the CRCM animal core facility for support.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement n°259735 (ENS@T-CANCER) to Judith Favier and Enzo Lalli, and by the French National Research Agency (ANR) through the LOCALDO (ANR-15-CE14-0017-01) and “Investments for the Future” LABEX SIGNALIFE (ANR-11-LABX-0028-01) grants to Enzo Lalli. Carmen Ruggiero was supported by a Fondation de France grant (00057927).
REFERENCES
1. Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, Rene-Corail F, Elarouci N, Sbiera S, Kroiss M, et al. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet. 2014; 46:607-612.
2. Pinto EM, Chen X, Easton J, Finkelstein D, Liu Z, Pounds S, Rodriguez-Galindo C, Lund TC, Mardis ER, Wilson RK, Boggs K, Yergeau D, Cheng J, et al. Genomic landscape of paediatric adrenocortical tumours. Nat Commun. 2015; 6:6302.
3. Zheng S, Chemiack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, Lerario AM, Else T, Knijnenburg TA, Ciriello G, Kim S, Assié G, Morozova O, et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell. 2016; 29:723-736.
4. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006; 12:895-904.
5. Terzolo M, Angeli A, Fassnacht M, Daffara F, Tauchmanova L, Conton PA, Rossetto R, Buci L, Sperone P, Grossrubatscher E, Reimondo G, Bollito E, Papotti M, et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med. 2007; 356:2372-2380.
6. Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, Welin S, Schade-Brittinger C, Lacroix A, Jarzab B, Sorbye H, Torpy DJ, Stepan V, et al. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med. 2012; 366:2189-2197.
7. Luconi M, Mannelli M. Xenograft models for preclinical drug testing: implications for adrenocortical cancer. Mol Cell Endocrinol. 2012; 351:71-77.
8. Hantel C, Beuschlein F. Xenograft models for adrenocortical carcinoma. Mol Cell Endocrinol. 2016; 421:28-33.
9. Barlaskar FM, Spalding AC, Heaton JH, Kuick R, Kim AC, Thomas DG, Giordano TJ, Ben-Josef E, Hammer GD. Preclinical targeting of the type I insulin-like growth factor receptor in adrenocortical carcinoma. J Clin Endocrinol Metab. 2009; 94:204-212.
10. Doghman M, El Wakil A, Cardinaud B, Thomas E, Wang J, Zhao W, Peralta-Del Valle MH, Figueiredo BC, Zambetti GP, Lalli E. Regulation of insulin-like growth factor – mammalian target of rapamycin signalling by microRNA in childhood adrenocortical tumors. Cancer Res. 2010; 70:4666-4675.
11. Luconi M, Mangoni M, Gelmini S, Poli G, Nesi G, Francalanci M, Pratesi N, Cantini G, Lombardi A, Pepi M, Ercolino T, Serio M, Orlando C, Mannelli M. Rosiglitazone impairs proliferation of human adrenocortical cancer: preclinical study in a xenograft mouse model. Endocr Relat Cancer. 2010; 17:169-177.
12. Doghman M, Lalli E. Efficacy of the novel dual PI3-kinase/mTOR inhibitor NVP-BEZ235 in a preclinical model of adrenocortical carcinoma. Mol Cell Endocrinol. 2012; 364:101-104.
13. Hantel C, Lewrick F, Reincke M, Süss R, Beuschlein F. Liposomal doxorubicin-based treatment in a preclinical model of adrenocortical carcinoma. J Endocrinol. 2012; 213:155-161.
14. Doghman M, Lalli E. Lack of long-lasting effects of mitotane adjuvant therapy in a mouse xenograft model of adrenocortical carcinoma. Mol Cell Endocrinol. 2013; 381:66-69.
15. Hantel C, Jung S, Mussack T, Reincke M, Beuschlein F. Liposomal polychemotherapy improves adrenocortical carcinoma treatment in a preclinical rodent model. Endocr Relat Cancer. 2014; 21:383-394.
16. Nagy Z, Baghy K, Hunyadi-Gulyás É, Micsik T, Nyírő G, Rácz G, Butz H, Perge P, Kovalszky I, Medzihradszky KF, Rácz K, Patócs A, Igaz P. Evaluation of 9-cis retinoic acid and mitotane as antitumoral agents in an adrenocortical xenograft model. Am J Cancer Res. 2015; 5:3645-3658.
17. Beuschlein F, Jakoby J, Mentz S, Zambetti G, Jung S, Reincke M, Süss R, Hantel C. IGF1-R inhibition and liposomal doxorubicin: progress in preclinical evaluation for the treatment of adrenocortical carcinoma. Mol Cell Endocrinol. 2016; 428:82-88.
18. Poli G, Cantini G, Armignacco R, Fucci R, Santi R, Canu L, Nesi G, Mannelli M, Luconi M. Metformin as a new anti-cancer drug in adrenocortical carcinoma. Oncotarget. 2016; 7:49636-49648. doi: 10.18632/oncotarget.10421.
19. Hantel C, Shapiro I, Poli G, Chiapponi C, Bidlingmaier M, Reincke M, Luconi M, Jung S, Beuschlein F. Targeting heterogeneity of adrenocortical carcinoma: evaluation and extension of preclinical tumor models to improve clinical translation. Oncotarget. 2016; 7:79292-79304. doi: 10.18632/oncotarget.12685.
20. Beuschlein F, Galac S, Wilson DB. Animal models of adrenocortical tumorigenesis. Mol Cell Endocrinol. 2012; 351:78-86.
21. Basham KJ, Hung HA, Lerario AM, Hammer GD. Mouse models of adrenocortical tumors. Mol Cell Endocrinol. 2016; 421:82-97.
22. Doghman M, Karpova T, Rodrigues GA, Arhatte M, De Moura J, Cavalli LR, Virolle V, Barbry P, Zambetti GP, Figueiredo BC, Heckert LL, Lalli E. Increased steroidogenic factor-1 dosage triggers adrenocortical cell proliferation and cancer. Mol Endocrinol. 2007; 21:2968-2987.
23. Elkin M, Vlodavsky I. Tail vein assay of cancer metastasis. Curr Protoc Cell Biol. 2001; Chapter 19:Unit19.2.
24. Pinto EM, Morton C, Rodriguez-Galindo C, McGregor L, Davidoff AM, Mercer K, Debelenko LV, Billups C, Ribeiro RC, Zambetti GP. Establishment and characterization of the first pediatric adrenocortical carcinoma xenograft model identifies topotecan as a potential chemotherapeutic agent. Clin Cancer Res. 2013; 19:1740-1747.
25. Tissier F, Cavard C, Groussin L, Perlemoine K, Fumey G, Hagneré AM, René-Corail F, Jullian E, Gicquel C, Bertagna X, Vacher-Lavenu MC, Perret C, Bertherat J. Mutations of β-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005; 65:7622-7627.
26. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011; 147:275-292.
27. Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, Groom AC. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998; 153:865-873.
28. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013; 19:1423-1437.
29. Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. 2015; 6:10697-10711. doi: 10.18632/oncotarget.4037.
30. Salomon A, Keramidas M, Maisin C, Thomas M. Loss of β-catenin in adrenocortical cancer cells causes growth inhibition and reversal of epithelial-to-mesenchymal transition. Oncotarget. 2015; 6:11421-11433. doi: 10.18632/oncotarget.3222.
31. Khorram-Manesh A, Ahlman H, Jansson S, Nilsson O. N-cadherin expression in adrenal tumors: upregulation in malignant pheochromocytoma and downregulation in adrenocortical carcinoma. Endocr Pathol. 2002; 13:99-110.
32. Wang T, Rainey WE. Human adrenocortical carcinoma cell lines. Mol Cell Endocrinol. 2012; 351:58-65.
33. Malleter M, Tauzin S, Bessede A, Castellano R, Goubard A, Godey F, Levêque J, Jézéquel P, Campion L, Campone M, Ducret T, MacGrogan G, Debure L, et al. CD95L cell surface cleavage triggers a prometastatic signaling pathway in triple-negative breast cancer. Cancer Res. 2013; 73:6711-6721.
34. Menara M, Oudijk L, Badoual C, Bertherat J, Lepoutre-Lussey C, Amar L, Iturrioz X, Sibony M, Zinzindohoué F, de Krijger R, Gimenez-Roqueplo AP, Favier J. SDHD immunohistochemistry: a new tool to validate SDHx mutations in pheochromocytoma/paraganglioma. J Clin Endocrinol Metab. 2015; 100:E287-E291.